Introduction
BRCA1 and BRCA2 are the most
frequently altered susceptibility genes (∼20%) in familial breast
cancer (BC) (1,2). Women with a deleterious mutation in
BRCA1 or BRCA2 have approximately a 70% probability
of developing BC by the age of 70 (3). The difference in incidence of
particular mutations in different ethnic groups is due to a
‘founder’ effect. Recent evidence has indicated that disease risk
in carriers of high-risk mutations (e.g. BRCA1 and
BRCA2) could vary also due to other genetic and
environmental factors. Genome-wide association studies (GWAS) have
identified several common alleles able to modify breast or ovarian
cancer risk for BRCA1 and BRCA2 mutation carriers,
even if more modifiers still remain to be identified (4). Furthermore, other genes or
chromosomal regions with moderate and low penetrance have been
shown to confer risk of BC (i.e. CHECK2, p53, PALB2, 2q35,
11q13.3) (5,6). However, currently our knowledge of
genetic patterns is not sufficient to allow us to identify all
hereditary BC.
Due to their role in cell metabolism, mitochondria
have long been suspected to be involved in metabolic alterations of
tumors (7). In particular,
mitochondrial DNA (mtDNA) is highly susceptible to molecular
alterations by environmental carcinogens because of a lack of
introns and histone-like proteins. Moreover, mtDNA has a high
mutation rate due to the close proximity to sources of reactive
oxygen species (ROS), complex I (NADH-ubiquinone oxidoreductase, EC
1.6.5.3) and III (ubiquinol: cytochrome c oxidoreductase; EC
1.10.2.2), and also due to the low efficiency of repair systems.
These nucleotide modifications have been hypothesized to promote a
cascade of events that increases the ROS production thus
encouraging cancer development (8,9).
Recent studies suggested that several somatic mtDNA mutations,
especially in the mitochondrial respiratory complex I genes and in
the D-Loop region, may predispose individuals to cancer (10–12)
and, in particular, to BC (8,13).
The D-Loop is the main regulatory region of mitochondrial
transcription and replication that encompasses essential and
strongly conserved consensus sequence elements (14) as well as loci that rapidly
accumulate mutations (i.e., the two hypervariable regions,
HV1 and HV2) (15). On the
other hand, the high frequency of mutation rate as well as the lack
of recombination and the mtDNA poly-ploid status makes it
possible to classify individuals through specific mtDNA variants
that make up ‘haplogroups’ (16).
Haplogroups, groups of mtDNA haplotypes sharing the same mutational
motifs by descent from a common female ancestor, have been shown to
possess both geographical- and ethnic-specific differences in
prevalence (17). They are
extremely common in a continent or even in a single geographic
area/population group or completely absent in others (18). Some studies have tried to focus on
the hypothetical association between a haplogroup and an increasing
BC risk, but without definitive results (19,20).
So far, studies have not been carried out to investigate the role
of mtDNA alterations in BC heredity. Thus, the role of mtDNA
variation needs to be further investigated.
Starting from all these observations, we aimed to
verify whether mtDNA alterations and/or a specific mtDNA haplogroup
could be used as a susceptibility marker for familial BC or as a
BRCA1/BRCA2 gene modifier.
Materials and methods
Subject collection and DNA
extraction
After signing informed consent, 56 Caucasian
subjects from the Region Puglia were enrolled at the National
Cancer Research Centre ‘Istituto Tumori Giovanni Paolo II’ of Bari,
Italy; 32 familial BC patients, 14 sporadic and 10 healthy subjects
unrelated to familial BC. Among the 32 familial cases, 22 were
affected by BC and 10 were healthy relatives from high risk
families. Patients were classified as ‘familial’ when they
satisfied clinical characteristics as previously reported (21). Among the familial cases, 10
subjects were BRCA1 mutation carriers, 10 BRCA2
mutation carriers and 12 wild-type for both genes (BRCAX).
The BRCA molecular screening of patients was performed as
reported in our previous study (21). Patients were characterized
according to pathological features: TNM classification, nuclear
grading, estrogen (ER) and progesterone receptor (PgR) tumor
content (Table I).
 | Table I.Clinicopathological characteristics
of patients with respect to the different subgroups. |
Table I.
Clinicopathological characteristics
of patients with respect to the different subgroups.
| Familial breast
cancer | |
|---|
|
|---|
| Variables | BRCA1
mutated n=10 | BRCA2
mutated n=10 | BRCAX
n=12 | Sporadic BC
n=14 |
|---|
| Gender | | | | |
| F | 7 | 8 | 12 | 14 |
| M | 3 | 2 | 0 | 0 |
| Age | | | | |
| ≤45 yrs. | 7 | 6 | 2 | 2 |
| >45 yrs. | 3 | 4 | 10 | 12 |
| IDC | 90% (n=9) | 100% (n=10) | 75% (n=9) | 78% (n=11) |
| T | | | | |
| 1–2 | 4 | 7 | 8 | 11 |
| 3–4 | 0 | 3 | 4 | 3 |
| N+ | 30% (n=3) | 40% (n=4) | 90% (n=10) | 64% (n=9) |
| G3 | 60% (n=6) | 70% (n=7) | 55% (n=5) | 64% (n=9) |
| ER+ | 30% (n=3) | 100% (n=10) | 67% (n=8) | 28% (n=4) |
|
PgR+ | 30% (n=3) | 80% (n=8) | 67% (n=8) | 36% (n=5) |
|
Mib+ | 80% (n=8) | 80% (n=8) | 58% (n=7) | 71% (n=10) |
|
ErbB2+ | 50% (n=5) | 50% (n=5) | 17% (n=2) | 28% (n=4) |
Members of four different Caucasian families
affected by familial breast/ovarian cancer transmitted along the
maternal lineage were also analyzed to study the transmission of
mitochondrial alterations into each family and their association
with the presence of BRCA mutations.
BRCA1 and BRCA2 mutations were
identified in DNA extracted from stored blood by using the DNAeasy
Blood and Tissue kit (Qiagen, Valencia, CA, USA) according to the
manufacturer’s protocol.
Amplification and sequencing
Total genomic DNA (1/(g) was extracted from the
blood of 36 patients and 20 healthy subjects enrolled at the
National Cancer Research Centre of Bari (Italy) and used for
amplification with the MitoALL Resequencing kit according to the
manufacturer’s instructions (Applied Biosystems, Foster City, CA,
USA). The MTND4 (1 of the 7 mitochondrial complex I genes)
and the D-Loop mitochondrial region (spanning nucleotides
10760–12137 and 16024-576, respectively) were selectively amplified
by PCR. D-loop and MTND4 were selected because they present
the highest mutational rate among the mtDNA genes (22). PCR products were purified with
ExoSAP-IT™ (Affymetrix, OH, USA) and then both strands directly
sequenced using the BigDye Deoxy Terminator Cycle Sequencing kit
(v. 3.1, Applied Biosystems Inc.) in an automated sequencer (3130×l
Genetic Analyzer; Applied Biosystems Inc.). To evaluate if mtDNA
presented different polymorphisms in BC-affected individuals, the
sequences were compared for similarities against the revised
Cambridge Reference Sequence (NC_012920). The quality of sequences
and the coherence of the variants were compared to the
international HmtDB database (http://www.hmtdb.uniba.it:8080/hmdb). Mitochondrial
haplogroups were identified using several markers as reported at
http://www.phylotree.org.
To confirm the data on haplogroup association with
the presence of BRCA1 or BRCA2 mutations, a second
group including 25 subjects, 8 carrying BRCA1, 10 carrying
BRCA2 alterations and 7 BRCAX, were investigated at
13 loci to define to which haplogroup they belonged.
In silico analysis
All mtDNA sequencing results were analyzed in
silico to verify the possible pathogenetic role of each
variant. Mutational analysis was carried out in HmtDB: if the
variability index (v.i.) was close to ‘0’ the substitution was
considered as a polymorphism; if v.i. was near to ‘ 1’, the
substitution was considered frequent in the population. Polyphen
software (http://genetics.bwh.harvard.edu/pph/) was used to
predict a possible impact of the evidenced amino acid substitutions
on the structure and function of the encoded proteins.
Statistical analysis
Mutations in subgroups were summarized using
cross-tabulations and analyzed by the Chi-square test. Associations
between mitochondrial alterations or haplogroups and
clinicopathological characteristics were assessed using Chi-square
tests for categorical variables. All analyses were conducted using
SPSS version 17.0 and were considered statistically significant
when p≤0.05.
Results
Identification of mtDNA variants in BC
patients compared to healthy subjects
To evaluate if mtDNA presented different
polymorphisms in BC affected individuals, mtDNA from the blood of
36 patients and 20 healthy subjects were aligned to the revised
Cambridge Reference Sequence (rCRS) and nucleotide variants
recorded. DNA sequencing of the D-Loop and MTND4 regions
identified a total of 136 nucleotide variants in all the subjects:
107 in the D-Loop region and 29 in MTND4. The
over-represented as well as the novel variants are listed in
Table II. To assess a potential
role in increasing the risk of onset of BC, we searched the
variability of the identified SNPs in HmtDB. We found that 17 SNPs
presented a variability index (v.i.) equal to ‘0’; 54 SNPs showed a
v.i. ranging from 0.001 to 0.010 when compared to the genome of
healthy individuals whereas a v.i. ranging from 0.002–0.026 was
revealed when comparing them to subjects affected by any pathology;
58 SNPs presented a v.i. above the highest value of the range. The
variants were retrieved also in Mitomap (http://www.mitomap.org/MITOMAP). All resulted as
haplogroup markers and no significant difference was shown between
BC and healthy subjects. However, 7 variants (149insC, 452delT and
m.16026T>C in the D-Loop and m.10799C>T, m.11279C>A,
m.11524A>C and m.11925C>A in MTND4) were not reported
either in HmtDB or Mitomap but were all found at very low frequency
(Table III). Among the novel
variants retrieved in MTND4, the two non-synonymous,
m.11279C>A (p.L174M) and m.11524A>C (p.K255N), were analysed
by Polyphen (http://genetics.bwh.harvard.edu/pph2/) and were
predicted as ‘benign’, not determining structural variations at
protein level.
| Table II.mtDNA variants and relative
frequencies in the D-loop and MTND4 in BC subjects. |
Table II.
mtDNA variants and relative
frequencies in the D-loop and MTND4 in BC subjects.
| D-loop | % |
|---|
| D310 | 97.2 |
| m.263A>G | 94.4 |
| m.73A>G | 61.0 |
| m.16519T>C | 41.7 |
| m.16126T>C | 27.8 |
| m.462C>T | 17.0 |
| m.150C>T;
m.295C>T; m.16069C>T; m.16311T>C | 16.7 |
| m.195T>C;
m.228G>A; m.515delAC; m.16189T>C; m.16224C>T | 13.9 |
| m.152T>C;
m.185G>A | 11.0 |
| m.93A>G;
m.T146C; m.153A>G; m.G207G>A; m.C285C>T; m.T489T>C;
m.A16183A>C; | 8.3 |
| m.16278C>T,
m.16292C>T; m.16294C>T; m.16298T>C; m.16362T>C | |
| m.494delC | 6.3 |
| m.200A>G;
m.452delT; m.499G>A; m.557insC; m.568–573; m.16017T>C;
m.16093T>C; | 5.6 |
| m.16129G>A;
m.16209T>C, m.16223C>T; m.16304T>C; m.16343A>G;
m.16527T>C | |
| m.456C>T | 2.9 |
| m.56insT;
m.64C>T; m.72T>C; m.149insC; m.151C>T; m.199T>C;
m.203G>A; m.204T>C; | 2.8 |
| m.225G>A;
m.226T>C; m.239T>C; m.250T>C; m.285insA; m.336T>C;
m.385A>G; m.497C>T; | |
| m.514C>T;
m.514insC; m.515insA; m.16026T>C; m.16067C>T; m.16092T>C;
m.16114C>T; | |
| m.16162A>G;
m.16172T>C; m.16179C>T; m.16192C>T; m.16249T>C;
m.16261C>T; | |
| m.16265A>T;
m.16270C>T; m.16287C>T; m.16293A>G; m.16326T>C;
m.16354C>T; | |
| m.16356T>C;
m.16399A>G; m.16482A>G | |
|
| MTND4 | % |
|
| m.11719G>A | 47.2 |
| m.11251A>G | 25.0 |
| m.11467A>G | 22.2 |
| m.10822C>T;
m.11299T>C; m.11812A>G | 8.3 |
| m.10799C>T;
m.10915T>C; m.11176G>A; m.10927T>C; m.10907T>C;
m.11009T>C; | 2.8 |
| m.11017T>C;
m.11167A>G; m.11253T>C; m.11279C>A; m.11332C>T;
m.11353T>C; | |
| m.11362A>G;
m.11377G>A; m.11476C>T; m.11485T>C; m.11524A>C;
m.11840C>T; | |
| m.11869C>A;
m.G11887G>A; m.G11914A; m.11925C>A; m.12127G>A | |
| Table III.D-Loop and MTND4 SNPs in
BRCA1 mutation carrier and non-carrier patients. |
Table III.
D-Loop and MTND4 SNPs in
BRCA1 mutation carrier and non-carrier patients.
| mtDNA SNPs | BRCA1
carrier vs. BRCA1 non-carrier | P-value | OR (95% IC) |
|---|
| m.153A>G | 50% vs.5% | 0.009 | 19 (1.8–201.9) |
| m.195T>C | 60% vs.40% | 0.04 | 6 (1.12–31.99) |
| m.225G>A | 40% vs.5% | 0.03 | 12.7
(1.18–136.28) |
| m.226T>C | 40% vs.5% | 0.03 | 12.7
(1.18–136.28) |
| m.16183A>C | 40% vs.5% | 0.03 | 12.7
(1.18–136.28) |
| m.16278C>T | 40% vs.8% | 0.03 | 7.3 (1.3–41.4) |
| m.16519T>C | 90% vs.30% | 0.003 | 21
(2.15–204.6) |
| m.11719G>A | 70% vs.15% | 0.005 | 13.2
(2.13–82.13) |
When we evaluated a possible association between
specific SNPs and the BC status, we found that the only variant
exclusively present in BC and not in healthy subjects was the
m.150C>T (16.7% vs. 0%, P=0.06), whereas four variants resulted
more frequently present in healthy than in BC subjects: m.195T>C
(55% vs. 13.9%, P=0.002), m.225G>A (20 vs. 2.8%, P=0.05),
m.16519T>C (75% vs. 41.7%, P=0.016), m.16189T>C (40% vs.
13.9%, P=0.03).
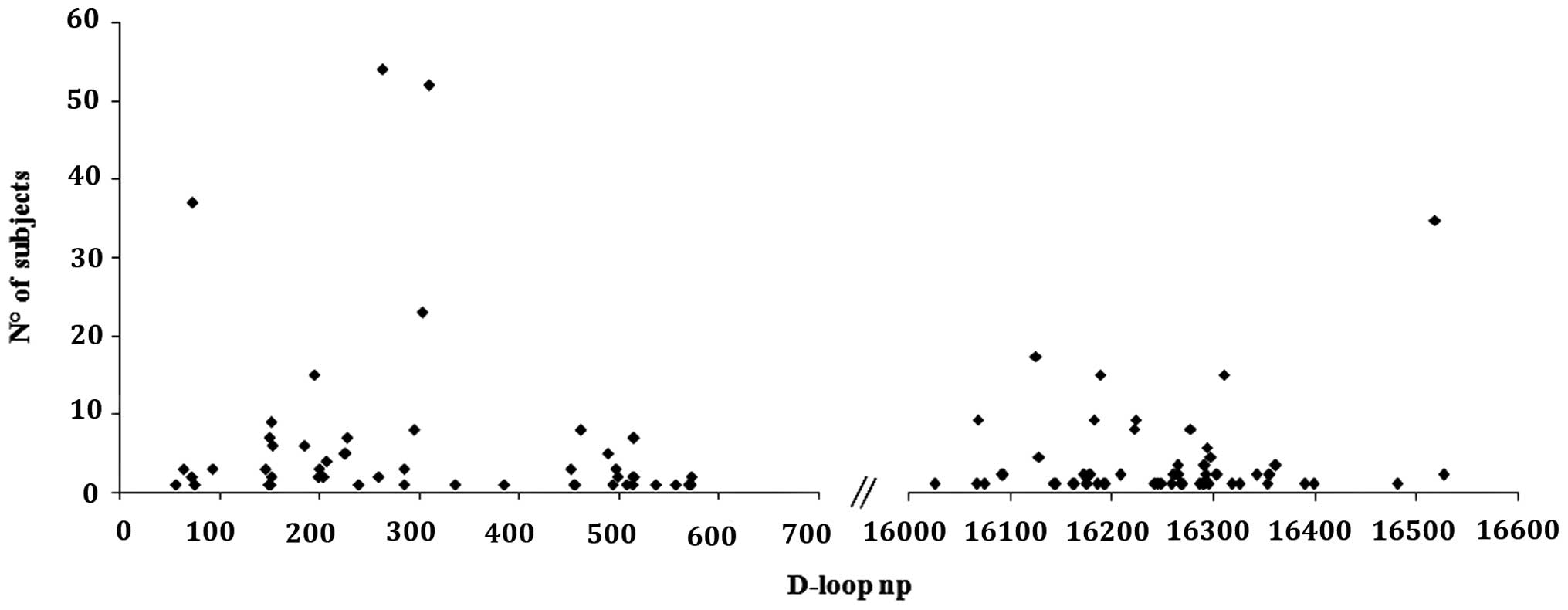
Microsatellity instability in the D-Loop region,
including the two common poly-C stretch variations at nucleotide
positions (np) 303–315 (D310) and 568–573, in addition to the
poly-CA stretch at 514–523, was also detected (Table II). We found that the 36 BC
subjects showed length variations at 514–523 and 568–573 with a
higher frequency than in healthy subjects: 16.7 and 5% vs. 1 and
0%, respectively; on the other hand, D310 resulted the most
frequently polymorphic site in the D-Loop both in BC and in healthy
subjects. When considering the variability of the D-loop region, as
expected, most of the variants were found in the D-Loop
hypervariable regions; 48/107 variants (45%) in HV1; 31/107 (29%)
in HV2 and 20/107 (18.7%) in HV3 (Fig. 1). No differences between BC and
healthy subjects were found.
Haplogroup analysis of the overall 36 BC patients
showed that H and J/T were the most represented haplogroups (33 and
25%, respectively) whereas haplogroup X was absent in our series of
healthy subjects.
D-Loop and MTND4 variants with respect to
BC features
In order to evaluate a possible association between
mtDNA alterations and some BC characteristics, we correlated mtDNA
variants with tumor grade differentiation (G1, G2 and G3). Several
D-Loop and MTND4 alterations indeed resulted to be linked to
specific cancer pathological features. Particularly, m.11719G>A
and m.16519T>C appeared to be linked to scarcely differentiated
tumors: 57% in G3 vs. 12.5% in G2 (P=0.006) and 57% in G3 vs. 12.5%
in G2 (P=0.05), respectively. m. 16126T>C frequently occurred in
hormone receptor-negative subjects: 47% in ER− and
PgR− vs. 12% in ER+ and PgR+
(P=0.035). Regarding haplogroup analysis, haplogroup H was more
represented in G2 than in G1 patients (75 vs. 24%, P=0.02), thus
suggesting a possible correlation between the mtDNA background and
tumor differentiation grade.
Some mtDNA variants enhanced the risk of
carrying BRCA1 mutations
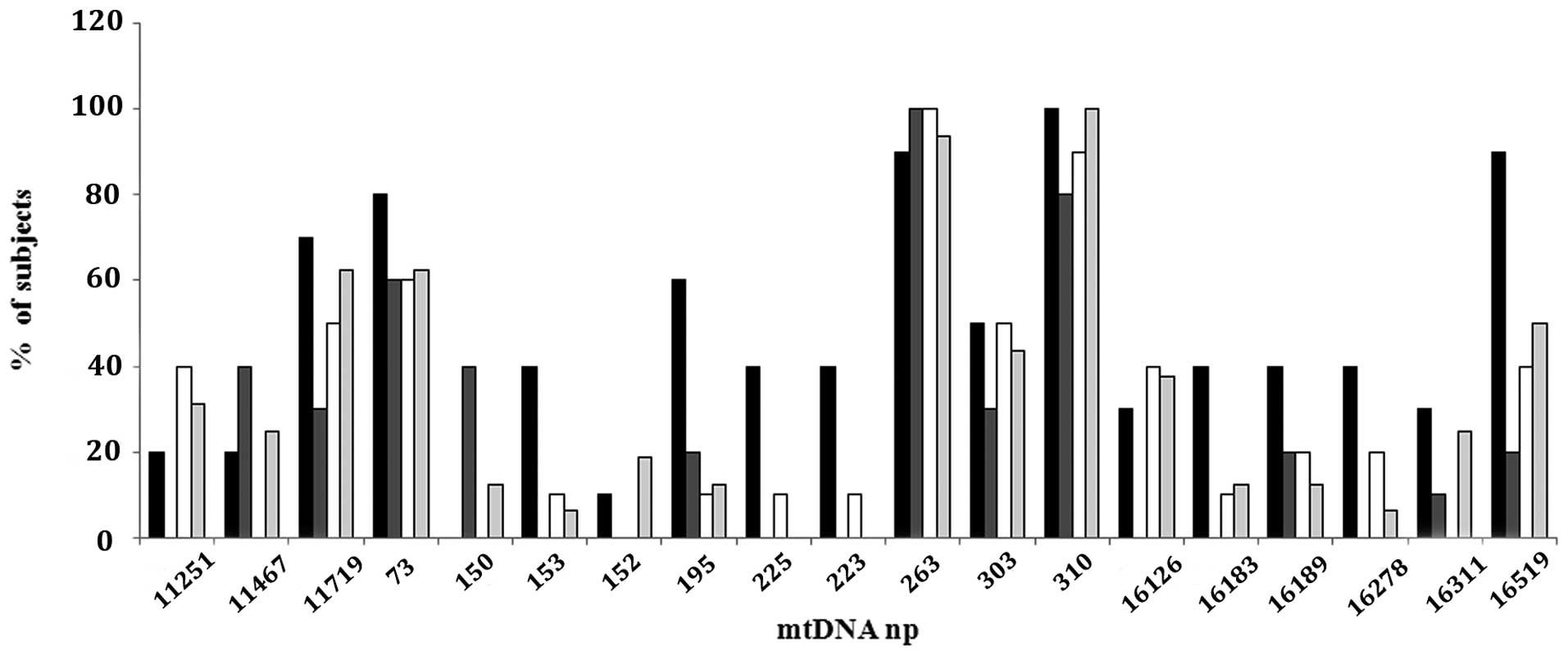
Familial and sporadic BC subjects did not show
differences in number or type of D-loop and MTND4
alterations (Fig. 2). The sporadic
patients presented 14 substitutions in the MTND4 gene, and
60 substitutions in the D-Loop region: 100% showed a variation at
D310, 62% at 11719, 50% at 16519, and 44% at 303 np. Among the
familial subjects, the BRCA1 mutated group presented 9
substitutions in the MTND4 gene, and 38 substitutions in the
D-Loop region: 100% of patients showed D310 variation, 90% at
16519, 70% at 11719, 60% at 195, 50% at 303 and 40% at 153, 225,
226, 16183, 16189 and 16278 np. BRCA2 mutated subjects
showed 9 substitutions in the MTND4 gene, and 35
substitutions in the D-Loop region: 80% presented variation at
D310, 40% at 11467 and 150, and 30% at 303 and 11719 np. The
BRCAX group revealed 4 substitutions in the MTND4
gene, and 37 substitutions in the D-Loop region: 90% presented
variation at D310, 50% at 303 and 11719, and 40% at 11251, 16126
and 16519 np (Fig. 2).
When we considered only BRCA1 mutation
carriers compared to non-carriers we found a significant
association with 7 D-Loop SNPs (m.153A>G, m-195T>C,
m.225G>A, m.226T>C, m.16183A>C, m.16519T>C,
m.16278C>T) and one SNP in the MTND4 gene
(m.11719G>A), as reported in Table
III. The mutations conferring BRCA2 inactivation were
not associated with any of the mtDNA variants detected, with the
exception of the variation at positions 514–523 that was found at
higher frequency (60%) in the BRCA2 carriers. Furthermore,
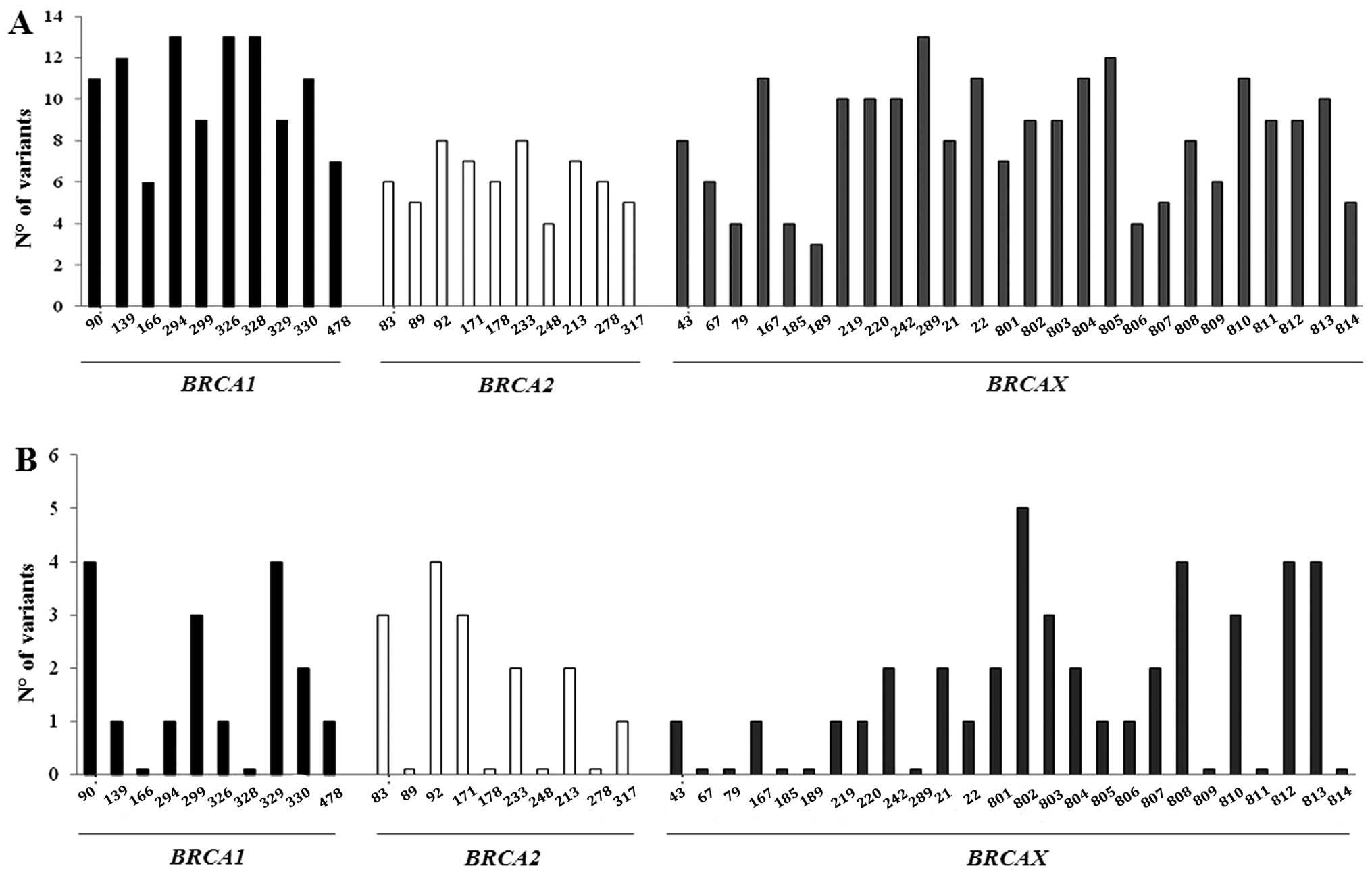
when considering the contemporary alterations of the two mtDNA
regions in each patient, all subjects showed mutations in the
D-Loop with at least 3 changes up to 16, while 72.5% of subjects
had alterations in the MTND4 gene up to 6 alterations at the
same time. As expected, the D-Loop was much more variable than the
MTND4 gene; however, while mutation distribution in the
D-Loop was independent of the presence of BRCA mutations,
non-carrier familial BCs presented fewer changes in the
MTND4 gene. This result was not, however, statistically
significant (Fig. 3).
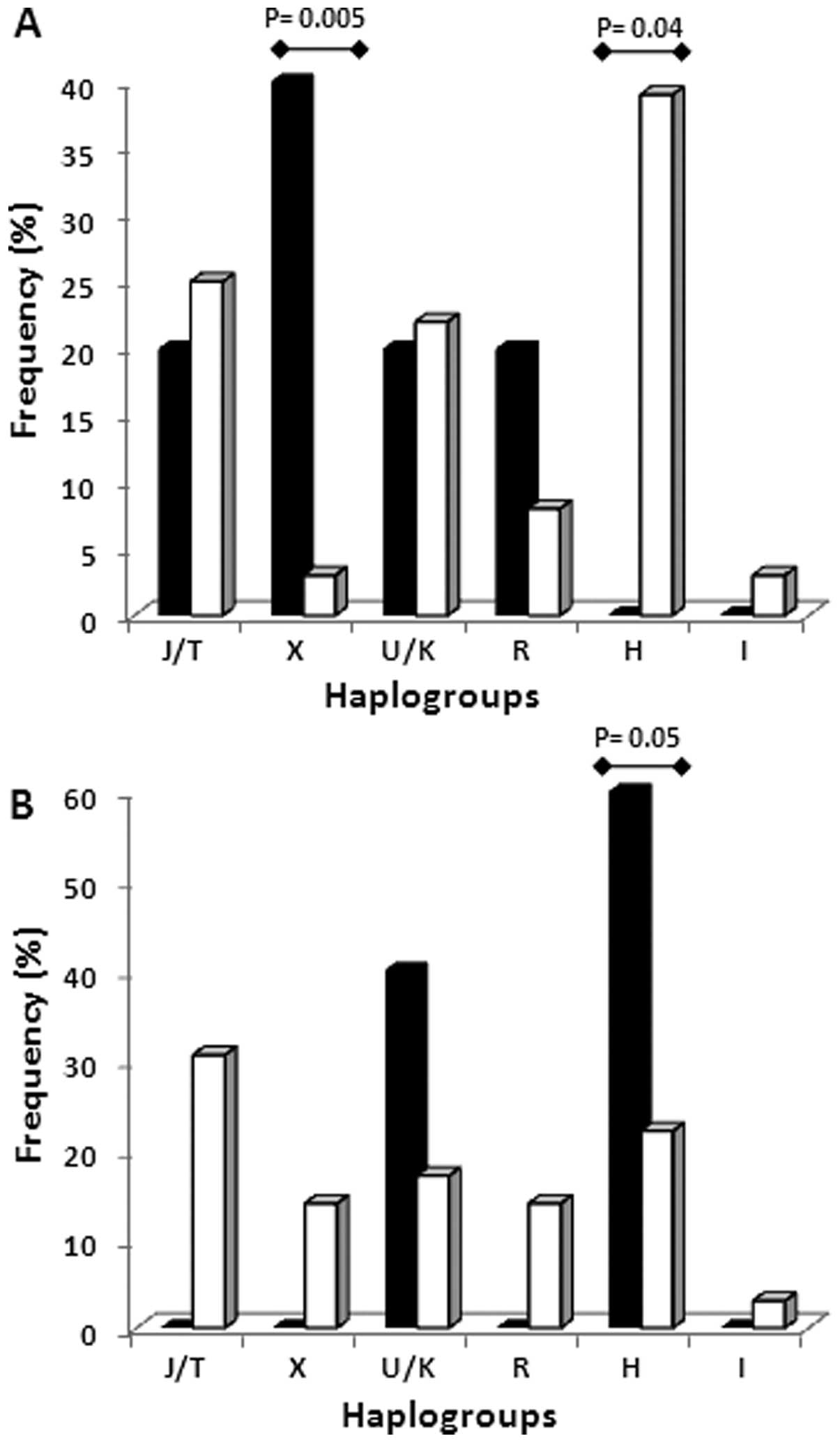
Haplogroup analysis in patients carrying and not
carrying BRCA mutations showed that the X and H haplogroups
are able to discriminate between BRCA1 and BRCA2
carriers. Haplogroup X was significantly more frequent in
BRCA1 carriers and completely absent in BRCA2
carriers (P=0.005), while haplogroup H was significantly less
represented in BRCA1 carriers (P=0.04) and mostly present in
BRCA2 carriers (P=0.05) (Fig.
4). To confirm the association between a specific haplogroup
and BRCA1 or BRCA2 mutations, a further subset of 25
subjects were analyzed. The results confirmed the association
demonstrating that seven BRCAX subjects belonged to
haplogroup X (n=2) and H (n=5). Haplogroup X resulted in 100% of
eight BRCA1 carriers and haplogroup H in 100% of ten
BRCA2 carriers.
Mitochondrial DNA variants in the BRCA
families
Members of four different families affected by
familial breast/ovarian cancer transmitted along the maternal
lineage were also analyzed (Table
IV). In each family, all the analyzed members presented, as
expected, specific variants in both the D-Loop and MTND4
regions independently of the presence of a tumor. However, some
alterations were associated to BRCA mutation type being
present in members of different BRCA1 carrier families while
others were specific of a familial group (m.64C>T,
m.16183A>C, m.16189T>C, m.11812A>G). In addition, these
results were consistent with the association between specific
haplogroups and BRCA mutations as found in the above 25
subjects.
| Table IV.Mutation clustering in members of
hereditary BC families. |
Table IV.
Mutation clustering in members of
hereditary BC families.
| Families | Pathological
status | BRCA
mutation | D-Loop
mutations | MTND4
mutation | Haplogroup |
|---|
| DA | | | | | |
| Sister #1 | H | B1 | C64T, A73G,
A153G, T195C, | - | X |
| Sister #2 | H | B1 | G225A, T226C, D310,
A16183C, | - | |
| Brother | H | B1 | C16226T, C16278T,
T16519C | - | |
| PA | | | | | |
| Aunt | A | B1 | A73G, A153G, T195C,
G225A, | A11812G | X |
| Nephew | H | B1 | T226C, D310,
A16183C, T16189C, | | |
| Niece | H | B1 | C16223T,
C16278T, T16519C | | |
| PR | | | | | |
| Sister #1 | A | BX | A73G, G185A, G228A,
C295T, | T11253C | J |
| Sister #2 | A | BX | D310, C462T,
514–523, C16069T | | |
| SS | | | | | |
| Sister #1 | A | B2 | G207A, 514–523,
G16129A | - | H |
| Sister #2 | A | B2 | | - | |
Discussion
In this study, we have shown that the mitochondrial
regions D-Loop and MTND4 display a panel of mtDNA
loci significantly associated with the presence of mutations
in the BRCA1 gene, and that specific mitochondrial
haplogroups are present in patients with mutations in BRCA1
or BRCA2. As expected, the vast majority (72.7%) of the
mtDNA mutations identified in our study were in the D-Loop region
where microsatellite instability (MSI) was also detected.
Conflicting hypotheses have been proposed about the frequency of
various MSI in the mitochondrial genome (23), being identified in various types of
cancer (24,25). D310, which is located in one of the
two highly conserved regions (Conserved Sequence Block I, CSB I)
and regulates mtDNA replication (26), has been previously suggested to be
involved in carcinogenesis and included in a set of SNPs that could
be considered as cancer susceptibility biomarkers (27). Recently, the relatively high
frequency of alterations (32.5%) detected in D310 has been
associated with clinicopathological parameters of breast cancer
(BC). Specifically, 66.7% of the alterations were observed in stage
II BC, indicating that D310 instability may be a critical feature
for early progression of BC (28).
Moreover, other recent results highlighted D310 in some unrelated
BC harboring BRCA1 mutations (29). These data would support an
implication of this mitochondrial allelic variant in cell
assessment associated with BRCA1 mutations if we had not
found a high percentage of mutations even in healthy controls.
Similarly, no significant association between alterations
frequently retrieved in the D-loop (m.263A>G) or previously
described as a highly polymorphic marker (m.73A>G,
m.16519T>C) (19) and risk of
developing BC were found. When analyzing MTND4, we found
that similarly to that detected in BC cell lines (30) m. 11251A>G and m. 11719G>A
were highly frequent in BC subjects but equally represented in
healthy controls. However, considering the clinicopathological
features of the patients, this is the first report on the
association between the presence of specific mitochondrial
polymorphisms and tumor differentiation grade (m.11719G>A and
m.16519T>C) or hormone receptor status (m.16126T>C).
Hypothesizing that the mitochondrial localization of
BRCA1 protein could have a role in regulating both mtDNA damage and
apoptotic activity and that its mislocalization could cause cancer,
this study focused on a possible relationship between mtDNA
variants and germinal BRCA1 mutations. In literature, a
diagnostic algorithm showed that BRCA mutations accelerate
the onset of mtDNA alterations, leading to phenotypic expression of
premature aging and BC (31). We
have shown that seven D-Loop and one MTND4 variations are
associated with an increased risk of harboring also BRCA1
mutations. Of note, among these, m.16183A>C is located at the 3’
sequence of the Termination Associated Sequence (TAS) and the DNA
7S binding site which is involved in the regulation of mtDNA
synthesis (32). The alteration
m.16519T>C, retrieved in 90% of BRCA1 carriers compared
to non-carriers (30%), was previously found in literature to
increase familial BC risk (19).
Our result confirms that m. 16519T>C is a hot spot mutation
playing a relevant role in developing BC. All the other alterations
associated with BRCA1 significant mutation risk have also
been described in literature in different cancer tissues, such as
oral, prostate, thyroid, breast, ovarian cancer and melanoma
(29,33). Conversely, variants m.153A>G and
m.225G>A, previously associated with breast carcinogenesis
(34) have been found as
mitochondrial polymorphisms.
In literature, scant data are available on the
relationship of specific mitochondrial haplogroups and an
increasing BC risk in Europe. Although haplogroup H is common in
Europe, representing 40% of European mtDNAs (35,36),
our haplogroup analysis revealed that X and H significantly
discriminate BRCA1 or BRCA2 mutated subjects. These
data suggested a possibly higher susceptibility role for these
mitochondrial haplogroups to present specific alterations in the
genes predicting familial BC risk. When determining the nucleotide
sequence in all the available members of two families, the
mutations detected in the D-Loop resulted significantly associated
with the presence of BRCA1 mutations since they are
transmitted in all family members and identify the X haplogroup.
Consequently, they could be considered as dominantly inherited
alterations and as representing predicting factors in terms of
susceptibility to familial BC risk.
The identification of significant mtDNA variants
associated with BC suggests the involvement of mtDNA as a genetic
modifier considering both single polymorphisms and specific
haplogroups. To better understand the intrinsic clinical
implications of the specific mitochondrial biomarkers and
haplogroups herein identified, further data involving subjects with
and without cancer need to be collected and analyzed. The
intriguing results presented in this study can contribute to
facilitating the possibility of their application in identifying
high risk individuals for developing a specific screening approach
for early diagnosis of BC.
Acknowledgements
The authors would like to thank
Caroline Oakley for manuscript revision. This work was supported by
2010 funds from the University of Bari (Fondi ex-60%, 2009–2011)
and by Fondazione Cassa di Risparmio di Puglia, Bari, Italy.
References
|
1.
|
Welsch PL and King MC: BRCA1 and BRCA2 and
the genetics of breast and ovarian cancer. Hum Mol Genet.
10:705–713. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Wideroff L, Vadaparampil ST, Greene MH,
Taplin S, Olson L and Freedman AN: Hereditary breast/ovarian and
colorectal cancer genetics knowledge in a national sample of US
physicians. J Med Genet. 42:749–755. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Evans DG, Shenton A, Woodward E, Lalloo F,
Howell A and Maher ER: Penetrance estimates for BRCA1 and BRCA2
based on genetic testing in a Clinical Cancer Genetics service
setting: risks of breast/ovarian cancer quoted should reflect the
cancer burden in the family. BMC Cancer. 8:1552008. View Article : Google Scholar
|
|
4.
|
Barnes DR and Antoniou AC: Unravelling
modifiers of breast and ovarian cancer risk for BRCA1 and BRCA2
mutation carriers: update on genetic modifiers. J Int Med.
271:331–343. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Tang LL, Chen FY, Wang H, et al: Haplotype
analysis of eight genes of the monoubiquitinated FANCD2-DNA
damage-repair pathway in breast cancer patients. Cancer Epidemiol.
37:311–317. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Fanale D, Amodeo V, Corsini LR, Rizzo S,
Bazan V and Russo A: Breast cancer genome-wide association studies:
there is strength in numbers. Oncogene. 31:2121–2128. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Chatterjee A, Mambo E and Sidransky D:
Mitochondrial DNA mutations in human cancer. Oncogene.
25:4663–4674. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Imanishi H, Hattori K, Wada R, et al:
Mitochondrial DNA mutations regulate metastasis of human breast
cancer cells. PLoS One. 6:e234012011. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Martinez-Outschoorn UE, Lin Z, Trimmer C,
Flomenberg N and Wang C: Cancer cells metabolically ‘fertilize’ the
tumor microenvironment with hydrogen peroxide, driving the Warburg
effect: implications for PET imaging of human tumors. Cell Cycle.
10:2504–2520. 2011.
|
|
10.
|
Akouchekian M, Houshmand M, Akbari MH,
Kamalidehghan B and Dehghan M: Analysis of mitochondrial ND1 gene
in human colorectal cancer. J Res Med Sci. 16:50–55.
2011.PubMed/NCBI
|
|
11.
|
Ding C, Li R, Wang P, Jin P, Li S and Guo
Z: Identification of sequence polymorphisms in the D-Loop region of
mitochondrial DNA as a risk factor for lung cancer. Mitochondrial
DNA. 23:251–254. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Fendt L, Niederstätter H and Huber G:
Accumulation of mutations over the entire mitochondrial genome of
breast cancer cells obtained by tissue microdissection. Breast
Cancer Res Treat. 128:327–336. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Sultana GN, Rahman A, Shahinuzzaman AD,
Begum RA and Hossain CF: Mitochondrial DNA mutations - candidate
biomarkers for breast cancer diagnosis. Chin J Cancer. 31:449–454.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Taanman JW: The mitochondrial genome:
structure, transcription, translation and replication. Biochim
Biophys Acta. 1410:103–123. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Suzuki M, Toyooka S, Miyajima K, Iizasa T,
Fujisawa T, Bekele NB and Gazdar AF: Alterations in the
mitochondrial displacement Loop in lung cancers. Clin Cancer Res.
9:5636–5641. 2003.PubMed/NCBI
|
|
16.
|
van Oven M and Kayser M: Updated
comprehensive phylogenetic tree of global human mitochondrial DNA
variation. Hum Mutat. 30:E386–E394. 2009.PubMed/NCBI
|
|
17.
|
Torroni A, Achilli A, Macaulay V, Richards
M and Bandelt HJ: Harvesting the fruit of the human mtDNA tree.
Trends Genet. 22:339–345. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Underhill PA and Kivisild T: Use of y
chromosome and mitochondrial DNA population structure in tracing
human migrations. Annu Rev Genet. 41:539–564. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Bai RK, Leal SM, Covarrubias D, Liu A and
Wong LJ: Mitochondrial genetic background modifies breast cancer
risk. Cancer Res. 67:4687–4894. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Fang H, Shen L, Chen T, He J and Ding Z:
Cancer type-specific modulation of mitochondrial haplogroups in
breast, colorectal and thyroid cancer. BMC Cancer. 10:4212010.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Tommasi S, Crapolicchio A, Lacalamita R,
Bruno M and Monaco A: BRCA1 mutations and polymorphisms in a
hospital-based consecutive series of breast cancer patients from
Apulia, Italy. Mutat Res. 578:395–405. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Gasparre G, Romeo G, Rugolo M and Porcelli
AM: Learning from oncocytic tumors: Why choose inefficient
mitochondria? Biochim Biophys Acta. 1807:633–642. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Peng Z, Xie C, Wan Q, Zhang L, Li W and Wu
S: Sequence variations of mitochondrial DNA D-Loop region are
associated with familial nasopharyngeal carcinoma. Mitochondrion.
11:327–333. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Liu VW, Shi HH, Cheung AN, et al: High
incidence of somatic mitochondrial DNA mutations in human ovarian
carcinomas. Cancer Res. 61:5998–6001. 2001.PubMed/NCBI
|
|
25.
|
Liu VW, Yang HJ, Wang Y, et al: High
frequency of mitochondrial genome instability in human endometrial
carcinomas. Br J Cancer. 89:697–701. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Pham XH, Farge G, Shi Y, Gaspari M,
Gustafsson CM and Falkenberg M: Conserved sequence box II directs
transcription termination and primer formation in mitochondria. J
Biol Chem. 281:24647–24652. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Lin CS, Wang LS, Chang SC, Chou TY and Hsu
WH: Associated microsatellite alterations in mitochondrial DNA and
in TP53 in thoracic esophageal squamous cell carcinoma. Oncol Rep.
28:69–76. 2012.PubMed/NCBI
|
|
28.
|
Alhomidi MA, Vedicherla B, Movva S, Rao
PK, Ahuja YR and Hasan Q: Mitochondrial D310 instability in Asian
Indian breast cancer patients. Tumour Biol. 34:2427–2732. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Troudi W, Loueslati B, Baccar A, Ben Ayed
F, Ben Ammar El and Gaaied A: Penetrance of BRCA1 gene mutation and
DNA mitochondrial in Tunisian breast cancer occurrence. Tunis Med.
87:494–498. 2009.(In French).
|
|
30.
|
Ma Y, Bai RK, Trieu R and Wong LJ:
Mitochondrial dysfunction in human breast cancer cells and their
transmitochondrial cybrids. Biochim Biophys Acta. 1797:29–37. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Hossein R and Houshmand M: Diagnostic
algorithm for identification of individuals with hereditary
predisposition to breast cancer. Lik Sprava. 1–2:103–108.
2008.PubMed/NCBI
|
|
32.
|
Fernandez-Silva P, Enriquez JA and Montoya
J: Replication and transcription of mammalian mitochondrial DNA.
Exp Physiol. 88:41–56. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Ebner S, Lang R, Mueller EE, Eder W,
Oeller M, et al: Mitochondrial haplogroups, control region
polymorphisms and malignant melanoma: a study in middle European
Caucasians. PLoS One. 6:e271922011. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Czarnecka AM, Krawczyk T, Plak K, et al:
Mitochondrial genotype and breast cancer predisposition. Oncol Rep.
24:1521–1534. 2010.PubMed/NCBI
|
|
35.
|
Torroni A, Huoponen K, Francalacci P,
Petrozzi M and Morelli L: Classification of European mtDNAs from an
analysis of three European populations. Genetics. 144:1835–1850.
1996.PubMed/NCBI
|
|
36.
|
Torroni A, Richards M, Macaulay V, Forster
P and Villems R: mtDNA haplogroups and frequency patterns in
Europe. Am J Hum Genet. 66:1173–1177. 2000. View Article : Google Scholar : PubMed/NCBI
|