Introduction
Arachidonic acids are converted into prostaglandin
(PG) H2, which is the precursor of eicosanoids,
including PGs, prostacyclin (PGI2), and thromboxanes
(TXs), in a reaction that is catalyzed by cyclooxygenases (COXs).
There are three COX isoenzymes: COX-1, −2, and −3. COX-1 is
constitutively expressed in most tissues, and plays an important
role in protecting the gastric mucosa, regulating platelet
aggregation, and maintaining renal blood flow (1). COX-3 was initially identified as an
alternatively spliced variant of COX-1 in dogs (2). Although COX-3 is a potential target
of the antipyretic and analgesic effects of acetaminophen, its
detailed function remains unclear (2). In contrast, it is known that
cyclooxygenase-2 (COX-2) is induced by various stimuli such as
pro-inflammatory cytokines during inflammation or the initiation
and progression of cancer. In cancer, it is suspected that COX-2
plays an important role in angiogenesis, invasion, apoptosis
resistance, immune evasion, and drug resistance (3–9).
Therefore, it might play a role in the antitumor effects of
non-steroidal anti-inflammatory drugs (NSAIDs). Selective COX-2
inhibitors have been developed to reduce gastrointestinal
dysfunction, and for chemotherapy or chemoprevention in various
human cancers. The antitumor effects of selective COX-2 inhibitors
are exerted via diverse means, including COX-2-dependent and
-independent mechanisms and the activation of intrinsic and
extrinsic apoptotic pathways. However, the detailed mechanism of
action of the antitumor effects of selective COX-2 inhibitors in
various cancers remains controversial.
Canine mammary tumors are the most common tumors in
female dogs without contraception, and approximately half of all
cases are malignant. However, it is difficult to diagnose malignant
canine mammary tumors histopathologically (10). Accordingly, several studies have
explored molecular markers to diagnose or treat malignant canine
mammary tumors. Among these, COX-2 has received significant
attention as a diagnostic and therapeutic target (11–13).
Furthermore, previous reports suggested that canine mammary tumors
could be a suitable model for studying human breast cancer
(14).
COX-2 overexpression has been reported in human
breast cancer (15). Similarly,
COX-2 expression was elevated in canine mammary tumors compared
with normal mammary tissue (16).
In particular, a previous study demonstrated that although no
expression was detected in the normal mammary gland, COX-2 was
expressed in 56 and 24% of adenocarcinoma and adenoma samples,
respectively (16). This report
also suggested that COX-2-positive tumor cells might have a higher
malignant tendency (16).
Furthermore, a previous study reported a correlation between
vascular endothelial growth factor (VEGF) and COX-2 levels; the
enhanced production of VEGF resulted in increased intra-tumoral
microvessel density (17). These
findings suggest that COX-2 might be a potential marker for poor
prognosis and a target for chemotherapy in canine mammary tumors.
We previously demonstrated the usefulness of selective COX-2
inhibitors as therapeutic agents in canine mammary tumor (CF33)
cells, which express high levels of COX-2 (18). However, the detailed mechanism of
action of celecoxib in canine mammary tumor is still not completely
understood.
Our previous study suggested that selective COX-2
inhibitors exert potential antitumor effects via both
COX-2-dependent and -independent mechanisms (18). Accordingly, the aim of this study
was to explore the detailed mechanism of action of selective COX-2
inhibitors in canine mammary tumor cells. We analyzed their
antitumor effects in AZACB canine mammary tumor cells, which
express low levels of COX-2, to minimize the effect of COX-2.
Furthermore, we used three different inhibitors (celecoxib,
etodolac, and meloxicam) that are highly selective for COX-2; these
inhibitors have potential clinical utility as analgesics and
anti-inflammatory agents in osteoarthritic dogs (19,20).
Materials and methods
Chemicals
We used meloxicam, etodolac, and celecoxib to assess
the antitumor effect of selective COX-2 inhibitors. Celecoxib and
etodolac were purchased from Sigma-Aldrich (Tokyo, Japan), and
meloxicam was obtained from Wako Pure Chemicals Industries, Ltd.,
(Osaka, Japan). 2,5-Dimethyl-celecoxib (DMC), a structural analog
of celecoxib, was purchased from Sigma-Aldrich. All the drugs were
dissolved in 100% DMSO (Wako Pure Chemicals Industries, Ltd.) at
different concentrations and stored at −20°C. Control cells were
treated with DMSO at a final concentration of 0.1%, whereas parent
cells were untreated. The following antibodies were used in the
current study: anti-COX-2 (Abcam, Tokyo, Japan), anti-β-actin
(Sigma-Aldrich), anti-p27 kip1 (BD Transduction Laboratories,
Tokyo, Japan), anti-Bax (Millipore, Billerica, MA, USA), and
anti-Bid (Abnova, Taipei, Taiwan). All other antibodies were
purchased from Cell Signaling Technology, Inc. (Tokyo, Japan). The
inhibitors caspase-8 (Z-IETD-FMK) and caspase-9 (Z-LEHD-FMK) were
obtained from R&D Systems (Minneapolis, MN, USA), and were
dissolved in 100% DMSO and stored at −20°C.
Cell lines and culture conditions
AZACB cells were purchased from Primary Cell Co.,
Ltd. (Hokkaido, Japan), CF33, and CF41.MG cells were purchased from
American Type Culture Collection (Manassas, VA, USA). The cells
were cultured in Dulbecco’s modified Eagle’s medium (Nissui
Pharmaceutical Co., Ltd., Tokyo, Japan) containing 10%
heat-inactivated fetal bovine serum (FBS), 4 mM L-glutamine, 10
mg/ml streptomycin, and 10,000 U/ml penicillin G at 37°C in a 5%
CO2 incubator. AZACB, CF33, and CF41.MG cells were
cultured as described previously (18,21,22).
Western blotting
Cells were lysed in radioimmunoprecipitation assay
buffer containing 25 mM Tris-HCl (pH 7.6), 150 mM NaCl, 1% NP-40,
0.1% SDS, 1% sodium deoxycholate, and various protease inhibitors
(1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 μg/ml aprotinin, 1 mM
dithiothreitol, 1 mM NaVO4, and 0.5 mM
phenylmethylsulfonyl fluoride). Whole cell lysates were prepared as
described previously (23). The
protein concentrations of the cell lysates were then quantified
using the Bradford method with a Pierce® BCA Protein
Assay kit (Pierce Biotechnology, Inc., Rockford, IL, USA). Total
cell lysates (15–25 μg) were boiled for 5 min in 2X Laemmli sample
loading buffer, and were then separated by SDS-PAGE on 12% (p27,
Bax, Bim, and Bid) and 10% (COX-2) gels. The gels were then
transferred to polyvinylidene difluoride membranes (Bio-Rad
Laboratories, Tokyo, Japan). The expression of specific proteins
was detected by electrochemiluminescence using WesternBright
Quantum or WesternBright Sirius (Advansta, Menlo Park, CA, USA),
and observed using ChemiDoc XRS (Bio-Rad Laboratories).
Cell viability assays
AZACB and CF33 cells were plated into 96-well plates
(BD Falcon; Nippon Becton Dickinson, Tokyo, Japan) at a density of
2.5×103 and 1×103 cells/well, respectively.
After 24 h, the cells were treated with different concentrations of
selective COX-2 inhibitors or DMC. Twenty-four hours after
treatment, cell number was determined using a WST-8 assay (Cell
Counting kit-8; Dojindo Laboratories, Kumamoto, Japan) according to
the manufacturer’s instructions. The absorbance was measured at 450
nm using a Benchmark Plus microplate reader (Bio-Rad Laboratories).
The experiment was performed using five replicates. As a control,
it was confirmed that there were no changes in cell viability
before drug treatment (day 0).
Measurements of prostaglandin
E2 (PGE2)
AZACB cells were plated into 100-mm tissue culture
dishes (BD Falcon; Nippon Becton Dickinson) at a density of
9.0×105 cells/dish. After 24 h, the cells were treated
with 100 μM celecoxib in culture medium containing 2 or 10% FBS.
Twenty-four hours later, culture media samples were collected. They
were then centrifuged immediately at 500 × g for 5 min at 4°C to
remove cells or debris, and the supernatants were harvested. The
concentration of PGE2 in the culture medium was measured
using a PGE2 enzyme immunoassay kit - Monoclonal (Cayman
Chemical Co., Ann Arbor, MI, USA) following the manufacturer’s
instructions. The absorbance at 405 nm was measured using a
Benchmark Plus microplate reader (Bio-Rad Laboratories). The
experiment was performed in triplicate.
Flow cytometric cell cycle analysis
AZACB cells were seeded at a density of
5.0×105 cells in 100-mm tissue culture dishes (BD
Falcon; Nippon Becton Dickinson). After 24 h of exposure to
selective COX-2 inhibitors (10, 25, 50 and 100 μM meloxicam and
etodolac, and 10, 25, 45, 50, 75 and 100 μM celecoxib), AZACB cells
were harvested and washed with PBS, resuspended in 70% ethanol in
PBS, and frozen at −30°C overnight. Before analysis, the cells were
incubated for 15 min in propidium iodide (PI)/RNase Staining Buffer
(BD Pharmingen, San Diego, CA, USA) in the dark. The suspension was
then filtered into a 5-ml polystyrene round-bottomed tube with a
cell-strainer cap and was analyzed using FACSCanto (both from
Becton-Dickinson, Franklin Lakes, NJ, USA). Data analyses were
performed using FlowJo 7 (Tree Star, Inc., Ashland, OR, USA).
Assessing changes in mitochondrial
potential
Mitochondrial permeability and membrane
depolarization were measured using a MitoPT®
tetramethylrhodamine ethyl ester (TMRE) assaykit (Immuno Chemistry
Technologies, LLC, Bloomington, MN, USA) according to the
manufacturer’s instructions. AZACB cells were seeded into 100-mm
dishes (BD Falcon; Nippon Becton Dickinson) at a density of
3×105 cells/dish. After 24 h of treatment with selective
COX-2 inhibitors, the cells were exposed to TMRE. The mitochondrial
fluorescence intensity was measured using FACSCanto (488 nm
excitation and 574 nm emission), and data were analyzed using
FlowJo 7 (Tree Star, Inc.).
Annexin V/PI staining and flow
cytometry
The different stages of apoptosis were analyzed
using the ApoAlert® Annexin V-fluorescein isothiocyanate
(FITC) Apoptosis kit (Clontech Laboratories, Mountain View, CA,
USA) following the manufacturer’s instructions. Both adherent and
non-adherent cells were harvested using 0.25% of trypsin, and then
centrifuged at 1,200 rpm for 5 min. The cell pellets were then
washed and resuspended in binding buffer, and then incubated with
Annexin V-FITC and PI for 15 min in the dark at room temperature.
The samples were analyzed using FACSCanto, and the data were
analyzed using FlowJo 7.
Real-time reverse
transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated from AZACB cells using TRIzol
reagent (Life Technologies, Carlsbad, CA, USA), and was reverse
transcribed to cDNA using PrimeScript™ RT kit (Takara Bio, Inc.,
Shiga, Japan) following the manufacturer’s instructions as
described previously (16,19–21).
Real-time PCR was performed using SYBR Premix Ex Taq™ II (Takara
Bio, Inc.), an ABI Prism 7500 Real-Time PCR system (Applied
Biosystems, Inc., Foster City, CA, USA) and the following
conditions: 95°C for 30 sec, and 40 cycles of 95°C for 5 sec and
60°C for 34 sec. Glyceraldehyde-3-phosphate dehydrogenase
(GAPDH) expression was used as an internal control. The
primer sequences used to amplify p21, p27,
Bcl-2, and GAPDH are shown in Table I. The primers for Bcl-2 were
purchased from Takara Bio, Inc., and the other primers were
obtained from Operon Biotechnology (Tokyo, Japan). All samples were
amplified in triplicate in each experiment. The relative expression
levels of mRNA were calculated using the comparative threshold
cycle (Ct) method.
 | Table IReal-time RT-PCR primer
sequences. |
Table I
Real-time RT-PCR primer
sequences.
| Gene | Forward
(5′→3′) | Reverse
(5′→3′) |
|---|
| GAPDH |
ATTCTATCCACGGCAAATCC |
GGACTCCACAACATACTCAG |
| p21 |
CCTAATCTGCTCACCGGAAG |
GGTGGCAAGCAGGGTATGTA |
| p27 |
CTCAGGCCAACTCAGAGGAC |
TCTTAGGCGTCTGCTCCACT |
| Bcl-2 |
TGAACCGGCATCTGCACAC |
GAGCAGCGCCTTCAGAGACA |
Measuring caspase-3/7, −8, and −9
activity
Caspase-3/7, −8, and −9 activity was analyzed using
Caspase-Glo® 3/7, 8 and 9 assay kits (Promega Corp.,
Madison, WI, USA), respectively, following the manufacturer’s
instructions. Cells were cultured in white opaque tissue culture
plates (BD Falcon; Nippon Becton Dickinson) at a density of
2.5×103 cells/well. Twenty-four hours after drug
treatment (100 μM meloxicam and etodolac, or 10, 25, 40, 50, 75,
and 100 μM celecoxib), the fluorescence was measured every 30 min
for up to 180 min using the LB 960 Microplate Luminometer Centro
(Berthold Japan K.K., Tokyo, Japan). All samples were measured in
triplicate in each experiment.
Statistical analysis
Data are presented as means ± SD. Statistical
analyses were performed using the Bonferroni test or Mann-Whitney
test to identify significant differences between the selective
COX-2 inhibitor-treated cells and control cells. P<0.05 was
considered statistically significant.
Results
Celecoxib inhibits the proliferation of
AZACB cells
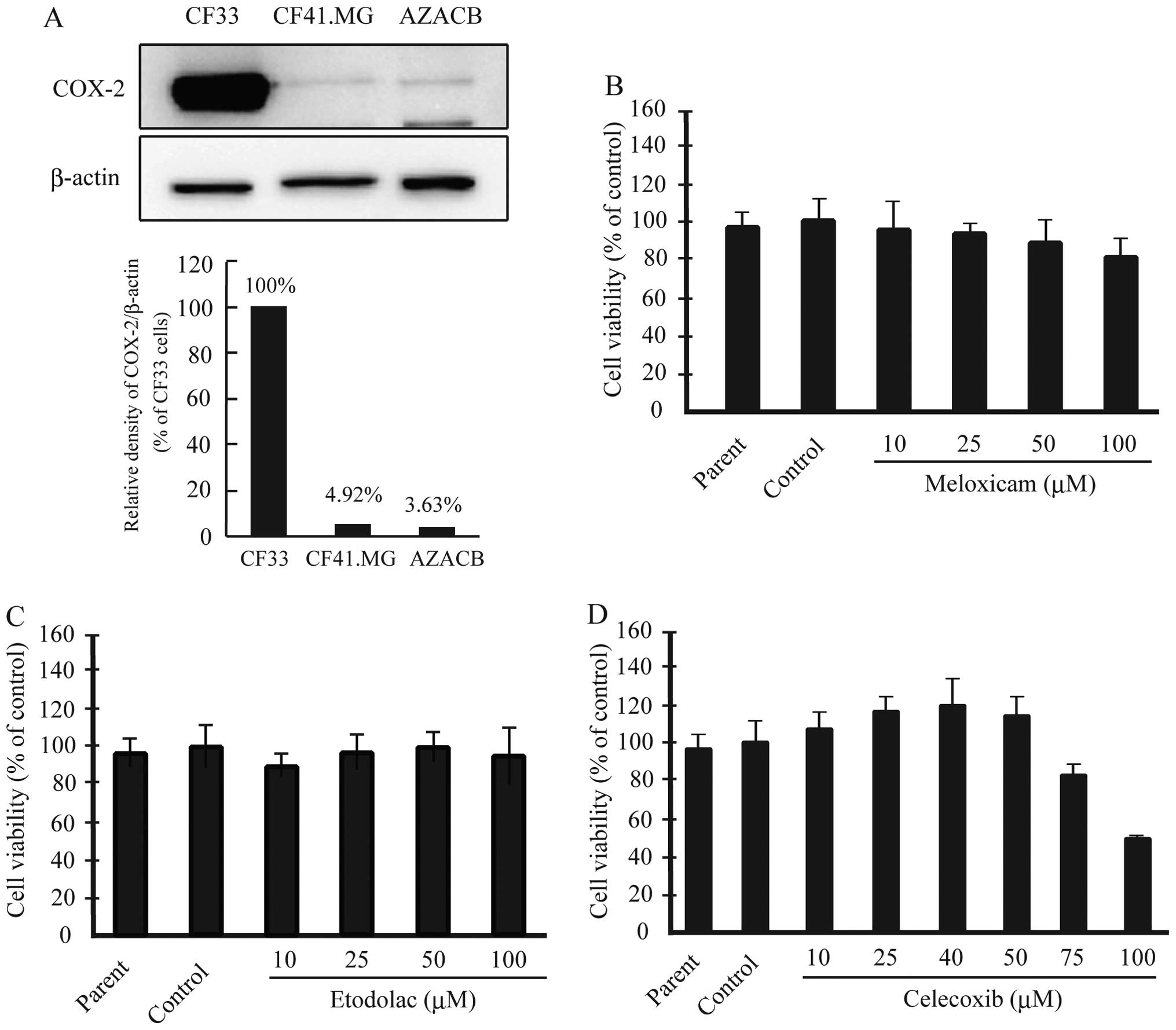
We reported previously that AZACB cells expressed
lower levels of COX-2 protein than CF33 cells (18). In the current study, we confirmed
that AZACB cells expressed the lowest levels of COX-2 among various
canine mammary tumor cell lines, including CF33 and CF41.MG cells
(Fig. 1A). Next, we assessed the
effect of the selective COX-2 inhibitors meloxicam, etodolac, and
celecoxib on cell viability to determine whether they inhibited the
proliferation of AZACB cells. We measured cell proliferation using
WST-8 assays 24 h after treatment with the selective COX-2
inhibitors. There was no difference in proliferation between the
parent and control cells. As shown in Fig. 1D, 75 and 100 μM celecoxib
significantly induced growth arrest 24 h after treatment compared
with control cells; however, meloxicam and etodolac had no
significant effect (Fig. 1B and
C). These results suggest that celecoxib markedly inhibited the
proliferation of AZACB cells.
Celecoxib inhibits the proliferation of
AZACB cells mainly via COX-2-independent mechanisms
Numerous reports have suggested that NSAIDs exert
antitumor effects on human cancer cells via COX-2-independent
mechanisms (24). Next, we
evaluated whether celecoxib inhibited cell proliferation in a
COX-2-dependent or -independent manner by examining the effect of
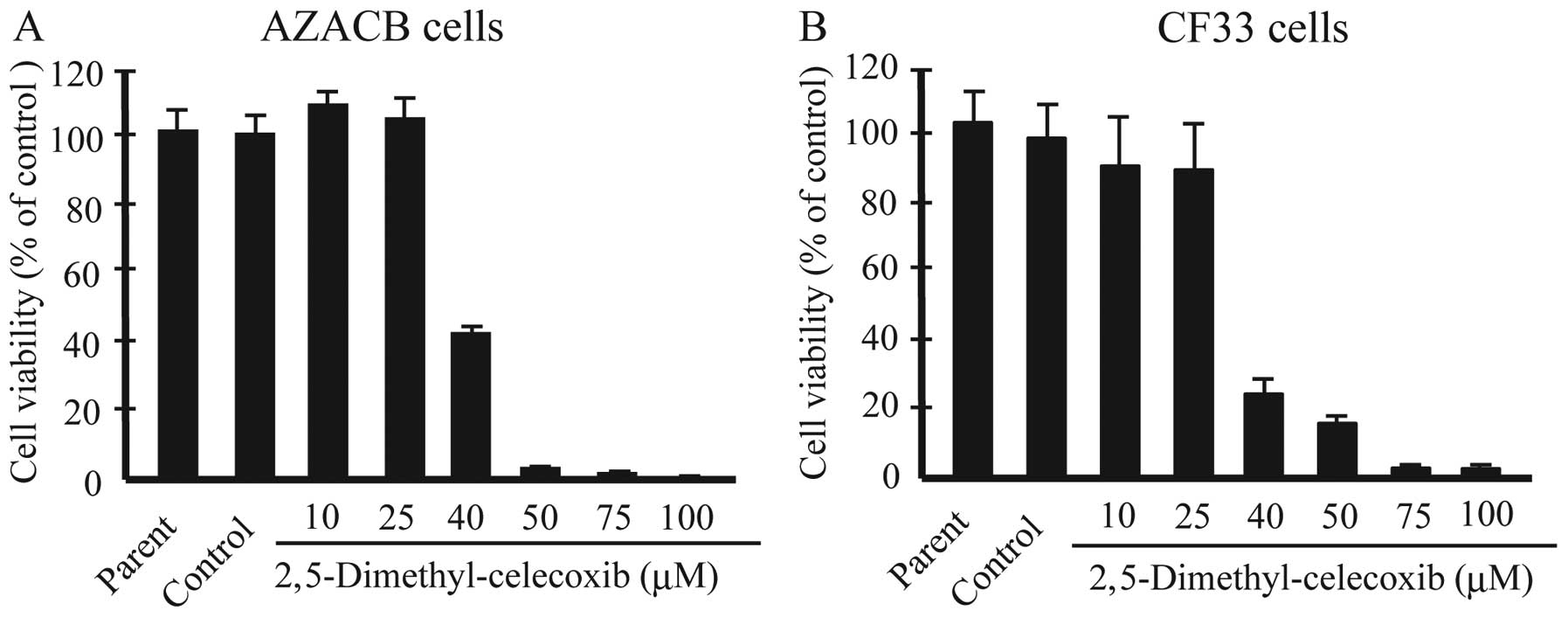
DMC on the proliferation of canine mammary tumor cells. DMC is a
structural isomer of celecoxib that lacks COX-2-inhibitory activity
(25). As shown in Fig. 2A, DMC inhibited the proliferation
of AZACB and CF33 cells, which express low and high levels of
COX-2, respectively (Fig. 2B).
This suggests that celecoxib inhibited the proliferation of canine
mammary tumor cells via COX-2-independent mechanisms.
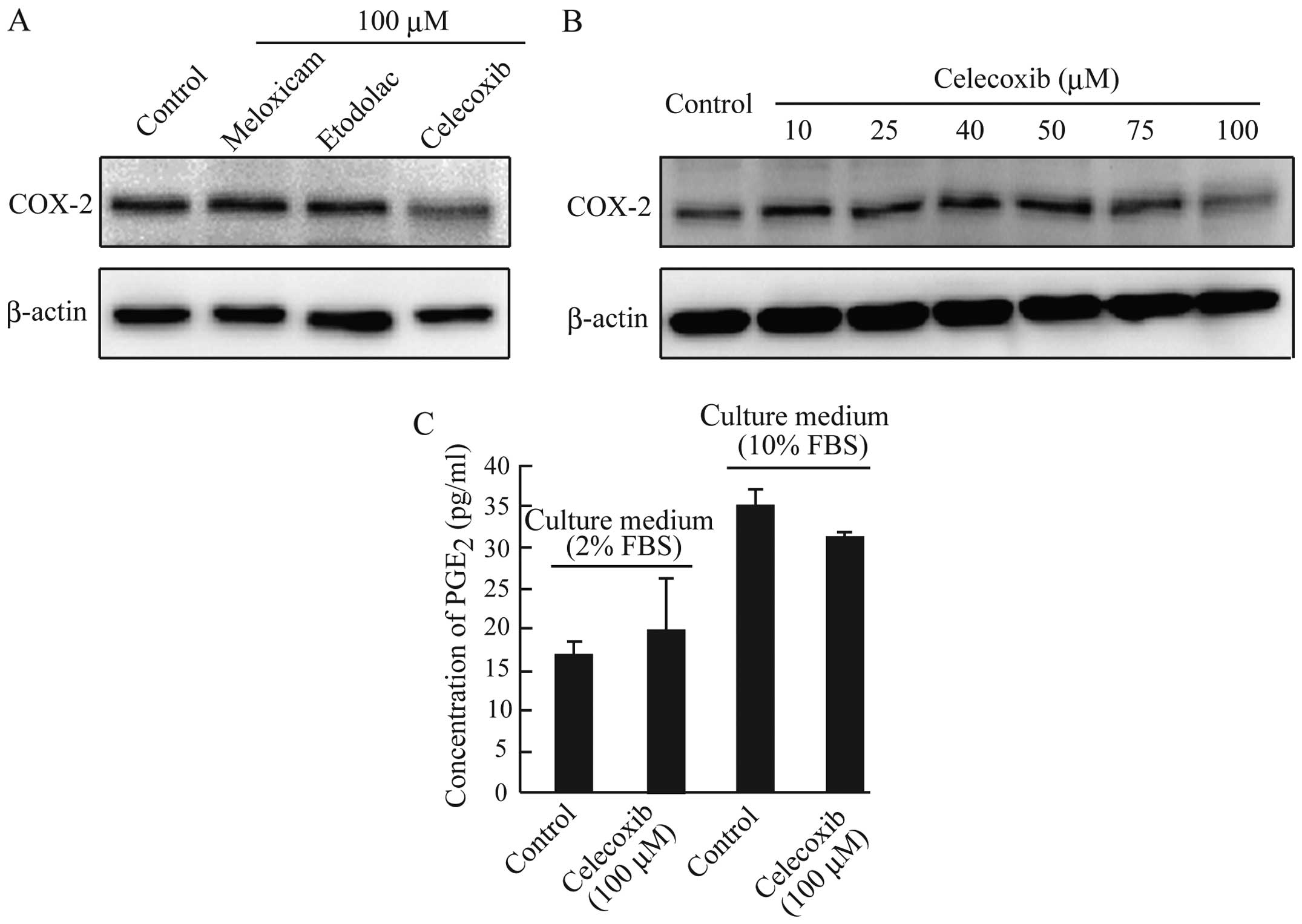
We demonstrated previously that celecoxib
downregulated the expression of COX-2 in CF33 cells (16). As shown in Fig. 3A and B, COX-2 protein expression
was reduced only in AZACB cells treated with 100 μM celecoxib. It
is well known that COX-2 catalyzes the production of PGs such as
PGE2. However, despite the reduced expression of COX-2,
no significant changes in PGE2 production were observed
in celecoxib-treated AZACB cells compared with control cells
(Fig. 3C). These results suggest
that celecoxib-induced growth inhibition in AZACB cells is mediated
mainly by COX-2-independent mechanisms. Furthermore, the unchanged
PGE2 secretion after celecoxib treatment might be caused
by low levels of COX-2 expression compared with other canine
mammary tumor cells. These results suggest that celecoxib might
affect the expression and/or activation of proteins such as NF-κB,
which regulates COX-2 expression (24,26–29).
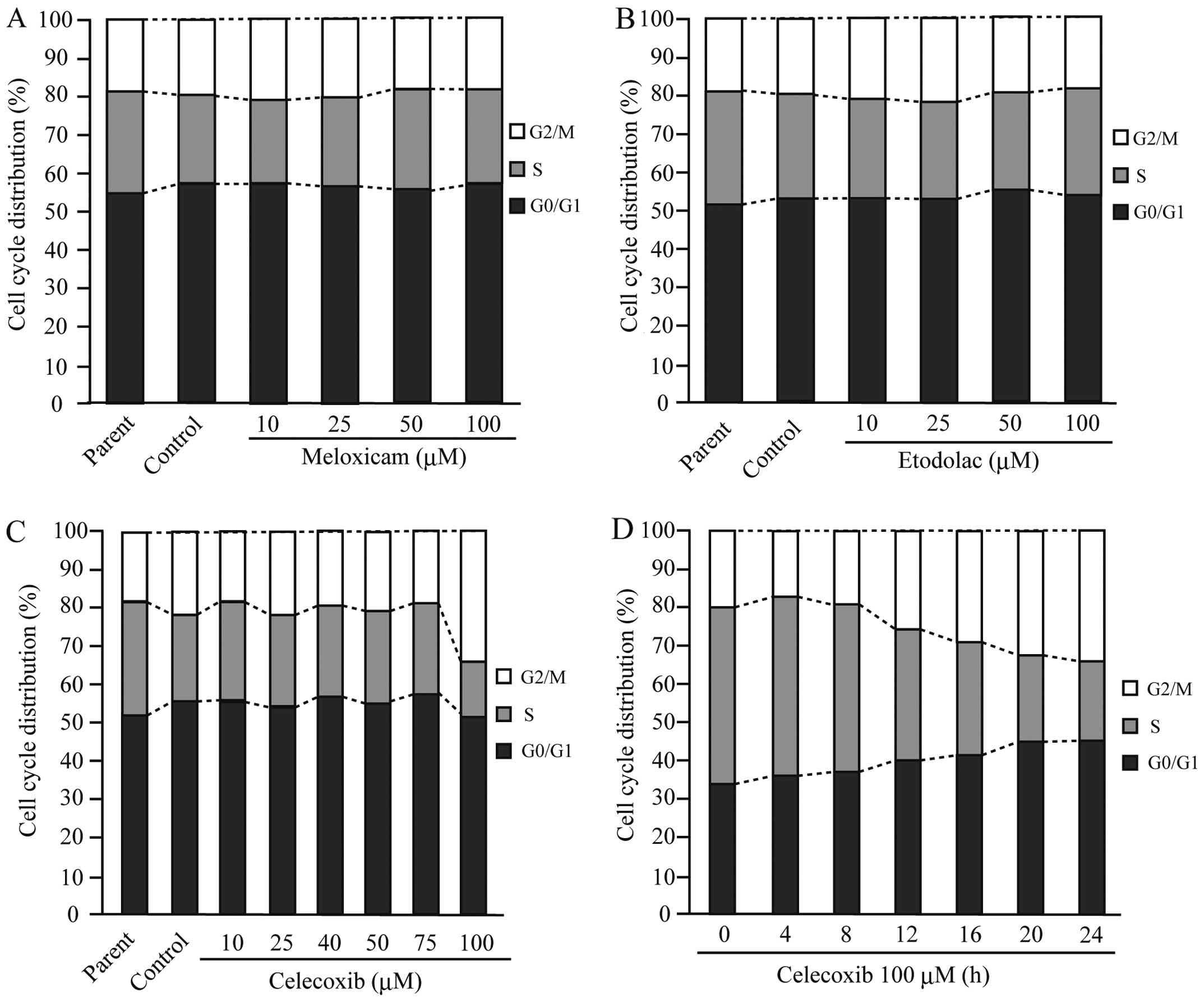
Celecoxib decreased the number of cells
in S phase and increased G2/M arrest by upregulating p21 and
p27
We demonstrated previously that celecoxib treatment
reduced the number of CF33 cells in S phase and increased those in
G0/G1 (18). In addition,
meloxicam and etodolac slightly induced G0/G1 arrest (18). As shown in Fig. 4A and B, there was no significant
change in the cell cycle distribution patterns in AZACB cells
treated with etodolac and meloxicam. However, treatment with 100 μM
celecoxib markedly induced G2/M arrest and decreased the number of
cells in S phase (Fig. 4C).
Furthermore, this effect occurred after 12 h of treatment (Fig. 4D). To confirm that celecoxib
induced cell cycle arrest at the G2/M phase, we next evaluated the
expression of the cyclin-dependent kinase inhibitors (CDKIs)
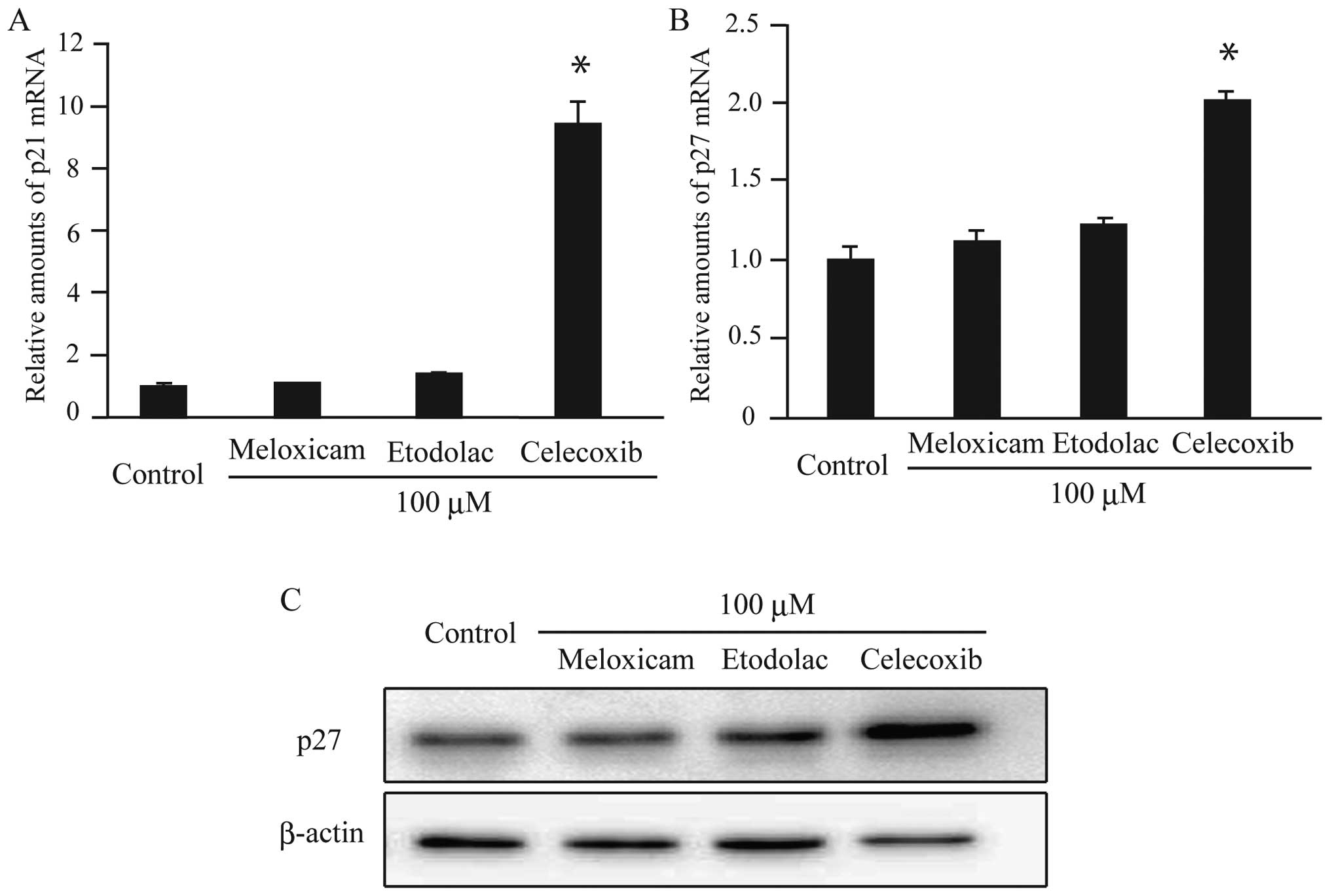
p21 and p27. There were no changes in the expression
of p21 and p27 in meloxicam- or etodolac-treated
cells (Fig. 5A–C). In contrast,
treatment with celecoxib increased the levels of both p21
and p27 in AZACB cells (Fig.
5A–C). Therefore, these data suggest that celecoxib induced
G2/M arrest and reduced the number of AZACB cells in S phase by
increasing the expression of CDKIs, including p21 and
p27.
Treatment with 100 μM celecoxib markedly
induced AZACB cell apoptosis
We recently reported that celecoxib markedly
inhibited the proliferation of CF33 canine mammary tumor cells by
inducing apoptosis (18).
Therefore, we next assessed the effects of meloxicam, etodolac, and
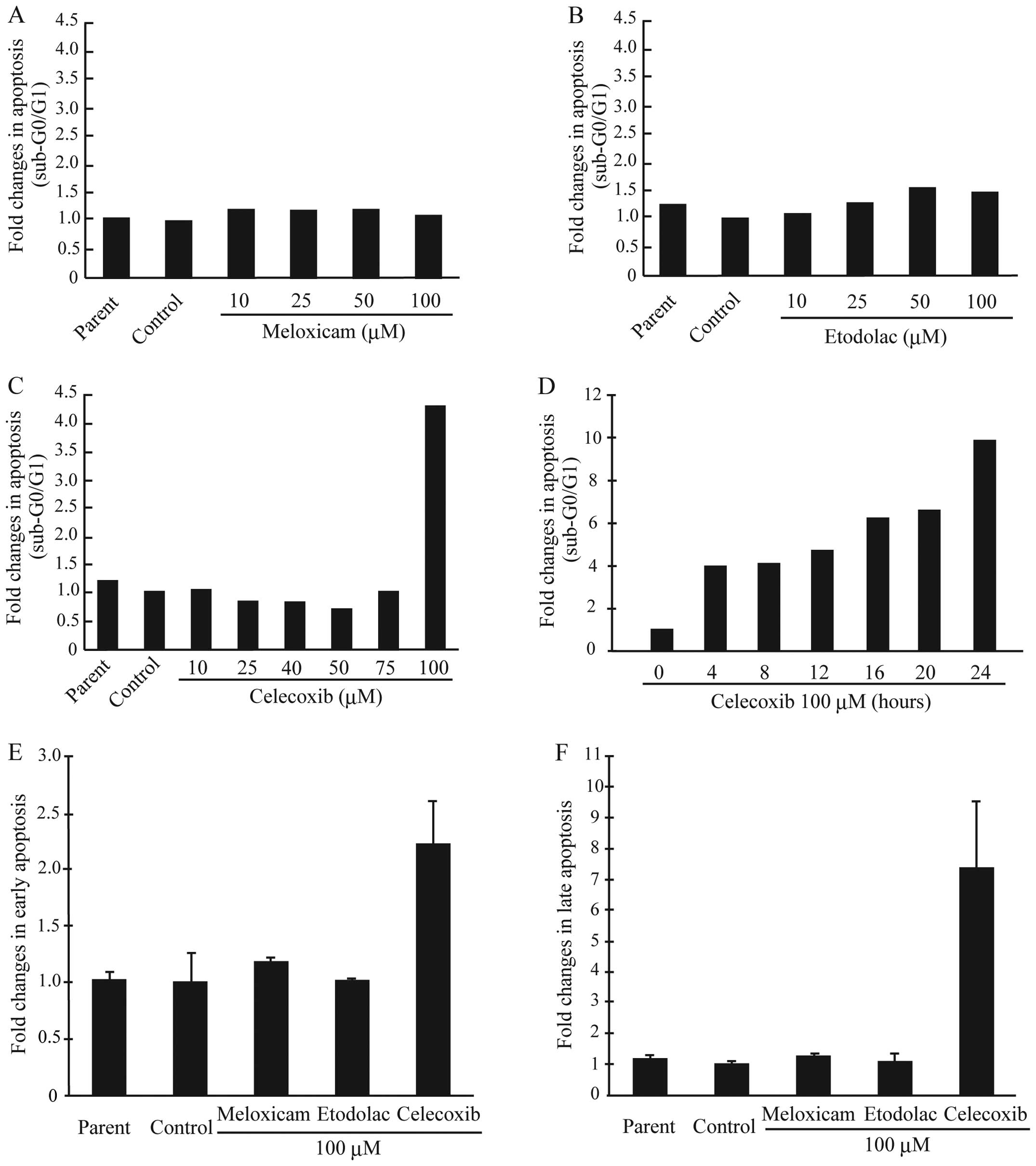
celecoxib on apoptosis in AZACB cells. As shown in Fig. 6A and B, there were no changes in
apoptosis in meloxicam- or etodolac-treated AZACB cells. However,
as expected, treating cells with 100 μM celecoxib induced apoptosis
(Fig. 6C); these effects were
time-dependent (Fig. 6D). To
confirm these observations, we next analyzed apoptosis using
Annexin V/PI double staining. Data revealed that treatment with 100
μM celecoxib for 24 h induced both early and late apoptosis
(Fig. 6E and F). As expected,
treating AZACB cells with 100 μM meloxicam or etodolac had no
effect on either early or late apoptosis (Fig. 6E and F).
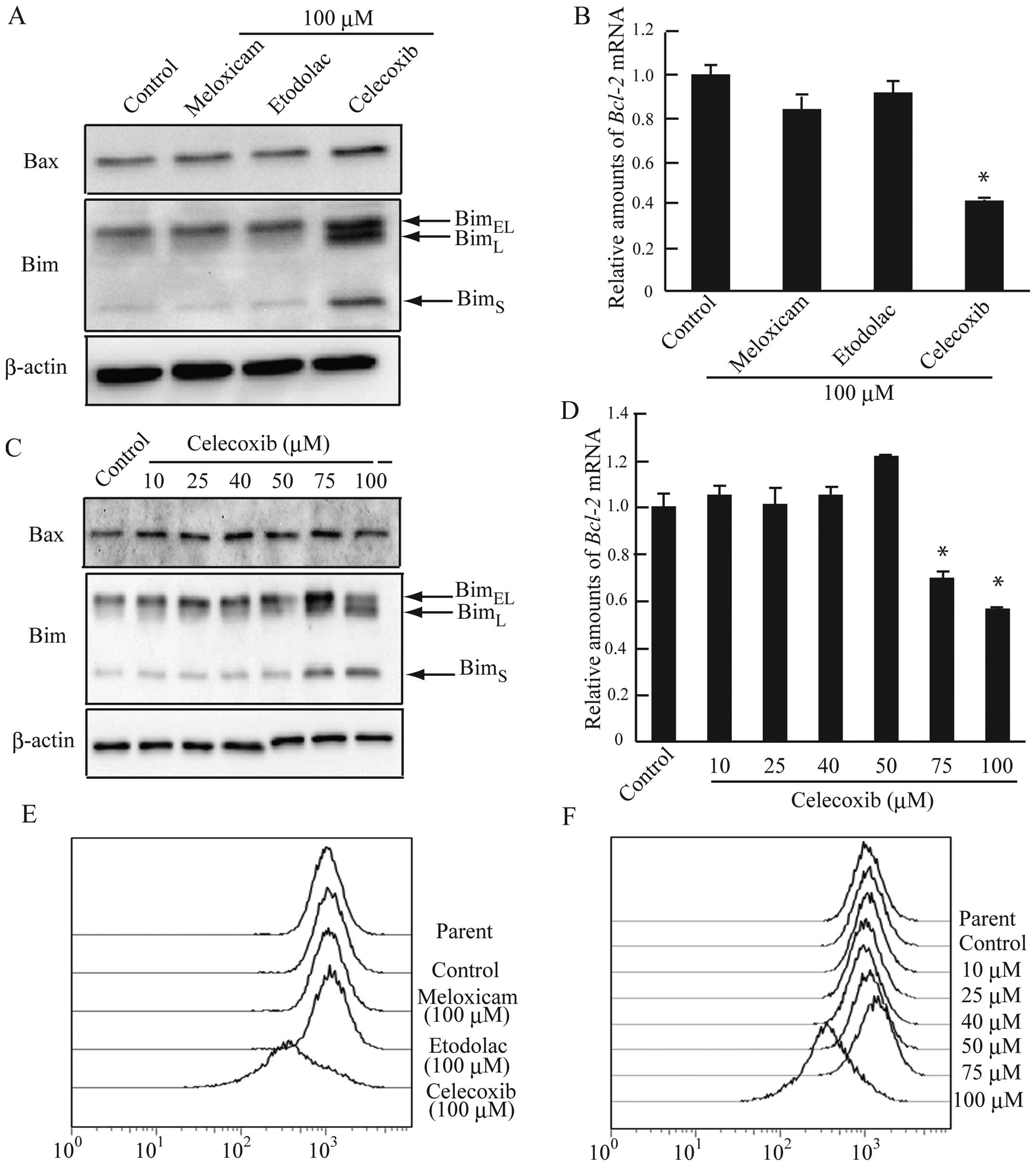
The imbalance between pro-apoptotic (Bax, Bim, and
Bak) and anti-apoptotic proteins (Bcl-2 and Bcl-xL) leads to
apoptosis by stimulating mitochondrial outer membrane
permeabilization (MOMP) (30).
Accordingly, we assessed changes in the expression of apoptotic
proteins in celecoxib-treated AZACB cells. Celecoxib-treated AZACB
cells exhibited elevated Bax and Bim expression, and reduced
Bcl-2 expression (Fig.
7A–D). However, levels of Bax were only slightly elevated in
celecoxib-treated AZACB cells (Fig. 7A
and C). These changes were induced by treatment with ≥75 μM
celecoxib (Fig. 7C and D). It is
known that MOMP leads to a decrease in mitochondrial membrane
potential (31). Therefore, we
analyzed changes in mitochondrial membrane potential using flow
cytometric analysis of TMRE-stained cells to directly measure
whether celecoxib induced MOMP. Our results showed that treating
cells with 100 μM celecoxib, but not meloxicam and etodolac,
decreased mitochondrial membrane potential (Fig. 7E). Moreover, the decrease in
mitochondrial membrane potential was observed only in AZACB cells
treated with 100 μM celecoxib, and not lower doses (Fig. 7F). These findings confirm that
celecoxib induces apoptosis in AZACB cells, which express low
levels of COX-2.
Celecoxib induces apoptosis in AZACB
cells by activating both the intrinsic and extrinsic apoptotic
pathways
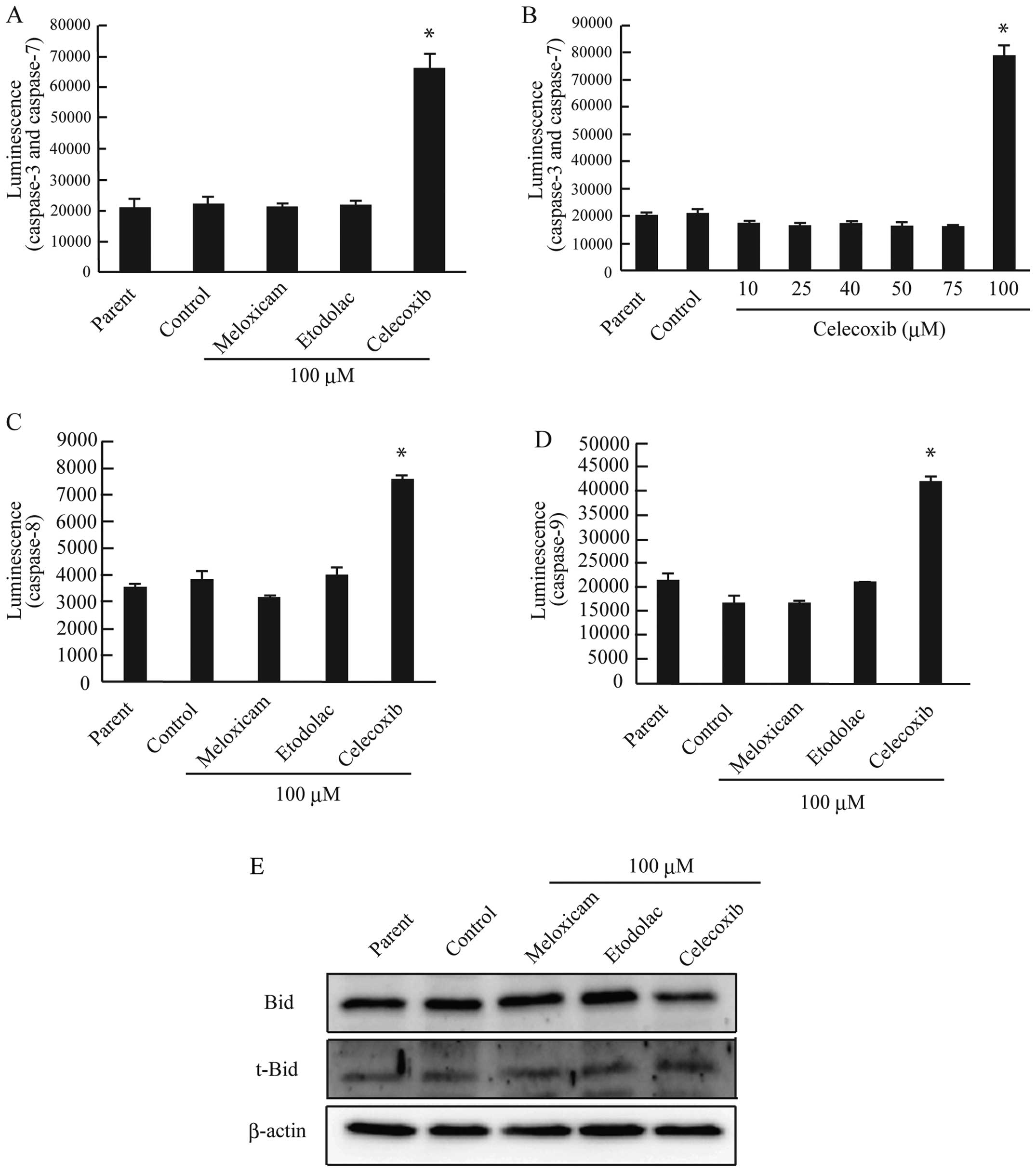
To further clarify the effect of selective COX-2
inhibitors on apoptosis, we analyzed the caspase-3/7 activity. Only
celecoxib induced the activation of caspase-3/7 (Fig. 8A), and this effect required
treatment with a dose of 100 μM celecoxib (Fig. 8B). Caspase-dependent apoptosis is
divided into the intrinsic and extrinsic pathways, which are
induced by the activation of initiator caspase-8 or −9,
respectively (32–34). The subsequent activation of the
effector caspase-3 and −7 then leads to apoptosis (35). To determine whether celecoxib
induced apoptosis via the intrinsic or extrinsic apoptotic pathway,
we measured the activity of caspase-8 and −9 in AZACB cells treated
with selective COX-2 inhibitors. As shown in Fig. 8C and D, celecoxib induced the
activation of both caspase-8 and −9 in AZACB cells. Celecoxib also
induced the cleavage of Bid to truncated Bid (Fig. 8E). These data suggest that
celecoxib-induced apoptosis is mediated by both the intrinsic and
extrinsic apoptotic pathways.
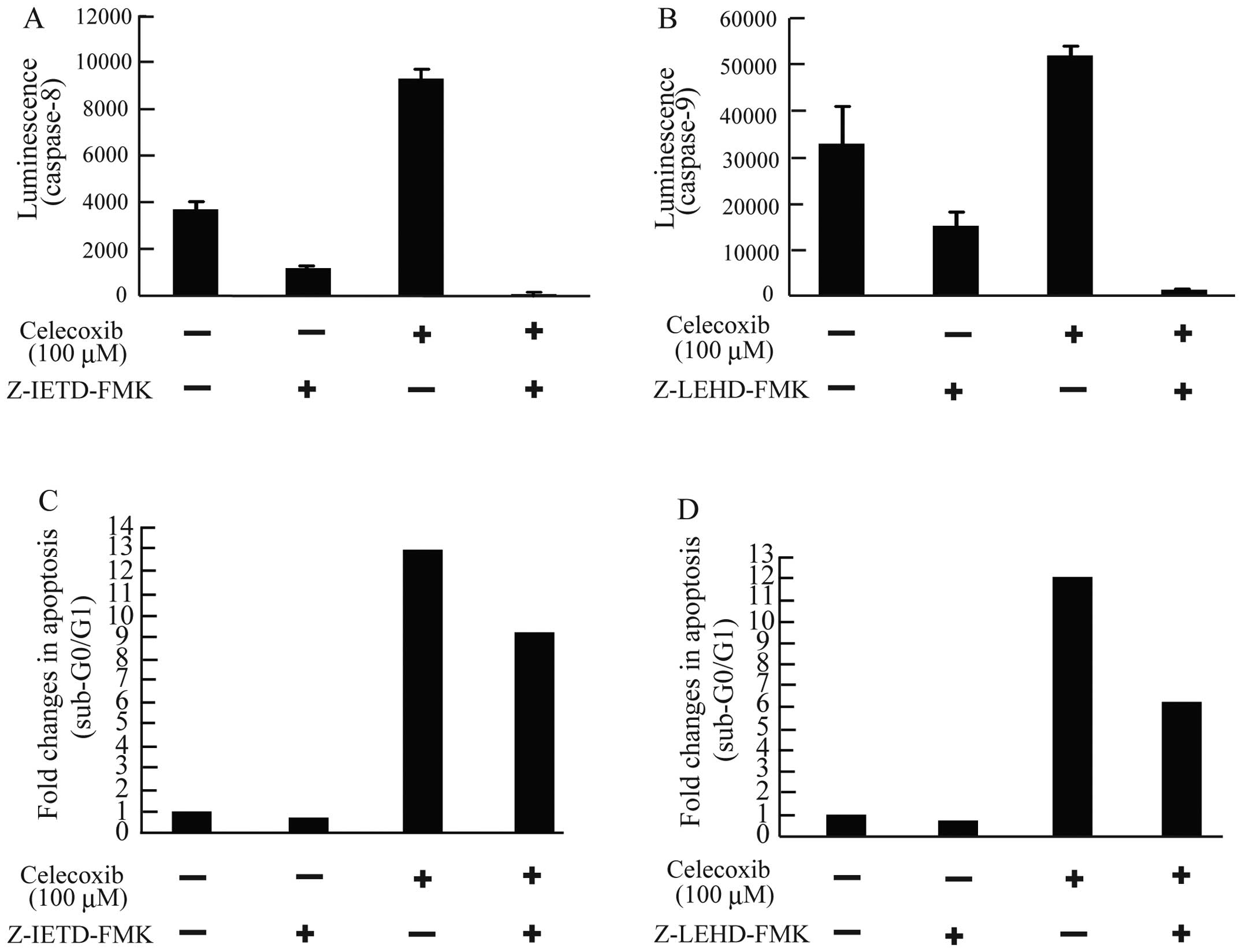
To confirm the activation of these apoptotic
pathways in celecoxib-treated cells, we next assessed the effect of
caspase-8 and −9 inhibitors on celecoxib-induced apoptosis in AZACB
cells. The caspase-8 inhibitor Z-IETD-FMK completely inhibited
celecoxib-induced caspase-8 activation (Fig. 9A). However, it did not completely
block celecoxib-induced apoptosis (Fig. 9C). Similarly, the caspase-9
inhibitor Z-LEHD-FMK significantly inhibited celecoxib-induced
caspase-9 activation in AZACB cells (Fig. 9B). However, it did not completely
inhibit celecoxib-induced apoptosis (Fig. 9D). These results strongly support
the notion that celecoxib-induced apoptosis is mediated by both the
intrinsic and extrinsic apoptotic pathways in AZACB cells.
Discussion
In humans, the regular intake of NSAIDs such as
aspirin and selective COX-2 inhibitors is associated with a
decreased risk of cancer incidence, distant recurrence, and
cancer-related deaths in various cancers, including breast and
colon cancers (36,37). However, it largely remains unclear
whether NSAIDs might be useful chemotherapy or chemopreventative
agents in canine mammary tumors. Our findings revealed that
celecoxib inhibited cell proliferation by decreasing the number of
cells in S phase and increasing G2/M arrest by stimulating the
expression of the CDKIs p21 and p27 in AZACB cells. Furthermore,
our findings suggest that celecoxib might exert antitumor effects
mainly via COX-2-independent mechanisms in canine mammary tumor
cells. In addition, celecoxib might induce apoptosis by activating
both the intrinsic and extrinsic apoptotic pathways in AZACB cells.
We demonstrate, for the first time, that celecoxib shows potential
be a useful chemotherapy agent in canine mammary tumor cells,
regardless of COX-2 expression levels.
Several studies have reported that the aberrant
overexpression of COX-2 is observed in human cancers such as breast
cancer, prostate cancer, lung cancer, and colorectal adenomas and
carcinomas (38). In addition,
elevated COX-2 levels were associated with unfavorable outcome,
lymph node metastasis, and distant metastasis (39–41).
Therefore, various studies have suggested that selective COX-2
inhibitors might be useful chemopreventative and chemotherapeutic
agents in human breast cancer. Similar to human breast cancer, some
reports have identified correlations between COX-2 expression and
angiogenesis, poor prognosis, and the development of distant
metastases in canine mammary tumors (17,42,43).
Consistent with this, we demonstrated previously that selective
COX-2 inhibitors, particularly celecoxib, had powerful antitumor
activity in CF33 cells that was mediated by the induction of
apoptosis (18). Furthermore, the
current study suggests that celecoxib exhibited antitumor effects
in canine mammary tumor cells regardless of COX-2 expression. These
results strongly support the hypothesis that celecoxib might be a
useful chemotherapeutic agent in canine mammary tumor cells.
Furthermore, celecoxib might be a potent adjunctive therapeutic
agent for intractable canine mammary tumors.
Apoptosis (programmed cell death), plays a key role
in the development and regulation of tissue homeostasis. Apoptosis
can be triggered by at least two major pathways: the intrinsic
(mitochondrial) and the extrinsic (death-receptor) pathway. In the
extrinsic pathway, caspase-8 is activated after the interaction of
death receptors, including CD95, tumor necrosis factor-related
apoptosis-inducing ligand (TRAIL)-R1 and -R2, as well as
TNF-receptor-1, with their cognate ligands CD95, TRAIL, and TNF,
respectively (32,34). In contrast, in the intrinsic
pathway, caspase-9 is activated by MOMP followed by an imbalance
between pro- and anti-apoptotic proteins (30,32–34).
These distinct pathways then converge to activate the effector
caspase-3 and −7 (30). However,
it remains controversial whether celecoxib-induced apoptosis is
mediated by the intrinsic or extrinsic pathway. Celecoxib-induced
apoptosis occurred via the intrinsic pathway and a Bak-dependent,
Bcl-2-independent pathway in Jurkat T-lymphoma cells (44,45).
In contrast, Liu et al reported that celecoxib-induced
apoptosis in human non-small cell lung cancer cell lines was
mediated by activation of the extrinsic pathway following increased
TRAIL-R2 expression, enhanced TRAIL-induced apoptosis, and the
downregulation of cellular FADD-like interleukin-1 β-converting
enzyme-inhibitory protein (46,47).
The current study suggests that celecoxib activated
both the intrinsic and extrinsic pathways in AZACB cells, which was
mediated by the activation of the initiator caspase-8 and −9. In
the intrinsic pathway, our data suggest that an imbalance between
pro-apoptotic (Bax and Bim) and anti-apoptotic (Bcl-2) proteins
leads to breakdown of mitochondrial membrane potential, which
sequentially activates caspase-9 and −3/7. In the extrinsic
pathway, celecoxib activates caspase-3/7 via −8 in AZACB cells.
Interestingly, Bid plays a key role in crosstalk between the
intrinsic and extrinsic apoptotic pathways (34). Therefore, the findings of the
current study support the notion that the cleavage of Bid by active
caspase-8 might induce mitochondrial depolarization in
celecoxib-treated AZACB cells. In addition, specific inhibitors of
caspase-8 and −9 could not completely block celecoxib-induced
apoptosis in AZACB cells. The present study showed, for the first
time, that activation of both the intrinsic and extrinsic apoptotic
pathways play a critical role in celecoxib-induced apoptosis in
canine mammary tumors. A recent study reported that Bim, a
pro-apoptotic Bcl-2 homology domain 3-only protein, is a critical
mediator of anoikis in MCF10A cells (48). The current study demonstrated that
a high dose of celecoxib significantly upregulated Bim protein
expression in AZACB cells. These results suggest that celecoxib
induced anoikis in AZACB cells.
The interaction of COX-2-derived PGs (e.g.,
PGE2) with their receptors (e.g., EP1, 2, 3, and 4)
induces apoptosis resistance, cell proliferation, angiogenesis,
invasion, and metastasis (49–54).
However, it was revealed previously that NSAIDs exert antitumor
activities in cancer cells via both COX-2-dependent and
-independent mechanisms (41). For
COX-2-independent mechanisms, it was proposed that NSAIDs affect
gene expression patterns or the activation of various molecules
such as NF-κB, pyruvate dehydrogenase lipoamide kinase isozyme 1
(PDK1)/Akt, p21, and peroxisome proliferator-activated receptors
(24). In particular, NF-κB plays
a critical role in cell survival and regulates the expression of
various genes in cancer cells. NF-κB is generally localized in the
cytoplasm in its inactive form bound to its inhibitor protein IκBα.
The phosphorylation and subsequent proteasomal degradation of IκBα
leads to the nuclear translocation of NF-κB (55). Furthermore, COX-2 expression is
regulated by NF-κB in various cells (26–29).
Therefore, it is possible that NF-κB might be a key molecule in
both the celecoxib-induced downregulation of COX-2 and
COX-2-independent antitumor effects in canine mammary tumor cells.
Our data also demonstrated that celecoxib enhanced the expression
of both p21 and p27. Therefore, our observations suggest that both
p21 and p27 might play critical roles in celecoxib-induced
COX-2-independent anti-tumor mechanisms in canine mammary
tumors.
Recently, Seo et al reported that celecoxib
exerted antitumor effects in both COX-2-expressing and
-non-expressing canine melanoma cells (56). Consistent with this, the current
study demonstrated that a high dose of celecoxib induced growth
arrest and apoptosis in canine mammary tumor cells, regardless of
COX-2 expression. Taken together, these data suggest that celecoxib
might act via both COX-2-dependent and -independent mechanisms in
various canine cancers. Accordingly, selective COX-2 inhibitors
might cause more favorable therapeutic responses in various canine
cancers compared with human or other cancers.
In conclusion, our findings support the hypothesis
that celecoxib might be a viable chemotherapy or chemopreventative
agent in canine mammary tumors, regardless of COX-2 expression. In
particular, celecoxib exerts antitumor activity via
COX-2-independent mechanisms in canine mammary tumors. In the
future, it might be possible to use a combination of celecoxib and
other antitumor drugs to treat canine mammary tumors, regardless of
their COX-2 expression status.
Acknowledgements
We thank Ms. Mitsune Suzuki and Ms. Aiko Sato (Nihon
University, Kanagawa, Japan) for help in performing the
experiments. This study was supported in part by a Grant-in-Aid
from Nihon University (to T. Saito).
References
|
1
|
Hawkey CJ: COX-2 inhibitors. Lancet.
353:307–314. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chandrasekharan NV, Dai H, Roos KL,
Evanson NK, Tomsik J, Elton TS and Simmons DL: COX-3, a
cyclooxygenase-1 variant inhibited by acetaminophen and other
analgesic/antipyretic drugs: cloning, structure, and expression.
Proc Natl Acad Sci USA. 99:13926–13931. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tsujii M and DuBois RN: Alterations in
cellular adhesion and apoptosis in epithelial cells overexpressing
prostaglandin endoperoxide synthase 2. Cell. 83:493–501. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tsujii M, Kawano S and DuBois RN:
Cyclooxygenase-2 expression in human colon cancer cells increases
metastatic potential. Proc Natl Acad Sci USA. 94:3336–3340. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tsujii M, Kawano S, Tsuji S, Sawaoka H,
Hori M and DuBois RN: Cyclooxygenase regulates angiogenesis induced
by colon cancer cells. Cell. 93:705–716. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sheng H, Shao J, Morrow JD, Beauchamp RD
and DuBois RN: Modulation of apoptosis and Bcl-2 expression by
prostaglandin E2 in human colon cancer cells. Cancer Res.
58:362–366. 1998.PubMed/NCBI
|
|
7
|
Saikawa Y, Sugiura T, Toriumi F, Kubota T,
Suganuma K, Isshiki S, Otani Y, Kumai K and Kitajima M:
Cyclooxygenase-2 gene induction causes CDDP resistance in colon
cancer cell line, HCT-15. Anticancer Res. 24:2723–2728.
2004.PubMed/NCBI
|
|
8
|
Liu B, Qu L and Tao H: Cyclo-oxygenase 2
up-regulates the effect of multidrug resistance. Cell Biol Int.
34:21–25. 2009.PubMed/NCBI
|
|
9
|
Rolle CE, Sengupta S and Lesniak MS:
Mechanisms of immune evasion by gliomas. Adv Exp Med Biol.
746:53–76. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Goldschmidt M, Peña L, Rasotto R and
Zappulli V: Classification and grading of canine mammary tumors.
Vet Pathol. 48:117–131. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Spugnini EP, Porrello A, Citro G and Baldi
A: COX-2 over-expression in canine tumors: potential therapeutic
targets in oncology. Histol Histopathol. 20:1309–1312.
2005.PubMed/NCBI
|
|
12
|
Klopfleisch R, von Euler H, Sarli G, Pinho
SS, Gärtner F and Gruber AD: Molecular carcinogenesis of canine
mammary tumors: news from an old disease. Vet Pathol. 48:98–116.
2011. View Article : Google Scholar
|
|
13
|
Doré M: Cyclooxygenase-2 expression in
animal cancers. Vet Pathol. 48:254–265. 2011. View Article : Google Scholar
|
|
14
|
Pinho SS, Carvalho S, Cabral J, Reis CA
and Gärtner F: Canine tumors: a spontaneous animal model of human
carcinogenesis. Transl Res. 159:165–172. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Howe LR: Inflammation and breast cancer.
Cyclooxygenase/prostaglandin signaling and breast cancer. Breast
Cancer Res. 9:2102007. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Doré M, Lanthier I and Sirois J:
Cyclooxygenase-2 expression in canine mammary tumors. Vet Pathol.
40:207–212. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Queiroga FL, Pires I, Parente M, Gregório
H and Lopes CS: COX-2 over-expression correlates with VEGF and
tumour angiogenesis in canine mammary cancer. Vet J. 189:77–82.
2011. View Article : Google Scholar
|
|
18
|
Saito T, Tamura D and Asano R: Usefulness
of selective COX-2 inhibitors as therapeutic agents against canine
mammary tumors. Oncol Rep. 31:1637–1644. 2014.PubMed/NCBI
|
|
19
|
Kato M, Nishida S, Kitasato H, Sakata N
and Kawai S: Cyclooxygenase-1 and cyclooxygenase-2 selectivity of
non-steroidal anti-inflammatory drugs: investigation using human
peripheral monocytes. J Pharm Pharmacol. 53:1679–1685. 2001.
View Article : Google Scholar
|
|
20
|
Sanderson RO, Beata C, Flipo RM, Genevois
JP, Macias C, Tacke S, Vezzoni A and Innes JF: Systematic review of
the management of canine osteoarthritis. Vet Rec. 164:418–424.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Saito T, Dai T and Asano R: The hyaluronan
synthesis inhibitor 4-methylumbelliferone exhibits antitumor
effects against mesenchymal-like canine mammary tumor cells. Oncol
Lett. 5:1068–1074. 2013.PubMed/NCBI
|
|
22
|
Saito T, Tamura D, Nakamura T, Makita Y,
Ariyama H, Komiyama K, Yoshihara T and Asano R:
4-Methylumbelliferone leads to growth arrest and apoptosis in
canine mammary tumor cells. Oncol Rep. 29:335–342. 2013.
|
|
23
|
Saito T, Kawana H, Azuma K, Toyoda A,
Fujita H, Kitagawa M and Harigaya K: Fragmented hyaluronan is an
autocrine chemo-kinetic motility factor supported by the
HAS2-HYAL2/CD44 system on the plasma membrane. Int J Oncol.
39:1311–1320. 2011.PubMed/NCBI
|
|
24
|
Gurpinar E, Grizzle WE and Piazza GA:
COX-independent mechanisms of cancer chemoprevention by
anti-inflammatory drugs. Front Oncol. 3:1812013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kardosh A, Wang W, Uddin J, Petasis NA,
Hofman FM, Chen TC and Schönthal AH: Dimethyl-celecoxib (DMC), a
derivative of celecoxib that lacks cyclooxygenase-2-inhibitory
function, potently mimics the anti-tumor effects of celecoxib on
Burkitt’s lymphoma in vitro and in vivo. Cancer Biol Ther.
4:571–582. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Luque I and Gélinas C: Rel/NF-kappa B
factors in oncogenesis. Semin Cancer Biol. 8:103–111. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Surh YJ, Chun KS, Cha HH, Han SS, Keum YS,
Park KK and Lee SS: Molecular mechanisms underlying chemopreventive
activities of anti-inflammatory phytochemicals: down-regulation of
COX-2 and iNOS through suppression of NF-kappa B activation. Mutat
Res. 480–481:243–268. 2001. View Article : Google Scholar
|
|
28
|
Takada Y, Bhardwaj A, Potdar P and
Aggarwal BB: Nonsteroidal anti-inflammatory agents differ in their
ability to suppress NF-kappaB activation, inhibition of expression
of cyclooxygenase-2 and cyclin D1, and abrogation of tumor cell
proliferation. Oncogene. 23:9247–9258. 2004.PubMed/NCBI
|
|
29
|
Baeuerle PA and Baltimore D: I kappa B: a
specific inhibitor of the NF-kappa B transcription factor. Science.
242:540–546. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tait SW and Green DR: Mitochondria and
cell death: outer membrane permeabilization and beyond. Nat Rev Mol
Cell Biol. 11:621–632. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Darzynkiewicz Z, Bruno S, Del Bino G,
Gorczyca W, Hotz MA, Lassota P and Traganos F: Features of
apoptotic cells measured by flow cytometry. Cytometry. 13:795–808.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hengartner MO: The biochemistry of
apoptosis. Nature. 407:770–776. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ola MS, Nawaz M and Ahsan H: Role of Bcl-2
family proteins and caspases in the regulation of apoptosis. Mol
Cell Biochem. 351:41–58. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Igney FH and Krammer PH: Death and
anti-death: tumour resistance to apoptosis. Nat Rev Cancer.
2:277–288. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li P, Nijhawan D, Budihardjo I,
Srinivasula SM, Ahmad M, Alnemri ES and Wang X: Cytochrome c and
dATP-dependent formation of Apaf-1/caspase-9 complex initiates an
apoptotic protease cascade. Cell. 91:479–489. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Menter DG, Schilsky RL and DuBois RN:
Cyclooxygenase-2 and cancer treatment: understanding the risk
should be worth the reward. Clin Cancer Res. 16:1384–1390. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Holmes MD, Chen WY, Li L, Hertzmark E,
Spiegelman D and Hankinson SE: Aspirin intake and survival after
breast cancer. J Clin Oncol. 28:1467–1472. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Howe LR, Subbaramaiah K, Brown AM and
Dannenberg AJ: Cyclooxygenase-2: a target for the prevention and
treatment of breast cancer. Endocr Relat Cancer. 8:97–114. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ristimäki A, Sivula A, Lundin J, Lundin M,
Salminen T, Haglund C, Joensuu H and Isola J: Prognostic
significance of elevated cyclooxygenase-2 expression in breast
cancer. Cancer Res. 62:632–635. 2002.PubMed/NCBI
|
|
40
|
Ranger GS, Thomas V, Jewell A and Mokbel
K: Elevated cyclooxygenase-2 expression correlates with distant
metastases in breast cancer. Anticancer Res. 24:2349–2351.
2004.PubMed/NCBI
|
|
41
|
Reader J, Holt D and Fulton A:
Prostaglandin E2 EP receptors as therapeutic targets in breast
cancer. Cancer Metastasis Rev. 30:449–463. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Queiroga FL, Pires I, Lobo L and Lopes CS:
The role of Cox-2 expression in the prognosis of dogs with
malignant mammary tumours. Res Vet Sci. 88:441–445. 2010.
View Article : Google Scholar
|
|
43
|
Queiroga FL, Perez-Alenza MD, Silvan G,
Peña L, Lopes C and Illera JC: Cox-2 levels in canine mammary
tumors, including inf lammatory mammary carcinoma:
clinicopathological features and prognostic significance.
Anticancer Res. 25:4269–4275. 2005.PubMed/NCBI
|
|
44
|
Müller AC, Handrick R, Elsaesser SJ,
Rudner J, Henke G, Ganswindt U, Belka C and Jendrossek V:
Importance of Bak for celecoxib-induced apoptosis. Biochem
Pharmacol. 76:1082–1096. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jendrossek V, Handrick R and Belka C:
Celecoxib activates a novel mitochondrial apoptosis signaling
pathway. FASEB J. 17:1547–1549. 2003.PubMed/NCBI
|
|
46
|
Liu X, Yue P, Zhou Z, Khuri FR and Sun SY:
Death receptor regulation and celecoxib-induced apoptosis in human
lung cancer cells. J Natl Cancer Inst. 96:1769–1780. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Liu X, Yue P, Schönthal AH, Khuri FR and
Sun SY: Cellular FLICE-inhibitory protein down-regulation
contributes to celecoxib-induced apoptosis in human lung cancer
cells. Cancer Res. 66:11115–11119. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Reginato MJ, Mills KR, Paulus JK, Lynch
DK, Sgroi DC, Debnath J, Muthuswamy SK and Brugge JS: Integrins and
EGFR coordinately regulate the pro-apoptotic protein Bim to prevent
anoikis. Nat Cell Biol. 5:733–740. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sun Y, Tang XM, Half E, Kuo MT and
Sinicrope FA: Cyclooxygenase-2 overexpression reduces apoptotic
susceptibility by inhibiting the cytochrome c-dependent apoptotic
pathway in human colon cancer cells. Cancer Res. 62:6323–6328.
2002.PubMed/NCBI
|
|
50
|
He Q, Luo X, Huang Y and Sheikh MS:
Apo2L/TRAIL differentially modulates the apoptotic effects of
sulindac and a COX-2 selective non-steroidal anti-inflammatory
agent in Bax-deficient cells. Oncogene. 21:6032–6040. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Shao J, Evers BM and Sheng H:
Prostaglandin E2 synergistically enhances receptor tyrosine
kinase-dependent signaling system in colon cancer cells. J Biol
Chem. 279:14287–14293. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sonoshita M, Takaku K, Sasaki N, Sugimoto
Y, Ushikubi F, Narumiya S, Oshima M and Taketo MM: Acceleration of
intestinal polyposis through prostaglandin receptor EP2 in
Apc(Delta 716) knockout mice. Nat Med. 7:1048–1051. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Nagatsuka I, Yamada N, Shimizu S, Ohira M,
Nishino H, Seki S and Hirakawa K: Inhibitory effect of a selective
cyclooxygenase-2 inhibitor on liver metastasis of colon cancer. Int
J Cancer. 100:515–519. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Timoshenko AV, Xu G, Chakrabarti S, Lala
PK and Chakraborty C: Role of prostaglandin E2 receptors in
migration of murine and human breast cancer cells. Exp Cell Res.
289:265–274. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Chen Z, Hagler J, Palombella VJ, Melandri
F, Scherer D, Ballard D and Maniatis T: Signal-induced
site-specific phosphorylation targets I kappa B alpha to the
ubiquitin-proteasome pathway. Genes Dev. 9:1586–1597. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Seo KW, Coh YR, Rebhun RB, Ahn JO, Han SM,
Lee HW and Youn HY: Antitumor effects of celecoxib in COX-2
expressing and non-expressing canine melanoma cell lines. Res Vet
Sci. 96:482–486. 2014. View Article : Google Scholar : PubMed/NCBI
|