Introduction
The majority of CRC are well- to
moderately-differentiated adenocarcinomas (WMD-CRC) with apparent
glandular morphology. Pathogenesis of such a differentiated-type
CRC is a multistep process, recognized as an adenoma-carcinoma
sequence, starting from a polypoid lesion such as adenoma, with
subsequent progression to adenocarcinoma, and finally metastatic
adenocarcinoma (1). Throughout
this process, a series of genetic alterations such as mutation in
APC, β-catenin, KRAS, p53 and
TGF-βR-II and microsatellite instability (MSI) are known to
be accumulated with disease progression (2). Among these changes, mutations in Wnt
signaling cascade such as APC and β-catenin occur at
the initial step and play a key role in carcinogenesis of WMD-CRC
(3). In contrast,
poorly-differentiated adenocarcinomas (PD-CRC), defined by minimal
glandular structure (1), are
relatively rare accounting for only 5–10% of CRCs (4), and molecular pathogenesis and
clinicopathological features are reportedly distinct from WMD-CRC
(5). PD-CRC is heterogeneous and
has been subclassified into several types (6). One relatively rare subset of PD-CRC
is ‘medullary type adenocarcinomas’ which are characterized by
MSI-positive, right-sided, scant fibrous stroma and relatively
favorable prognosis (7). However,
most PD-CRCs are aggressive and highly metastatic, with a less
favorable prognosis than that of WMD-CRC (8–10).
As for genetic alterations, several investigators reported that PD
showed a lower incidence of KRAS mutation, a higher
incidence of BRAF mutation, MSI, a higher promoter
methylation of P16 than WMD-CRC (11–13).
However, the molecular pathogenesis and therapeutic target specific
for PD still remain largely unknown.
The aberrantly activated HER family of receptor
tyrosine kinases including EGFR, HER2, HER3 and HER4 are associated
with carcinogenesis and tumor progression, and therefore are major
molecular targets in various epithelial malignancies including
gastrointestinal cancers (14,15).
Among them, EGFR overexpression such as gene amplification was
reported in 12–17% of CRC patients and activation of EGFR results
in acceleration of growth and survival of tumor cells through the
MAPK or PI3K/Akt pathways and correlates with a poor patient
outcome (16). Molecular targeting
therapy with monoclonal antibody to EGFR (cetuximab) has been
clinically used in patients with metastatic CRC in combination with
chemotherapy (17). However, EGFR
expression as assessed by immunohistochemistry proved to be
insufficient to predict cetuximab sensitivity (18). Alternatively, KRAS mutation
at codons 12 or 13, and more recently BRAF mutation (V600E)
have been reported as promising new predictive markers for
cetuximab-resistance in patients with metastatic CRC (19–21).
Unlike EGFR, HER3 has a unique kinase-inactive
nature because of the amino acid substitutions in critical residue
in the tyrosine kinase domain, and is therefore not a direct target
for HER tyrosine kinase inhibitor. Upon binding with heregulin
ligand, HER3 can be transphosphorylated and then successfully
activate PI3K, via its multiple docking sites for the p85
regulatory subunit of PI3K and dimerization with other HER members
such as HER2 (22). Therefore,
HER3 plays a key regulatory role in transducing signals downstream
to PI3K/Akt pathways. Recent studies demonstrated that HER3
overexpression and consequent activation of PI3K/Akt signaling lead
to resistance to tyrosine kinase inhibitors such as gefitinib in
HER2 overexpressing breast cancer, because of reactivation of HER3
signaling after transient inhibition by the drugs and the
difficulty of consistent blocking HER3/PI3K/Akt pathway (23). HER3/PI3K/Akt pathway also plays a
negative regulatory role in p27Kip1-mediated G1 cell
cycle arrest by trastuzumab in breast cancers (24). Furthermore, HER3 is frequently
overexpressed in CRC (25). These
findings suggest a potential role of HER3 in responsiveness of CRC
to EGFR targeting therapy and that HER3 is therefore a potential
predictive marker for drug resistance or susceptibility. To date,
however, the functional and clinical significance of HER3
expression in PD-CRC is not entirely known.
In the present study, we found that HER3 expression
in a clinical specimen of primary PD-CRC is significantly lower
compared with that of WMD-CRC. Furthermore, we investigated the
molecular mechanism of gefitinib sensitivity using a HER3-deficient
and metastatic PD-CRC cell line that exhibits high sensitivity to
gefitinib. Our findings raise the possibility of EGFR-targeting
therapeutics as a new strategy against highly malignant PD-CRC.
Materials and methods
Compounds
Gefitinib (ZD1839; Iressa) was provided by
AstraZeneca (Macclesfield, UK). Human recombinant heregulin was
obtained from R&D Systems (Minneapolis, MN). For western blot
analysis, rabbit polyclonal anti-Akt, phospho-Akt (Ser473) (Cell
Signaling, Beverly, MA), PI3Kp85 (Upstate, Lake Placid, NY), mouse
monoclonal anti-EGFR, HER2, p21WAF1/CIP (all from Cell
Signaling), HER3 (clone 2F12, Upstate), p27Kip1 (BD
Pharmingen, San Diego, CA), Skp2 (Zymed, Carlsbad, CA), β-actin (BD
Pharmingen) and rabbit monoclonal anti-phospho-HER3 (Tyr1289) (Cell
Signaling) were used. For immunohistochemistry, mouse monoclonal
anti-E cadherin, vimentin (Dako, Glostrup, Denmark), β-catenin (BD
Pharmingen), EGFR (Novocastra, Newcastle, UK), rabbit polyclonal
anti-HER2 (Dako), p27Kip1 (Thermo Scientific, Fremont,
CA), rabbit monoclonal anti-HER3 (D22C5, Cell Signaling) were used.
For flow cytometry, mouse monoclonal anti-EGFR (NeoMarkers), HER2
(44E7, Cell Signaling), and HER3 (Ab-10, NeoMarkers) were used.
Patients
A total of 62 colorectal cancer patients consisting
of 38 WMD-CRC and 24 PD-CRC patients were enrolled in this study.
PD-CRC (1998–2002) and WMD-CRC patients (1998) underwent operation
at the Department of Gastroenterological Surgery, Aichi Cancer
Center Central Hospital. Analysis using these clinical samples was
carried out under approval by the institutional ethics review board
of Aichi Cancer Center.
Immunohistochemical analysis of primary
colorectal cancers
Surgically resected specimens were fixed in 10%
buffered formalin and embedded in paraffin. For antigen retrieval,
the sections were treated with microwave at 98°C for 10 min in
citrate buffer pH 6.0. After blocking of nonspecific reactions by
normal serum for 30 min, these sections were incubated at 4°C
overnight with primary antibodies, thoroughly washed in PBS, then
incubated with biotinylated secondary antibodies. The sections were
washed again with PBS and incubated with streptavidin-peroxidase
complex (Vectastain ABC kit, Vector Laboratories, Burlingame, CA)
for 60 min. The sites of peroxidase binding were visualized using
0.01% diaminobenzidine (DAB) as a chromogen.
Expression of EGFR and HER2 protein was scored for
each of the 3+, 2+, 1+ and 0 categories based on the membrane
staining according to the criteria of HercepTest. In this study, we
subclassified cases into 3 classes as negative, 0; weakly positive
staining, 1+; strongly positive staining, 2+ and 3+. For HER3, the
staining pattern of the CRC was both membranous and cytoplasmic,
and the latter cytoplasmic pattern was more prominent compared with
that of EGFR/HER2, but we evaluated cases based on the membranous
staining as negative, 0; weakly positive staining, 1+; strongly
positive staining, 2+ and 3+, like EGFR/HER2.
Cell lines
COLM-5 cell line was established from liver
metastasis of PD-CRC Japanese patient, and COLM-2 was established
from WMD-CRC Japanese patient as described previously (26). These cell lines were cultured in
Dulbecco’s modified Eagle’s medium (DMEM; Nissui Pharmaceutical
Co., Tokyo, Japan) supplemented with 10% fetal bovine serum (FBS)
(Gibco, Grand Island, NY), 100 U/ml penicillin and 100 μg/ml
streptomycin (Falcon, BD Labware, Franklin Lakes, NJ) in a
humidified 5% CO2 incubator at 37°C. To evaluate
metastasis clearly with green fluorescence, these cells were
transfected with the pEGFP-C1 plasmid (Clontech Laboratories, Palo
Alto, CA) using FuGENE6 transfection reagent (Roche Diagnostics,
Basel, Switzerland).
In vitro growth inhibition assay
The cells were plated at a density of
1×104 cells/well in 96-well plastic plates (Falcon) and
treated with increasing doses of gefitinib (0.1, 1.0 and 10 μM).
The number of viable cells was assessed with a trypan-blue dye
exclusion test on day 3 by counting using a hemocytometer in
quadruplicate.
Measurement of caspase-3/7 activity
Cells were plated at 1×104 cells in
96-well plates, and cultured in the presence of gefitinib (0.1, 1.0
and 10 μM) for 24 h. Caspase-3/7 activity was then measured using
Apo-One Homogenous caspase-3/7 Assay Kit (Promega Co., Madison, WI)
according to the manufacturer’s instructions.
FISH analysis
Amplification of the EGFR and HER2
gene was determined by dual-color FISH method using Passvision EGFR
or HER2 DNA probe kit (Vysis Inc., Downers Grove, IL) according to
the manufacturer’s protocol. The nucleus was counterstained with
4′,6-diamidino-2-phenylindole (DAPI). The slides were observed
under BX60 fluorescence microscope equipped with a DP50 digital
camera (Olympus, Tokyo, Japan). A cell was considered to have
amplification when a signal ratio of specific probe to the
centromere region of the corresponding chromosome was more than
2.
Flow cytometry
Cells were suspended in PBS and then incubated with
mouse monoclonal antibodies to human EGFR, HER2 and HER3 for 30 min
on ice. After washing with PBS containing 0.5% bovine serum albumin
(BSA) and 5 mM EDTA, they were exposed to FITC (PE)-conjugated
anti-mouse IgG (Molecular Probes) for 30 min. The intensity of
fluorescence was measured by FACSCalibur (BD Biosciences, San
Diego, CA).
Cell cycle analysis
Cells were treated with gefitinib at a concentration
of 10 μM for 24 h, and stained with propidium iodide using
CycleTest Plus Kit (Becton Dickinson, San Jose, CA) according to
the manufacturer’s instructions. Flow cytometric analysis was done
in FACSCalibur (BD Biosciences). Data collected from 10,000 cells
for each experiment were analyzed by ModFit software (Verity
Software House, Topsham, ME).
Western blot analysis
To evaluate the effects of gefitinib under
ligand-mediated stimulation, cells were incubated in serum-free
medium for 24 h, and exposed to gefitinib for 2 h at 37°C. Cells
were then incubated for 5–10 min with heregulin (10 ng/ml). Cells
were then lysed at 4°C in lysis buffer [25 mM Tris-HCl, pH 7.5, 150
mM NaCl, 0.1% NP-40, 1 mM EDTA and protease inhibitor Cocktail plus
phosphatase inhibitor Cocktail (Roche Diagnostics, Mannheim,
Germany)]. A total of 10 μg of the samples were separated by
SDS-PAGE, transferred to Immune-Blot PVDF Membrane (Bio-Rad),
immunoblotted with antibodies described above, and visualized using
Super Signal West Pico (Dura) Chemiluminescence Substrate (Thermo
Scientific).
RT-PCR assay
Total RNA was extracted using Isogen (Nippon Gene,
Tokyo, Japan) and cDNA was then synthesized with SuperScript II
reverse transcriptase (Invitrogen, Carlsbad, CA) according to the
manufacturer’s protocol. A reverse transcription- polymerase chain
reaction assay (RT-PCR) for p27Kip1 and HER3 was
performed using specific oligonucleotide primer pairs and probes
#60, #37, respectively (Universal Probe Library, Roche
Diagnostics), on the LightCycler instrument (Roche Diagnostics). As
an internal control, glyceraldehyde-3-phosphate dehydrogenase
(GAPDH) was used.
Xenograft studies for anti-tumor and
anti-metastatic activity
All animal experiments were carried out under the
approval of the Institutional Ethics Committee for Animal
Experiment of Aichi Cancer Center Research Institute. Five millions
cells were injected subcutaneously into the left abdominal flanks
of 8-week-old male nude mice of KSN strain (Shizuoka Laboratory
Animal Center, Hamamatsu, Japan). For gefitinib treatment, mice
(n=5) were orally administered gefitinib with a gastric tube at a
dose of 0 or 150 mg/kg/day from 1–2 weeks post-injection, five
times per week for 3–4 weeks. In the control groups, mice were
orally administered vehicle (0.5% polysorbate, Merck, Darmstadt,
Germany). Tumor maximum diameter (L) and the right angle diameter
to that axis (W) were measured with a slide caliper every week.
Tumor volume was calculated by the following formula, L × W × W ×
1/2. To assess lung metastasis, we pretreated nude mice 4 times
with intraperitoneal injection of rabbit antiserum to bovine asialo
GM1 (400 μg/0.2 ml/mouse) (Wako Pure Chemical, Osaka, Japan) before
(once) and after (3 times) transplantation, which resulted in a
substantial increase in lung metastasis via depletion of NK cells.
A million of GFP-tagged cells were then injected intravenously, and
macroscopic lung metastases were evaluated by counting visible
parietal nodules in mice with or without gefitinib treatment at 3
weeks after injection. For peritoneal metastasis, 3×106
cells were injected intraperitoneally. At 4 weeks post-injection,
mice with or without gefitinib treatment were sacrificed, and
peritoneal metastases were detected macroscopically and quantified
as described previously (27).
Overexpression of HER3 in COLM-5
cells
A cDNA of HER3 open reading frame was purchased from
Origene (Rockville, MD), and inserted into the pcDNA3.2-DEST vector
(Invitrogen). The HER3 cDNA was confirmed by sequencing. The
expression vector was introduced in COLM-5 cells by FuGENE-6
reagent according to manufacturer’s instruction. Stable HER3
expressing COLM-5 cells were established by a selection with G418
reagent.
Statistical analysis
The statistical significance of differences in
growth was analyzed using the Student’s t-test. Differences in the
incidence between groups were analyzed with Fisher’s exact test. A
p-value <0.05 was considered significant.
Results
Deficient or low HER3 expression in the
primary PD-CRC
We examined EGFR, HER2 and HER3 expression of 24
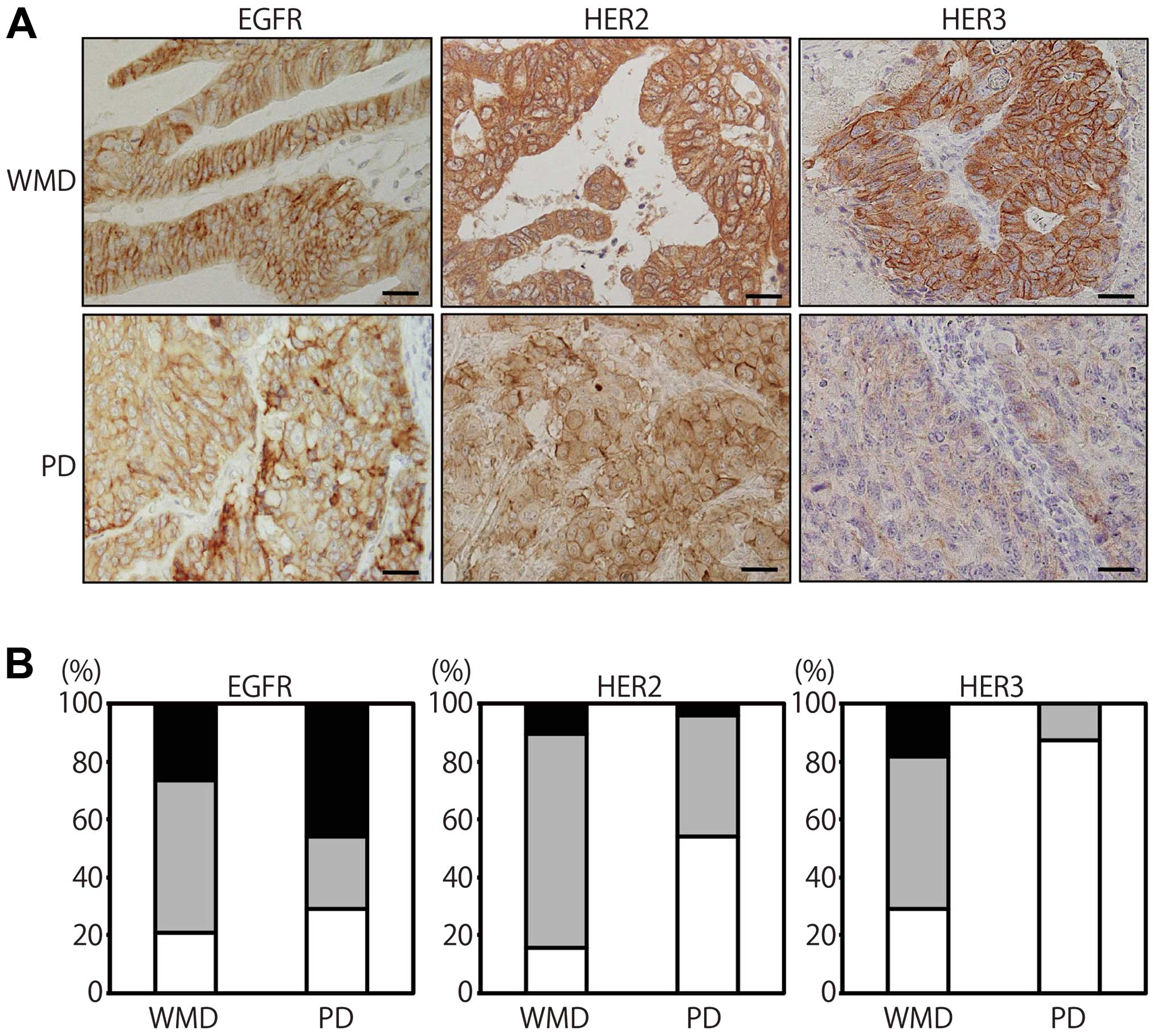
PD-CRC and 38 WMD-CRC by an immunohistochemical analysis (Fig. 1A). EGFR positivity rate (weakly
positive and strongly positive) of PD-CRC (71%) was almost
comparable to that of WMD-CRC (79%), but for the EGFR strong
membranous staining, the positivity rate of PD (46%) was higher
than WMD-CRC (26%) (Fig. 1B). HER2
positivity rate is higher in WMD-CRC (84%) than PD-CRC (46%).
However, circumferential strong membranous staining (2+ and 3+)
like EGFR was relatively rare even in WMD-CRC (11%). HER3
expression localized both in the cytoplasm and the membrane and the
positivity rate was significantly lower in PD-CRC (12%) than
WMD-CRC (71%), indicating that the majority of PD-CRC is
HER3-negative. HER3 positivity rate was significantly different
between WMD-CRC and PD-CRC (p<0.01, Fisher’s exact test)
(Fig. 1A and B). Tumors with
EGFR-positive, HER2-positve and HER3-negative phenotype, designated
by EGFR+/HER2+/HER3−, accounted
for 37% of total PD cancers.
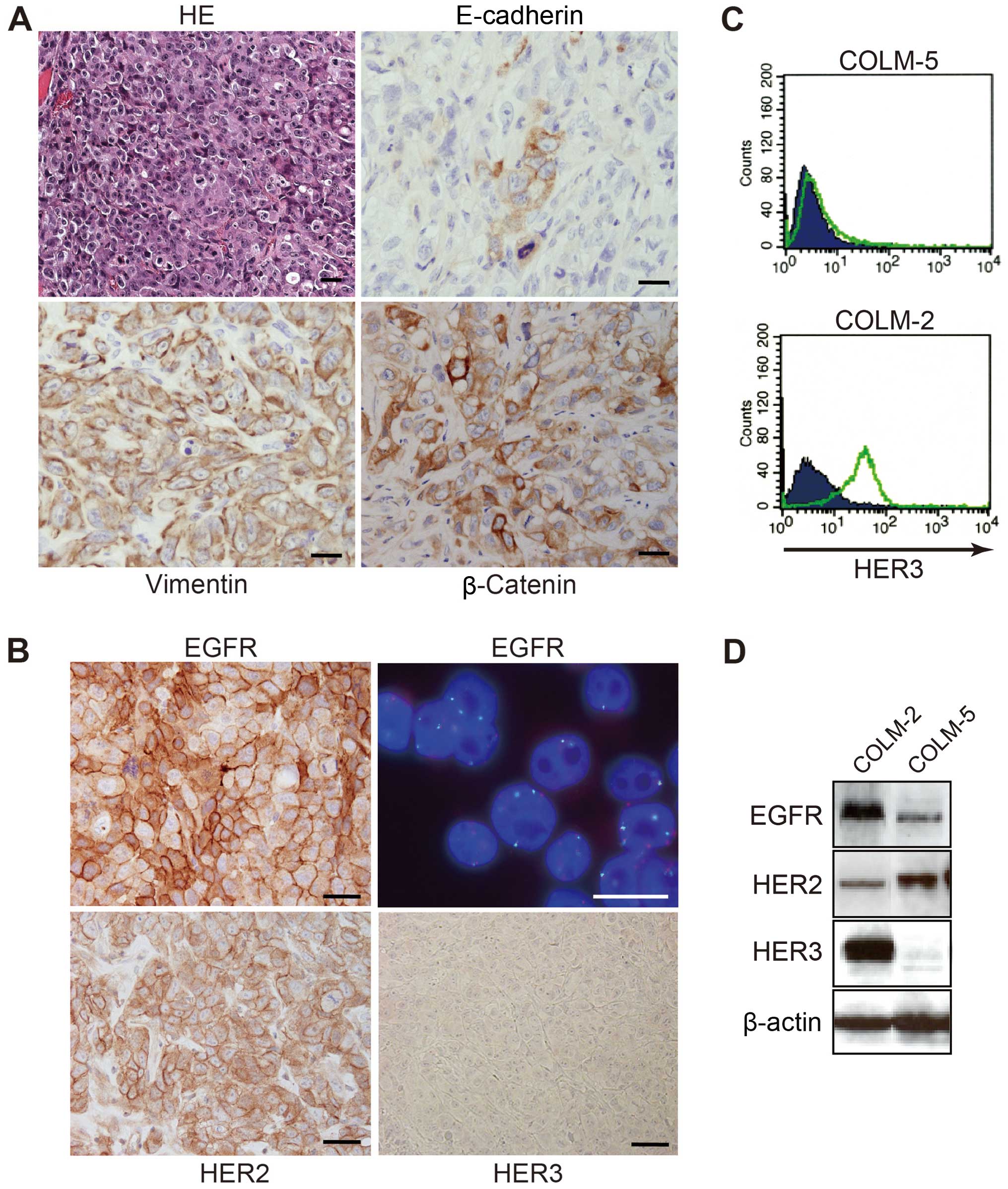
COLM-5 PD-CRC cells express EGFR and
HER2, but not HER3
COLM-5 cells established from a resected liver
metastasis of a PD-CRC patient formed subcutaneous tumors with
histological features of poorly-differentiated adenocarcinoma in
nude mice (Fig. 2A). COLM-5 tumors
were strongly stained for vimentin and slug, but still maintained
epithelial characteristics such as scant cytokeratin and E-cadherin
staining, indicating their feature of epithelial-mesenchymal
transition (EMT). COLM-5 tumors exhibited nuclear staining of P53
(data not shown), but not β-catenin (Fig. 2A) and no KRAS and
BRAF mutation was observed (data not shown). In contrast,
COLM-2 xenografts exhibited typical features of
moderately-differentiated adenocarcinoma with glandular formation,
which were strongly positive for cytokeratin and E-cadherin. The
COLM-2 cells showed nuclear accumulation of β-catenin like the
majority of WMD-CRC (data not shown).
Immunohistochemical analysis of transplanted tumors
in nude mice showed that the COLM-5 cells were strongly positive
(2+ and 3+) for both EGFR and HER2, respectively, based on the
membrane staining, but negative for HER3 on their surface. However,
FISH analysis demonstrated neither EGFR nor HER2 gene amplification
in COLM-5 cells (Fig. 2B). Flow
cytometric analysis confirmed that COLM-5 cells were deficient of
HER3 expression on their surface (Fig.
2C). Western blot analyses of the cultured cells further
confirmed overexpression of EGFR and HER2 and deficient HER3
expression of COLM-5 cells, whereas COLM-2 cells overexpressed
EGFR, HER2 and HER3 (Fig. 2D).
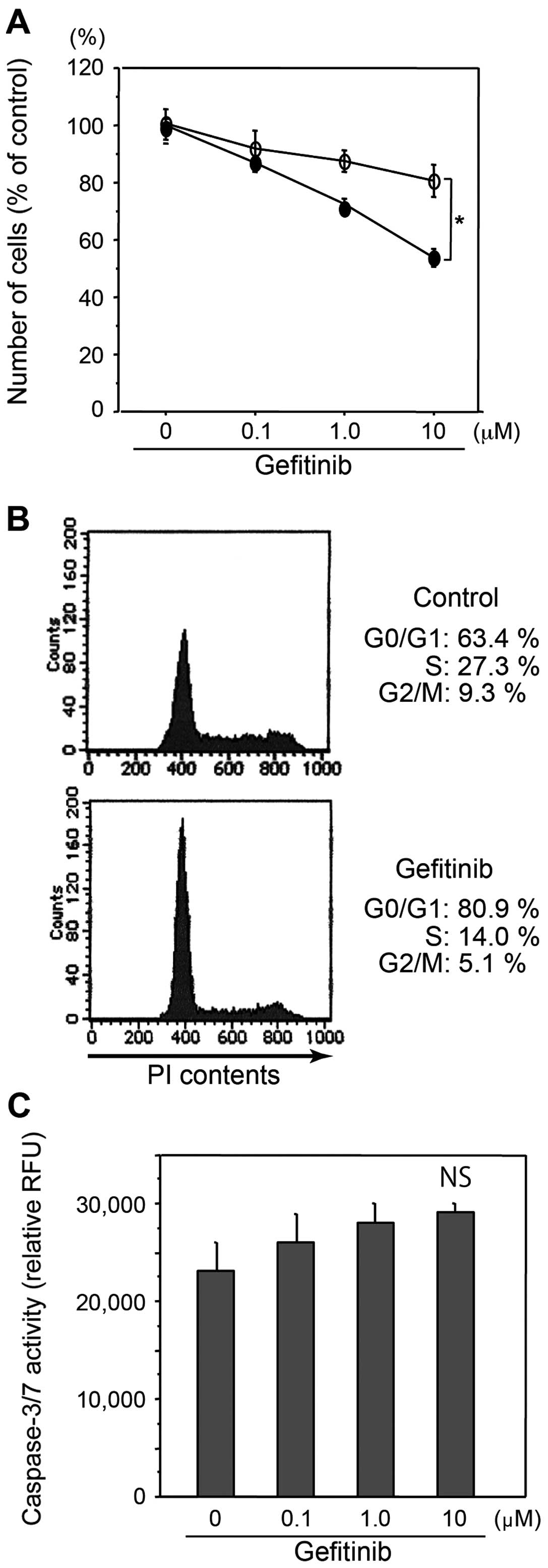
COLM-5 cells exhibit high sensitivity to
gefitinib
Gefitinib significantly inhibited growth of COLM-5
cells in vitro more strongly than COLM-2 cells (p<0.05)
(Fig. 3A). Cell cycle analysis by
flow cytometry revealed that gefitinib treatment decreased S phase
cells from 27.3 to 14.0% and increased G0–G1 phase cells from 63.4
to 80.9% in COLM-5 cells, indicating significant induction of G1
arrest of cell cycle (Fig. 3B). In
contrast, no G1 cell cycle arrest by gefitinib in COLM-2 cells was
observed (data not shown). Caspase-3/7 assay showed that
significant apoptosis was not induced by gefitinib in vitro
in COLM-5 or COLM-2 cells even at high concentrations (Fig. 3C).
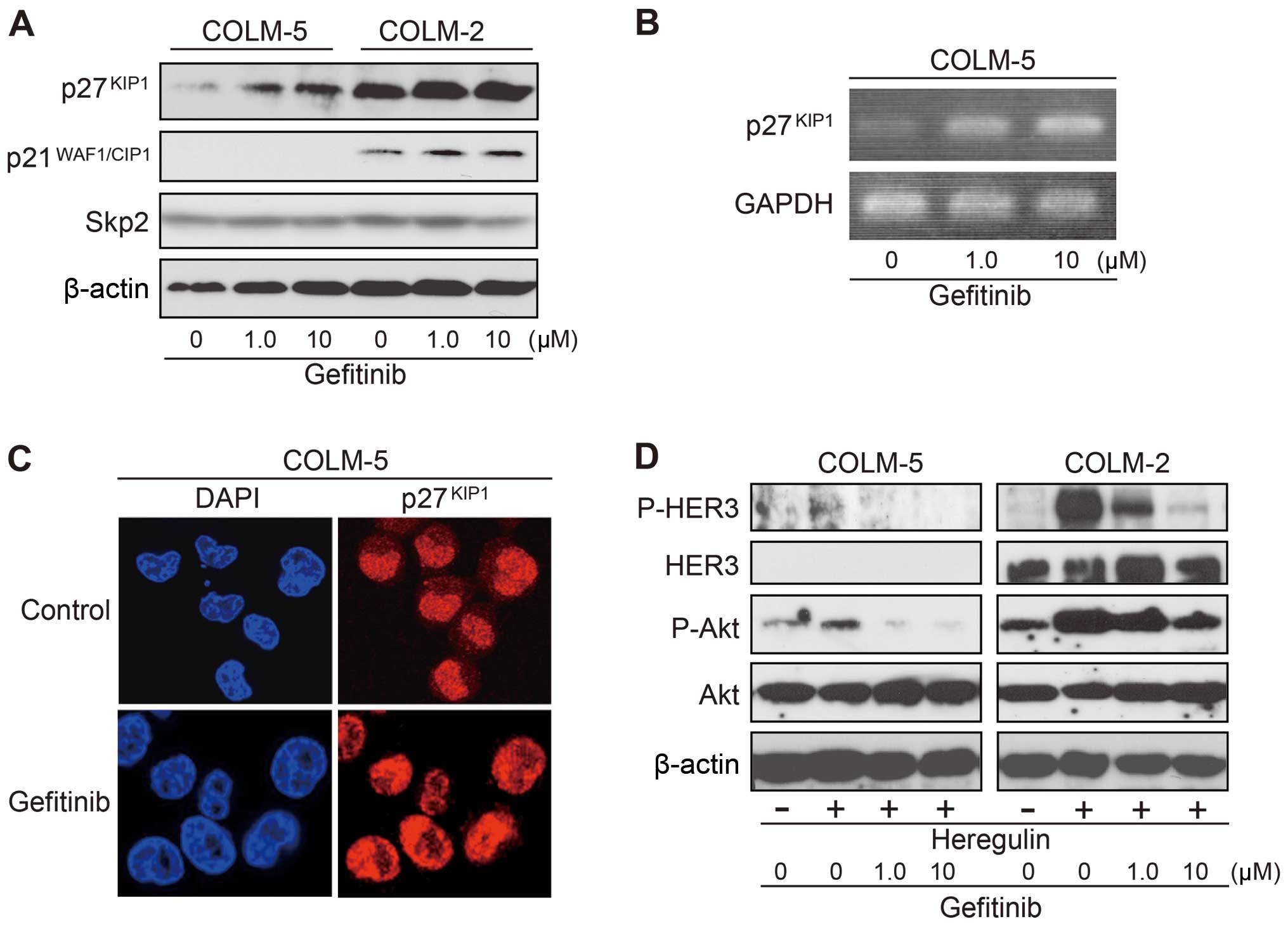
Gefitinib upregulates p27Kip1
and attenuates the HER3/PI3K/Akt signaling pathway in COLM-5
cells
To gain insights into molecular mechanisms
underlying the sensitivity to gefitinib of COLM-5 cells, we
examined the expression of p27Kip1 and
p21WAF1/CIP1. In COLM-5 cells, p27Kip1
protein was weakly expressed under basal condition, and
significantly increased by gefitinib treatment in a dose-dependent
manner (Fig. 4A). We confirmed
that gefitinib treatment upregulates p27Kip1 also at
mRNA level by RT-PCR analysis (Fig.
4B). In contrast, COLM-2 cells highly expressed both
p27Kip1 and p21WAF1/CIP1, but the expression
was unchanged after gefitinib treatment (Fig. 4A). No p21WAF1/CIP1
expression was detected in COLM-5 cells. Expression level of Skp2,
which regulates p27Kip1 expression at protein level
(28,29), did not change by gefitinib
treatment (Fig. 4A), suggesting
that p27Kip1 was induced at a transcriptional level. An
immunofluorescence analysis confirmed nuclear relocalization of
p27Kip1 from cytoplasm after gefitinib treatment
(Fig. 4C). Upon stimulation with
heregulin, increase in phosphorylation of HER3 and Akt was
detectable in COLM-5 cells, but much weaker than in COLM-2 cells.
The slight increase in phophorylation of Akt by heregulin was
almost completely reversed by gefitinib in COLM-5 cells, whereas
such a suppression of phophorylation of Akt by gefitinib was slight
in COLM-2 cells (Fig. 4D).
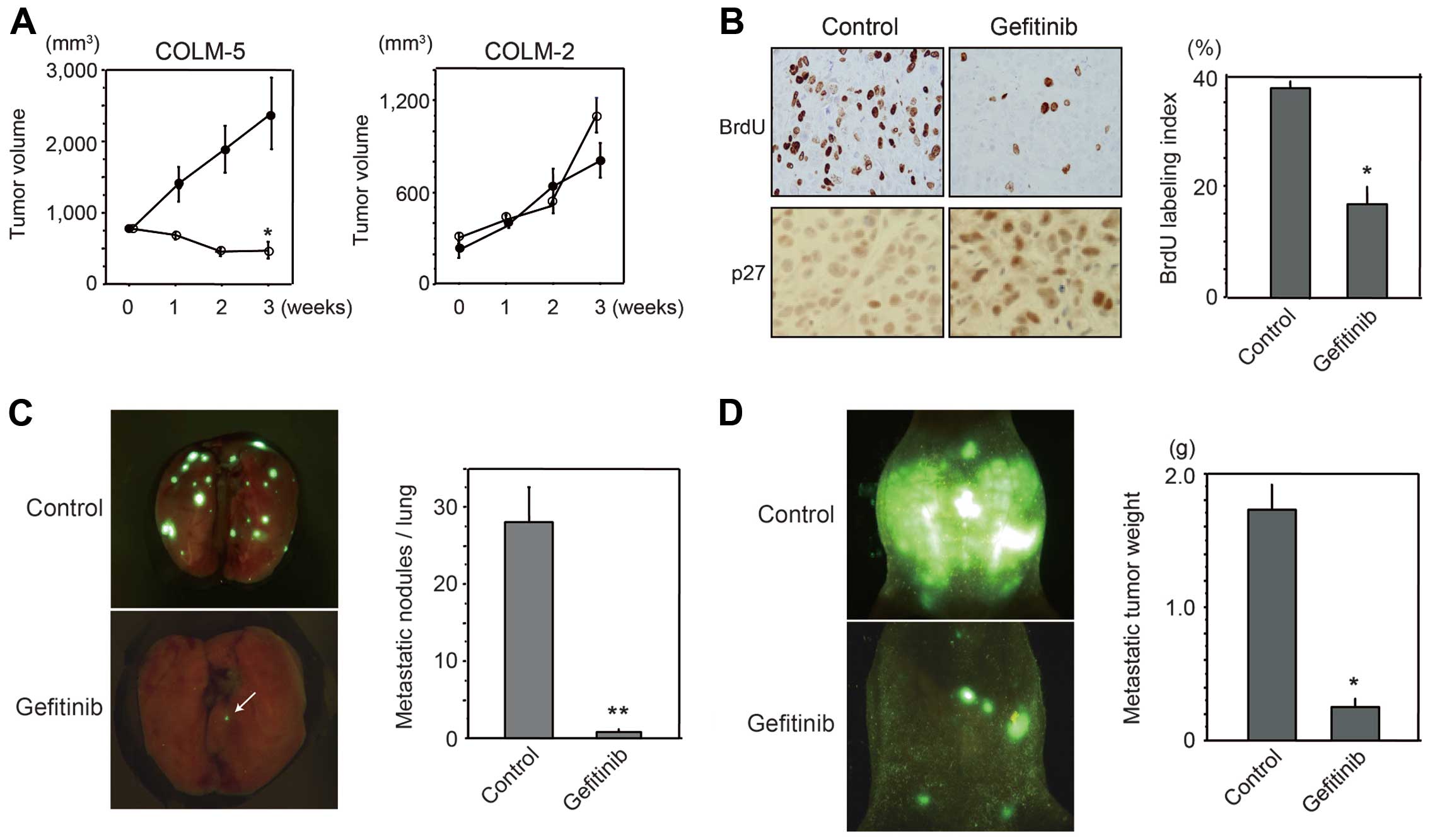
Gefitinib inhibits COLM-5 xenografts and
metastases in vivo
Antitumor effect of gefitinib in vivo was
examined using subcutaneous (sc) transplanted tumors in nude mice.
Treatment with gefitinib markedly suppressed sc tumor growth of
COLM-5 xenografts (p<0.01), but did not significantly suppress
COLM-2 xenografts (Fig. 5A). BrdU
incorporation was significantly reduced in gefitinib-treated COLM-5
tumors (Fig. 5B). Furthermore,
increase in nuclear accumulation of p27Kip1 protein
after gefitinib treatment for 3 weeks was observed in COLM-5 tumors
(Fig. 5B). Such remarkable
antitumor effects of gefitinib was not observed in COLM-2 sc tumors
in nude mice (data not shown). Furthermore, we demonstrated that
gefitinib markedly inhibited lung metastasis of COLM-5 cells by
more than 90% (p<0.001), and 2 of 5 mice tested became
metastasis-free after 4 weeks of gefitinib treatment (Fig. 5C). We also assessed the effects of
gefitinib using an experimental peritoneal dissemination model of
COLM-5 cells. In control mice, peritoneal metastases progressively
form peritoneal carcinomatosis with massive ascites within 2 months
following injection, whereas in gefitinib-treated mice, peritoneal
metastasis localized in limited areas such as the omentum and
mesentery without ascites formation (Fig. 5D). Metastatic tumor weight in the
peritoneal cavity was significantly decreased in gefitinib-treated
mice as compared with non-treatment control (p<0.01).
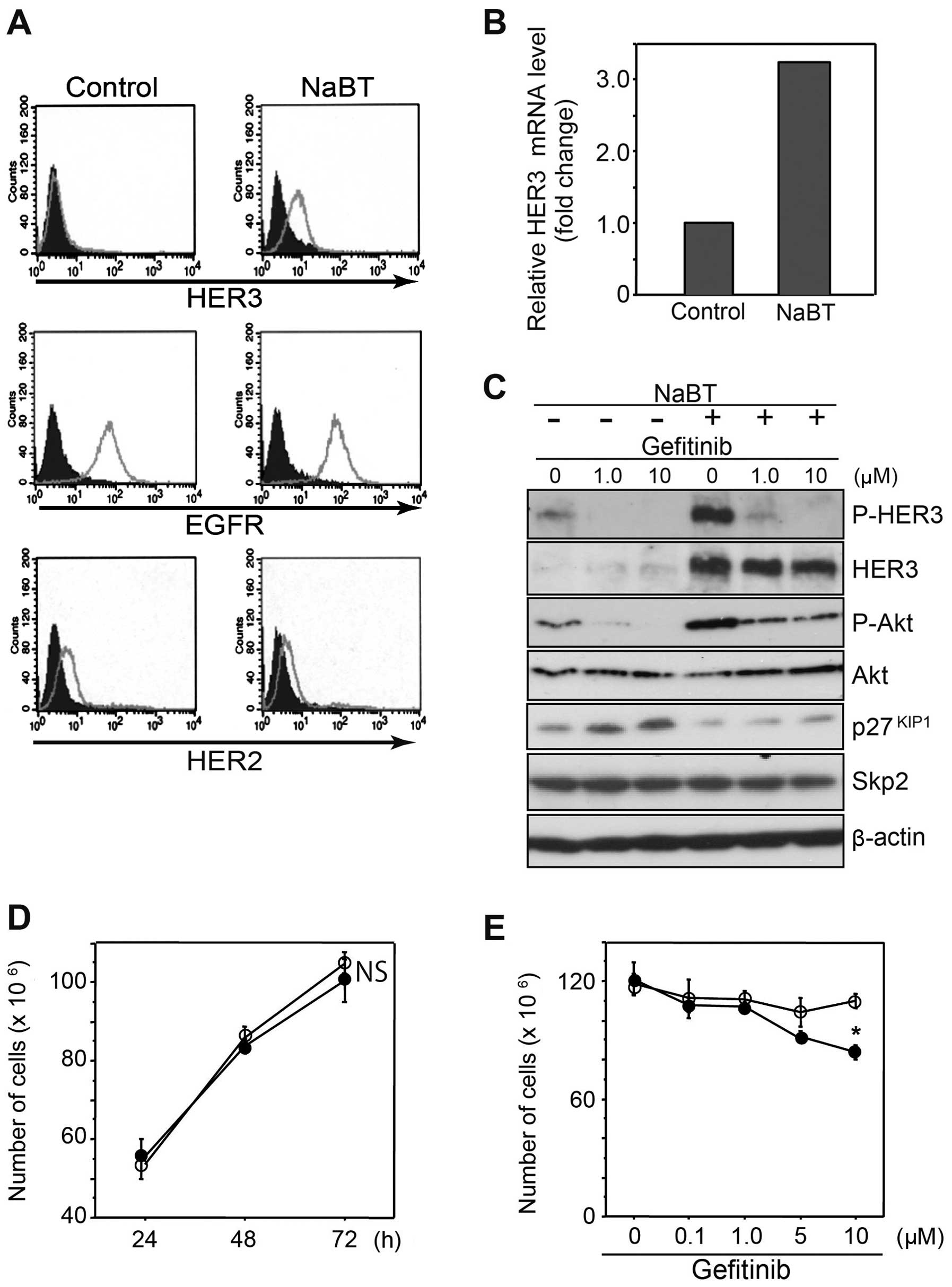
HDAC inhibitors upregulate HER3
expression in COLM-5 cells
Next we hypothesized that HER3 deficiency of COLM-5
cells might reflect their undifferentiated phenotype. To test this,
we treated COLM-5 cells with sodium butyrate (NaBT), an HDAC
inhibitor, an established inducer of differentiation. NaBT
treatment significantly induced expression of E-cadherin in COLM-5
cells, indicating enhancement of epithelial differentiation,
although MUC2 and villin were not significantly affected (data not
shown). A flow cytometric analysis demonstrated that NaBT increased
HER3 expression, but not EGFR and HER2 expression on the surface of
COLM-5 cells (Fig. 6A). HER3 mRNA
was also increased by NaBT treatment (Fig. 6B). A western blot analysis
confirmed significant upregulation of HER3, P-HER3 and P-Akt in
COLM-5 cells, indicating activation of HER3/PI3K/Akt signaling by
NaBT. Though gefitinib almost completely inhibited phosphorylation
of HER3 and Akt in the control, it failed to suppress NaBT-mediated
upregulation of P-Akt (Fig. 6C).
Interestingly, p27Kip1 expression was increased by
gefitinib treatment with concomitant decrease in P-Akt. Such a
gefitinib-mediated upregulation of p27Kip1 was reduced
with activation of HER3/Akt signaling by NaBT treatment (Fig. 6C), indicating the presence of a
HER3/PI3K/Akt-mediated negative regulatory mechanism of
p27Kip1 in COLM-5 cells. Treatment with NaBT at a lower
concentration (0.25 mM) for 6–12 h induced HER3 expression, without
phenotypic change such as growth retardation (Fig. 6D). Such an upregulation of HER3 by
NaBT resulted in significant restoration of gefitinib-mediated
growth inhibition in COLM-5 cells (p<0.05) (Fig. 6E).
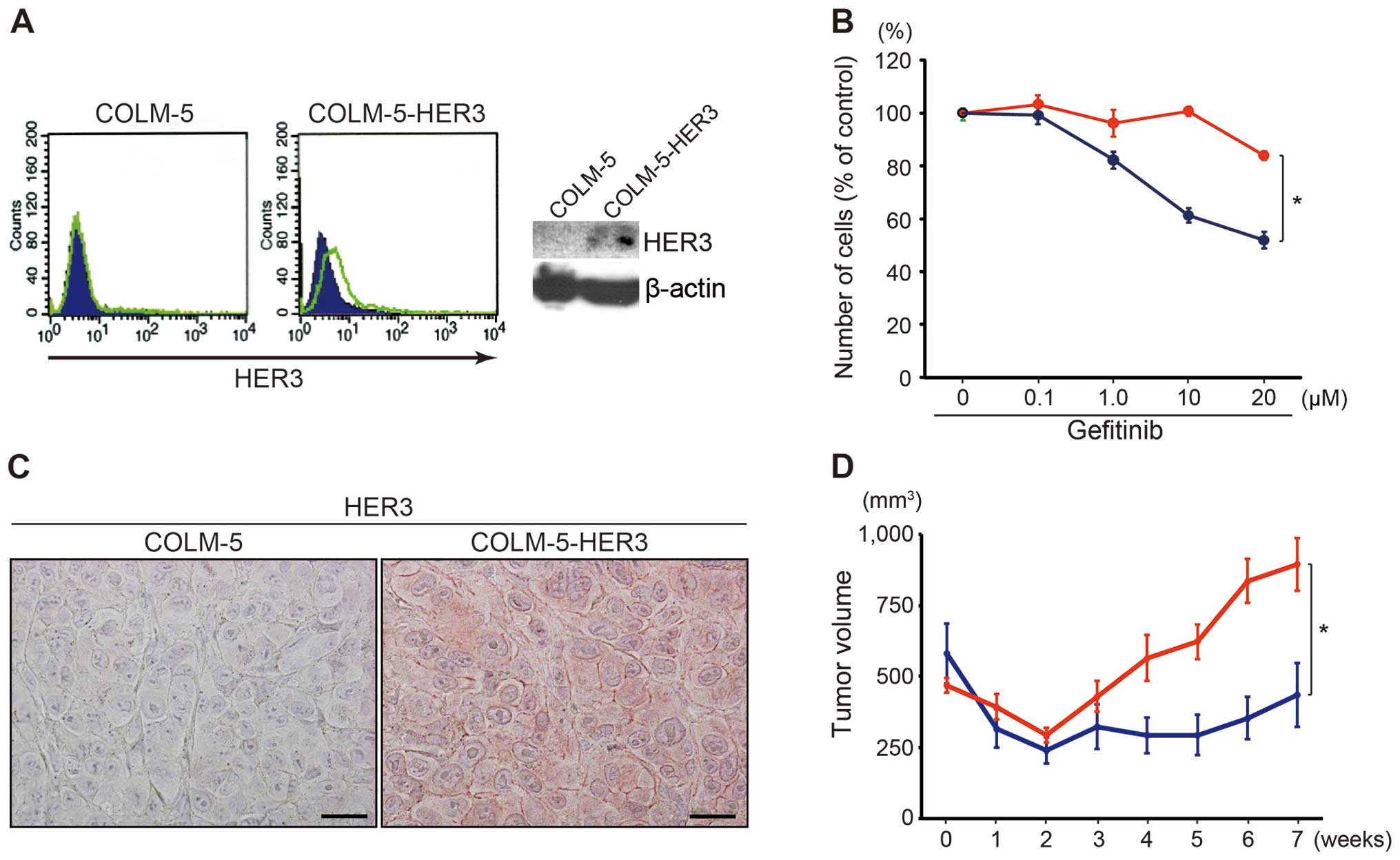
HER3 overexpression attenuates
gefitinib-sensitivity in COLM-5 cells
In order to test whether HER3 would affect
gefitinib-sensitivity, we established COLM-5 cells that stably
overexpress HER3. Increased expression of more than 4-fold median
intensity of HER3 protein on cellular surface was confirmed by FACS
analysis and also by western blot analysis (Fig. 7A). The forced expression of HER3
induced significant resistance toward gefitinib in COLM-5 cells
in vitro (Fig. 7B).
Furthermore, we examined efficacy of gefitinib for COLM-5
xenografts in mice. HER3 overexpression was also confirmed by an
immunohistochemical analysis of sc tumors in mice (Fig. 7C). Such an increased expression of
HER3 resulted in a significant induction of gefitinib-resistance of
the COLM-5 sc xenografts in mice (Fig.
7D).
Discussion
In the present study, we demonstrated that the
incidence of deficient HER3 expression in PD-CRC cases was much
higher than WMD-CRC, indicating that HER3 expression in CRC
correlates with their degree of histological differentiation. To
our knowledge, there are four immunohistochemical studies that
investigated HER3 expression in CRC tissues, but only one study
reported HER3 expression in PD-CRC cases, even in a limited number
of PD cases (n=8). They showed no HER3 expression (0/8) in PD-CRC
cases, supporting our present findings (30). Because of the relatively rare
incidence of PD-CRC, our present immunohistochemical study of HER3
expression focusing on PD-CRC (n=24) is, to our knowledge, the
first detailed analysis to be reported in this respect. The other
three studies did not report HER3 expression of CRC according to
their histological differentiation. Two studies reported the
relationship between HER3 overexpression and patient’s outcome, but
there is some controversy. One study showed HER3 overexpression was
associated with a poor outcome of CRC patients (31), whereas two other studies including
a recent meta-analysis in 12 studies showed that HER3
overexpression could not be linked with poor prognosis in CRC,
despite association with short survival in patients with several
types of solid tumors, including breast, ovarian cancers and
melanoma (32,33). The reason for this discrepancy
between studies may be primarily due to the difference in the
antibody used in these studies. HER3 staining pattern differed from
cytoplasmic predominant staining to partially membranous staining
and the positivity rate varies greatly from 17 to 70% in colorectal
cancers depending on the antibody used. A problem with HER3
immunohistochemistry is that the protocol has not been standardized
internationally between the study groups, unlike
immunohistochemical staining of EGFR and HER2. In this respect, the
HER3 antibody used in our study was selected in terms of membrane
staining among several commercially available antibodies and
therefore, has a rationale for investigating functional aspects of
HER3 expression by immunohistochemistry.
To investigate the effect of HER3 in the molecular
targeting therapy against PD-CRC, we used a metastatic cell line
(COLM-5) from a PD-CRC patient established in our laboratory
without artificial gene transfection. To date, PD cell lines
available are only a few such as RKO and SW620 cells. To our
knowledge, COLM-5 is the first PD cell line showing high
sensitivity to EGFR targeting drugs such as cetuximab and
gefitinib. COLM-5 cells have the following unique characteristics:
i) they exhibit no β-catenin nuclear accumulation, unlike the
majority of CRC, indicating their process of carcinogenesis is
independent of constitutive activation of Wnt/β-catenin signaling;
ii) COLM-5 cells show significant HER family expression (score 2+)
such as EGFR and HER2, but no expression of HER3 (score 0). iii)
COLM-5 cells strongly express vimentin and Slug and show high
invasive capability, but some epithelial characteristics still
remain such as E-cadherin and CK20 expression in a limited cell
population, indicating an EMT-like feature. iv) COLM-5 cells have a
high metastatic potential to multiple organs such as lung,
peritoneal cavity and lymph nodes in xenograft models. These
features are in sharp contrast to a typical differentiated-type CRC
cell line (COLM-2), which shows activation of Wnt/β-catenin
signaling and gefitinib-resistance and is non-metastatic. Analysis
of clinical samples as described above shows that a subset of PD
with EGFR+/HER2+/HER3− phenotype
like a COLM-5 cell line accounts for approximately 37% and is not
uncommon in clinical PD cases, suggesting the COLM-5 cell line does
not represent only a very rare CRC case, but reflects a relatively
common PD-CRC subset.
COLM-5 cells showed high sensitivity to gefitinib
when assessed both in vitro and in vivo. In fact, a
subcutaneous tumor cannot substantially grow beyond initial tumor
size by the treatment, and lung and peritoneal metastases were
abolished by the early onset treatment with gefitinib. The COLM-5
cell line is also very sensitive to cetuximab, a chimeric
monoclonal antibody to EGFR (data not shown). Concerning the
mechanism underlying high gefitinib sensitivity of COLM-5 cells, we
found that growth inhibition of COLM-5 cells by gefitinib was
primarily due to the cell cycle arrest at G1 phase rather than
induction of apoptosis. G1 arrest of the cell cycle is induced by
p21WAF1/CIP1 and p27Kip1, the members of the
Cip/Kip family of cyclin-dependent kinase inhibitors, which
suppress CDK2, thereby inhibiting phosphorylation of Rb protein
(34). In head and neck squamous
cancer cells, it is shown that gefitinib induces both
p21WAF1/CIP1 and p27Kip1, accompanied by G1
cell cycle arrest (35). In the
present study, however, we found that cell cycle arrest by
gefitinib in COLM-5 cells was mainly mediated by the upregulation
of p27Kip1, but not by p21WAF1/CIP1. The
latter p21WAF1/CIP1, a main target of P53, was not
expressed at all in COLM-5 cells, possibly due to the lack of
induction by the mutated P53 (36). We found a significant increase in
p27Kip1 mRNA expression after gefitinib treatment.
Furthermore, we observed increased nuclear relocalization of
p27Kip1 from the cytoplasm after gefitinib treatment,
which possibly results from a decrease in phosphorylation of
p27Kip1 at Thr157 by Akt. These results strongly suggest
that both transcriptional upregulation and nuclear relocalization
of p27Kip1 through blockade of PI3K/Akt pathway are
responsible for G1 cell cycle arrest and therefore gefitinib
sensitivity of COLM-5 cells.
HER3 is known to be a key player regulating signal
transduction upstream to PI3K/Akt pathways (22). In fact, with immunoprecipitation
experiments, we confirmed enhancement of HER2/HER3 heterodimer
formation, phosphorylation of HER3, and association of p85
regulatory subunit of PI3K within the dimer in response to
stimulation with heregulin ligand in HER3 overexpressing COLM-2
cells, but such dimer formations were not evident in HER3-deficient
COLM-5 cells (data not shown). Furthermore, we found that
HER3/PI3K/Akt signaling can be modestly activated only when
stimulated with ligand and could be completely suppressed by the
gefitinib in COLM-5 cells. In contrast, HER3/PI3K/Akt signaling was
markedly activated by the ligand stimulation and therefore was only
partially inhibited by gefitinib in COLM-2 cells. Upregulation of
p27Kip1 by gefitinib was found to closely correlate with
such an abolishment of phosphorylation of HER3 and downstream Akt,
suggesting that p27Kip1 expression is tightly, and
negatively regulated by HER3/PI3K/Akt signaling in COLM-5 cells.
These results suggest that deficient HER3 expression plays an
important role in the high gefitinib sensitivity of COLM-5
cells.
The upregulation of HER3, but not EGFR and HER2 in
COLM-5 cells by sodium butyrate, an HDAC inhibitor, suppressed
gefitinib-mediated p27Kip1 upregulation and resulted in
reduced gefitinib sensitivity. These findings further support the
idea that HER3-deficiency is responsible for the high gefitinib
sensitivity of COLM-5 cells by enhancing gefitinib-mediated
blockade of Akt phosphorylation and subsequent upregulation of
p27Kip1. In contrast, HER3 overexpression in COLM-2
cells might circumvent inhibitory activity of gefitinib by the
constitutive activation of PI3K/Akt pathway, which results in
impaired upregulation of p27Kip1. Consistent with this,
we confirmed that forced expression of HER3 in COLM-5 cells by the
transfection attenuated gefitinib sensitivity both in vivo
and in vitro, confirming a direct regulatory effect of HER3
in gefitinib sensitivity of PD-CRC cells. These results are in line
with the recently reported findings that siRNA- and
antibody-mediated blockade of HER3 reduced proliferation and
induced apoptosis in established colon cancer cell lines (31).
In conclusion, we showed a high incidence of
deficient HER3 expression in PD-CRC cases, and by using a highly
metastatic PD-CRC cell line with high sensitivity to gefitinib, we
demonstrated for the first time that HER3-deficiency conferred
gefitinib sensitivity by enhancing P27Kip upregulation
and subsequent G1 cell cycle arrest through inactivation of
PI3K/Akt signaling. To date, molecular targeting therapy for PD
remains an essentially unresolved area in CRC. The present
preclinical study together with analysis of clinical specimens cast
new insight into molecular targeting therapy against a particular
type of CRC and raised the possibility that a malignant subset of
PD with EGFR+/HER2+/HER3−
phenotype may be a promising new candidate as a therapeutic target
for EGFR targeting drugs.
Acknowledgements
This study was supported in part by a grant from the
Ministry of Health, Labor and Welfare, Japan and the Ministry of
Education, Science, Sports, Culture and Technology.
References
|
1
|
Hamilton SR, Rubio CA, Vogelstein B, et
al: Carcinoma of the colon and rectum. World Health Organization
Classification of Tumours. Pathology and Genetics of Tumours of the
Digestive System. Hamilton SR and Aaltonen LA: IARC Press; Lyon:
pp. 105–119. 2000
|
|
2
|
Fearon ER and Vogelstein B: A genetic
model for colorectal tumorigenesis. Cell. 61:759–767. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fodde R, Smits R and Clevers H: APC,
signal transduction and genetic instability in colorectal cancer.
Nat Rev Cancer. 1:55–67. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Arai T, Takubo K, Sawabe M and Esaki Y:
Pathologic characteristics of colorectal cancer in the elderly: a
retrospective study of 947 surgical cases. J Clin Gastroenterol.
31:67–72. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kazama Y, Watanabe T, Kanazawa T, Tanaka
J, Tanaka T and Nagawa H: Poorly differentiated colorectal
adenocarcinomas show higher rates of microsatellite instability and
promoter methylation of p16 and hMLH1: a study matched for T
classification and tumor location. J Surg Oncol. 97:278–283. 2008.
View Article : Google Scholar
|
|
6
|
Xiao H, Yoon YS, Hong SM, Roh SA, Cho DH,
Yu CS and Kim JC: Poorly differentiated colorectal cancers:
correlation of microsatellite instability with clinicopathologic
features and survival. Am J Clin Pathol. 140:341–347. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hinoi T, Tani M, Lucas PC, et al: Loss of
CDX2 expression and microsatellite instability are prominent
features of large cell minimally differentiated carcinomas of the
colon. Am J Pathol. 159:2239–2248. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chung CK, Zaino RJ and Stryker JA:
Colorectal carcinoma: evaluation of histologic grade and factors
influencing prognosis. J Surg Oncol. 21:143–148. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kawabata Y, Tomita N, Monden T, et al:
Molecular characteristics of poorly differentiated adenocarcinoma
and signet-ring-cell carcinoma of colorectum. Int J Cancer.
84:33–38. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim JH, Rhee YY, Bae JM, Cho NY and Kang
GH: Loss of CDX2/CK20 expression is associated with poorly
differentiated carcinoma, the CpG island methylator phenotype, and
adverse prognosis in microsatellite-unstable colorectal cancer. Am
J Surg Pathol. 37:1532–1541. 2013. View Article : Google Scholar
|
|
11
|
Seshimo I, Yamamoto H, Mishima H, et al:
Expression and mutation of SMAD4 in poorly differentiated carcinoma
and signet-ring cell carcinoma of the colorectum. J Exp Clin Cancer
Res. 25:433–442. 2006.PubMed/NCBI
|
|
12
|
Rad R, Cadiñanos J, Rad L, et al: A
genetic progression model of Braf (V600E)-induced intestinal
tumorigenesis reveals targets for therapeutic intervention. Cancer
Cell. 24:15–29. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Coffee EM, Faber AC, Roper J, et al:
Concomitant BRAF and PI3K/mTOR blockade is required for effective
treatment of BRAF (V600E) colorectal cancer. Clin Cancer Res.
19:2688–2698. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hynes NE and Lane HA: ERBB receptors and
cancer: the complexity of targeted inhibitors. Nat Rev Cancer.
5:341–354. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Oshima Y, Tanaka H, Murakami H, Ito Y,
Furuya T, Kondo E, Kodera Y and Nakanishi H: Lapatinib sensitivity
of two novel trastuzumab-resistant HER2 gene-amplified gastric
cancer cell lines. Gastric Cancer. 17:450–462. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mayer A, Takimoto M, Fritz E, Schellander
G, Kofler K and Ludwig H: The prognostic significance of
proliferating cell nuclear antigen, epidermal growth factor
receptor, and mdr gene expression in colorectal cancer. Cancer.
71:2454–2460. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cunningham D, Humblet Y, Siena S, et al:
Cetuximab monotherapy and cetuximab plus irinotecan in
irinotecan-refractory metastatic colorectal cancer. N Engl J Med.
351:337–345. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chung KY, Shia J, Kemeny NE, et al:
Cetuximab shows activity in colorectal cancer patients with tumors
that do not express the epidermal growth factor receptor by
immunohistochemistry. J Clin Oncol. 23:1803–1810. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Moroni M and Veronese S: Gene copy number
for epidermal growth factor receptor (EGFR) and clinical response
to anti-EGFR treatment in colorectal cancer. A cohort study. Lancet
Oncol. 6:279–286. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lievre A, Bachet JB, Le Corre D, et al:
KRAS mutation is predictive of response to cetuximab therapy in
colorectal cancer. Cancer Res. 66:3992–3995. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yokota T, Ura T, Shibata N, et al: BRAF
mutation is a powerful prognostic factor in advanced and recurrent
colorectal cancer. Br J Cancer. 104:856–862. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Menendez JA and Lupu R:
Transphosphorylation of kinase-dead HER3 and breast cancer
progression: a new standpoint or an old concept revisited? Breast
Cancer Res. 9:1112007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sergina NV, Rausch M, Wang D, Blair J,
Hann B, Shokat KM and Moasser MM: Escape from HER family tyrosine
kinase inhibitor therapy by the kinase inactive HER3. Nature.
445:437–441. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yakes FM, Chinratanalab W, Ritter CA, King
W, Seelig S and Arteaga CL: Herceptin-induced inhibition of
phosphatidylinositol- 3 kinase and Akt is required for
antibody-mediated effects on p27, cyclin D1, and antitumor action.
Cancer Res. 62:4132–4141. 2002.PubMed/NCBI
|
|
25
|
Maurer CA, Friess H, Kretschmann B, et al:
Increased expression of erbB3 in colorectal cancer is associated
with concomitant increase in the level of erbB2. Hum Pathol.
29:771–777. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ito Y, Nakanishi H, Kodera Y, Hirai T,
Nakao A and Kato T: Characterization of a novel lymph node
metastasis model from human colonic cancer and its preclinical use
for comparison of anti-metastatic efficacy between oral S-1 and
UFT/LV. Cancer Sci. 101:1853–1860. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yokoyama H, Nakanishi H, Kodera Y, et al:
Biological significance of isolated tumor cells and micrometastasis
in lymph nodes evaluated using a green fluorescent protein
(GFP)-tagged human gastric cancer cell line. Clin Cancer Res.
12:361–368. 2006. View Article : Google Scholar
|
|
28
|
Carrano AC, Eytan E, Hershko A and Pagano
M: Skp2 is required for ubiquitin-mediated degradation of the CDK
inhibitor P27. Nat Cell Biol. 3:193–199. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pagano M: Control of DNA synthesis and
mitosis by the Skp2-p27-Cdk1/2 axis. Mol Cell. 14:414–416. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kountourakis P, Pavlakis K, Psyrri A, et
al: Prognostic significance of HER3 and HER4 protein expression in
colorectal adenocarcinomas. BMC Cancer. 6:462006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Beji A, Horst D, Engel J, et al: Toward
the prognostic significance and therapeutic potential of HER3
receptor tyrosine kinase in human colon cancer. Clin Cancer Res.
18:956–968. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ocana A, Vera-Badillo F, Seruga B,
Templeton A, Pandiella A and Amir E: HER3 overexpression and
survival in solid tumors: A meta-analysis. J Natl Cancer Inst.
105:266–273. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Baiocchi G, Lopes A, Coudry RA, et al:
ErbB family immunohistochemical expression in colorectal cancer
patients with higher risk of recurrence after radical surgery. Int
J Colorectal Dis. 24:1059–1068. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Coqueret O: New roles for p21 and p27
cell-cycle inhibitors: a function for each cell compartment? Trends
Cell Biol. 13:65–70. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Di Gennaro E, Barbarino M, Bruzzese F, et
al: Critical role of both p27KIP1 and
p21CIP1/WAF1 in the antiproliferative effect of ZD1839
(‘Iressa’), an epidermal growth factor receptor tyrosine kinase
inhibitor, in head and neck squamous carcinoma cells. J Cell
Physiol. 195:139–150. 2003.
|
|
36
|
Varna M, Lehmann-Che J, Turpin E, et al:
p53 dependent cell-cycle arrest triggered by chemotherapy in
xenografted breast tumors. Int J Cancer. 124:991–997. 2009.
View Article : Google Scholar : PubMed/NCBI
|