Introduction
MicroRNAs (miRNAs) are a class of small, endogenous,
non-coding RNAs which act as post-trancriptional regulators via
binding to the 3′ untranslated regions (3′-UTRs) of the target gene
mRNA, and have vital functions in many of physiological and
pathological processes, as well as in carcinogenesis (1–5). It
has been reported that miRNAs can act as oncogene or tumor
suppressor gene in various cancers, including lung, colorectal, and
liver cancer (6,7).
Gallbladder cancer (GBC) with poor prognosis is one
of the most prevalent and aggressive malignant types of cancer in
China. GBC is the tenth most common cancer in Shanghai and the
average survival of patients is ∼9 months (8). Many patients have no chance to accept
radical operation when they are diagnosed. Despite its poor
prognosis and high morbidity, the miRNA function in GBC remains
largely unknown.
The miRNA microarray assays performed on 4 pairs of
GBC and paracancerous tissues, revealed that miR-26a is
significantly downregulated in GBC tissues. It has been reported
that miR-26a was downregulated in pancreatic, nasopharyngeal,
breast and lung cancer, and miR-26a can acts as tumor suppressor
gene via directly targeting enhancer of Zeste homolog 2 (EZH2)
(9-12). Also in hepatocellular carcinoma
cells, miR-26a can inhibit cell proliferation by regulating cyclin
D2, E2 and IL-6 (13,14). It has been demonstrated that
expression levels of miR-26a are lower in the tumors of renal cell
carcinoma patients who developed tumor relapse, moreover, the
lowest levels of miR-26a are observed in primary metastatic tumors
(15). These findings suggested
that miR-26a might play a vital role in GBC physiological and
pathological processes.
In the present study, we demonstrated that miR-26a
is significantly downregulated in GBC tissues compared with
paracancerous tissues and miR-26a is closely correlated with the
histologic grade of the neoplasm. Enforced, highly expressed
miR-26a was able to inhibit GBC cell proliferation via cell cycle
G1/S arrest. We verified that the high mobility group AT-hook 2
(HMGA2) was the direct and functional target of miR-26a.
Materials and methods
Clinical specimens
GBC and the matched paracancerous gallbladder
tissues (2 cm from the tumor) were obtained from surgical specimen
from Huashan Hospital (Fudan University, Shanghai, China).
Diagnosis of each case was confirm by pathological examination.
Informed consent was obtained from each patient who provided a
specimen, and the research protocol was approved by the Ethics
Committee of Huashan Hospital.
Cell culture and reagent
HEK-293T, GBC-SD, EH-GB1 and SGC-996 cells were
cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented
with 10% fetal bovine serum (FBS) and 1% antibiotics.
RNA extraction, reverse transcription and
quantitative real-time PCR
Total RNA was extracted from tissues or cells with
TRIzol reagent (Invitrogen) according to the manufacturer’s
protocol. Complementary DNA was synthesized with the PrimeScript RT
reagent kit (Takara). Quantitative real-time PCR analyses were
performed with SYBR Premix Ex Taq II (Takara). The primers used are
listed in Table I. TaqMan microRNA
assays (Applied Biosystems) were used to quantify the expression
levels of miR-26a.
 | Table I.The primers used in the
experiment. |
Table I.
The primers used in the
experiment.
| Gene | Use | (5′-3′) |
|---|
| Pri-miR-26a | PCR | F:
ACTAGTCAGAGCAAGACTCGGCAGGGTGTCTG
R: GTCGACCACCAGGCTTCCAATGGATCAGTGGTC |
| HMGA2-ORF | PCR | F:
CCGGAATTCATGAGCGCACGCGGTGAGG
R: CGCGGATCCCTAGTCCTCTTCGGCAGACTCTTGT |
| HMGA2-3′-UTR | PCR | F:
CCGCTCGAGGTGCCCTTTGTGTGTTCCAGGAGGA
R: ATGTACGCGGCCGCCAAGATGTGTGCAAGGGGTG |
|
HMGA2-3′-UTR-MUT1 | PCR | F:
TTTTCGTATAATCTTGTAGACACTTTGAACTTGATTTTTAACT
R: TTAGAAATAAAAAGTTAAAAATCAAGTTCAAAGTGTCTACAAG |
|
HMGA2-3′-UTR-MUT2 | PCR | F:
GCTTCTCTGCTAGATTTCTACATTATGAACTAAATTTTTTAACCA
R: GGAGCGACTTGGTTAAAAAATTTAGTTCATAATGTAGAAATCT |
|
HMGA2-3′-UTR-MUT3 | PCR | F:
AAATAAAAGCCAACCTTCAAAGAATGAACTAGCTTTGTAGGTG
R: CTTGTTGCATCTCACCTACAAAGCTAGTTCATTCTTTGAAGGT |
| HMGA2 | Real-time PCR | F:
GAAGCAGCAGCAAGAACC
R: CCAGTGGCTTCTGCTTTC |
| GAPDH | Real-time PCR | F:
TCTGACTTCAACAGCGACAC
R: GCCAAATTCGTTGTCATACC |
| HMGA2 | siRNA |
CAAGAGGCAGACCUAGGAAdTdT (sense) |
Oligonucleotide transfection
miR-26a mimics were synthesized by GenePharma. The
sequences are 5′-UUCAAGUAAUC CAGGAUAGGCU-3′ (sense) and
5′-CCUAUCCUGGAUUA CUUGAAUU-3′ (antisense). Small interfering RNAs
(siRNAs) targeting HMGA2, E2F7 and PIK3R3 were synthesized by
RiboBio. The sequences are listed in Table I. Cells were transfected with
oligonucleotides using Lipofectamine 2000 reagent (Invitrogen) at a
final concentration of 50 nM and collected for assays at 48 h
post-transfection.
Cell proliferation and colony formation
assays
The cell proliferation was measured with the Cell
Counting Kit-8 (CCK-8) (Dojindo Laboratories) following the
manufacturer’s instruction and cell numbers were recorded by the
optical density at 450 nm (OD450). For colony formation
assay, 1×103 cells were plated in each well of a 6-well
plate and incubated at 37°C for 2 weeks. Cells were fixed with 4%
paraformaldehyde and stained with 1% crystal violet
(Sigma-Aldrich). Megascopic cell colonies were counted and
analyzed.
In vivo tumor formation assay
GBC-SD cells stably expressing miR-26a or vector
control were collected and suspended in serum-free DMEM. Each mouse
(male BALB/c-nu/nu, 6-weeks old) was injected subcutaneously in the
upper back with 2.0×106 GBC-SD cells in 200 μl
DMEM. The mice were sacrificed after 6 weeks, and the tumor weight
was recorded. The whole experiment was carried out according to the
protocols approved by the Shanghai Medical Experimental Animal Care
Commission.
Vector constructs
The human pri-miR-26a sequence was amplified from
normal human genomic DNA and cloned into the lentivirus expression
vector pWPXL (a generous gift from Dr Didier Trono) to generate
pWPXL-miR-26a. The 3′-UTR of HMGA2 was amplified and
inserted (XhoI + NotI) downstream of the stop codon
of Renilla luciferase in psiCHECK2 vector (Promega). The
open reading frame (ORF) of HMGA2 was amplified and cloned
into another lentiviral vector, pLVX-IRES-Neo (Clontech
Laboratories), to generate pLVX-HMGA2. The primers used are listed
in Table I.
Lentivirus production and
transduction
Lentivirus particles were harvested 48 h after
pWPXL-miR-26a (or pLVX-HMGA2) transfection with the packaging
plasmid psPAX2 and VSV-G envelope plasmid pMD2.G (a gift from Dr
Didier Trono) into HEK293T cells by using Lipofectamine 2000
reagent (Invitrogen). GBC-SD and EH-GB1 cells were infected with
recombinant lentivirus plus 6 μg/ml polybrene
(Sigma-Aldrich).
Luciferase assay
HEK-293T cells were cultured in 96-well plates and
co-transfected with 20 ng psiCHECK-2-HMGA2-3′-UTR and 5 pmol
miR-26a mimic or negative control. After 48 h of incubation, the
firefly and Renilla luciferase activities were measured
using the Dual-Luciferase reporter assay system (Promega).
Cell cycle analysis
Cells were collected and fixed in 75% ethanol at
−20°C overnight. The fixed cells were washed three times with
phosphate-buffered saline (PBS) and stained with 25 μg/ml
propidium iodide (PI) (Kaiji Biotech, Nanjing, China), 10
μg/ml RNase A (Sigma-Aldrich), 0.05 mM ethylene diamine
tetraacetic acid (EDTA) and 0.2% Triton X-100 in PBS for 30 min.
DNA content was analyzed with FACSCalibur flow cytometer (BD
Biosciences). The results were analyzed using ModFit software (BD
Biosciences).
Western blot analysis
Proteins were separated on 10% SDS-PAGE and
transferred to nitrocellulose membrane (Bio-Rad Laboratories). Then
the membrane was blocked with 5% non-fat milk and incubated with
rabbit anti-E2F7 antibody (Abgent) (1:400), rabbit anti-PI3 kinase
p85 (PIK3R3) antibody (C-term) (Abgent) (1:400), mouse anti-GAPDH
antibody (Sigma-Aldrich) (1:3,000), mouse anti-Rb (4H1) (Cell
Signaling Technology) (1:1,000), rabbit anti-Phospho-Rb
(Ser807/811) antibody (Cell Signaling Technology) (1:1,000), rabbit
anti-Phospho-Rb (Ser780) antibody (Cell Signaling Technology)
(1:1,000), and rabbit anti-HMGA2 antibody (Cell Signaling
Technology) (1:1,000). The proteins were detected with enhanced
chemiluminescence reagents (Thermo Fisher Scientific).
Statistical analysis
The results are shown as the means and standard
deviation (SD). The data were analyzed with Student’s t-test
(two-tailed); The clinical significance of miR-26a was analyzed by
the way of Fisher’s exact test in GraphPad Prism 5. P<0.05 was
considered to indicate a statistically significant result.
Results
miR-26a is frequently downregulated in
GBC and associated with the histologic grade of the neoplasm
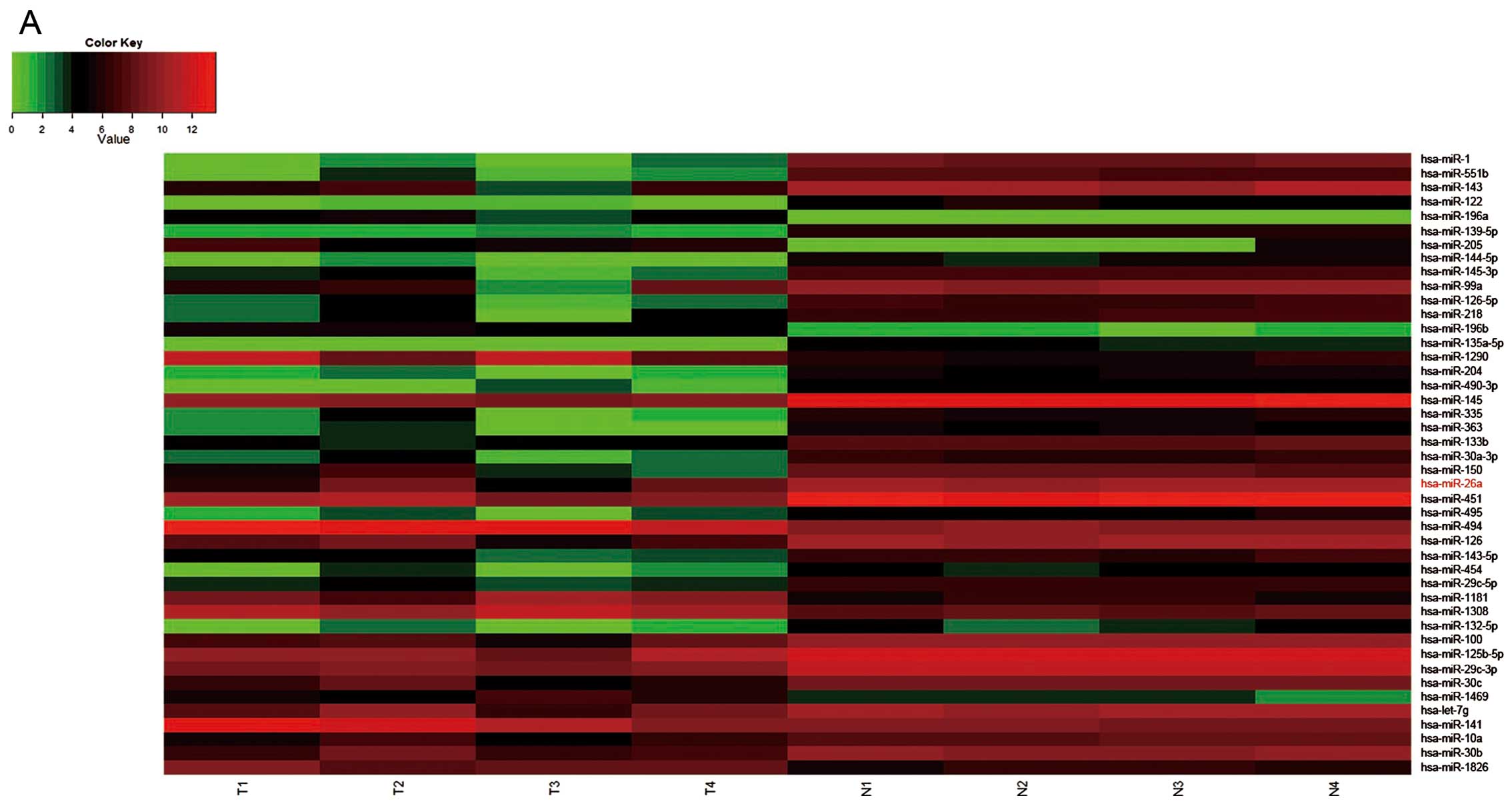
From the results of miRNA expression profiling chip
in 4 paired GBC and paracancerous tissues, we found that the
miR-26a was obviously downregulated (Fig. 1A). And then, we detected mature
miR-26a in 23 pairs of GBC and paracancerous tissues and GBC cell
lines by real-time PCR. The results confirmed that miR-26a
expression was obviously downregulated in GBC tissues compared with
the paired paracancerous tissues (Fig.
1B). Based on the 23 paired GBC and paracancerous tissues, the
miR-26a expression was associtated with the neoplasm histological
grade (Table II).
| Table II.The clinical significance of
miR-26a. |
Table II.
The clinical significance of
miR-26a.
| miR-26a high | miR-26a low | Total | P-value |
|---|
| Age (years) | | | | |
| >55 | 4 | 8 | 12 | |
| ≤55 | 8 | 3 | 11 | 0.0995 |
| Total | 12 | 11 | 23 | |
| Gender | | | | |
| Male | 9 | 7 | 16 | |
| Female | 3 | 4 | 7 | 0.6668 |
| Total | 12 | 11 | 23 | |
| Grade | | | | |
| I+II | 8 | 2 | 12 | |
| III+IV | 4 | 9 | 11 | 0.0361 |
| Total | 12 | 11 | 23 | |
| CA199 | | | | |
| High | 5 | 7 | 12 | |
| Low | 7 | 4 | 11 | 0.4136 |
| Total | 12 | 11 | 23 | |
miR-26a inhibits GBC cell proliferation
in vitro and in vivo
The expression of miR-26a is downregulated in GBC
and in pancreatic, nasopharyngeal, breast and lung cancer, miR-26a
has been shown to act as a tumor suppressor to inhibit tumor cells
proliferation by directly targeting EZH2 (9–12),
therefore we hypothesized that miR-26a might have influence on GBC
proliferation.
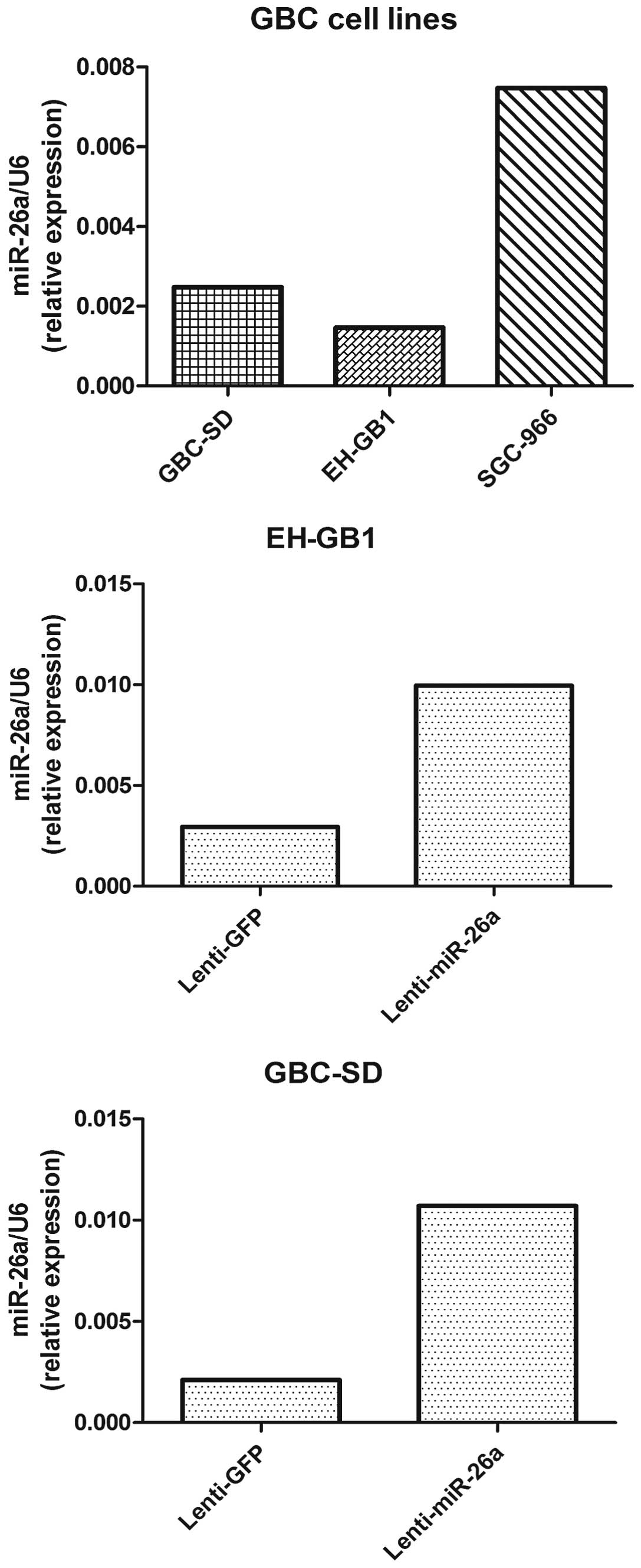
To examine the biological function of miR-26a in
GBC, we established miR-26a stably expressing cells. Both GBC-SD
cells and EH-GB1 cells were infected with the lentivirus containing
the miR-26a expression sequence, and the expression of miR-26a was
confirmed by real-time PCR (Fig.
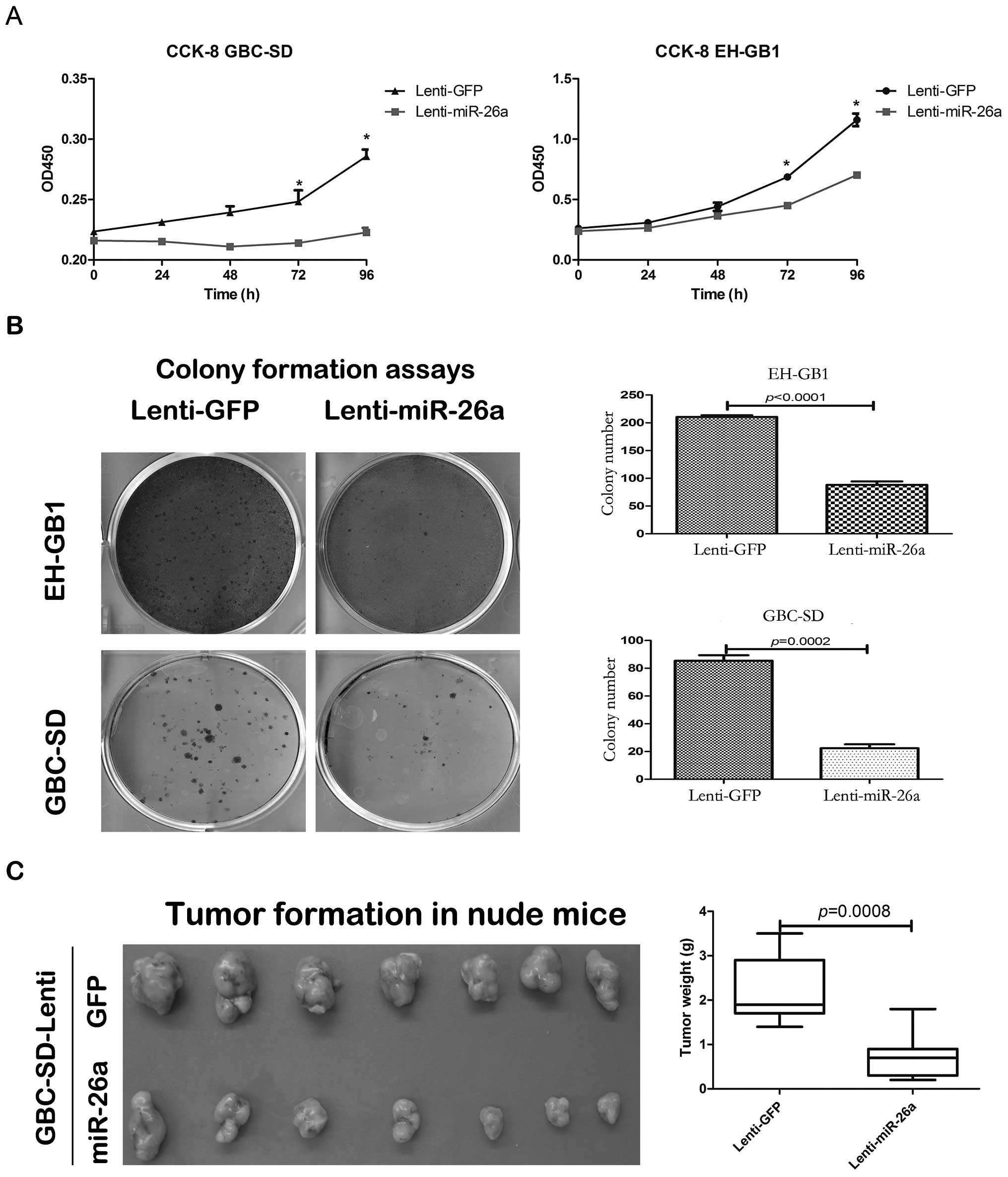
2). The cell proliferation of GBC-SD and EH-GB1 infected with
lenti-miR-26a was significantly decreased compared with those
infected with lenti-GFP based on the CCK-8 and colony formation
assays (Fig. 3A and B). We
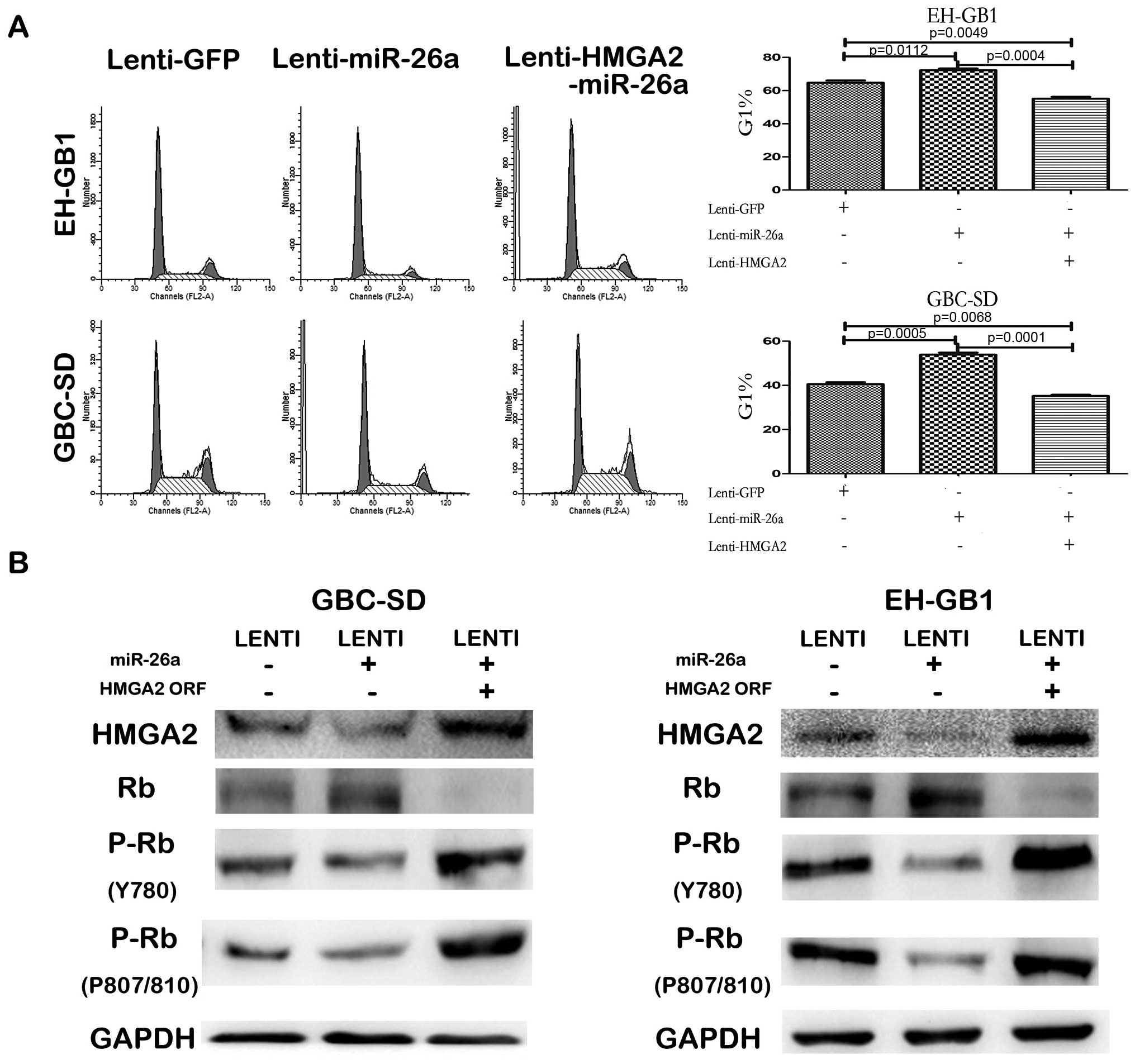
investigated the effect of miR-26a in cell cycle progression by
fluorescence-activated cell sorting analysis. It is shown that the
miR-26a blocked G1/S transition in both GBC-SD and EH-GB1 cells
(Fig. 7A). Since the expression of
miR-26a was extremely low in all GBC cell lines, we did not use the
miR-26a silencing method for functional experiments (Fig. 2).
To further determine the effect of miR-26a in GBC
cell proliferation in vivo, the GBC-SD cells stably
expressing miR-26a or GFP were subcutaneously injected into nude
mice. After 6 weeks, the mice were sacrificed to weigh the tumors.
The tumor weight of GBC-SD cells stably expressing miR-26a was
significantly lower than that of the stably expressing GFP
(Fig. 3C).
The above results indicate that miR-26a inhibited
the proliferation of GBC cells both in vitro and in
vivo.
HMGA2 is a direct downstream target of
miR-26a
To identify the direct downstream target responsible
for the effect of miR-26a on GBC cell proliferation, mRNA
microarray assays were carried out to find the genes that were
downregulated by miR-26a in EH-GB1 cells infected with
lenti-miR-26a or lenti-GFP. Potential targets were predicted by
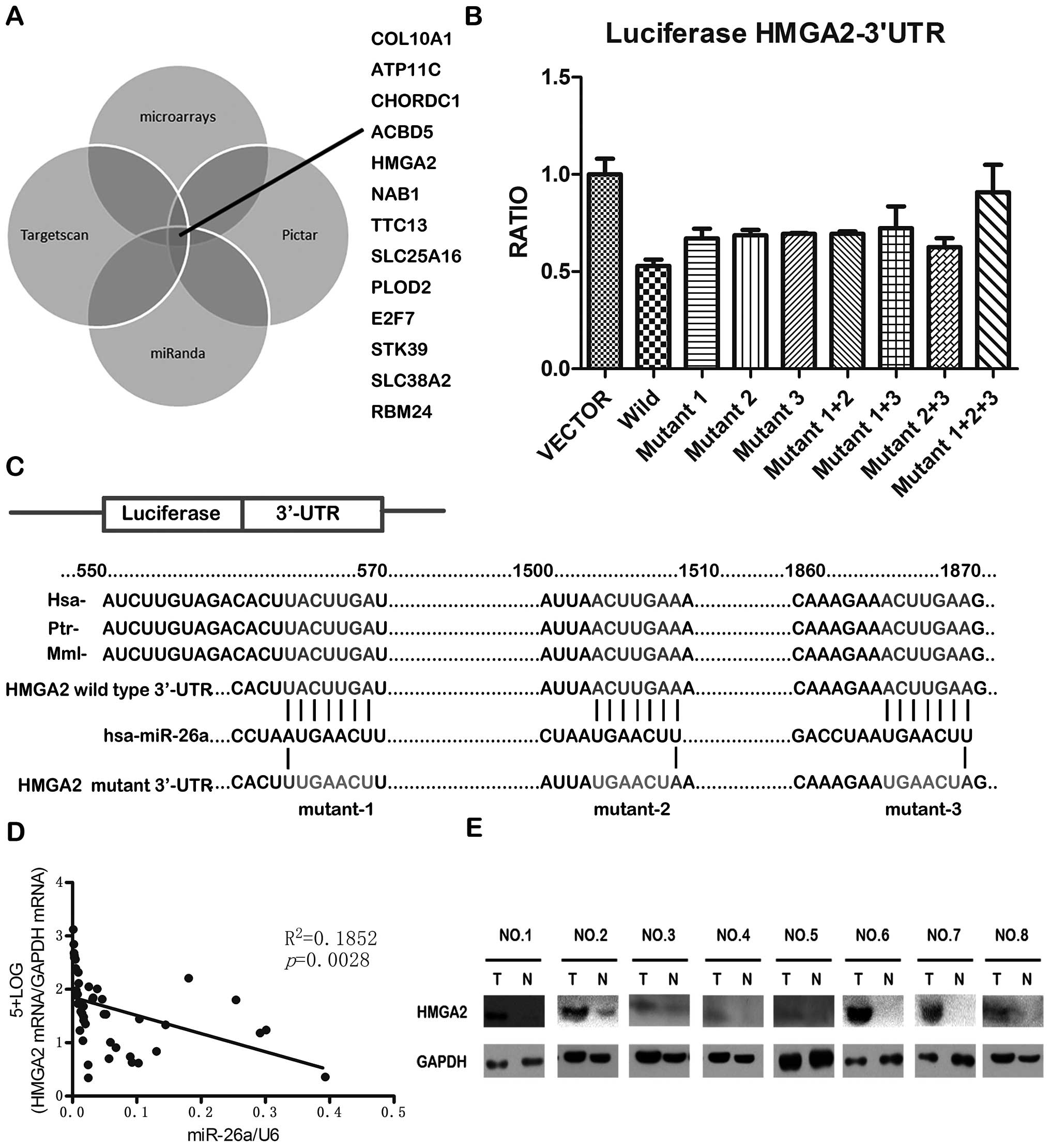
using TargetScan (http://www.targetscan.org), miRanda (http://www.miranda-im.org) and PicTar (http://pictar.mdc-berlin.de). By integrating the
results of the four strategies, 13 genes were found to be potential
targets of miR-26a (Fig. 4A). In
these 13 genes, phosphoinositide-3-kinase, regulatory subunit 3
(gamma) (PIK3R3), E2F transcription factor 7 (E2F7) and high
mobility group AT-hook 2 (HMGA2) have been reported as having
effects on the cell cycle inhibiting cell proliferation (16–21).
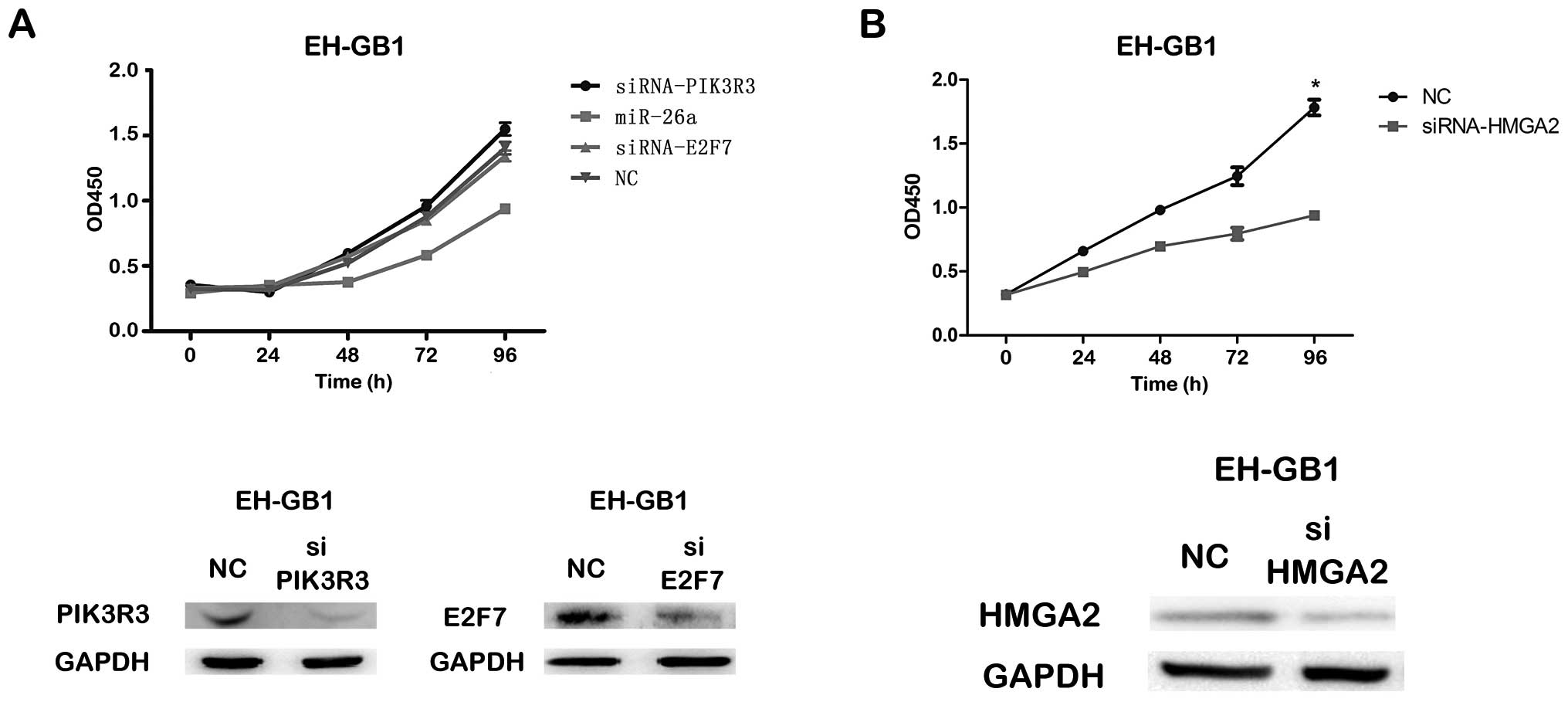
By CCK-8 and colony formation assays, we found that the siRNA
against PIK3R3 or E2F7 could not significantly inhibit
proliferation of EH-GB1 cell compared to NC, but the proliferation
of both EH-GB1 and GBC-SD cells transfected with siRNAs targeting
HMGA2 was significantly inhibited compared to that transfected with
NC (Fig. 5).
TargetScan indicated that HMGA2 contains 3 miR-26a
binding sites on its 3′-UTR, and the sequence of the binding site
is highly conserved in different species (Fig. 4C). We constructed vectors
containing wild-type or mutant 3′-UTR of HMGA2 directly inserting
downstream of the firely luciferase gene (Fig. 4C), and cotransfected wild-type or
mutant vector into HEK-293T cells with miR-26a expression plasmid
or vector control. The result showed that miR-26a could
significantly decrease the relative luciferase activity of
wild-type HMGA2-3′-UTR, while the luciferase activity in mutant
HMGA2-3′-UTR was not reduced as much as in wild-type HMGA2-3′-UTR
(Fig. 4B). It is suggested that
miR-26a could directly bind to the 3′-UTR of HMGA2. Furthermore, it
was found that the HMGA2 mRNA level and miR-26a level was
negatively correlated in the 23 pairs of GBC and paracancerous
tissues (Fig. 4D). In addition,
the HMGA2 protein levels were often higher in GBC tissues than in
the paired paracancerous tissues (Fig.
4E). Taken together, these results indicate that HMGA2 is a
direct downstream target for miR26a in GBC cells.
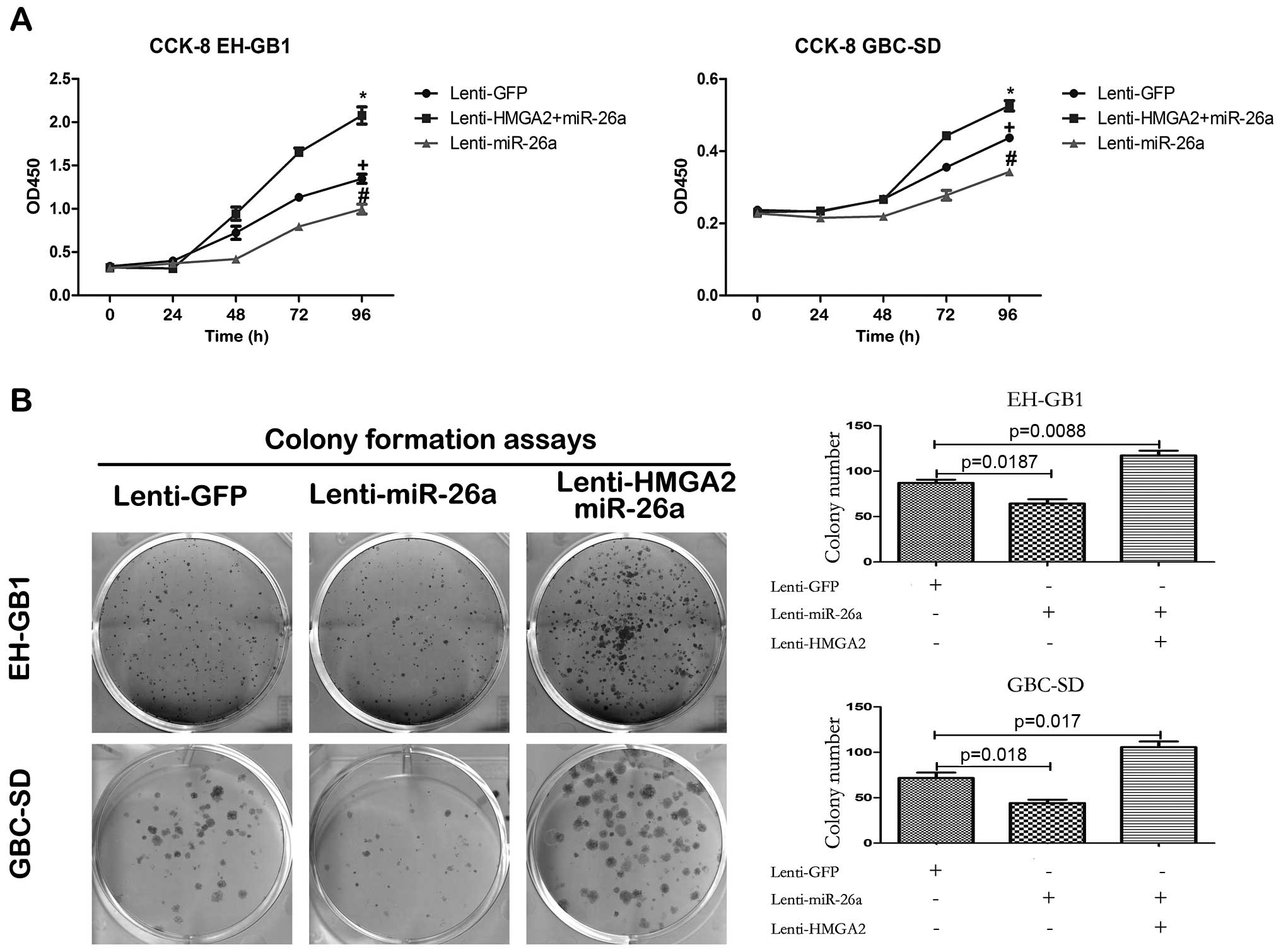
Reintroduction of HMGA2 abrogates
miR-26a-induced proliferation suppression
If HMGA2 indeed is a functional target of miR-26a in
GBC cells, reintroduction of HMGA2 without 3′-UTR into miR-26a
expressing GBC cells should be able to antagonize the effects of
miR-26a. In order to test this hypothesis, we constructed a
lentiviral expression vector of HMGA2 ORF (open reading frame)
without 3′-UTR and infected miR-26a stably expressing GBC cells
with this vector. Western blot assay after HMGA2 ORF lentivirus
infection, showed that the protein of HMGA2 expression recovered
(Fig. 7B). CCK-8 and colony
formation assays demonstrated that reintroduction of HMGA2
antagonized the inhibition of miR-26a to GBC cell proliferation
(Fig. 6). Furthermore, enforced
expression of HMGA2 counteracted the G1 arrest induced by miR-26a
(Fig. 7A). HMGA2 can bind the
complex formed by pRB and HDAC1 to dissociate pRB from genomic
promoter regions and promotes the transcriptional activity of E2F1
and blocks the G1/S transition to inhibit the cell proliferation.
In addition (22), we detected the
protein level of Rb and its phosphorylation level in the GBC cells
infected with GFP lentivirus, miR-26a lentivirus or both miR-26a
lentivirus and HMGA2 lentivirus. The results showed that in both
GBC-SD and EH-GB1 cells, Rb protein level was negatively correlated
with HMGA2 protein level but its phosphorylation level was
positively correlated with HMGA2 (Fig.
7B).
In summary, these results suggest that the HMGA2 is
a functional target of miR-26a in GBC cells.
Discussion
In the present study, we found low expression of
miR-26a in GBC by using microRNA microarray and RT-PCR and the
expression of miR-26a in GBC tissue is correlated with the
histological grade of the neoplasm. Furthermore, it was found that
miR-26a can inhibit the GBC cell proliferation in vivo and
in vitro. We identified HMGA2 which is upregulated in GBC
tissues to be the direct and functional target gene of miR-26a. In
addition, we demonstrated that the miR-26a inhibits HMGA2
expression and influence the proliferation of GBC cells through G1
arrest.
We verified that the expression of miR-26a is
significantly lower in GBC tumors than the paired normal
paracancerous tissues in 23 pairs of tissue samples, and the result
is consistent with the microRNA microarray chip data. In previous
studies it has been demonstrated that miR-26a is involved in
various malignant pathological processes. In pancreatic,
nasopharyngeal, breast and lung cancer, miR-26a can act as a tumor
suppressor to inhibit tumor cells proliferation by directly
targeting EZH2 (9–12). It has been also demonstrated that
in hepatocellular carcinoma cells, miR-26a can inhibit cell
proliferation via targeting cyclin D2, cyclin E2 and IL-6, and the
patients with high level of miR-26a in the tumor have better
prognosis (13,14). These findings, as well as our new
results, suggest that miR-26a plays a vital role in the tumor
proliferation and could act as a predictor of the treatment in
certain types of cancer. To date, the function and clinical
significance of miR-26a in GBC is still unclear. In the present
study, we demonstrated that miR-26a inhibited the proliferation of
GBC cells, and the level of miR-26a expression was associated with
the histological grade of the neoplasm.
To identify the mechanism of the miR-26a inhibition
effect on the proliferation of GBC cells, we investigated the
target gene HMGA2 mediating the function of miR-26a in GBC. HMGA2
is a member of the HMG (23).
HMGA2 proteins are abundant in pluripotent embryonic stem cells and
malignant human neoplasm, but it can be not detected in normal
somatic cells (21). HMGA2 acts as
an activator or a repressor of target gene expression and
facilitates DNA structural changes by the formation of the
specialized nucleoprotein structures at special promoter regions
(24). It is reported that HMGA2
binds the complex formed by pRB and HDAC1 to dissociate pRB from
genomic promoter regions and promotes the transcriptional activity
of E2F1 and blocks the G1/S transition to inhibit the cell
proliferation (22). It has been
discovered that HMGA2 is upregulated in many benign and malignant
tumors, such as colorectal, breast, pancreatic, ovarian, and lung
cancer (25–28). In the present study, it was found
that the mRNA and protein levels of HMGA2 were often upregulated in
GBC tissues. We also found that the HMGA2 siRNA inhibited the
proliferation of GBC cells and enforced HMGA2 protein expression
promoted the proliferation of GBC cells. In GBC samples, the levels
of miR-26a and HMGA2 mRNA were inversely associated, which
suggested the upregulation of HMGA2 may be partially due to the
downregulation of miR-26a. Furthermore, we found the G1 arrest
could be promoted by enforcing miR-26a expression and the arrest by
enforcing HMGA2 protein expression. Then, we confirmed the effect
by detecting the protein level of RB and pRB. It is suggested that
the HMGA2 is the direct and functional target of miR-26a and
involved the miR-26a effect on the proliferation of GBC cells.
Collectively, we found that miR-26a, which is
associated with the neoplasm histologic grade, is often
downregulated in GBC and it can inhibit the proliferation of GBC
cells by directly targeting HMGA2 through G1 arrest, thus miR-26a
shows potential as a therapeutic in GBC patients.
Acknowledgements
We thank Dr Didier Trono (School of
Life Sciences, Ecole Polytechnique Fédérale de Lausanne, 1015
Lausanne, Switzerland) for providing the psPAX2, pMD2.G and pWPXL
plasmids. The present study was supported by a grant from the
Science and Technology Commission of Shanghai Municipality (no.
12nm0501502 and no. 11nm0503900).
References
|
1.
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Bartel DP: MicroRNAs: target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Croce CM and Calin GA: miRNAs, cancer, and
stem cell division. Cell. 122:6–7. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Friedman RC, Farh KK, Burge CB, et al:
Most mammalian mRNAs are conserved targets of microRNAs. Genome
Res. 19:92–105. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Hernando E: microRNAs and cancer: role in
tumorigenesis, patient classification and therapy. Clin Transl
Oncol. 9:155–160. 2007. View Article : Google Scholar
|
|
7.
|
Negrini M, Ferracin M, Sabbioni S, et al:
MicroRNAs in human cancer: from research to therapy. J Cell Sci.
120:1833–1840. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Chen YL, Huang ZQ, Zhou NX, et al:
Clinical analysis of 110 patients with primary gallbladder
carcinoma. Zhonghua Zhong Liu Za Zhi. 29:704–706. 2007.(In
Chinese).
|
|
9.
|
Bao B, Ali S, Banerjee S, et al: Curcumin
analogue CDF inhibits pancreatic tumor growth by switching on
suppressor microRNAs and attenuating EZH2 expression. Cancer Res.
72:335–345. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Lu J, He ML, Wang L, et al: MiR-26a
inhibits cell growth and tumorigenesis of nasopharyngeal carcinoma
through repression of EZH2. Cancer Res. 71:225–233. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Zhang B, Liu XX, He JR, et al:
Pathologically decreased miR-26a antagonizes apoptosis and
facilitates carcinogenesis by targeting MTDH and EZH2 in breast
cancer. Carcinogenesis. 32:2–9. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Dang X, Ma A, Yang L, et al: MicroRNA-26a
regulates tumorigenic properties of EZH2 in human lung carcinoma
cells. Cancer Genet. 205:113–123. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Kota J, Chivukula RR, O’Donnell KA, et al:
Therapeutic microRNA delivery suppresses tumorigenesis in a murine
liver cancer model. Cell. 137:1005–1017. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Yang X, Liang L, Zhang XF, et al:
MicroRNA-26a suppresses tumor growth and metastasis of human
hepatocellular carcinoma by targeting interleukin-6-Stat3 pathway.
Hepatology. 58:158–170. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Slaby O, Redova M, Poprach A, et al:
Identification of MicroRNAs associated with early relapse after
nephrectomy in renal cell carcinoma patients. Genes Chromosomes
Cancer. 51:707–716. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Xia X, Cheng A, Akinmade D, et al: The
N-terminal 24 amino acids of the p55 gamma regulatory subunit of
phosphoinositide 3-kinase binds Rb and induces cell cycle arrest.
Mol Cell Biol. 23:1717–1725. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Deng Q, Wang Q, Zong WY, et al: E2F8
contributes to human hepatocellular carcinoma via regulating cell
proliferation. Cancer Res. 70:782–791. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Sozzani R, Maggio C, Giordo R, et al: The
E2FD/DEL2 factor is a component of a regulatory network controlling
cell proliferation and development in Arabidopsis. Plant Mol Biol.
72:381–395. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Hazar-Rethinam M, Cameron SR, Dahler AL,
et al: Loss of E2F7 expression is an early event in squamous
differentiation and causes derepression of the key differentiation
activator Sp1. J Invest Dermatol. 131:1077–1084. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Sirma H, Kumar M, Meena JK, et al: The
promoter of human telomerase reverse transcriptase is activated
during liver regeneration and hepatocyte proliferation.
Gastroenterology. 141:326–337. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Li O, Vasudevan D, Davey CA, et al:
High-level expression of DNA architectural factor HMGA2 and its
association with nucleosomes in human embryonic stem cells.
Genesis. 44:523–529. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Fusco A and Fedele M: Roles of HMGA
proteins in cancer. Nat Rev Cancer. 7:899–910. 2007. View Article : Google Scholar
|
|
23.
|
Ashar HR, Chouinard RA Jr, Dokur M, et al:
In vivo modulation of HMGA2 expression. Biochim Biophys Acta.
1799:55–61. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Eda A, Tamura Y, Yoshida M, et al:
Systematic gene regulation involving miRNAs during neuronal
differentiation of mouse P19 embryonic carcinoma cell. Biochem
Biophys Res Commun. 388:648–653. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Ahmed KM, Tsai CY and Lee WH: Derepression
of HMGA2 via removal of ZBRK1/BRCA1/CtIP complex enhances mammary
tumorigenesis. J Biol Chem. 285:4464–4471. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Cleynen I and Van de Ven WJ: The HMGA
proteins: a myriad of functions (Review). Int J Oncol. 32:289–305.
2008.PubMed/NCBI
|
|
27.
|
Sgarra R, Zammitti S, Lo Sardo A, et al:
HMGA molecular network: from transcriptional regulation to
chromatin remodeling. Biochim Biophys Acta. 1799:37–47. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Wang X, Liu X, Li AY, et al:
Overexpression of HMGA2 promotes metastasis and impacts survival of
colorectal cancers. Clin Cancer Res. 17:2570–2580. 2011. View Article : Google Scholar : PubMed/NCBI
|