Introduction
Acute myeloid leukemia (AML) and non-small cell lung
cancer (NSCLC) are known to be associated with high lethality
despite intensive treatment. With overall 5-year survival rates of
25.7 and 16.6%, respectively (1)
the treatment of AML and NSCLC remains an important field of
experimental and clinical investigation.
In the last decade, the knowledge and understanding
of tumorigenesis increased rapidly. Besides genetic mutations and
alterations, epigenetic changes, especially alterations in
methylation patterns, have been found in tumor development and
progression (2,3). The term ‘epigenetics’ is defined as
any change of gene expression that is not mediated by alterations
of the primary nucleotide sequence. Changes of the methylation
pattern occur during normal mammalian development and play an
important role in tissue-specific gene regulation, genomic
imprinting and X-chromosome inactivation by gene silcening
(4). In cancer cells, the
5-methyl-cytosine distribution is altered in two directions: while
the global methylation level is decreased (5), CpG islands appear to be
hypermethylated (6). These regions
often are associated with gene promoters and, therefore, might
contribute to cancer-specific silencing of tumor-suppressor-genes
(reviewed in ref. 7).
In normal as well as tumor cells, DNA methylation is
mainly regulated by the DNA methyltransferase (DNMT) 1, 3a and 3b
(8). These enzymes catalyze
maintenance (DNMT1) (9,10) and de novo methylation
(DNMT3a and DNMT3b) (11,12) by adding a methyl group to the
cytosine ring (8). As DNMT play an
important role in tumorigenesis (13,14),
they appear as promising targets for anticancer therapy.
5-azacytidine (Aza) is a nucleoside analogue that is
incorporated into DNA and RNA (15). While high doses result in
cytotoxicity (16), low dose Aza
leads to DNA hypomethylation by irreversible binding and
degradation of DNA methyltransferases (DNMT) (17,18).
In vitro and in vivo studies indicate that Aza is
able to reactivate epigenetically silenced tumorsuppressor-and
chemosensitivity-genes and alter cancer phenotypes by its
demethylating action (19,20). To date, it is approved for the
treatment of the myelodysplastic syndrome and acute myeloid
leukemia with 20–30% blasts in the bone marrow. There are few
reports concerning the treatment of solid tumors (21). Though Aza has proven clinical
activity, it does not work curatively.
We hypothesize that DNMT inhibitory drugs may
enhance the antitumor activity of cytotoxic agents. Therefore, we
exposed AML cell lines to cytarabine (AraC) or etoposide (Eto) and
NSCLC cell lines to cisplatin (Cis) or gemcitabine (Gem) alone and
in combination with low dose Aza in order to find synergistic drug
interactions.
Materials and methods
Cell culture
We used human leukemic cell lines U937 and HL60 and
the NSCLC cell lines A549 (adenocarcinoma) and HTB56 (anaplastic
carcinoma). Leukemic cells were maintained in RPMI (Gibco,
Carlsbad, CA, USA) and A549 cells in Dulbecco’s modified Eagle’s
medium (DMEM; Sigma-Aldrich, St. Louis, MO, USA), both supplemented
with 10% fetal calf serum and 1% penicillin/streptomycin. HTB56
cells were maintained in Minimum Essential Medium (MEM; Gibco),
supplemented with 10% fetal calf serum, 1% penicillin/streptomycin,
1% non-essential amino acids (Gibco), 1% L-glutamine
(Sigma-Aldrich) and 1% sodium pyruvate (Gibco). All cell cultures
were maintained in a humified atmosphere of 5% CO2 at
37°C. Cells were counted by the test of trypan blue exclusion.
Reagents
All drugs were purchased from Sigma-Aldrich. Aza was
diluted in 1X PBS to a final concentration of 50 mM,
gemcitabine-hydrochloride was diluted in 0.9% NaCl to a final
concentration of 5 mM, cis-diammineplatinum(II)-dichloride
in sterile ddH2O to a final concentration of 5 mM,
β-D-arabinofuranoside was diluted in 1X PBS to a final
concentration of 10 mM and etoposide in dimethylsulfoxide (Serva
Electrophoresis GmbH, Heidelberg, Germany) to 50 mM. Aza, AraC and
Eto were stored at −20°C, Cis at room temperature and protected
from light and Gem at 4°C.
Cell proliferation assays
Cell proliferation assays were performed by MTS
assays and by counting cells. For MTS assays, AML cells were
treated with Aza in a range from 50 to 2000 nM as well as AraC
(100–2000 nM) and Eto (30–3000 nM) alone or in combination with 25
and 100 nM of Aza. NSCLC cells were treated with Aza in a dose
range from 25 to 5000 nM as well as Cis (10–100000 nM) and Gem
(10–10000 nM) alone or in combination with 25 and 100 nM of Aza.
Cell proliferation was measured after 48-h treatment by reduction
of
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxy-methoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
according to the recommendations of the manufacturer (Promega
Corp., Fitchburg, WI, USA) at 490 nm with the Benchmark microplate
reader (Bio-Rad Laboratories, Hercules, CA, USA). MTS assays were
performed in triplicate.
Drug interactions were evaluated with CalcuSyn
Software (Biosoft, Cambridge, UK) according to the median-effect
method by Chou and Talalay (22).
A combination index (CI) <0.9 indicates synergistic drug
interactions, a CI of >1.1 is antagonistic and a CI between 0.9
and 1.1 stands for additive effects.
To analyze whether sequential or simultaneous
combination treatment is superior, low doses of Aza (25 and 100 nM)
were combined with AraC and Eto in AML cell lines as well as with
Gem in NSCLC cell lines. Sequentiell treatment consisted of 48-h
treatment with Aza followed by 48-h treatment with the respective
drug after removal of Aza. For simultaneous treatment the drugs
were exposed to both agents at the same time for 48 h.
Methylcellulose colony-formation
assay
After 24 h of drug treatment, drugs were washed out
and equal numbers of viable cells were plated in triplicates in
2.3% methylcellulose (Sigma-Aldrich). A549, HL60 and U937 colonies
containing >50 cells were quantified after 7 days of incubation
under an inverted microscope, HTB56 colonies after 11 days. AML
cell lines were treated with 25 nM of Aza, 25 nM of AraC, 50 nM of
Eto and the respective combinations of Aza and either AraC or Eto.
Both NSCLC cell lines were treated with 25 nM of Aza, 25 nM and 50
nM of Cis and the combination of Aza and both Cis doses. A549 cells
were additionally treated with 25 and 50 nM of Gem as well as the
combination of Aza and either 25 or 50 nM of Gem.
Bromodeoxyuridine incorporation and
fluorescence-activated cell sorting
AML cell lines were treated with 25 nM of Aza, 100
nM of AraC, 100 nM of Eto and the respective combinations of Aza
and AraC or Eto. NSCLC cell lines were treated with 25 nM of Aza,
100 nM of Cis, 100 nM of Gem as well as with the respective
combinations. After 22 h, bromodeoxyuridine was added for two
additional hours of incubation, then cells were stained according
to the manufacturer’s instructions (BD Biosciences, Franklin Lakes,
NJ, USA). BrdU incorporation was analyzed on a FACSCalibur flow
cytometer (Becton-Dickinson, San Jose, CA, USA) using BD CellQuest
Pro (BD Biosciences).
Active caspase-3 staining and
fluorescence-activated cell sorting
A549 and HTB56 cells were treated with 25 nM of Aza,
100 nM of Cis, 100 nM of Cis and the respective combinations for 24
h. After permeabilisation active caspase-3 was measured using
phycoerythrin-conjugated anti-active-caspase-3 antibody (BD
Biosciences) according to the manufacturer’s instructions and
analyzed by flow cytometry like described above.
DNA methylation analysis
Cells were treated according to colony formation
assays. After 24 h of treatment, DNA was isolated using the High
Pure PCR Template Preparation kit for genomic DNA (Roche Applied
Science, Penzberg, Germany) and DNA content was measured with the
NanoDrop spectrophotometer ND-1000™ (Peqlab Biotechnologie,
Erlangen, Germany). DNA methylation was analyzed by 450K bead
arrays using EZ-96 DNA Methylation™ kit (Zymo Research, Irvine, CA,
USA) with optimized PCR conditions according to the manual.
Analysis of methylation data was performed by BRB-ArrayTools
developed by Dr Richard Simon and the BRB-ArrayTools Development
Team.
Statistical analysis
All data are shown as mean plus standard deviation
(SD), if not otherwise specified. Statistical differences between
two groups were calculated by the Student’s t-test. A p-value of
<0.05 was considered as indicating significant differences.
Results
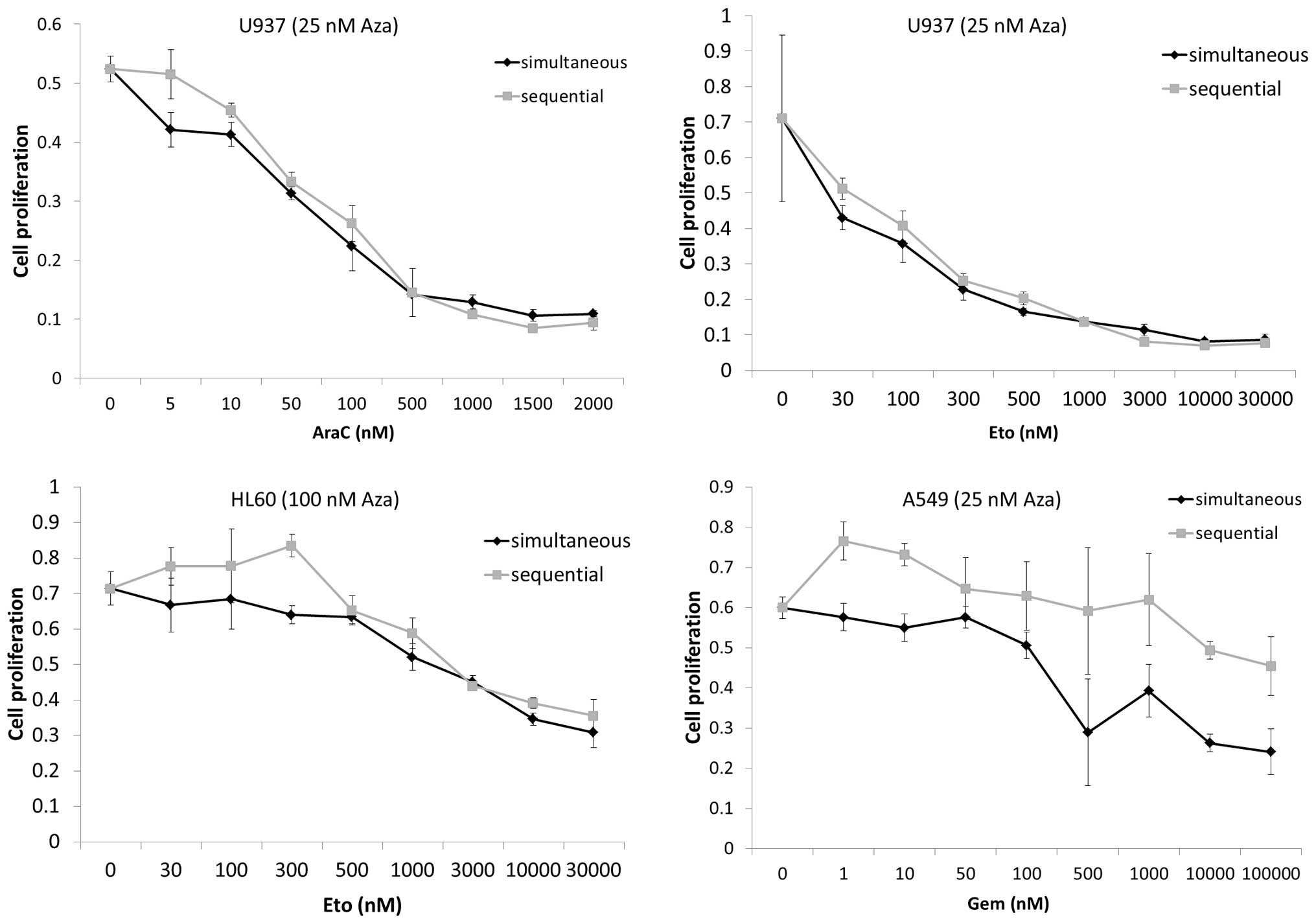
Simultaneous and sequential Aza
combinational treatment show similar effectiveness
Four human cancer cell lines (A549, HL60, HTB56 and
U937) were treated with simultaneous and sequential Aza containing
drug combinations to evaluate whether one form of application would
be superior.
Proliferation assays (Fig. 1) indicated no advantage of the
sequential over the simultaneous application. Considering these
results we chose the simultaneous application for further
experiments. The effects of Aza treatment alone on the different
cell lines are discussed below.
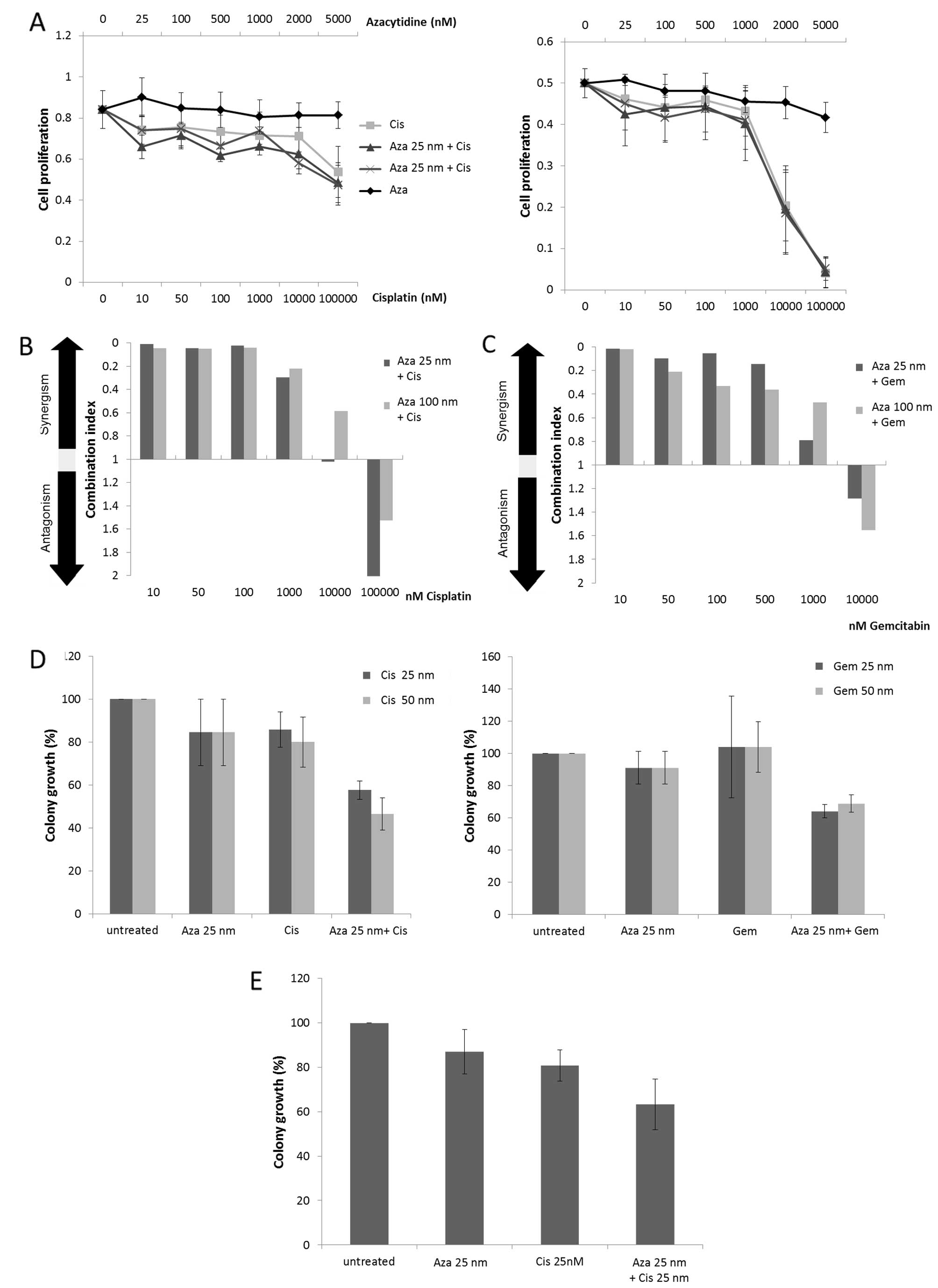
Low dose Aza shows synergistic effects
with Gem and Cis in NSCLC cell lines
Two human NSCLC cell lines (A549 and HTB56) were
assessed for their sensitivity to either Aza, Gem and Cis alone or
in combination by MTS-proliferation assay. Combinational treatment
consisted of low dose Aza (25 and 100 nM) combined with Gem or Cis
in a wide dose range. Effects were measured after 48-h treatment
and potential synergisms were calculated according to the method of
Chou and Talalay. Neither A549 cells (Fig. 2A, left panel) nor HTB56 cells
(Fig. 2A, right panel) showed
sensitivity to Aza alone. Nevertheless, low dose Aza enhanced the
activity of Gem and Cis in A549 (Fig.
2B and C) as well as the activity of Cis in HTB56 (data not
shown). We observed clear synergisms [Combination index (CI)
<0.9] in the proliferation assays for nearly every tested
combination. A549 cells revealed very strong (CI <0.1) and
strong synergisms (CI 0.1–0.3) for both 25 and 100 nM Aza combined
with 10–1000 nM Cis as well as for 25 nM Aza combined with 10–500
nM Gem. The combination of 100 nM with 10–500 nM Gem showed
slightly weaker synergism. HTB56 cells demonstrate very strong and
strong synergism at combinations of 25 nM Aza with 10–100 nM Cis
and 100 nM Aza with 10–50 nM Cis. Overall, the strength of the
calculated synergism decreased with increasing doses of Cis and Gem
(Fig. 2B and C). Colony formation
assays reaffirmed synergistic drug interactions after 24-h
treatment for both cell lines (Fig. 2D
and E). A549 cells indicate synergisms for the combination of
25 nM Aza with 25 nM (p=0.003) and 50 nM Cis (p=0.007) as well as
with 25 and 50 nM Gem (p=0.0046 and p=0.01). Clonal growth in HTB56
cells treated with 25 nM Aza and 25 nM Cis was synergistically
reduced as well (p=0.03) (Fig.
2E).
Effects of AraC and Eto in AML cell lines
are synergistically enhanced by low dose Aza
We treated two human AML cell lines (HL60 and U937)
with Aza, Eto and AraC alone or with combinations of 25 or 100 nM
Aza with varying doses of either Eto or AraC. The proliferation
assays revealed an enhancement of AraC and Eto activity by low dose
Aza in HL60 cells (Fig. 3A) and to
a lesser extent in U937 cells (data not shown). Calculation
according to Chou and Talalay indicated synergistic drug
interactions between these agents. In HL60 cells the combination of
25 nM Aza with 100–2000 nM AraC resulted mainly in very strong
synergisms (CI <0.1) whereas the combination of 100 nM Aza with
AraC revealed rather strong synergisms (CI 0.1–0.3). In U937 cells
these combinations effected weaker synergism and even additive
effects (CI 0.9–1.1) in higher AraC doses (Fig. 3C, left panel).
For the combination of 25 and 100 nM with 30–3000 nM
Eto we observed a clear dose dependency in regard to the quality of
synergistic interaction for both HL60 and U937 cells (Fig. 3B and C, right panel). The strength
of synergism decreased with increasing doses of Eto. However, the
lowest dose (30 nM Eto) constistantly showed antagonisms when
combined with 25 or 100 nM Aza in both cell lines. Colony formation
assays confirmed potential drug synergism after 24-h treatment. We
observed a distinct reduction of clonal growth by the combination
of 25 nM Aza and 25 nM AraC or 50 nM Eto in both cell lines
(Fig. 3D). However, although the
colony formation was observably inhibited, the Eto containing
combinations failed to reach statistical significance in HL60
(p=0.172) and U937 (p=0.079). Treatment with 25 nM Aza and 25 nM
AraC reached significance in HL60 cells (p=0.021), but not in U937
(p=0.051).
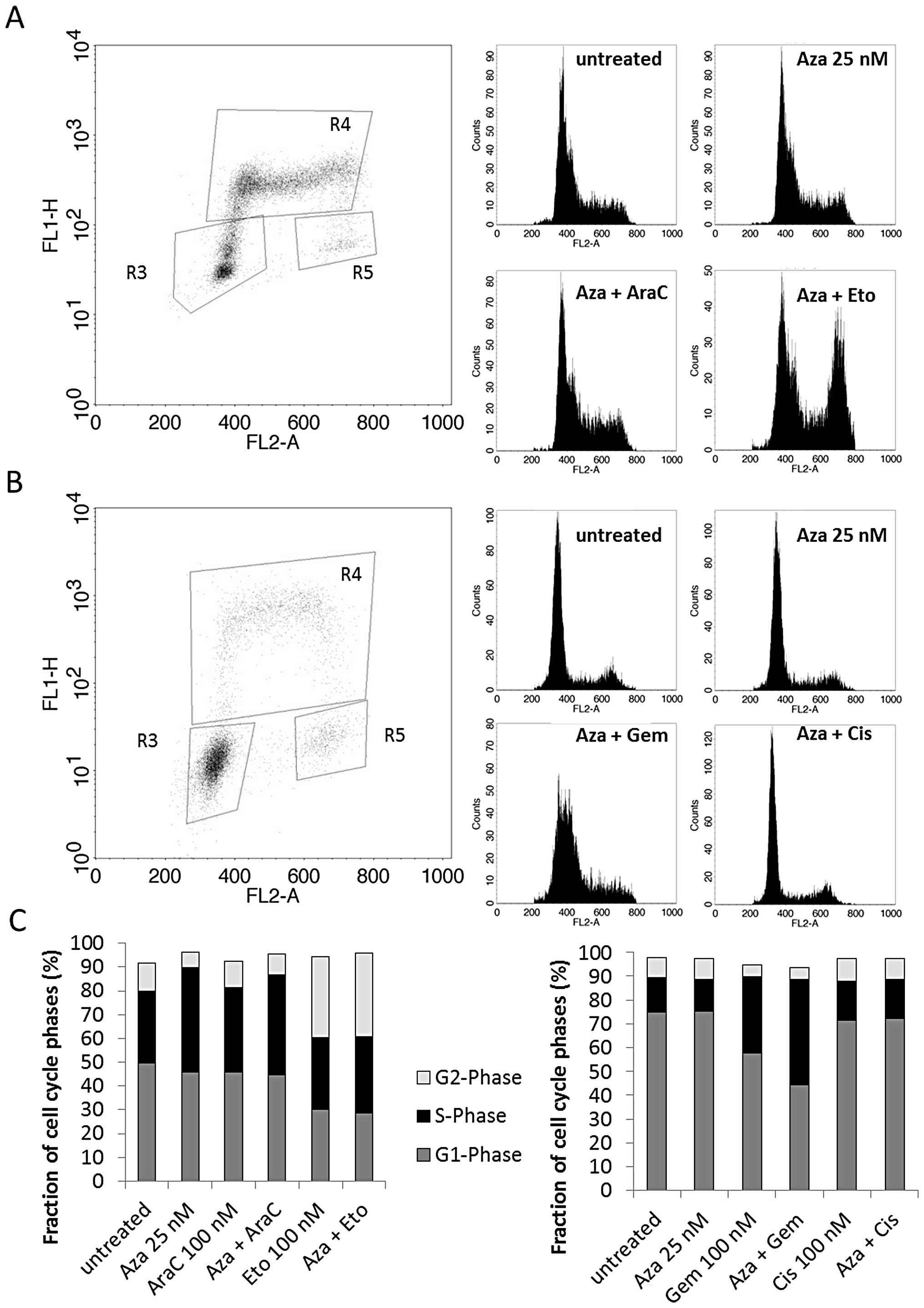
Low dose Aza treatment does not affect
the cell cycle nor induce increased activation of active
caspase-3
To further explore possible mechanisms behind the
observed synergisms we analyzed the cell cycle and the apoptosis
rate by flow cytometry. HL60 and U937 cells were treated with 25 nM
Aza, 100 nM AraC and 100 nM Eto alone and in combination for 24 h.
A549 and HTB56 cells were treated with 100 nM Cis and 100 nM Gem
along with the respective combinations. The BrdU incorporation flow
cytometry revealed no consistent trend towards a specific Aza
effect. Aza (25 nM) alone did not cause any alterations compared to
the untreated control in the four cell lines. We observed an
increase of the G2-phase by Aza plus Eto in both U937 (Fig. 4A) and HL60 cells (Fig. 4C, left). Aza plus Gem led to an
increase of the S-phase and a shift towards early S-phase in HTB56
cells (Fig. 4B and C, right
panel). The combination of Aza and AraC increased the S-phase in
U937 cells as well (Fig. 4A). All
alterations were consistent with the effects mediated by single
treatment with Eto, AraC and Cis in the respective cell lines.
However, the Gem-mediated increase of the S-phase in HTB56 cells
was enhanced by the addition of Aza. The cell cycle in A549 was not
altered by any treatment. The above results provide no evidence
that synergistic drug interactions in Aza containing combinations
might be explained by cell cycle alterations.
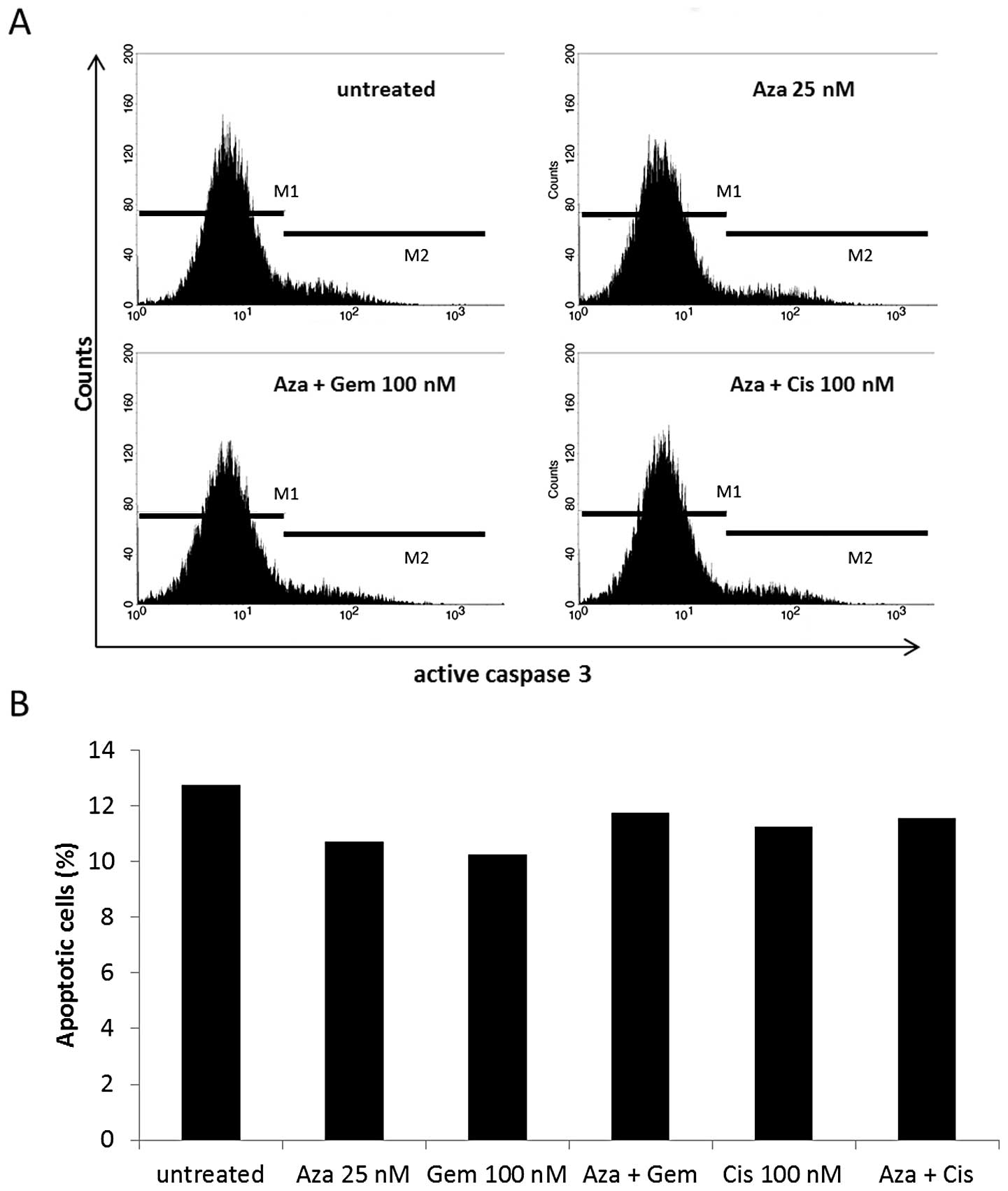
A549 and HTB56 were also analyzed by active
caspase-3 staining after 24-h treatment (Fig. 5). We observed no constant increase
in active caspase-3 levels in either cell line by any treatment.
Therefore, we found no indication of drug mediated early apoptosis
via this pathway after low dose treatment.
Global methylation patterns might not be
affected by 25 nM Aza or Aza containing combinations
The DNA methyltransferase inhibitor (DNMTi) Aza
leads to global DNA hypomethylation and is able to reactivate
silenced tumor suppressor genes by promoter demethylation. To
examine whether Aza induced alterations in DNA methylation might
give a possible explanation for the enhancement of cytotoxic drug
activity, thus we performed genome-wide methylation analysis using
450K bead arrays.
Cells were treated equally to those examined in
colony formation assays. We analysed HL60 and U937 cells treated
with 25 nM Aza and 25 nM AraC and two seperate samples of A549
cells treated with 25 nM Aza and 25 nM Cis alone and in combination
for 24 h.
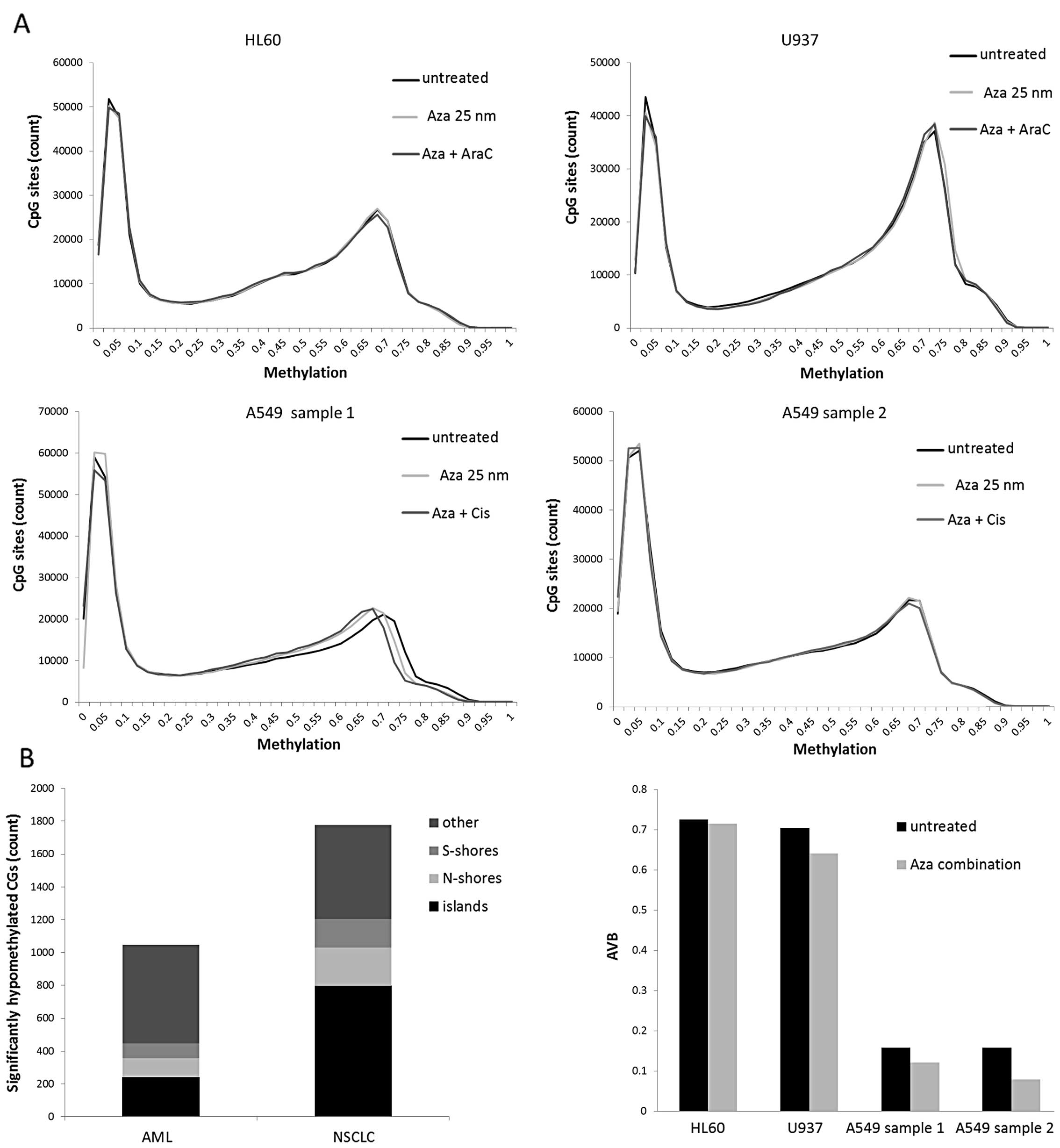
The methylation analysis revealed a bimodal
methylation distribution with a low methylation peak ranging from
average beta (AVB) values 0 to 0.15 and a high-methylation peak
ranging from AVB values 0.55 to 0.8 in every tested cell line
(Fig. 6A). In both A549 samples
the low-methylation peak contained 38.6–39.2% and the
high-methylation peak contained 31.1–32.2% of all CGs. The AML cell
lines showed a higher methylation level with a low-methylation peak
including 24.8–33.6% and a high-methylation peak including
37.3–48.7% of all CGs. In the histograms, we observed no shift of
these peaks, except for the A549 cells (sample 1). In the present
study, the treatment with 25 nM Aza caused a discrete shift to
lower methylation values which was slightly enhanced by the
addition of Cis.
We performed class comparisons to further evaluate
drug induced methylation changes. Our analysis revealed 1,046
significant hypomethylated (p<0.05) CGs in AML and 1,778
significant hypomethylated (p<0.05) CGs in NSCLC cell lines
after Aza and Aza combination treatment. Only 240 of 1,046 CGs in
AML and 798 of 1,778 CGs in NSCLC cells were found on islands
(Fig. 6B, left panel). Several of
these CGs were associated with candidate tumor suppressor genes,
e.g. MGMT, THRB, L3MBTL4, EDNRB, CDH13 and TGFBR3. However, the CGs
associated with these genes were methylated in untreated AML cells
but already hypomethylated in NSCLC cells as shown for MGMT
(Fig. 6B, right panel).
Low dose Aza induced CG-demethylation
increases in a time-dependent manner
To identify long-term effects of Aza and cytotoxic
drugs on DNA methylation patterns, we split some of the A549 cells
(samples 1 and 2) after the 24-h treatment for bead array analysis
and recultured them in Aza and Cis-free medium. After 6 days of
incubation we isolated the DNA and performed 450K bead arrays.
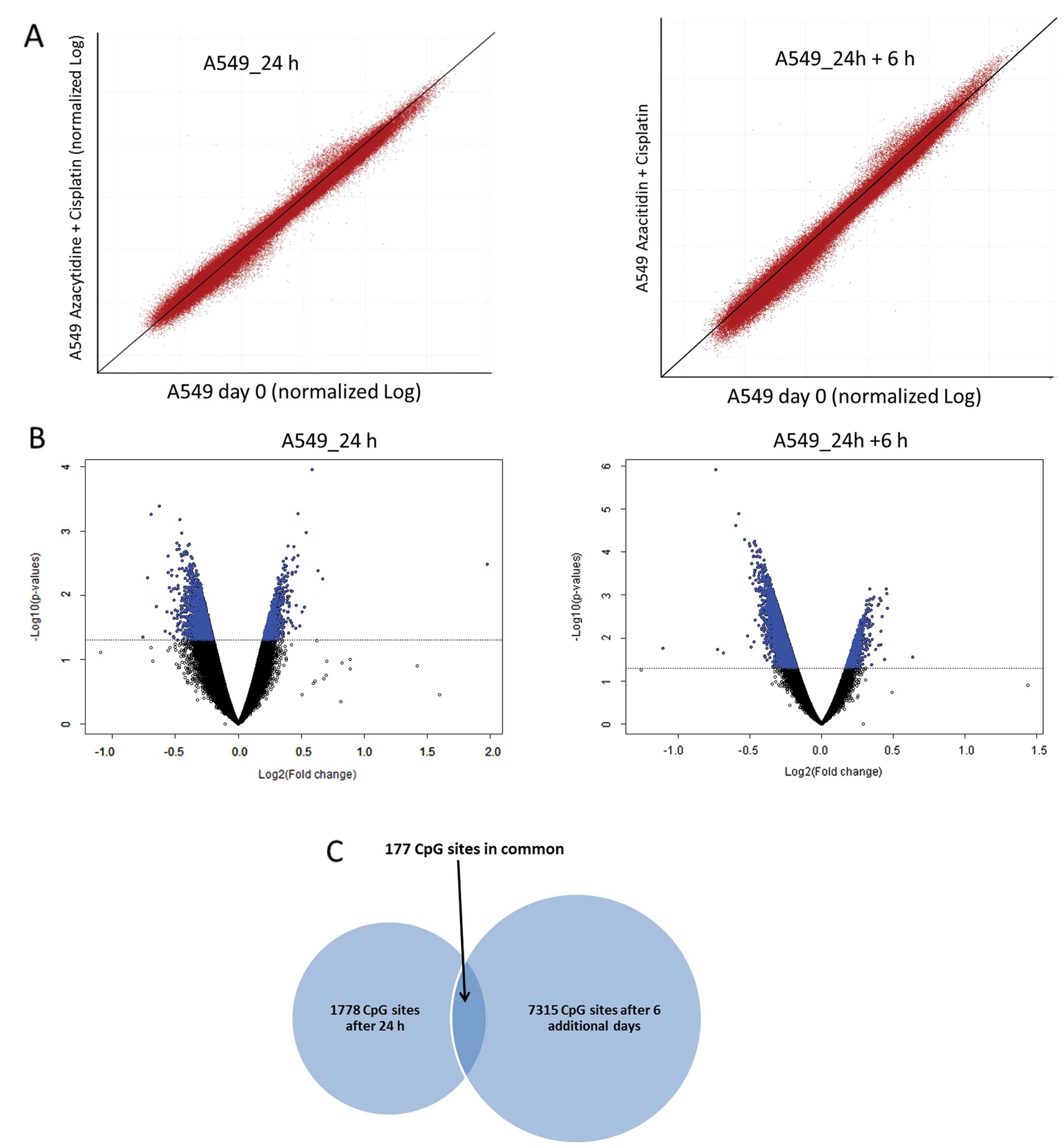
We compared the effects of Aza plus Cis combination
treatment after 24 h and after the 6-day incubation. The
scatterplots showed a shift especially of the low methylated CGs to
lower AVB values after the additional 6 days. However, the already
high methylated CGs shifted to even higher AVB values (Fig. 7A).
In class comparisons we found 7315 CGs significantly
hypomethylated (p<0.05) after Aza and Aza combinational
treatment with a 4.1-fold increase compared to analysis after 24-h
treatment. We further compared the DNA methylation of untreated and
combined treated cells with DNA isolated before the 24-h treatment
and found a significantly higher number of at least 10%
demethylated CGs after Aza plus Cis treatment compared to the
untreated cells (sample 1, 958–308 CGs; sample 2, 66–40 CGs,
Fisher’s exact test: p=0.035). The volcano plots also revealed an
increase in the number of significantly demethylated CGs through
Aza containing treatment. We observed only minor scattering in
methylation deviation (Fig.
7B).
To evaluate the consistency of the Aza induced
hypomethylation between these two time periods of measurement, we
compared the lists of significant hypomethylated CGs. Our findings
resulted in an overlap of over 177 CGs indicating that 10% of the
CGs being hypomethylated after 24-h treatment remain hypomethylated
after the longer incubation times (Fig. 7C). We also correlated the
significant hypomethylated CGs with their corresponding genes and
found 1,238 and 4,078 genes being affected by at least one
hypomethylated CG after 24-h treatment and the following 6-day
incubation. A comparison of these genes exposed an overlap of over
564 genes, so that nearly 45% of all genes that were affected by at
least one CG after 24-h treatment remained affected after 6 days of
incubation.
Discussion
Epigenetic agents might enhance cytotoxic drug
antitumor activity. In the present study we observed strong
synergisms according to the calculation by Chou and Talalay
(22) between low dose Aza and
AraC or Eto in AML, respectively, Cis and Gem in NSCLC cell lines.
These synergisms showed consistency over a high dose range and
different cell lines. Our findings go along with earlier studies
that indicate synergistic interactions between Aza and Cis in
multiple cancer types (23–25).
Furthermore, different studies describe increases in sensitivity to
AraC or Eto when combined with DNMT inhibitory drugs Aza and
decitabine (26–28). In comparison to these studies we
used much lower doses (25 nM Aza) and a shorter treatment time (24
h) but nevertheless observed distinct effects. These low drug
concentrations neither altered the cell cycle nor increased the
rate of apoptosis measured by active caspase-3 staining. Potential
synergisms between Aza and Gem have not been explored
previously.
In this study, treatment with Aza alone revealed
only discrete effects on cell proliferation. The maximal doses of 2
μM in AML and 5 μM in NSCLC cell lines decreased cell proliferation
in MTS assays by <17% after 48-h treatment. A549 cells even
appeared to be completely insensitive to Aza. These marginal
effects on cell proliferation by doses below 2 μM are consistent
with the findings of Hollenbach et al (15) and Murakami et al (29) who described a reduced cell
viability at concentrations ≥1 μM respectively >2 μM in four
different AML cell lines. Remarkably, colony formation was clearly
reduced by the very low dose of 25 nM of Aza for 24 h in three of
four cell lines. This decrease of >10% is comparable to the
inhibition of colony formation induced by 25 nM AraC, 25 nM and 50
nM cisplatin and even stronger than the effect of 25 nM
gemcitabine. It is possible, that the seven days of incubation
following the 24 h of treatment might enhance epigenetic Aza
effects that result in the observed reduction in colony formation.
Additionally, in colony formation assays the cells are not able to
create their preferred milieu of growth factors and other stimuli
(in contrast to cell suspensions) and therefore might be more
vulnerable to the applied drugs (30).
Using higher Aza doses, many authors describe a
dose-dependent induction of apoptosis (29,31)
and cell cycle aberrations. Hollenbach et al (15) observed an increase of the
sub-G1-fraction, while Hagemann et al (18) described an increase of the S phase
after doses below 10 μM, respectively a G2 phase arrest for doses
~10 μM Aza. As we found synergistic inhibition of cell
proliferation and colony formation after treatment with very low
doses of Aza, Cis, AraC, Eto and Gem, we focused on the effects of
these low drug concentrations in search for the mechanisms of
synergism. In contrast to higher doses, treatment with the
respective low doses neither altered the cell cycle nor increased
the rate of apoptosis measured by active caspase-3 staining in any
way that could be correlated with synergistic interaction.
An important question in regard to combination
treatment is the form of drug application schedule. We hypothesize
that Aza as an epigenetic agent might be able to sensitize cells
for cytotoxic drugs. As epigenetic effects such as demethylation
show time-dependence (31), it
seems likely that cytotoxic drug activity might be enhanced rather
by epigenetic pretreatment than simultaneous epigenetic-cytotoxic
therapy. Accordingly, many authors preferred sequential drug
application schedules (25–27,32).
Nevertheless, Quin et al (33) described synergistic interactions
between decitabine and AraC in both simultaneous and sequential
treatment. In the present study we observed no difference in
antitumor effectiveness between simultaneous and sequential Aza
combination treatment with all four cytotoxic drugs. Therefore, we
chose the simultaneous application for our further experiments.
There are several possible explanations for the
observed synergisms between Aza and cytotoxic agents. Aza is able
to reactivate silenced tumor suppressor genes by promoter
CpG-demethylation (34). As this
is the case, it is a rational hypothesis that synergistic drug-drug
interactions might be based on reactivated tumor suppressor and/or
chemo sensitivity genes that make cells more vulnerable for
cytotoxic treatment. Accordingly, Plumb et al (35) described that pretreatment with
decitabine sensitized ovarian carcinoma cells to subsequent
cisplatin treatment, most likely due to MLH-1 reactivation. DNMT
inhibitory drug-induced promoter demethylation does not necessarily
lead to increased gene transcription, but can affect decreased
expression (20,36). Efferth et al (26) observed that MDR-1 overexpressing
leukemic cells were sensitized to several cytotoxic drugs after Aza
treatment. Even though Aza led to a massive MDR1 promoter
demethylation, gene expression was rather decreased than increased.
Additionally, Kong et al (27) described in an early study that Aza
treatment increased the activity of 2′-deoxycytidine kinase (dCK)
in dCK-deficient HL60 cells and therefore partially reversed the
resistance to AraC. In the present study, we used a very low dose
of Aza over a short incubation time. Consequently, we found only a
relatively small number of significantly hypomethylated CG sites.
We found only a small overlap of CG sites that are significantly
hypomethylated in AML and NSCLC cells. Besides, we observed a
greater overlap of genes in AML and NSCLC that are associated with
significantly hypomethylated CG sites and that might indicate the
existance of several target genes. Some of these genes include
candidate tumor suppressor genes like MGMT (37,38),
THRB (39,40), L3MBTL4 (41), EDNRB (42), CDH13 (43) and TGFBR3 (44).
Aza treatment leads to genome-wide demethylation and
therefore alters the epigenetic landscape. Demethylation appears
preferentially at former high methylated CG sites and results in a
shift of the bimodal methylation distribution of the cell; the
high-methylation peak shifts to lower AVB values (18). Global hypomethylation appears to be
causally linked to chromosomal instability (45,46)
and might enhance cytotoxic drug activity, probably by DNA
damage-mediated apoptosis (31).
Accordingly, Quin et al (33) described that apoptotic cells showed
hypomethylation compared to non-apoptotic cells after combination
treatment with decitabine and AraC. Thus, hypomethylation might
sensitize cells to AraC treatment. Hascher et al (19) also hypothesize that Aza might act
rather by change of the global epigenome than by reactivation of
single tumor-suppressor genes. This idea is supported by the fact
that Aza induces a specific and reproducable methylation pattern,
that seems to be dominant over cell line-specific methylation
patterns (19,20). Additionally, although genes
regulated by Aza and decitabine only show slight overlap (15,20),
both agents act synergistically with several cytotoxic drugs, e.g.
cisplatin (23–25,47).
These data suggest that there might be other or additional
underlying mechanisms than specific promoter CG demethylation. In
the present study, we only observed small changes in methylation
patterns due to the very low dose of Aza. A shift of the bimodal
methylation distribution was found in one of four cell lines.
Additional incubation time after the 24-h treatment did not lead to
major changes in DNA methylation distribution, but resulted in a
4.1-fold increase of significantly hypomethylated CpG sites in A549
cells. Nearly 10% of those CpG sites that were significantly
hypomethylated after 24 h maintaining significantly hypomethylated
after the extended treatment schedule (177/1778 CpG sites). These
data indicate certain consistency as well as time-dependence of low
dose 5-azacytidine induced methylation changes. Although we only
found minor DNA methylation changes, we observed distinct
synergistic activity which raises the question whether these
minimal changes in methylation already make the cells more
susceptible for cytotoxic agents, or whether there are further
mechanisms independent of methylation.
Aza is incorporated to a much greater extent into
RNA than into DNA (15). This
distribution suggests that the RNA-incorporation might also be
involved in synergistic Aza interactions. The enzyme ribonucleotide
reductase (RNR) plays a key role in DNA synthesis, cell growth and
DNA repair (48). The catalytic
M2-subunit (RRM2) is significant for the enzymatic function
(48) and appears to be frequently
overexpressed in malignant cells (49,50).
Its overexpression leads to the typically increased DNA synthesis
in tumors and mediates resistance against nucleoside analoga
(51). Aimiuwu et al
(52) observed that Aza treatment
attenuated RRM2 mRNA as well as total RNA stability and resulted in
reduced RNR protein expression. A decrease of RNR activity
downregulates the cell’s stock of desoxyribonucleoside
triphosphates (48) and therefore,
might increase the incorporation of nucleoside analogs like AraC
and gemcitabine. Additionally, Rauscher et al (53) described in an earlier study that
RNR inhibition enhances AraC activity. Increased incorporation of
nucleoside analogs based on Aza mediated RNR inhibition might be a
mechanism for synergistic drug interaction. Furthermore, nucleoside
analogs decrease RNR activity themselves by allosteric inhibition
of the M2-subunit (48,54). Cis interferes with the M1 subunit
and therefore inhibits RNR activity as well (55). Additional inhibition on the RNA
level might potentiate the suppression of RNR activity by AraC,
gemcitabine and cisplatin. Earlier studies also indicate that
synergistic interactions between cisplatin and DNMT inhibitors
might be based on methylation-independent mechanisms (32,56).
These data suggest that there might be other or additional
mechanisms besides DNA hypomethylation enhancing cytotoxic drug
activity.
In conclusion, in the present study, we found that
Aza is a promising agent for combinational anticancer drug therapy.
We used very low Aza concentrations to exclude its cytotoxic
activity but concentrated on its potential as an epigenetic agent.
We analyzed drug interaction between Aza and four other cytotoxic
drugs of different modes of action in four different cell lines and
observed synergistic reduced cell proliferation and colony
formation. In search for the underlying mechanisms, we found
neither increased apoptosis nor cell cycle alterations. 450K bead
arrays revealed only few changes in global methylation but
nevertheless several CG-sites that were significantly
hypomethylated after Aza treatment. The number of significantly
hypomethylated CG-sites increased time-dependently. Thus, although
formally only observing a correlation between synergistic effects
of low-dose Aza with the cytotoxic activity of various anticancer
drugs and its demethylizing activity at low doses, a causal
relationship between the demethylizing effects of Aza and this
synergy is a valid hypothesis to be further studied preclinically
and clinically.
The results of the present study could have
relevance for the design of new drug combinations for AML or NSCLC
therapy. Especially the observed synergistic interactions between
Aza and gemcitabine as well as cisplatin in addition to the
well-known synergisms between gemcitabine and cisplatin (57) indicate that a triple combination of
these agents might be of clinical interest in NSCLC therapy.
Acknowledgements
The present study was supported by the German
Ministry for Education and Science (grant no. BMBF 01GS0873), the
‘Deutsche Forschungsgemeinschaft’ (grant no. DFG MU 1328/9-1) and
the ‘Deutsche Krebshilfe’ (grant no. 110262).
References
|
1
|
SEER cancer statistics review (CSR)
1975–2010 [Internet]. June 14–2013, cited March 6th 2014].
Available from: http://seer.cancer.gov/statfacts/.
|
|
2
|
Jones PA and Baylin SB: The fundamental
role of epigenetic events in cancer. Nat Rev Genet. 3:415–428.
2002.PubMed/NCBI
|
|
3
|
Baylin SB and Herman JG: DNA
hypermethylation in tumorigenesis: epigenetics joins genetics.
Trends Genet. 16:168–174. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jones PA and Baylin SB: The epigenomics of
cancer. Cell. 128:683–692. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gama-Sosa MA, Slagel VA, Trewyn RW,
Oxenhandler R, Kuo KC, Gehrke CW and Ehrlich M: The
5-methylcytosine content of DNA from human tumors. Nucleic Acids
Res. 11:6883–6894. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Herman JG, Merlo A, Mao L, et al:
Inactivation of the CDKN2/p16/MTS1 gene is frequently associated
with aberrant DNA methylation in all common human cancers. Cancer
Res. 55:4525–4530. 1995.PubMed/NCBI
|
|
7
|
Esteller M: Epigenetics in cancer. N Engl
J Med. 358:1148–1159. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Herman JG and Baylin SB: Gene silencing in
cancer in association with promoter hypermethylation. N Engl J Med.
349:2042–2054. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pradhan S, Bacolla A, Wells RD and Roberts
RJ: Recombinant human DNA (cytosine-5) methyltransferase. I
expression, purification, and comparison of de novo and maintenance
methylation. J Biol Chem. 274:33002–33010. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bird A: DNA methylation patterns and
epigenetic memory. Genes Dev. 16:6–21. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Okano M, Xie S and Li E: Cloning and
characterization of a family of novel mammalian DNA (cytosine-5)
methyltransferases. Nat Genet. 19:219–220. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Okano M, Bell DW, Haber DA and Li E: DNA
methyltransferases Dnmt3a and Dnmt3b are essential for de novo
methylation and mammalian development. Cell. 99:247–257. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ehrlich M: DNA hypomethylation in cancer
cells. Epigenomics. 1:239–259. 2009. View Article : Google Scholar
|
|
14
|
Damiani LA, Yingling CM, Leng S, Romo PE,
Nakamura J and Belinsky SA: Carcinogen-induced gene promoter
hypermethylation is mediated by DNMT1 and causal for transformation
of immortalized bronchial epithelial cells. Cancer Res.
68:9005–9014. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hollenbach PW, Nguyen AN, Brady H, et al:
A comparison of azacitidine and decitabine activities in acute
myeloid leukemia cell lines. PLoS One. 5:e90012010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cihak A: Biological effects of
5-azacytidine in eukaryotes. Oncology. 30:405–422. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tsai HC, Li H, Van Neste L, et al:
Transient low doses of DNA-demethylating agents exert durable
antitumor effects on hematological and epithelial tumor cells.
Cancer Cell. 21:430–446. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hagemann S, Heil O, Lyko F and Brueckner
B: Azacytidine and decitabine induce gene-specific and non-random
DNA demethylation in human cancer cell lines. PLoS One.
6:e173882011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hascher A, Haase AK, Hebestreit K, et al:
DNA methyltransferase inhibition reverses epigenetically embedded
phenotypes in lung cancer preferentially affecting polycomb target
genes. Clin Cancer Res. 20:814–812. 2014. View Article : Google Scholar
|
|
20
|
Flotho C, Claus R, Batz C, et al: The DNA
methyltransferase inhibitors azacitidine, decitabine and zebularine
exert differential effects on cancer gene expression in acute
myeloid leukemia cells. Leukemia. 23:1019–1028. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vidaza. EPAR - product information
(Internet). January 3–2014, cited February 6, 2014. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000978/WC500050239.pdf.
|
|
22
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: the combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Festuccia C, Gravina GL, D’Alessandro AM,
et al: Azacitidine improves antitumor effects of docetaxel and
cisplatin in aggressive prostate cancer models. Endocr Relat
Cancer. 16:401–413. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tikoo K, Ali IY, Gupta J and Gupta C:
5-azacytidine prevents cisplatin induced nephrotoxicity and
potentiates anticancer activity of cisplatin by involving
inhibition of metallothionein, pAKT and DNMT1 expression in
chemical induced cancer rats. Toxicol Lett. 191:158–166. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Francia G, Green SK, Bocci G, et al:
Down-regulation of DNA mismatch repair proteins in human and murine
tumor spheroids: implications for multicellular resistance to
alkylating agents. Mol Cancer Ther. 4:1484–1494. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Efferth T, Futscher BW and Osieka R:
5-azacytidine modulates the response of sensitive and
multidrug-resistant K562 leukemic cells to cytostatic drugs. Blood
Cells Mol Dis. 27:637–648. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kong XB, Tong WP and Chou TC: Induction of
deoxycytidine kinase by 5-azacytidine in an HL-60 cell line
resistant to arabinosylcytosine. Mol Pharmacol. 39:250–257.
1991.PubMed/NCBI
|
|
28
|
Asano T, Nakamura K, Fujii H, et al:
Altered expression of topoisomerase IIalpha contributes to
cross-resistant to etoposide K562/MX2 cell line by aberrant
methylation. Br J Cancer. 92:1486–1492. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Murakami T, Li X, Gong J, Bhatia U,
Traganos F and Darzynkiewicz Z: Induction of apoptosis by
5-azacytidine: drug concentration-dependent differences in cell
cycle specificity. Cancer Res. 55:3093–3098. 1995.PubMed/NCBI
|
|
30
|
Cross M and Dexter TM: Growth factors in
development, transformation, and tumorigenesis. Cell. 64:271–280.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Khan R, Schmidt-Mende J, Karimi M, et al:
Hypomethylation and apoptosis in 5-azacytidine-treated myeloid
cells. Exp Hematol. 36:149–157. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Abbruzzese JL and Frost P: Studies on the
mechanism of the synergistic interaction between
2′-deoxy-5-azacytidine and cisplatin. Cancer Chemother Pharmacol.
30:31–36. 1992. View Article : Google Scholar
|
|
33
|
Qin T, Youssef EM, Jelinek J, Chen R, Yang
AS, Garcia-Manero G and Issa JP: Effect of AraC and decitabine in
combination in human leukemic cell lines. Clin Cancer Res.
13:4225–4232. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Issa JP and Kantarjian HM: Targeting DNA
methylation. Clin Cancer Res. 15:3938–3946. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Plumb JA, Strathdee G, Sludden J, Kaye SB
and Brown R: Reversal of drug resistance in human tumor xenografts
by 2′-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene
promoter. Cancer Res. 60:6039–6044. 2000.PubMed/NCBI
|
|
36
|
Gius D, Cui H, Bradbury CM, et al:
Distinct effects on gene expression of chemical and genetic
manipulation of the cancer epigenome revealed by a multimodality
approach. Cancer Cell. 6:361–371. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gu C, Lu J, Cui T, et al: Association
between MGMT promoter methylation and non-small cell lung cancer: a
meta-analysis. PLoS One. 8:e726332013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Su Y, Xu H, Xu Y, Yu J, Xian Y and Luo Q:
Azacytidine inhibits the proliferation of human promyelocytic
leukemia cells (HL60) by demethylation of MGMT, DAPK and p16 genes.
Hematology. 17:41–46. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dmitriev AA, Kashuba VI, Haraldson K, et
al: Genetic and epigenetic analysis of non-small cell lung cancer
with NotI-microarrays. Epigenetics. 7:502–513. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dunwell TL, Hesson LB, Pavlova T, et al:
Epigenetic analysis of childhood acute lymphoblastic leukemia.
Epigenetics. 4:185–193. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Addou-Klouche L, Adelaide J, Finetti P, et
al: Loss, mutation and deregulation of L3MBTL4 in breast cancers.
Mol Cancer. 9:213PubMed/NCBI
|
|
42
|
Chen SC, Lin CY, Chen YH, et al: Aberrant
promoter methylation of EDNRB in lung cancer in Taiwan. Oncol Rep.
15:167–172. 2006.
|
|
43
|
Toyooka KO, Toyooka S, Virmani AK, et al:
Loss of expression and aberrant methylation of the CDH13
(H-cadherin) gene in breast and lung carcinomas. Cancer Res.
61:4556–4560. 2001.PubMed/NCBI
|
|
44
|
Finger EC, Turley RS, Dong M, How T,
Fields TA and Blobe GC: TbetaRIII suppresses non-small cell lung
cancer invasiveness and tumorigenicity. Carcinogenesis. 29:528–535.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Deng G, Nguyen A, Tanaka H, et al:
Regional hypermethylation and global hypomethylation are associated
with altered chromatin conformation and histone acetylation in
colorectal cancer. Int J Cancer. 118:2999–3005. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ehrlich M: DNA methylation in cancer: too
much, but also too little. Oncogene. 21:5400–5413. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shang D, Liu Y, Matsui Y, Ito N, Nishiyama
H, Kamoto T and Ogawa O: Demethylating agent 5-aza-2′-deoxycytidine
enhances susceptibility of bladder transitional cell carcinoma to
cisplatin. Urology. 71:1220–1225. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Eklund H, Uhlin U, Farnegardh M, Logan DT
and Nordlund P: Structure and function of the radical enzyme
ribonucleotide reductase. Prog Biophys Mol Biol. 77:177–268. 2001.
View Article : Google Scholar
|
|
49
|
Lu AG, Feng H, Wang PX, Han DP, Chen XH
and Zheng MH: Emerging roles of the ribonucleotide reductase M2 in
colorectal cancer and ultraviolet-induced DNA damage repair. World
J Gastroenterol. 18:4704–4713. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Morikawa T, Hino R, Uozaki H, et al:
Expression of ribonucleotide reductase M2 subunit in gastric cancer
and effects of RRM2 inhibition in vitro. Hum Pathol. 41:1742–1748.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Klisovic RB, Blum W, Wei X, et al: Phase I
study of GTI-2040, an antisense to ribonucleotide reductase, in
combination with high-dose cytarabine in patients with acute
myeloid leukemia. Clin Cancer Res. 14:3889–3895. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Aimiuwu J, Wang H, Chen P, et al:
RNA-dependent inhibition of ribonucleotide reductase is a major
pathway for 5-azacytidine activity in acute myeloid leukemia.
Blood. 119:5229–5238. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Rauscher F III and Cadman E: Biochemical
and cytokinetic modulation of L1210 and HL-60 cells by hydroxyurea
and effect on 1-beta-D-arabinofuranosylcytosine metabolism and
cytotoxicity. Cancer Res. 43:2688–2693. 1983.PubMed/NCBI
|
|
54
|
Gray SG, Baird AM, O’Kelly F, et al:
Gemcitabine reactivates epigenetically silenced genes and functions
as a DNA methyl-transferase inhibitor. Int J Mol Med. 30:1505–1511.
2012.PubMed/NCBI
|
|
55
|
Smith SL and Douglas KT: Stereoselective,
strong inhibition of ribonucleotide reductase from E. coli by
cisplatin. Biochem Biophys Res Commun. 162:715–723. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ellerhorst JA, Frost P, Abbruzzese JL,
Newman RA and Chernajovsky Y: 2′-deoxy-5-azacytidine increases
binding of cisplatin to DNA by a mechanism independent of DNA
hypo-methylation. Br J Cancer. 67:209–215. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Bergman AM, Ruiz van Haperen VW, Veerman
G, Kuiper CM and Peters GJ: Synergistic interaction between
cisplatin and gemcitabine in vitro. Clin Cancer Res. 2:521–530.
1996.PubMed/NCBI
|