Introduction
Hepatocellular carcinoma (HCC), the most common
primary liver tumor, is the fifth most common human malignancy and
the second leading cause of cancer death (695,900 deaths annually)
in men worldwide (1). Owing to
difficulty in early diagnosis and a lack of effective chemotherapy
and radiotherapy, the 5-year overall survival rate remains poor for
patients with advanced HCC (2–4).
Changes in growth factors, receptors and their downstream signaling
pathway components influence HCC development and malignancy. The
signaling pathways involved include IGF-IGF1R, hepatocyte growth
factor (HGF/MET), EGF-EGFR and TGFβ-TβR signaling (5). Ligands bind to their corresponding
receptors, which are usually overexpressed and/or dysregulated in
HCC, and mainly activate the PI3K-PKB (AKT) and MAPK pathways,
which affect tumor cell phenotypes (5–8). IGF
secreted by liver cells acts in an autocrine and/or paracrine
manner by binding to IGF1R (9).
This in turn initiates downstream cellular effectors including
insulin-receptor substrates and Src homology domain-containing
kinases, ultimately contributing to proliferation, anti-apoptosis,
invasiveness and metastasis (5,10–13).
Growth factors that are dysregulated in tumorigenesis and cancer
progression represent rational targets for HCC therapies.
Nevertheless, the precise oncogenic mechanisms by which growth
factors affect the transcriptional regulation of certain tumor
suppressor genes (TSGs) are unclear.
Epigenetic mechanisms play an important role in
driving tumorigenesis. DNA methylation catalyzed by DNA
methyltransferases (DNMTs), causes changes in chromatin structure,
DNA conformation and stability and patterns of DNA-protein
interaction. DNA hypermethylation leads to transcriptional
silencing of a considerable number of TSGs; thus, hypermethylation
has been proposed as the second hit in a model of TSG inactivation
(14). Overexpression of DNMTs has
been observed in various types of cancers (15–20),
and is significantly correlated with poor histological
differentiation and prognosis. These findings suggest that aberrant
DNA hypermethylation drives hepatocarcinogenesis. It is recognized
that many risk factors for HCC may upregulate DNMTs expression,
such as HBV-encoded protein X (21). The tobacco-specific carcinogen NNK
(nicotine-derived nitrosamine ketone) induces DNMT1 protein
upregulation and binding to the promoters of various TSGs, which
ultimately leads to tumorigenesis and poor prognosis in lung cancer
(22). These findings shed light
on the relevance of the expression of DNMTs in carcinogenesis.
However, there is little evidence on the delicate mechanism of
DNMTs overexpression in HCC. The findings presented here highlight
the mechanistic significance of aberrant DNMT1 expression induced
by IGF1 in HCC.
Materials and methods
Cell culture
HCC cell lines (HepG2, BEL-7402 and SMMC-7721) were
obtained from the Type Culture Collection of the Chinese Academy of
Sciences (Shanghai, China). The HL-7702 immortalized normal human
liver cell line was obtained from the China Center for Type Culture
Collection (Wuhan, China). Cell lines were cultured in Dulbecco’s
modified Eagle’s medium (DMEM; Gibco) or RPMI-1640 medium (Gibco)
supplemented with 10% fetal bovine serum (FBS; HyClone) and 1%
penicillin-streptomycin.
Quantitative RT-PCR (qRT-PCR)
qRT-PCR was performed as previously described
(23) using an ABI PRISM 7300
detection system. qRT-PCR reactions were performed using SYBR-Green
(Roche) technology and were repeated at least three times.
Flow cytometry and cell cycle
synchronization assays
HepG2 cells were seeded in DMEM (0.1% FBS) medium at
a density of 5×105cells/35-mm dish. The cells were
treated with DMSO or 15 μM 5-aza-2-deoxycytidine (5′-Aza;
Sigma-Aldrich) in time-course experiments. Cell suspensions in PBS
were prepared for analysis using a FlowCellect Bivariate Cell Cycle
kit (Millipore) according to the manufacturer’s protocol. At
various time-points after release, cells were pulsed with 10 μM
BrdU for 2 h immediately before harvesting and fixation. Cell
pellets were processed for double staining with an anti-BrdU
antibody (Millipore) and propidium iodide (PI, 10 μg/ml;
Millipore), followed by analysis on a flow cytometer (Partec CyFlow
Space). Gating was performed to focus on G1, S and G2/M
populations.
In vivo tumorigenicity assays and 5′-Aza
treatment
Female athymic nude mice (Vital River Animal Center,
Beijing, China) were housed in a temperature-controlled sterile
room in which humidity and light were carefully controlled and
monitored. Animal welfare and experimental procedures were in
strict accordance with the regulations approved by the
Institutional Animal Care Committee of the Medical College at
Xiamen University. HepG2 cells (5×106) were injected
subcutaneously into the flank of 6-week-old nude mice. When the
tumor size reached 0.5 cm3, the mice were randomly
divided into two groups and subjected to the following treatments:
i) i.p. injection of PBS as a control; ii) 1 mg/kg 5′-Aza was given
by i.p. injection every 2 days over a treatment time of 14 days
(seven injections). Tumor volume was calculated as volume =
width2 × length × 0.52 mm3.
ChIP-on-chip and target gene
verification
The protocol provided by NimbleGen was followed. In
brief, HepG2 cells were grown, cross-linked with formaldehyde and
sheared by sonication. Antibodies (DNMT1, DNMT3B and IgG; Santa
Cruz Biotechnology) were used for immunoprecipitation. A HG18
RefSeq Promoter microarray (NimbleGen) was used for hybridization
and analysis according to the manufacturer’s instructions. Data
extraction was performed using NimbleScan version 2.5 and
significant hybridization peaks (FDR <0.05) were identified. For
target gene verification we used real-time PCR and a chromatin
immunoprecipitation (ChIP) assay.
ChIP and DNA methylation assay
The two experiments were performed as previously
described with slight modification (24,25).
Statistical analysis
SPSS 15.0 software (SPSS, Inc., Chicago, IL, USA)
was used for all statistical analyses. Data from cell growth and
mouse tumorigenesis experiments were analyzed by one-way ANOVA. To
analyze differences in DNMT1 protein levels among various cell
treatments, a two-tailed Student’s t-test was used. Survival curves
were calculated by the Kaplan-Meier method, and comparison was
performed using a log-rank test. Results for parametric variables
are expressed as mean ± SE. In all cases, P<0.05 was considered
to indicate a statistically significant result.
Results
Identification of target genes of DNMT1
and DNMT3B
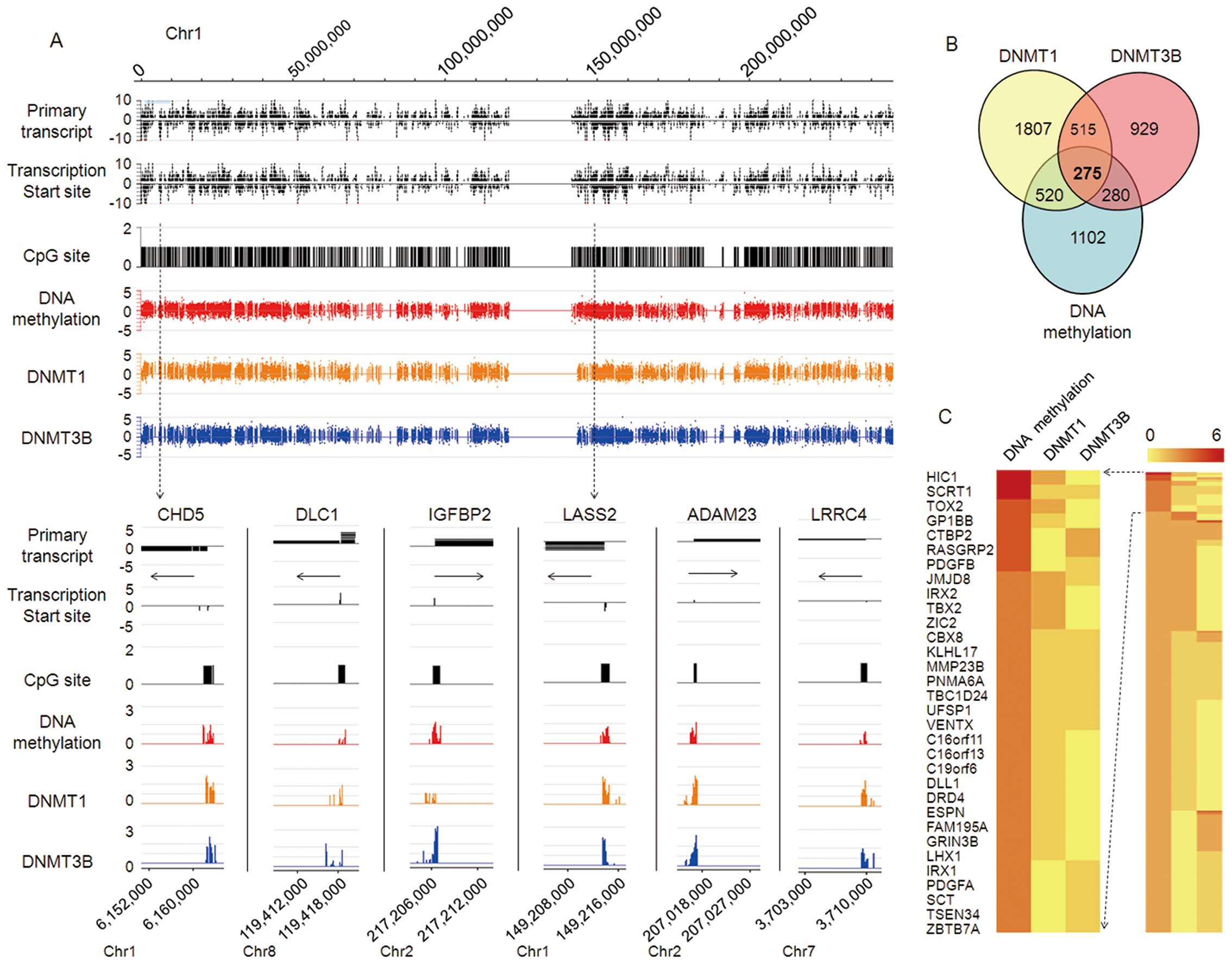
To screen for genomic DNA methylation profiles and
potential target genes in HCC, we carried out ChIP-on-chip
coupled with CpG island microarray (MCAM) analysis in HepG2 cells.
Extensive genome-wide DNA methylation of CpG islands of target
genes was determined, with high co-localization of DNMT1 and
DNMT3B; results for chromosome 1 are shown in Fig. 1A. We identified 2,177 genes with
promoter hypermethylation in HepG2 cells, and 3,117 and 1,999 genes
affected by DNMT1 and DNMT3B, respectively. Of these, 520 and 280
genes shared a methylation chip with DNMT1-ChIP or DNMT3B-ChIP,
respectively. Among all three groups, 275 genes were screened out
(Fig. 1B). Consistent with the
above results, unsupervised hierarchical clustering analysis
demonstrated that the top 32 genes with promoter hypermethylation
in ChIP-on-chip results are closely related to tumor
development and progression (Fig.
1C). However, we also found that DNA methylation of some gene
sites does not rely on DNMT1 or DNMT3B (data no shown), implying
the potential DNA methylation role of other DNMTs in HCC, such as
DNMT3A; this will be the subject of further investigation.
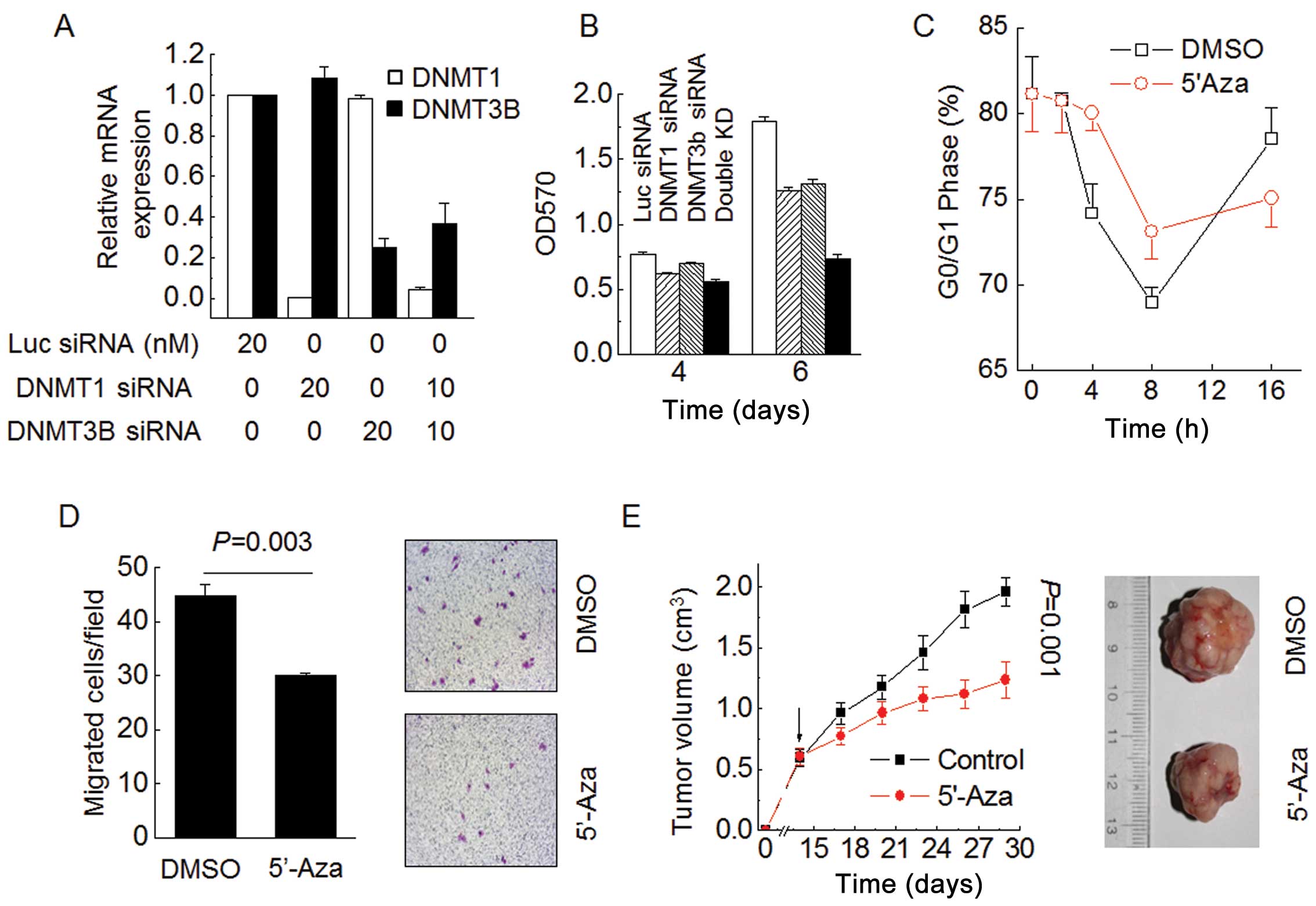
Knockdown of DNMTs inhibit HCC malignant
phenotype
We focused on the functional effect of DNMTs in
controlling HCC malignant phenotype. MTT assay results show that
transient inhibition of either DNMT1 or DNMT3B by siRNA was
sufficient to reduce HepG2 cell proliferation (Fig. 2A and B). Strikingly, both siRNAs
cooperatively suppressed HepG2 cell proliferation after 6 days and
had a more pronounced effect than either siRNA alone (Fig. 2B). Time-course cell cycle analysis
revealed that 5′-Aza inhibited HepG2 cell cycle progress from G1
phase to S phase (Fig. 2C).
Moreover, 5′-Aza treatment inhibited HepG2 cell migration (Fig. 2D; P=0.003). To delineate the role
of DNA methylation in HCC growth in vivo, we performed
xenograft experiments in which HepG2 cells were injected
subcutaneously into immunodeficient nude mice. The tumor growth
rate significantly decreased after treatment with 5′-Aza compared
to control animals. The average tumor volume was 1.96
cm3 in the control group on day 28 compared to only 1.23
cm3 in the group treated with 5′-Aza (Fig. 2E; P=0.001). These results argue
that DNMTs cooperatively mediate DNA methylation and broadly
participate in controlling the malignant phenotype of HCC
cells.
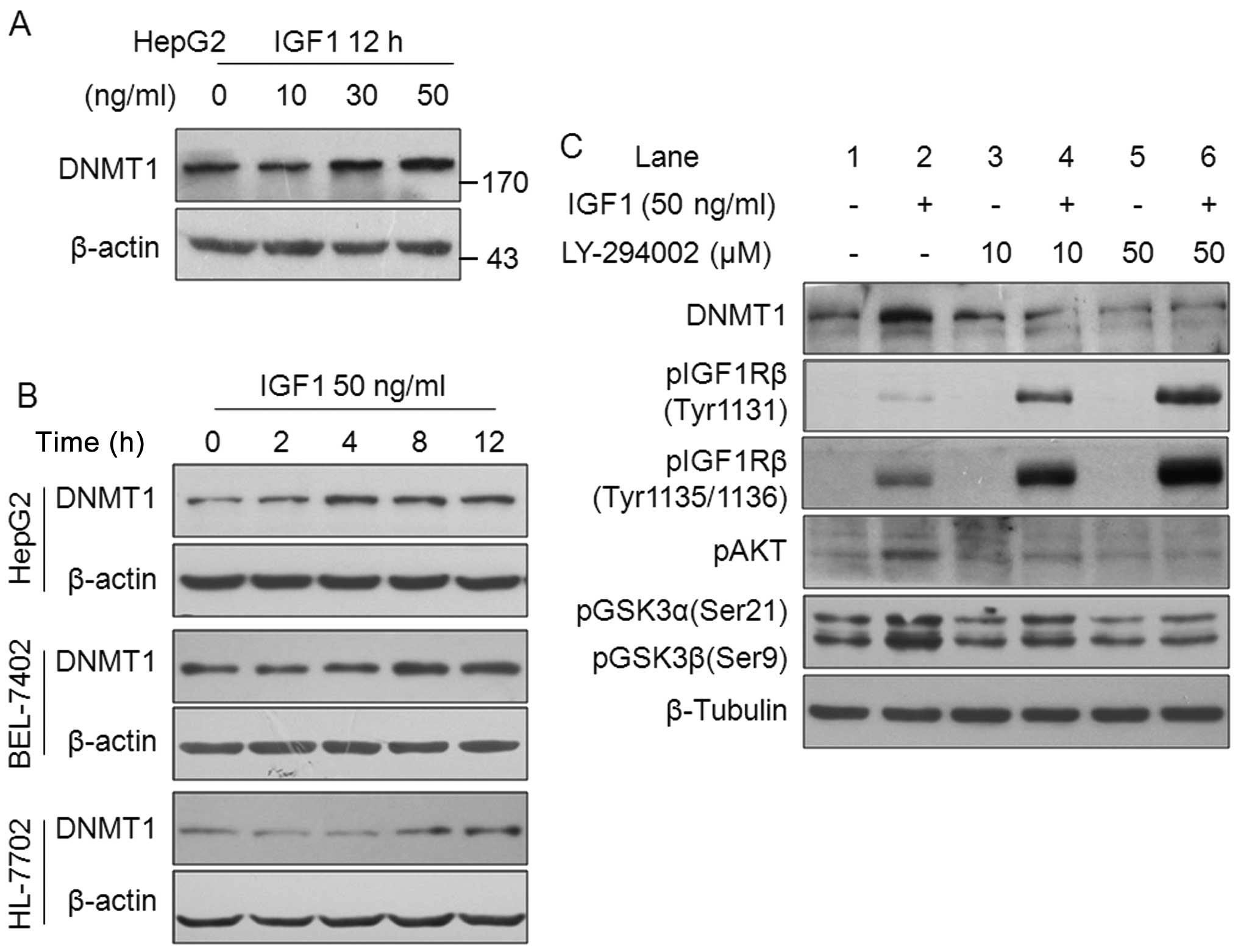
DNMT1 protein expression is upregulated
by growth factors
Epigenetic regulation is sensitive to the
environment, including extracellular environmental factors. We
hypothesized that DNA methylation is influenced by growth factor
pathways in HCC cells. To dissect the potential relationship
between HCC-related growth factors and DNMT1 expression, we
determined the effect of IGFs, HGF and EGF on DNMT1 expression in
HCC cells. The results from western blotting clearly show that
DNMT1 protein expression was upregulated by IGF1 stimulation in a
dose-dependent manner in HepG2 cells (Fig. 3A). Time course experiments revealed
that 50 ng/ml IGF1 increased DNMT1 expression in HepG2, BEL-7402
and HL-7702 cells (Fig. 3B).
Furthermore, HGF and EGF increased DNMT1 protein expression (data
no shown). These results point to a novel pathway of HCC-related
growth factors that act by upregulating DNMT1, and suggest that
DNMT1 has an important biological function in regulating HCC
malignant phenotype.
We investigated the involvement of the IGF-IGF1Rβ
pathway in the regulation of DNMT1 expression. The PI3K-AKT pathway
is a key downstream signal of the IGF-IGF1Rβ activation. To confirm
whether this pathway is responsible for DNMT1 upregulation by IGF1,
experiments to clarify which signaling pathway was blocked were
performed. No obvious inhibition of DNMT1 expression was observed
after treatment with 10 μM of LY-294002 (Fig. 3C, lane 3). However, 10 and 50 μM of
LY-294002 significantly suppressed IGF1-induced DNMT1 expression,
with downregulation of pAKT and pGSK3 (Fig. 3C, lanes 4 and 6). Notably, blocking
of the PI3K-AKT pathway by LY-294002 led to a marked increase in
IGF1-induced pIGF1Rβ phosphorylation (not necessarily activation)
in a dose-dependent manner (Fig.
3C, lanes 2, 4 and 6), indicating the pivotal role of the
PI3K-AKT pathway in IGF-IGF1Rβ activation signal transduction in
the HepG2 cells. Furthermore, we found that PI3K-AKT
phosphorylation was effectively stimulated by IGF2, HGF and EGF
(data no shown). Taken together, the results indicate the
transmission effects of the PI3K-AKT pathway on IGF-IGF1Rβ-induced
DNMT1 protein expression.
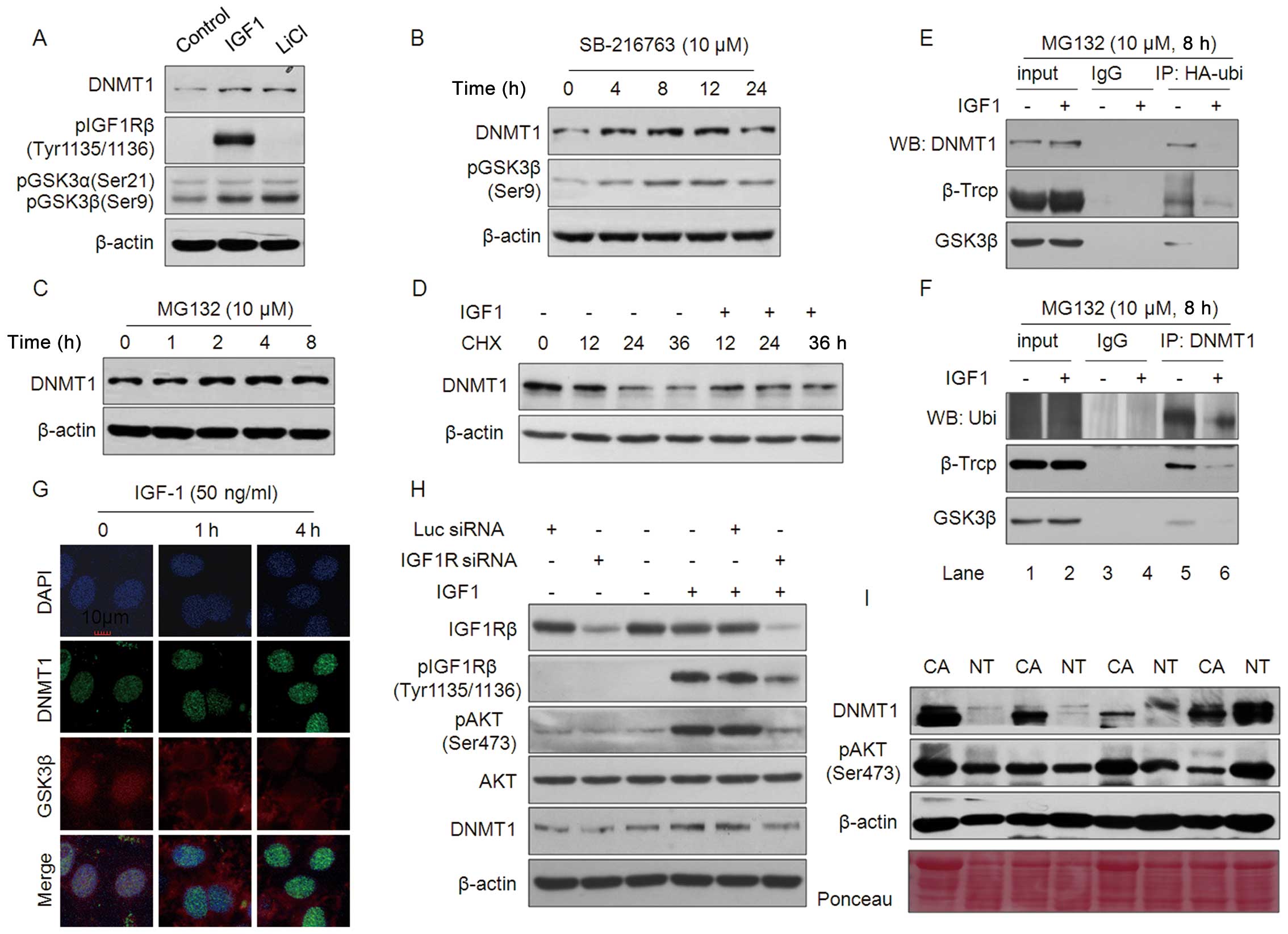
IGF1 inhibits
ubiquitin-proteasome-mediated degradation of DNMT1 protein by
GSK3β-βTrCP complexes
Next, we focused on how the PI3K-AKT pathway affects
IGF1-IGF1Rβ axis-induced DNMT1 expression. IGF1 and HGF did not
affect DNMT1 mRNA levels (data no shown), so it is likely
that its effect is on regulating the protein, including protein
stability. It has been reported that AKT phosphorylates and
inactivates GSK3β, which recruits βTrCP to degrade substrate
protein through a ubiquitin-proteasome pathway and also stabilize
DNMT1 and maintains DNA methylation (26,27).
To explore this possibility, HepG2 cells were treated with either
IGF1 or the GSK3β inhibitor LiCl. Similar to IGF1, 20 M LiCl
upregulated DNMT1 expression and GSK3β phosphorylation in a manner
independent of IGF1Rβ activation (Fig.
4A). pGSK3β (Ser9), an inactive form, gradually increased with
DNMT1 expression in HepG2 cells treated with SB-216763, a GSK3β
inhibitor, for 4 h or longer (Fig.
4B). MG132, a proteasome-specific inhibitor, also increased
DNMT1 protein levels in HepG2 cells (Fig. 4C). Fig. 4D shows that the synthesis inhibitor
cycloheximide (CHX) decreased DNMT1 protein levels in a
time-dependent manner; this decrease was rescued by IGF1 in HepG2
cells. These results indicate that IGF1-induced DNMT1 protein
upregulation involves the GSK3β-mediated protein degradation
pathway.
Next, HA-ubiquitin or DNMT1 plasmids were
transiently transfected into HepG2 cells and co-immunoprecipitation
(co-IP) was performed with either anti-HA or anti-DNMT1 antibodies,
respectively (Fig. 4E and F). As
expected, results from western blotting showed that ubiquitin
protein interacts with DNMT1, βTrCP and GSK3β (Fig. 4E, lane 5), and this interaction was
abrogated by treatment with IGF1 (Fig.
4E, lane 6). The reduced interaction between DNMT1 and the
ubiquitin-βTrCP-GSK3β complex induced by IGF1 treatment was
confirmed by reverse co-IP with an anti-DNMT1 antibody (Fig. 4F, lanes 5 and 6).
Immunofluorescence (IF) staining indicated that the active form of
GSK3β co-localized with DNMT1 in the nucleus, which was abrogated
by IGF1 treatment, resulting in DNMT1 upregulation (Fig. 4G). Consistent with this
observation, Fig. 4H shows that
siRNA-mediated IGF1R knockdown (KD) substantially blocked
IGF1-induced pI3K-AKT pathway activation and DNMT1 expression.
Finally, we evaluated the relationship between pAKT activation and
DNMT1 expression in clinical HCC samples. Results from western
blotting revealed that DNMT1 expression was markedly increased in
59.4% (19/32) samples compared with adjacent normal tissue. pAKT
was noticeably increased in 71.9% (23/32) of HCC samples (Fig. 4I). Among the samples, 16 cases
showed upregulation of both DNMT1 and pAKT. These results indicate
that IGF-IGF1Rβ activation upregulates DNMT1 expression via an
AKT-GSK3β-mediated ubiquitinproteasome pathway in HCC.
IGF1 suppresses TSGs transcription via
DNA hypermethylation
Finally, we focused on several potential TSGs
(DLC1, CHD5, IGFBP2, ADAM23,
LRCC4, LASS2, SCAI, HOXB13,
AXIN1 and CSRNP1) commonly methylated in HCC, and
performed real-time qPCR to detect their expression in HepG2 cells
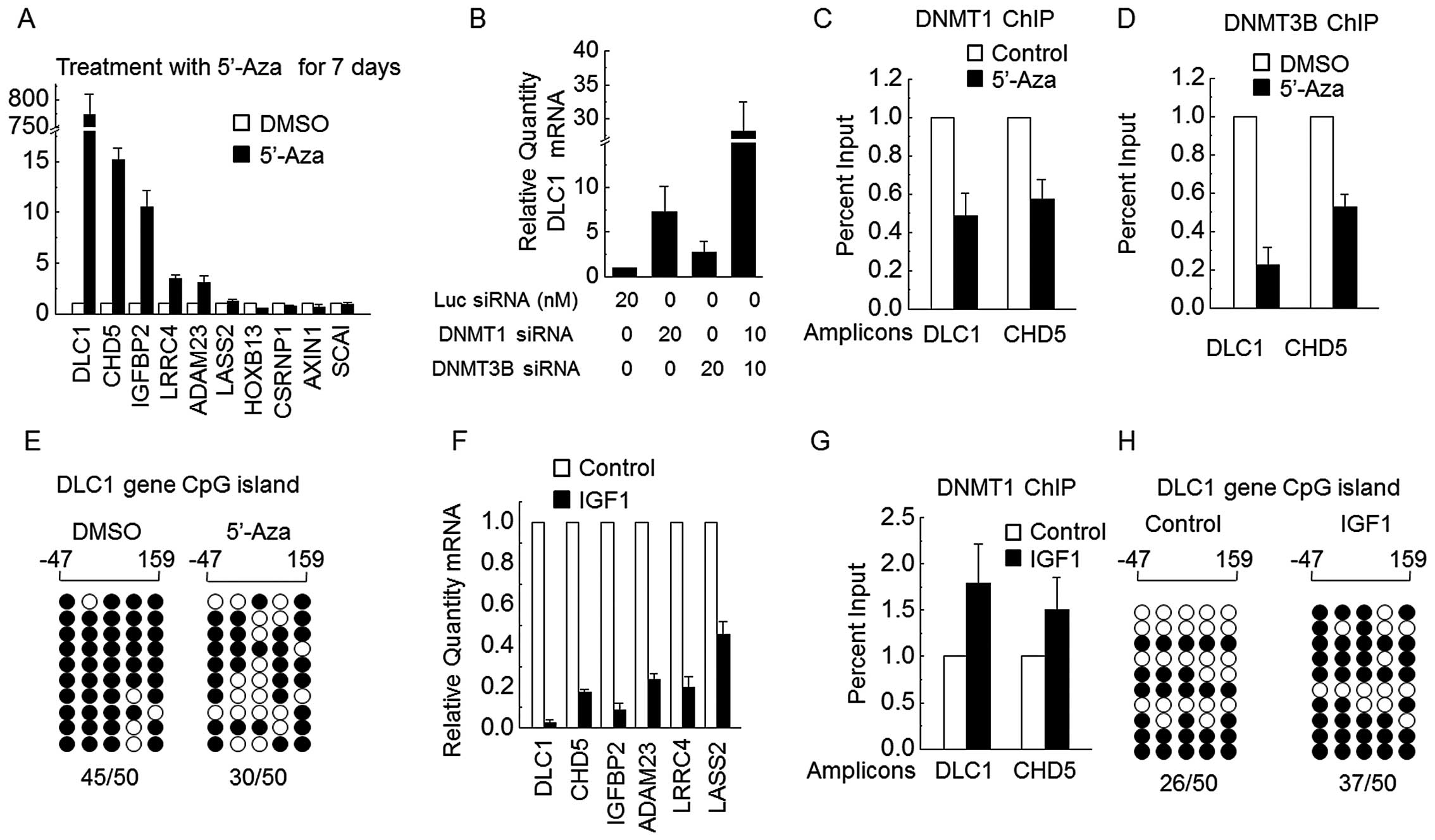
treated with 5′-Aza. For HepG2 cells exposed to 5′-Aza for 7 days,
DLC1, CHD5, IGFBP2, ADAM23 and
LRCC4 were markedly upregulated (Fig. 5A). We also found that either DNMT1
or DNMT3B siRNA obviously upregulated DLC1 expression, and
siRNAs for both DNMT1 and DNMT3B together dramatically increased
DLC1 mRNA levels (Fig. 5B).
ChIP assays showed that DLC1 and CHD5 promoters were
bound by DNMT1 and DNMT3B, and this binding was abrogated by 5′-Aza
(Fig. 5C and D). Bisulfate
sequencing assays showed that CpG loci of DLC1 are heavily
methylated in wild-type HepG2 cells and the DNA methylation level
was obviously reduced by 7 days of treatment with 5′-Aza (Fig. 5E). These results demonstrate that
DNMT1 and DNMT3B cooperate in methylation of the DLC1 and
CHD5 promoters.
As expected, the mRNA expression of certain DNMTs
target TSGs (DLC1, CHD5, IGFBP2,
ADAM23, LRRC4 and LASS2) was significantly
downregulated in HepG2 cells exposed to IGF1 for 7 days (Fig. 5F). Consistent with this
observation, binding of DNMT1 to either DLC1 or CHD5
CpG loci was enhanced in HepG2 cells treated with IGF1 for 7 days
(Fig. 5G). Fig. 5H shows that DNA methylation levels
for the DLC1 promoter were increased by 7 days of treatment
with IGF1. These results clearly show that IGF1 suppresses TSG
transcription, at least in part, through DNA methylation
mechanisms.
Discussion
Emerging evidence suggests a relationship between
DNA hypermethylation and hepatocarcinogenesis (28). However, the mechanistic and
biological significance of DNA hypermethylation in human HCC is
unclear. DNMTs promote liver cancer cell proliferation by
regulating the expression and activity of cell cycle proteins.
Moreover, siRNA interference of DNMT1 or/and DNMT3B significantly
inhibits liver cancer cell proliferation, and 5′-Aza can inhibit
HepG2 cell migration and the growth of established tumors in mice.
Overall, DNA methylation is a potential target for effective
treatment of HCC. The HCC control potential of chemotherapy is
limited by the tolerance of hepatoma cells to drugs. Kurita et
al (29) reported that double
knockdown of DNMT1 and DNMT3B sensitizes HCC cells to
TRAIL-mediated apoptosis, indicating the potential importance of
aberrant DNA hypermethylation in the chemotherapy tolerance of HCC.
This seems to be related to silencing of certain TSGs by DNA
hypermethylation, which controls resistance to drugs in the liver.
The function of 5′-Aza in eliminating methylation is explicit, but
its clinical use as an anti-neoplastic agent is limited by its
toxicity. However, demethylation could be a novel supplementary
therapy that might enhance the curative effect of conventional
chemotherapy in HCC; this will be an important issue for clinical
investigation.
It is now thought that relapse and metastasis
severely limit rehabilitation and HCC prognosis after traditional
treatments such as surgery and radio/chemotherapy. It has been
confirmed that tumor-derived growth factors are associated with
recurrence following radical hepatectomy in HCC. Our results reveal
a previously unknown epigenetic mechanism: as one component of the
tumor microenvironment, growth factors can promote HCC malignant
phenotype through DNMT1 upregulation, resulting in nuclear DNMT1
accumulation and hypermethylation of TSG promoters. The mechanism
and significance of DNMT1 overexpression induced by growth factors
remain poorly understood. We identified a novel growth factor
pathway that involves de novo DNA methylation and tumor
suppressor silencing via upregulation of DNMT1 protein expression.
The significance of IGF-IGF1R in hepatocarcinogenesis has been
confirmed (30) and involves
multiple stages and genes, as well as numerous signaling pathways,
such as MAPK and PI3K/AKT (5). The
present study provides direct evidence that activation of the
IGF1-IGF1Rβ axis induces DNMT1 protein accumulation in the nucleus,
primarily through PI3K-AKT signaling. Co-IP assays revealed that
DNMT1 interacts with βTrCP and GSK3β in HepG2 cells, which involves
the ubiquitin-mediated protein degradation pathway. IGF1 protects
DNMT1 stability through abrogation of the DNMT1-βTrCP-GSK3β
complex. These findings are in agreement with the notion that DNMT1
protein ubiquitination and proteasome-mediated degradation depend
on a destruction domain mapped to the N-terminal 120 amino acids
that is also important for βTrCP interaction (22,31).
In clinical samples, high correlation between PI3K-AKT activation
and DNMT1 upregulation was observed. This confirms the crucial role
of pI3K-AKT in IGF1-induced DNMT1 upregulation. This is similar to
the report that NNK activates AKT, inhibiting GSK3β-βTrCP complex
formation and thereby attenuating proteasomal degradation of DNMT1
in lung cancer cells (22). We
investigated whether a similar mechanism applies to other growth
factors and IGF1. The results show that IGF2, EGF and HGF activate
AKT phosphorylation. It suggests that protein stability may be the
common mechanism in growth factor-induced DNMT1 upregulation.
Although some studies have found increased
expression of DNMTs in HCC, the mechanistic significance is unknown
at present. Our studies answer this question by identifying a novel
mechanism in which DNMT1 overexpression depends on extracellular
cytokines, similar to the mechanism for NNK regulation of DNMT1
expression via the ubiquitin protein degradation pathway (22). These findings further confirm that
epigenetic modifications such as DNA methylation play an
indispensable role in regulating the expression of target genes and
controlling cell proliferation, differentiation and functional
specialization in hepatocarcinogenesis. We used a promoter array to
identify genome-wide methylation profiles in HCC cells and found
significant hypermethylation of CpG sites for a series of TSGs
(DLC1, CHD5, IGFBP2, LRRC4 and
LASS2) with notable biological significance in
hepatocarcinogenesis. A putative tumor suppressor gene,
DLC1, was identified and mapped to chromosome 8p21.3-22, a
region suspected to harbor tumor suppressor genes and recurrently
deleted in HCC, prostate, lung and breast cancers. The present
study provides evidence that DNMT1 and DNMT3B are directly
co-located on DLC1 CpG sites, resulting in promoter
hypermethylation. Significant downregulation of DLC1 mRNA
expression in HCC tissues was associated with enhanced CpG
hypermethylation. DNA methylation is another crucial mechanism for
DLC1 inactivation besides genetic regulation. Mapping to 1p36,
chromodomain helicase DNA binding protein 5 (CHD5) is a
tumor suppressor that serves as a master regulator of a tumor
suppressive network controlling proliferation, apoptosis and
senescence. The present study provides direct evidence that DNMT1
and DNMT3B bind to the CpG loci of DLC1 and CHD5,
leading to promoter hypermethylation and transcriptional silencing.
IGF1 significantly suppressed mRNA expression of certain TSGs,
accompanied by increased accumulation of DNMT1 at DLC1 and
CHD5 CpG loci in HepG2 cells. These results clearly show
that IGF1 suppresses TSGs transcription via DNA methylation, at
least in part.
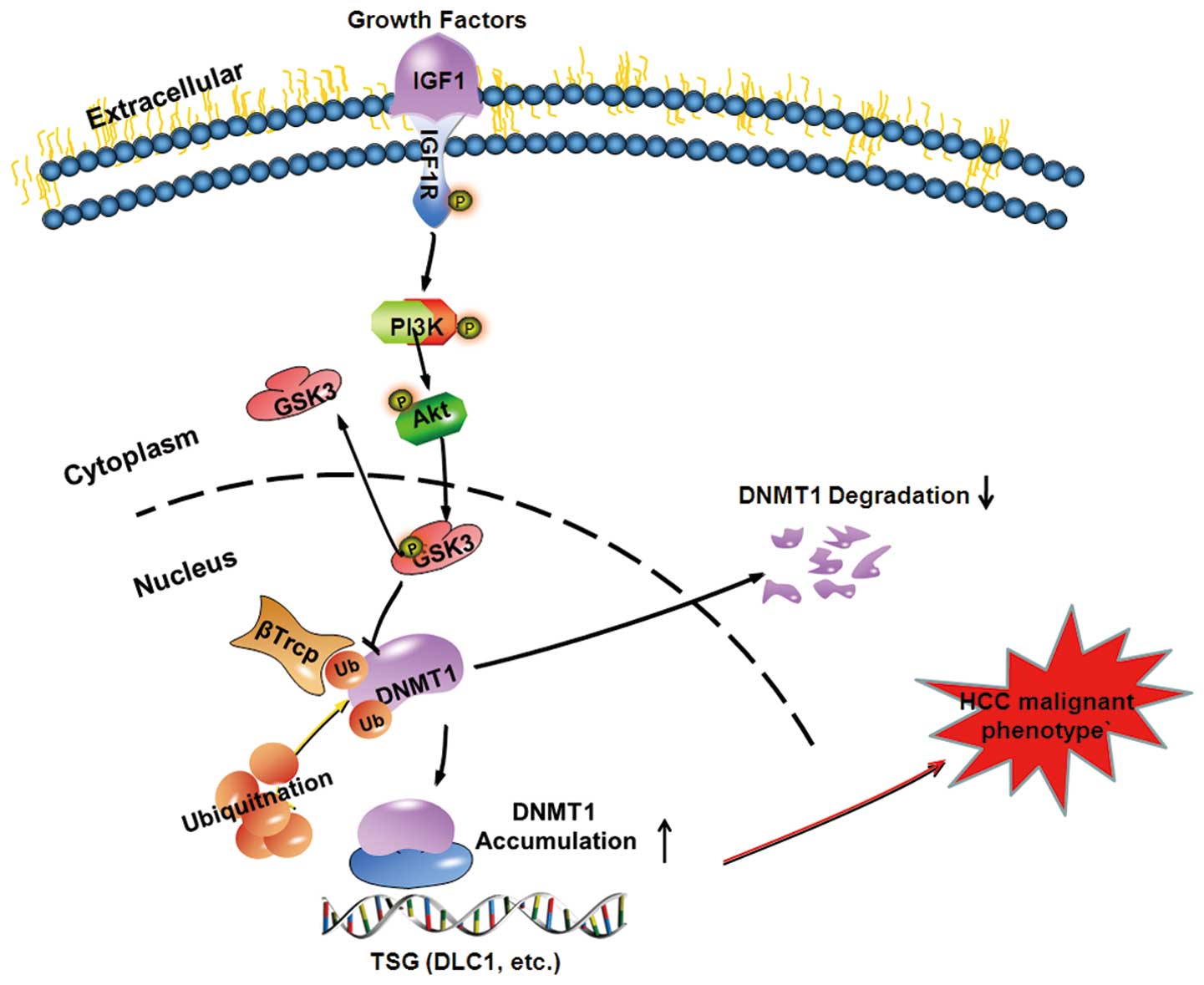
In conclusion, our results point to a novel
epigenetic mechanism for growth factor-mediated repression of TSG
transcription that involves DNA methylation (Fig. 6). IGF1 induces activation of AKT,
then promotes GSK3β phosphorylation at Ser9 to form inactive GSK3β,
which subsequently attenuates the ability of βTrCP to degrade DNMT1
protein. DNMT1 protein binds to the promoters of various TSGs and
results in promoter hypermethylation and transcriptional silencing,
which ultimately leads to tumorigenesis and poor prognosis. Our
results not only enriched our understanding of a novel epigenetic
mechanism of growth factor in the regulation of TSG transcription
by DNA methylation but also provided a novel target for HCC
diagnosis and prognosis.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (no. 81172286 and no.
81372618), the ‘973’Program (no. 2013CB910803) and the National Key
Sci-Tech Special Project of China (no. 2012ZX10002011-005). We
appreciate the valuable comments from other members of our
laboratories.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hayat MJ, Howlader N, Reichman ME and
Edwards BK: Cancer statistics, trends, and multiple primary cancer
analyses from the Surveillance, Epidemiology, and End Results
(SEER) Program. Oncologist. 12:20–37. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brenner H, Stegmaier C and Ziegler H:
Long-term survival of cancer patients in Germany achieved by the
beginning of the third millenium. Ann Oncol. 16:981–986. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kishi Y, Hasegawa K, Sugawara Y and Kokudo
N: Hepatocellular carcinoma: current management and future
development-improved outcomes with surgical resection. Int J
Hepatol. 2011:7281032011.PubMed/NCBI
|
|
5
|
Breuhahn K, Longerich T and Schirmacher P:
Dysregulation of growth factor signaling in human hepatocellular
carcinoma. Oncogene. 25:3787–3800. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Clendenen TV, Arslan AA, Lokshin AE, et
al: Temporal reliability of cytokines and growth factors in EDTA
plasma. BMC Res Notes. 3:3022010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhao B, Zhang C, Forsten-Williams K, Zhang
J and Fannon M: Endothelial cell capture of heparin-binding growth
factors under flow. PLoS Comput Biol. 6:e10009712010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Barbara L, Benzi G, Gaiani S, et al:
Natural history of small untreated hepatocellular carcinoma in
cirrhosis: a multivariate analysis of prognostic factors of tumor
growth rate and patient survival. Hepatology. 16:132–137. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chitnis MM, Yuen JS, Protheroe AS, Pollak
M and Macaulay VM: The type 1 insulin-like growth factor receptor
pathway. Clin Cancer Res. 14:6364–6370. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Whittaker S, Marais R and Zhu AX: The role
of signaling pathways in the development and treatment of
hepatocellular carcinoma. Oncogene. 29:4989–5005. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kurmasheva RT and Houghton PJ: IGF-I
mediated survival pathways in normal and malignant cells. Biochim
Biophys Acta. 1766:1–22. 2006.PubMed/NCBI
|
|
12
|
Shimizu M, Shirakami Y, Sakai H, et al:
EGCG inhibits activation of the insulin-like growth factor
(IGF)/IGF-1 receptor axis in human hepatocellular carcinoma cells.
Cancer Lett. 262:10–18. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pi-Chieh Wang K, Lee LM, Lin TJ, et al:
Gene transfer of IGF1 attenuates hepatocellular apoptosis after
bile duct ligation. J Surg Res. 167:237–244. 2011. View Article : Google Scholar
|
|
14
|
Jones PA and Baylin SB: The fundamental
role of epigenetic events in cancer. Nat Rev Genet. 3:415–428.
2002.PubMed/NCBI
|
|
15
|
Ting AH, Jair KW, Suzuki H, Yen RW, Baylin
SB and Schuebel KE: CpG island hypermethylation is maintained in
human colorectal cancer cells after RNAi-mediated depletion of
DNMT1. Nat Genet. 36:582–584. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Patra SK, Patra A, Zhao H and Dahiya R:
DNA methyltransferase and demethylase in human prostate cancer. Mol
Carcinog. 33:163–171. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Saito Y, Kanai Y, Nakagawa T, et al:
Increased protein expression of DNA methyltransferase (DNMT) 1 is
significantly correlated with the malignant potential and poor
prognosis of human hepatocellular carcinomas. Int J Cancer.
105:527–532. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Girault I, Tozlu S, Lidereau R and Bieche
I: Expression analysis of DNA methyltransferases 1, 3A, and 3B in
sporadic breast carcinomas. Clin Cancer Res. 9:4415–4422.
2003.PubMed/NCBI
|
|
19
|
Etoh T, Kanai Y, Ushijima S, et al:
Increased DNA methyltransferase 1 (DNMT1) protein expression
correlates significantly with poorer tumor differentiation and
frequent DNA hypermethylation of multiple CpG islands in gastric
cancers. Am J Pathol. 164:689–699. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lin RK, Hsu HS, Chang JW, Chen CY, Chen JT
and Wang YC: Alteration of DNA methyltransferases contributes to
5′CpG methylation and poor prognosis in lung cancer. Lung Cancer.
55:205–213. 2007. View Article : Google Scholar
|
|
21
|
Lambert MP, Paliwal A, Vaissiere T, et al:
Aberrant DNA methylation distinguishes hepatocellular carcinoma
associated with HBV and HCV infection and alcohol intake. J
Hepatol. 54:705–715. 2011. View Article : Google Scholar
|
|
22
|
Lin RK, Hsieh YS, Lin P, et al: The
tobacco-specific carcinogen NNK induces DNA methyltransferase 1
accumulation and tumor suppressor gene hypermethylation in mice and
lung cancer patients. J Clin Invest. 120:521–532. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dong C, Yuan T, Wu Y, et al: Loss of FBP1
by Snail-mediated repression provides metabolic advantages in
basal-like breast cancer. Cancer Cell. 23:316–331. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tiwari N, Tiwari VK, Waldmeier L, et al:
Sox4 is a master regulator of epithelial-mesenchymal transition by
controlling Ezh2 expression and epigenetic reprogramming. Cancer
Cell. 23:768–783. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Au SL, Wong CC, Lee JM, Wong CM and Ng IO:
EZH2-mediated H3K27me3 is involved in epigenetic repression of
deleted in liver cancer 1 in human cancers. PLoS One. 8:e682262013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Taketo MM: Shutting down Wnt
signal-activated cancer. Nat Genet. 36:320–322. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sun L, Zhao H, Xu Z, et al:
Phosphatidylinositol 3-kinase/protein kinase B pathway stabilizes
DNA methyltransferase I protein and maintains DNA methylation. Cell
Signal. 19:2255–2263. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ma X, Wang YW, Zhang MQ and Gazdar AF: DNA
methylation data analysis and its application to cancer research.
Epigenomics. 5:301–316. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kurita S, Higuchi H, Saito Y, et al: DNMT1
and DNMT3b silencing sensitizes human hepatoma cells to
TRAIL-mediated apoptosis via up-regulation of TRAIL-R2/DR5 and
caspase-8. Cancer Sci. 101:1431–1439. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu X, Jiang W, Aucejo F, et al:
Insulin-like growth factor I receptor beta expression in
hepatocellular carcinoma. Hum Pathol. 42:882–891. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Agoston AT, Argani P, Yegnasubramanian S,
et al: Increased protein stability causes DNA methyltransferase 1
dysregulation in breast cancer. J Biol Chem. 280:18302–18310. 2005.
View Article : Google Scholar : PubMed/NCBI
|