Introduction
Breast cancer is the most common cancer and the
second leading cause of cancer-related death among females in the
world, accounting for 23% of the total cancer cases and 14% of the
cancer deaths (1), indicating that
prevention and early therapy of breast cancer is needed urgently.
Currently, breast cancer is treated with surgery, chemotherapy,
endocrine therapy, radiation therapy and targeted therapy or
multidisciplinary synthetic therapy. Although these treatment
modalities are remarkable successful, a significant number of
patients either do not respond to therapy, or the tumor may recur
and metastasize during therapy. The unsatisfactory prognosis
strongly suggests that the evaluation of novel preventive agents is
urgently needed to decrease the incidence of breast cancer.
Studies have shown that ellagic acid, a dietary
flavonoid polyphenol which is abundant in pomegranate, muscadine
grapes, walnuts and strawberries, can inhibit cancer cells
proliferation and induce apoptosis (2–5).
However, the precise molecular mechanism by which EA inhibits
cancer cell growth is still unknown. Understanding the molecular
biological properties of EA may lead to the clinical development of
mechanism-based chemopreventive and therapeutic strategies for
breast cancer. The alterations of gene expression profiles by some
anticancer agents have been reported (6,7). In
the present study, cDNA microarray can detect the changes of gene
expression profiles and provide evidence for determining the
effects of anticancer agents on cancer cells. We used the
high-throughput gene chip, which contains 41,000+ known
genes to understand the potential molecular mechanism of EA on
MCF-7 breast cancer cells.
It is well known that many signal pathways play
important roles in the control of cell growth, differentiation,
apoptosis, inflammation, stress response, and many other
physiologic processes, respectively (8–11).
In the present study, several genes belonging to TGF-β/Smads
signaling pathway were regulated remarkably by EA treatment in
MCF-7 cells.
Materials and methods
EA and cell lines
Ellagic acid (EA) was purchased from Sigma Chemical
Co. (St. Louis, MO, USA). Stock solution of EA (2 mg/ml) was
prepared in dimethyl sulfoxide (DMSO), and filter sterilized before
use. The human breast cancer cell line MCF-7 was purchased from the
Cell Bank of Shanghai Institute of Biological Sciences, Chinese
Academy of Sciences (Shanghai, China). MCF-7 cells were cultured in
RPMI-1640 supplemented with 10% fetal bovine serum (FBS) and 1%
penicillin streptomycin solution in an atmosphere of 95% air and 5%
CO2 in a 37°C humidified incubator.
Cell proliferation assay
Cells were seeded in 96-well plates at a density
designed to reach 70–80% confluency. Cells were allowed to adhere
and 24 h later were treated with EA at 0, 10, 20, 30 and 40 μg/ml.
After 24, 48 or 72 h of treatment, 200 μl of MTT was added to each
well, and the cells were incubated for 4 h at 37°C. The medium was
discarded, and the dark blue formazan crystals were adequately
dissolved with DMSO for 10 min on a rocker platform. The absorbance
was measured at 570 nm using an ELISA reader (Tecan Sunrise,
Männedorf, Switzerland). The cell viability was calculated
according to: OD sample/OD control × 100%. The assay was performed
in triplicate.
Cell cycle analysis
MCF-7 cells (5×105) were seeded in T25
culture flasks and grew for 6 h to reach 50–60% confluency. Cells
were starved in serum-free medium for 24 h to achieve
synchronization. After returning to regular growth medium for 6 h,
cells were treated with EA at 0, 10, 20 and 30 μg/ml. DMSO (final
concentration <0.1%) was used as a negative control. After
treated for 24 h at 37°C, floating and adherent cells were
collected, washed with ice-cold PBS and fixed with 70% ethanol for
at least 12 h at 4°C. The cells were then treated with 80 mg/ml
RNase A and 50 μg/ml PI at a density of 1×106 cells/ml
for 30 min, and the stained cells were analyzed using a FACScan
cytometer (Becton-Dickinson, Franklin Lakes, NJ, USA).
Cell apoptosis analysis
The apoptotic rate was measured by FCM according to
the instructions provided by the Annexin V-FITC kit
(Becton-Dickinson). Briefly, following treatment with 0, 10, 20 and
30 μg/ml of EA for 24 h, cells were harvested after digestion with
0.25% trypsin. The collected cells were washed three times with
ice-cold PBS containing calcium and resuspended in binding buffer
(500 μl) at a density of 1×106 cells/ml, in which 500 μl
of cell suspension was added to a 5 ml FCM tube and Annexin V-FITC
(50 μg/ml, 5 μl) and PI (50 μg/ml, 5 μl) were added. Then the cells
were incubated for 30 min at room temperature in the dark. The
apoptotic percentage of 10,000 cells was determined using FCM.
Morphology and ultra structure of apoptotic cells
was observed by transmission electron microscopy (TEM). MCF-7 cells
were seeded at a density of 1×106 cells/flask and
treated with 15 μg/ml EA-supplemented medium for 24 h. DMSO was
used as a control. After incubation, cells were fixed with 2.5%
glutaraldehyde in 0.1 M sodium cacodylate buffer for 24 h at 4°C.
Then, the cells were washed in the same buffer three times, fixed
with 1% osmium tetroxide and dehydrated in graded ethanol. The 100%
ethanol solution was then replaced by propylene oxide and embedded
in epoxy resin, which was polymerized at 70°C for 8 h. Sections
were stained with uranyl acetate and lead citrate, and then
observed with a Hitachi H-7650 TEM (Hitachi High Technologies,
Tokyo, Japan).
The change of gene profiles in MCF-7
cells treated with EA
MCF-7 cells were plated at a density of
4×103 cells/cm2 in T75 culture flasks.
Synchronization was achieved as previously described. The cells
were harvested at 80–90% confluency. After exposure to EA (15
μg/ml) for 6, 12 and 24 h, total RNAs of the cells were harvested
using TRIzol reagent (Life Technologies, Carlsbad, CA, USA) and the
RNeasy kit (Qiagen, Dusseldorf, Germany) according to
manufacturer’s instructions. For each time-point, two preparations
of RNA samples were independently subjected to array hybridization.
After having passed RNA measurement using the NanoDrop ND-1000
(NanoDrop Technologies, Wilmington, DE, USA) and denaturing gel
electrophoresis, the samples were amplified and labeled using the
Agilent Quick Amp labeling kit and hybridized with Agilent whole
genome oligo-microarray in Agilent’s SureHyb hybridization
chambers. After hybridization and washing, the processed slides
were scanned with the Agilent DNA microarray scanner using settings
recommended by Agilent Technologies (Palo Alto, CA, USA).
The resulting text files extracted from Agilent
Feature extraction software (version 10.5.1.1) were imported into
the Agilent GeneSpring GX software (version 11.0) for further
analysis. The microarray data sets were normalized in Agilent
Feature extraction software, and then the genes recorded present in
all the samples were chosen for further analysis. Differentially
expressed genes were identified through fold-change screening.
The gene expression profiling experiment was
successfully completed on the samples. The profiling identified a
subset of the total number of probes analyzed by Agilent Whole
Genome Oligo microarray that are differentially expressed. GO
Analysis and Pathway Analysis were performed on this subset of
genes. More detailed information was found in Gene Ontology (GO)
report and Pathway Analysis report.
Real-time reverse transcription-PCR
analysis
Total RNA from MCF-7 cells exposed to 15 μg/ml EA
for 6, 12 or 24 h were used for transcriptomics analysis by
real-time reverse transcription-PCR (real-time RT-PCR) with
selected target genes. Each 2 μg of RNA was reversely transcribed
to cDNA using oligo(dT) primers and SuperScript II reverse
transcriptase kit (Life Technologies). Primers were designed using
Primer 5 software and synthesized by Sangon Biotech Co., Ltd.
(Shanghai, China). The primers are shown in Table I. Real-time RT-PCR reactions were
then performed in a total of 25 μl of reaction mixture using the
ABI Prism 7900HT sequence detection system (Applied Biosystems,
Foster City, CA, USA). Data were analyzed using the comparative Ct
method and was normalized by GAPDH expression in each sample.
 | Table IThe primer used for real-time RT-PCR
analysis. |
Table I
The primer used for real-time RT-PCR
analysis.
| Genes | Primer
sequenece |
|---|
| TGFβ1 |
TGGAAACCCACAACGAAATCTATG
GCTAAGGCGAAAGCCCTCA |
| TGFβR
II |
AAAGGTCGCTTTGCTGAGGTCTA
GTCGTTCTTCACGAGGATATTGGA |
| TGFβR I |
TTCAAACGTGCTGACATCTATGC
TTCCTGTTGACTGAGTTGCGATA |
| SMAD3 |
ATGGCCGGTTGCAGGTGTC
GGTTCATCTGGTGGTCACTGGTTTC |
|
p21Cip1 |
TTAGCAGCGGAACAAGGAGT
AGAAACGGGAACCAGGACA |
|
p15Ink4b |
TGGTGGCTACGAATCTTCCG
TCGTCGCTTGCACATCCTC |
|
p19Ink4d |
CACCCTGAAGGTCCTAGTGGAG
AGTGGGCAGGAGAAACAAGAAG |
|
p57Kip2 |
TCGGCTGGGACCGTTCA
TGTATGGCAGCTACAGCTTGTG |
| CCND1 |
ATGTTCGTGGCCTCTAAGATGAAG
GTGTTTGCGGATGATCTGTTTGT |
| CCNE |
CAGTTTGCGTATGTGACAGATGGA
GAGAAATGATACAAGGCCGAAGC |
| CCNA2 |
ATGATGAGCATGTCACCGTTCC
TCCATTGGATAATCAAGAGGGACC |
| p107 |
AGAACCACCAAAGTTACCACGAA
TCTTCAGAAGCCGTAAAGTCAGC |
| p130 |
AGAAGGGTGACTGAAGTTCGTG
CAACATTGACTTGGACAGGGAAG |
| RB1 |
AAAGGACCGAGAAGGACCAACT
CAGACAGAAGGCGTTCACAAAGT |
| E2F1 |
CCCCAACTCCCTCTACCCTT
CTCTCCCATCTCATATCCATCCTG |
| E2F2 |
CTGGAGTGCAGTGGCCTGAT
TGGCTCGTGCCTGTCATCTC |
| GAPDH |
GGTGAAGGTCGGAGTCAACGG
CCTGGAAGATGGTGATGGGATT |
Western blot analysis
Western blot analysis was performed using the lysis
buffer isolated protein. Briefly, 20 μg of protein was resolved in
10–15% SDS/PAGE and transferred to polyvinylidene fluoride
membrane. The blots were blocked in blocking buffer overnight at
4°C, and incubated with the primary antibody at 37°C for 1 h. The
antibody-antigen complexes were then detected with alkaline
phosphatase-conjugated anti-rabbit or anti-mouse IgG secondary
antibodies using a BCIP/NBT Alkaline Phosphatase Color Development
kit. The band was recorded by a digital camera and analyzed by
ImageJ software (NIH, Bethesda, MD, USA). The results were
normalized with β-actin. The following monoclonal antibodies were
used in the present study (source): anti-β-actin (Sigma Chemical),
anti-Smad3, anti-p21Cip1 (Boshide Co., Wuhan, China),
anti-phospho-Smad3 (Santa Cruz Biotechnology, Santa Cruz, CA, USA),
anti-Rb, anti-phospho-Rb, anti-TGFβ1, anti-TβR I and anti-TβR II
(Cell Signaling Technology, Beverly, MA, USA).
Statistical analysis
The Student’s t-test was used to determine the
significance between treatments and untreated controls, and
P<0.05 was considered significant.
Results
The effects of EA on cell proliferation
in MCF-7 cells
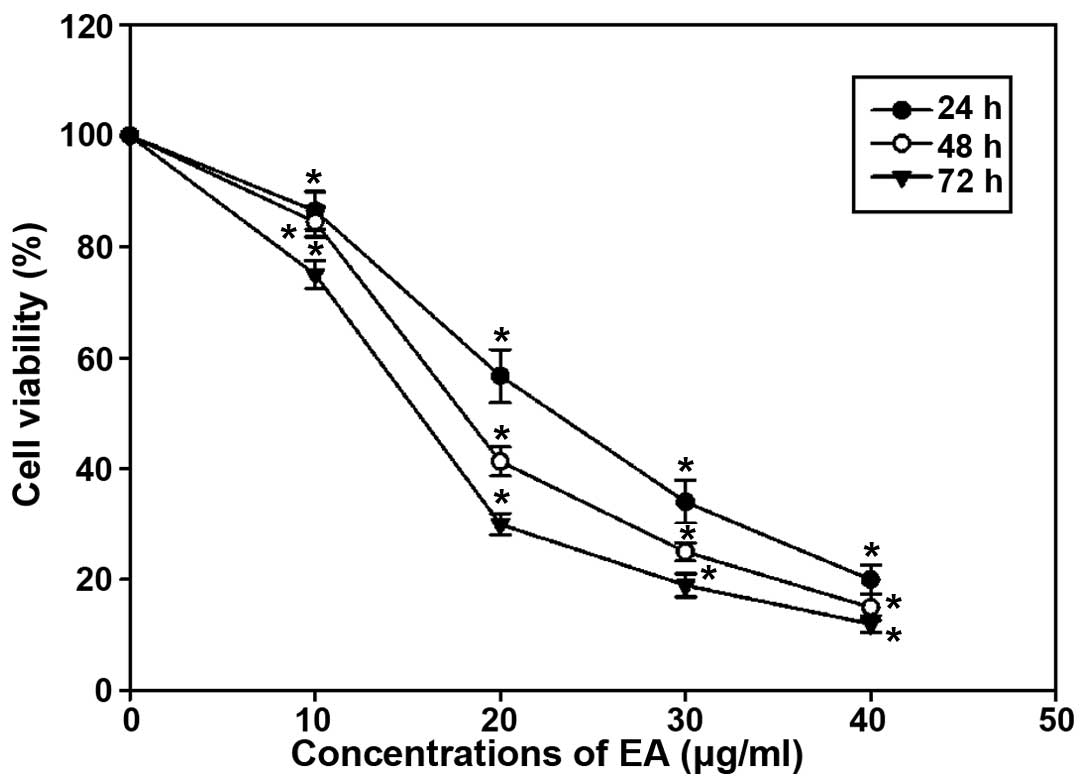
The inhibitory effect of EA on cell proliferation in
MCF-7 cells was assessed by MTT assay. As shown in Fig. 1, EA suppressed MCF-7 cell growth in
a time- and dose-dependent manner. The inhibitory response was
apparent as early as 24 h with a concentration range of EA from 10
to 40 μg/ml EA was found to reduce the cell number to 13.5 and 80%
of the untreated control at concentrations of 10 and 40 μg/ml,
respectively.
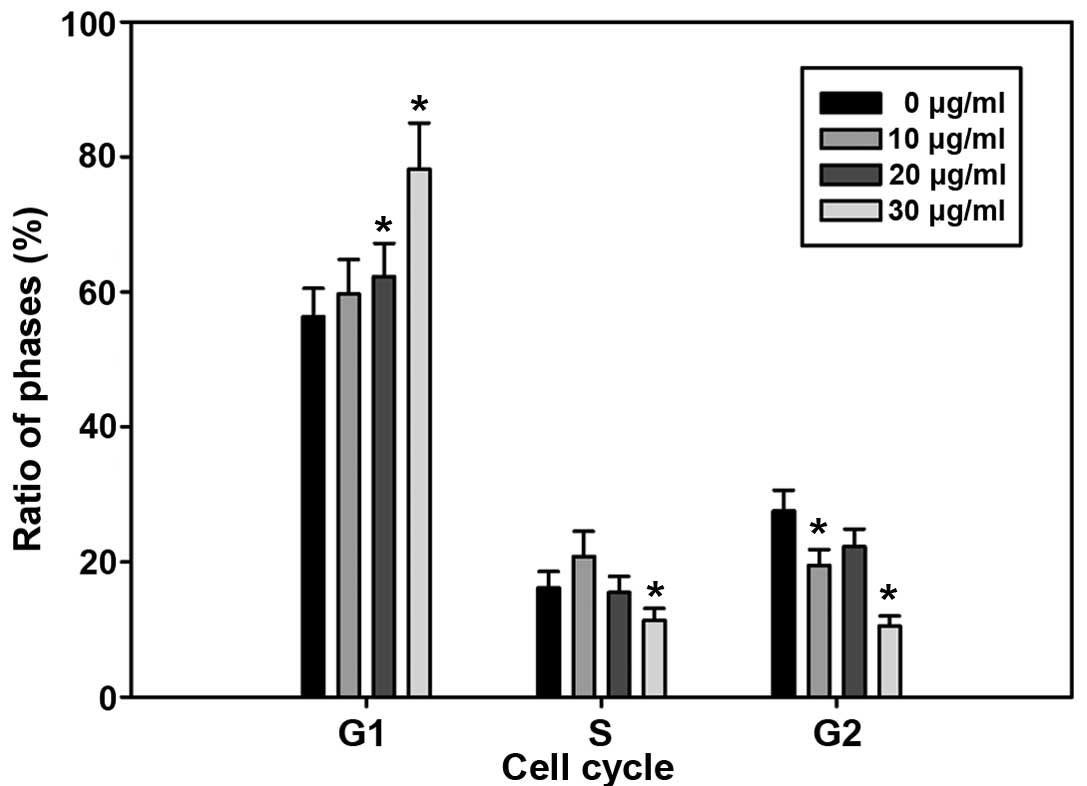
Cell cycle arrest at G0–G1 phase by
EA
To determine whether decreased cell number
accumulation was related to cell cycle arrest treated by EA, we
assessed the effect of EA on cell cycle perturbation by flow
cytometry. The data in Fig. 2
clearly showed a significant block in G0–G1 phase of the cell
cycle. The increase in the G0–G1 population was accompanied by a
delay of passage of cells to S phase. Synchronized control cells
that were not treated by EA moved into S phase.
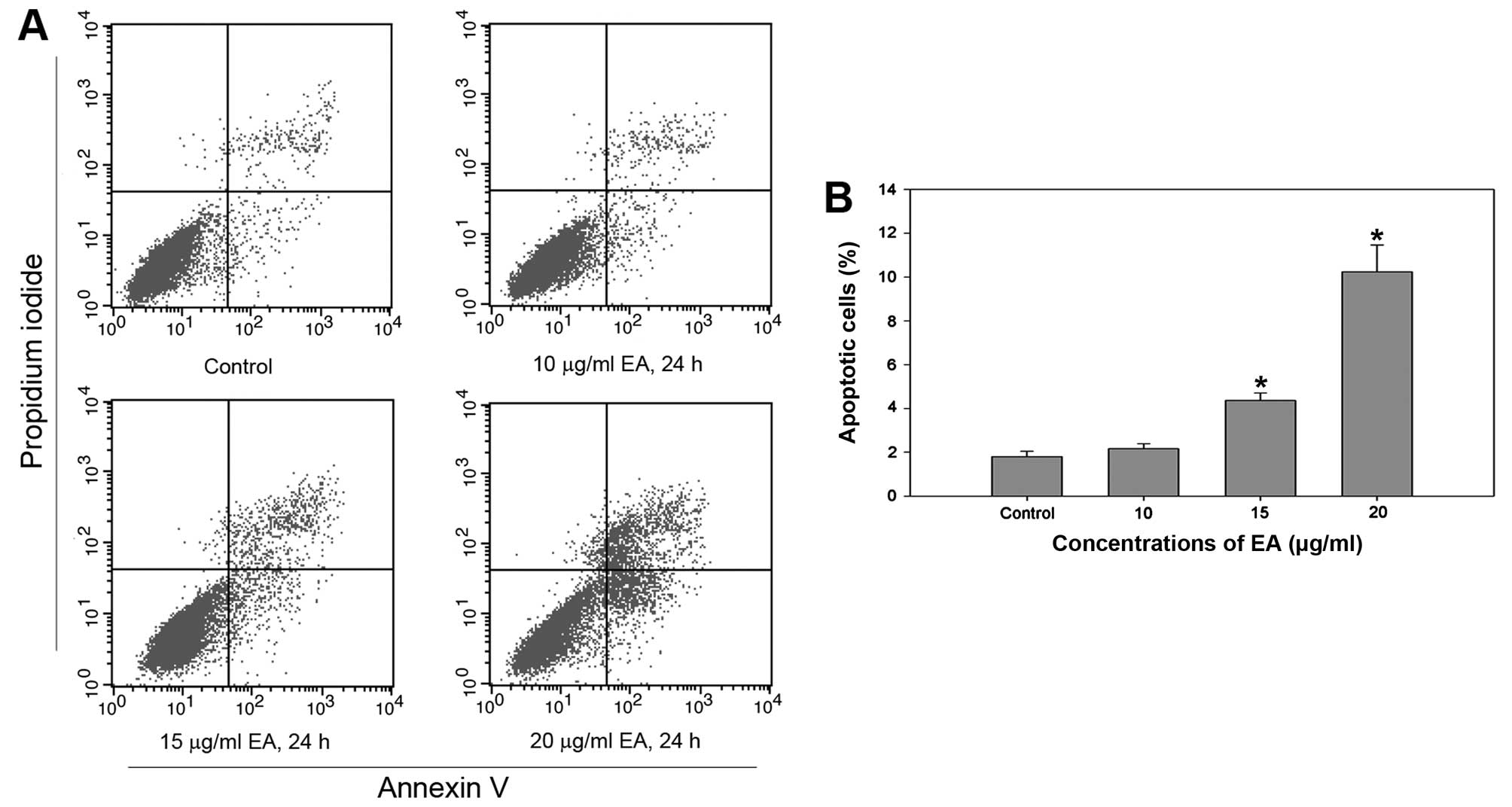
Effect of EA on cell apoptosis
In order to determine whether EA could induce cell
apoptosis, FCM and TEM were conducted in EA-treated MCF-7 cells and
the control. FCM showed that the cell apoptotic rates were
4.36±0.35 and 10.24±1.23 after treatment with 15 or 20 μg/ml of EA
for 24 h, respectively. MCF-7 cells treated with increasing
concentrations of EA exhibited a significant increase in the
apoptotic cell fraction, indicating apoptosis induction (Fig. 3).
Morphological and ultra-structural characteristics
of apoptosis in MCF-7 cells exposed to EA for 24 h were examined by
transmission electron microscope (TEM) (Fig. 4).
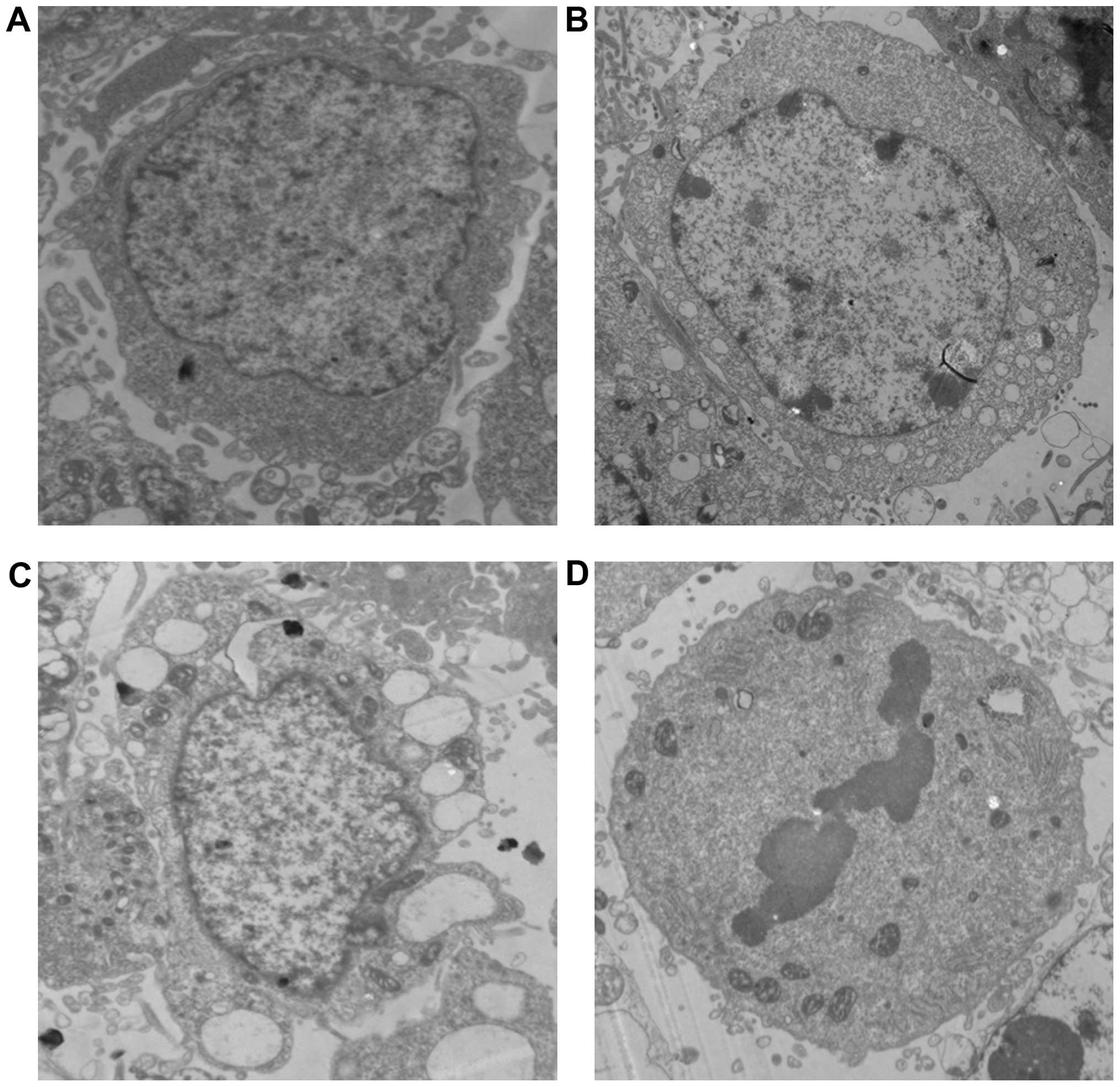 | Figure 4Morphology and ultrastructure of
MCF-7 cells were analyzed by TEM. MCF-7 cells were treated with EA
(15 μg/ml) for 24 h and examined by TEM at magnification ×10,000.
(A) In the control cells, the structure of the nucleus, abundance
of cytoplasm, as well as the size and shape of the mitochondria,
were all normal. (B) In the treated cells, the compaction and
margination of nuclear chromatin were quite obvious. (C) Cell
detachment, cell shrinkage, folded nuclear membrane, membrane
blebbing, increased nuclear heterochromatin, swelling of the
endoplasmic reticulum cisternae and vesicle formation (abundant
vacuoles with multivesicular bodies) were observed. (D) Nuclear
pyknosis and fragmentation occurred. |
Profiling of EA-responsive genes by cDNA
microarray analysis
We found a total of 4,738 genes that showed a
>2.0-fold change after 24 h of EA treatment. Among these genes,
2,547 genes were downregulated and 2,191 genes were upregulated in
EA-treated MCF-7 cells. The altered expressions of many genes did
not occurr after 6 h of EA treatment and changes were evident with
24 h of treatment (Table II).
| Table IIFold-changes of specific genes in
MCF-7 cells treated with EA. |
Table II
Fold-changes of specific genes in
MCF-7 cells treated with EA.
| MCF-7 |
|---|
|
|
|---|
| Genes | 6 h | 12 h | 24 h |
|---|
| NM_000660.4,
transforming growth factor, β1 (TGFβ1) | NC | NC | NC |
| NM_001024847,
transforming growth factor, β receptor II (TGFβRII) | NC | NC | NC |
| NM_004612.2,
transforming growth factor, β receptor 1 (TGFβRI) | NC | NC | NC |
| NM_005901.4, SMAD
family member 2 (SMAD2) | NC | NC | NC |
| NM_005902.3, SMAD
family member 3 (SMAD3) | NC | NC | 2.7 |
| NM_000389.1, CDK
inhibitor 1A (p21Cip1) | NC | 3.4 | 4.8 |
| NM_078487.3,
cyclin-dependent kinase inhibitor 2B
(p15Ink4b) | NC | NC | 4.8 |
| NM_001800.3,
cyclin-dependent kinase inhibitor 2D
(p19Ink4d) | NC | NC | 2.8 |
| NM_000076.2,
cyclin-dependent kinase inhibitor 1C
(p57Kip2) | NC | NC | 0.3 |
| NM_053056.2, cyclin
D1 (CCND1) | NC | 0.5 | 2.1 |
| NM_001238, cyclin E
1 (CCNE1) | 2.4 | NC | NC |
| NM_057749, cyclin E
2 (CCNE2) | 2.3 | NC | 0.3 |
| NM_001237.3, cyclin
A2 (CCNA2) | NC | NC | 0.3 |
| NM_002895.2,
retinoblastoma-like 1 (p107) | NC | NC | 0.4 |
| NM_005611.3,
retinoblastoma-like 2 (p130) | NC | NC | NC |
| NM_000321.2,
retinoblastoma 1 (RB1) | NC | NC | 0.5 |
| NM_005225.2, E2F
transcription factor 1 (E2F1) | NC | 0.5 | 0.2 |
| NM_004091.3, E2F
transcription factor 2 (E2F2) | NC | NC | 0.4 |
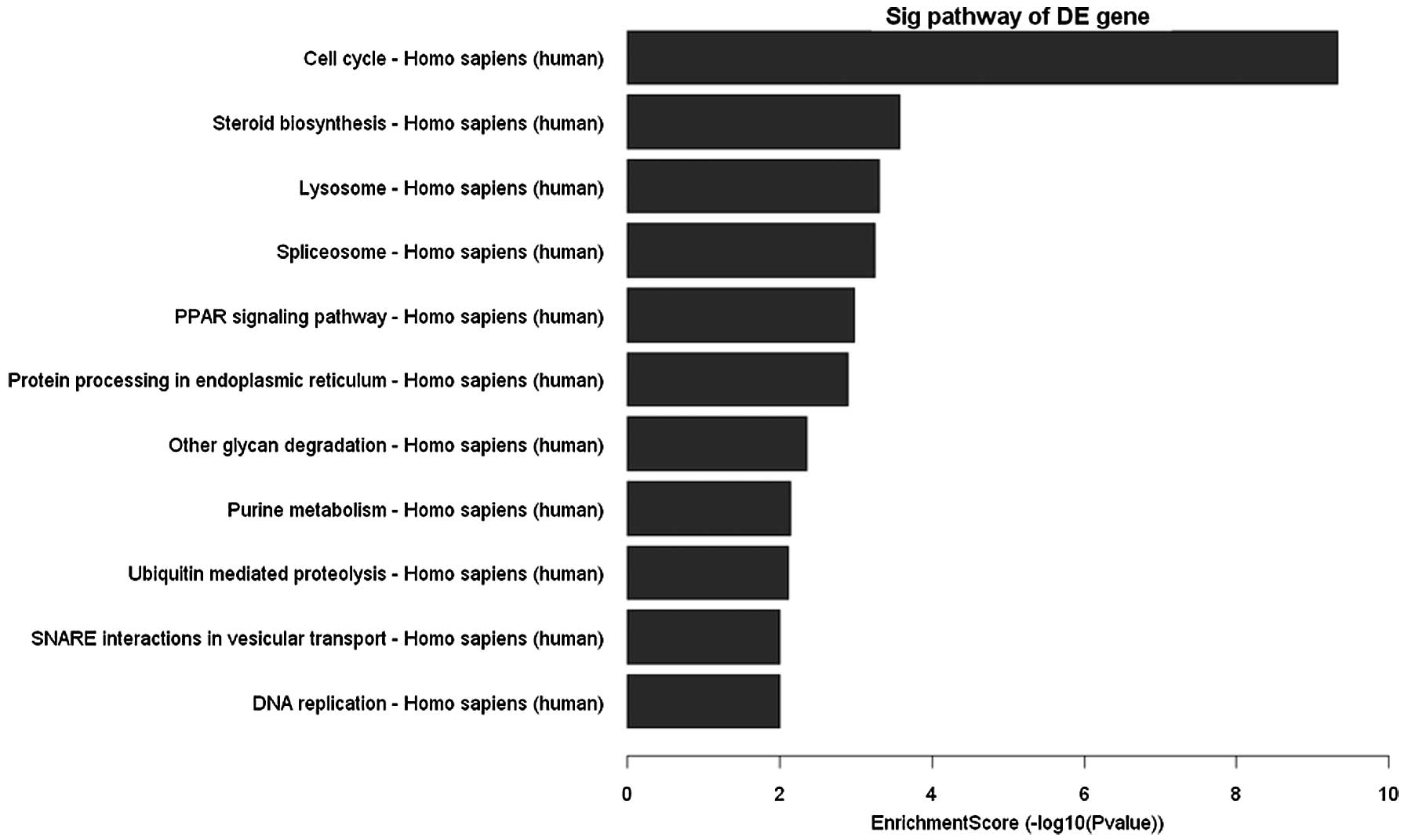
After clustering based analysis and pathway analysis
according to their biological functions, several genes, which are
related to cell cycle, apoptosis and DNA replication were found
increased or decreased (Fig. 5).
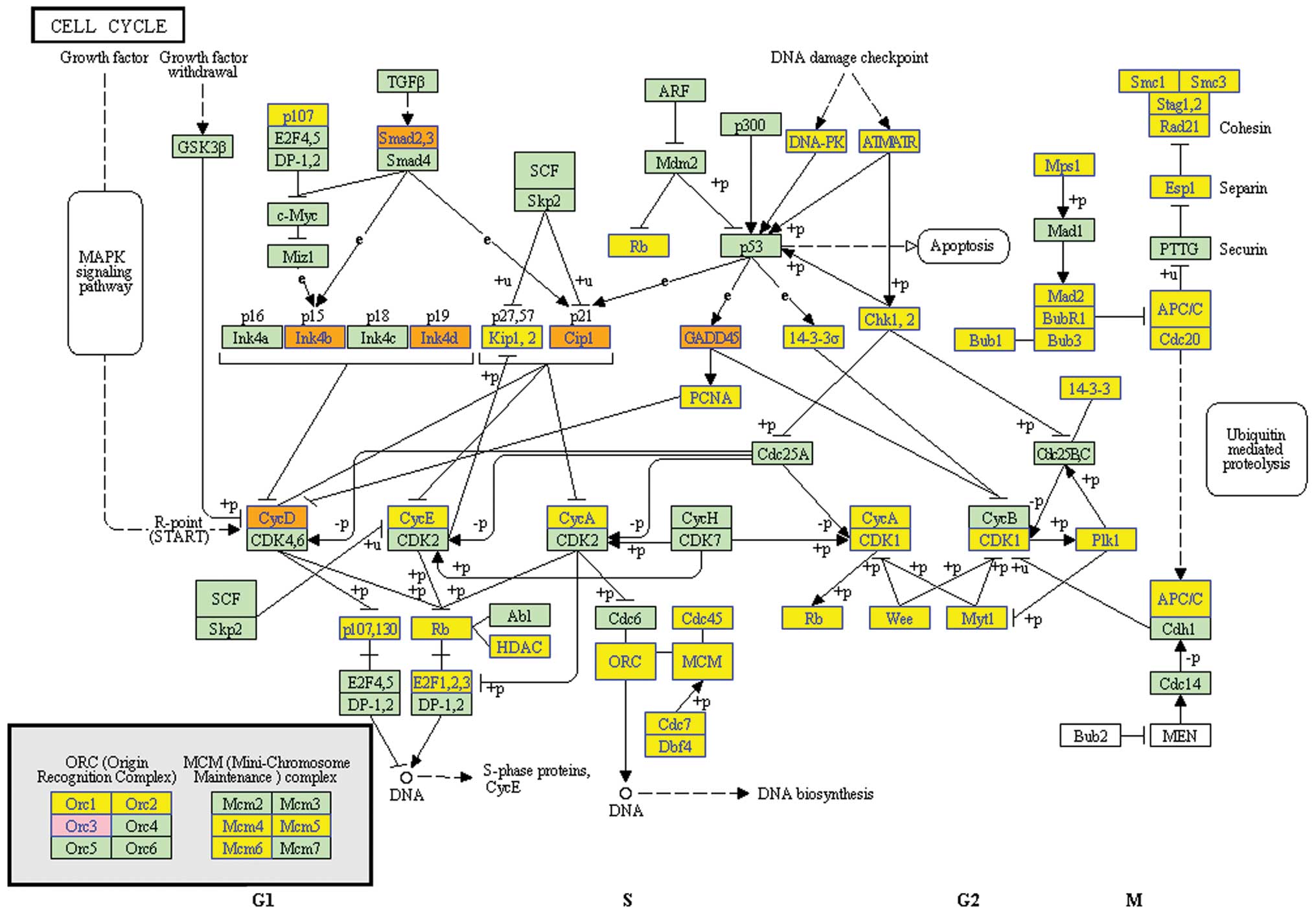
Especially, TGF-β/Smads pathway was found as the potential pathway,
by which EA induced cell cycle arrest in G0/G1 phase and educed the
inhibitory effect on MCF-7 cells (Fig.
6).
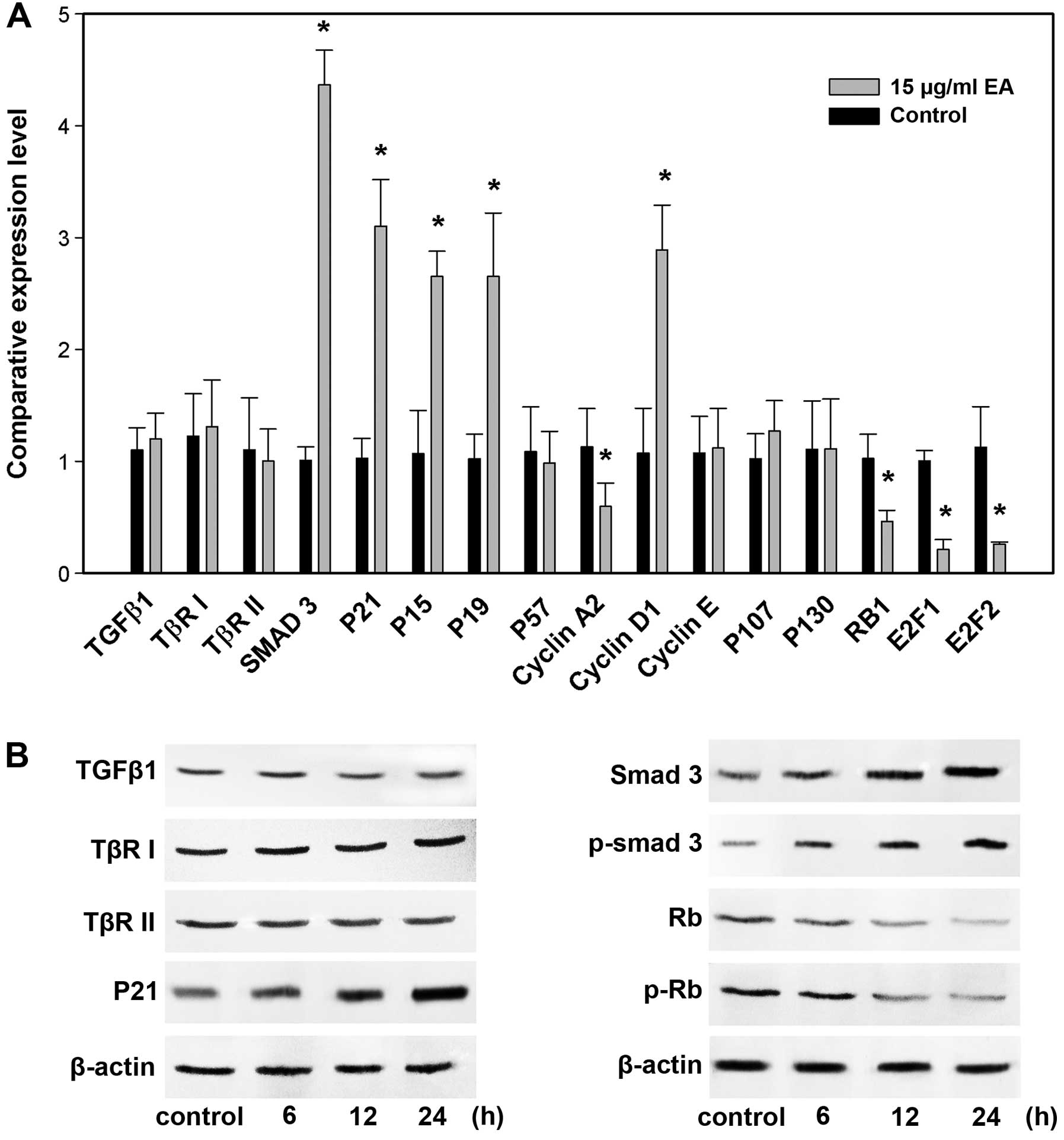
Detection of EA-responsive genes by
real-time RT-PCR and western blots
The next step was to confirm the changes of a subset
of 16 genes that showed pronounced regulation in TGF-β/Smads
pathway by real-time RT-PCR and/or western blot analysis. We
examined the genes (TGF-β1, TβR-I, TβR-II, Smad3,
p15Ink4b, p19Ink4d,
p21Cip1, p57Kip2, cyclin D1,
cyclin E, cyclin A2, E2F1, E2F2, Rb1, p107 and p130)
by real-time RT-PCR and some proteins (TGF-β1, TβR-I, TβR-II,
Smad3, p-Smad3, Rb1, p-Rb1 and
p21Cip1) by western blots. We were able to verify
eight proteins by western blot analysis and ten RNAs by real-time
RT-PCR. This represents a success rate of 75% (12 out of 16). The
results are shown in Fig. 7,
respectively. The lack of complete concordance could be
attributable to either false positive signals of the array data or
the discrepancy between transcript and protein expression. There
was a noteworthy observation. A decreased level of cyclin D1 was
found at 12 h, but an upregulation of cyclin D1 expression was
detected by cDNA microarray, real-time RT-PCR and western blot
analyses at 24 h. Cells lacking cyclin D1 are known to stay in cell
G0–G1 arrest (12,13). It is possible that at the early
time-point, the repression of cyclin D1 was necessary for
initiating EA-mediated cell cycle arrest, whereas at the later
time-point when G0–G1 arrest was firmly established, cells produced
more cyclin D1 for apoptosis to advance (14).
Discussion
To understand the molecular mechanisms leading to
EA-induced growth inhibition on MCF-7 cells, in this study, cDNA
microarray was used to elucidate the changes in gene expression
profile. On the basis of the collection of 41,000+ genes
screened by the whole human genome oligo microarray of Agilent,
4,738 genes were identified responsive to EA treatment for 24 h.
These genes are responsible for cell cycle arrest, apoptosis,
steroid biosynthesis, lysosome, splicesome, protein processing in
endoplasmic reticulum and DNA replication. TGF-β/Smads pathway was
found to be the target through pathway analysis, by which EA educed
the inhibitory effect on the growth of MCF-7 cells. In order to
confirm the result of cDNA microarray, 16 genes belonging to
TGF-β/Smads pathway were further characterized by either western
blots or real-time RT-PCR. On the basis of the results obtained
from the present study, we proposed that TGF-β/Smads signaling
pathways could mediate the outcome of EA-induced cell cycle
arrest.
Our results also showed that cyclins (cyclin A2 and
cyclin E2) were downregulated in EA-treated MCF-7 cells, whereas
cyclin-dependent kinase (Cdk) inhibitors
(p21Cip1, p15 and p19) were
upregulated. These results suggest that EA inhibited the growth of
breast cancer cell through the arrest of the cell cycle and
inhibition of proliferation. Driving of the cell cycle through one
phase to the other is tightly controlled by complex network events
of cyclins, Cdk and transcription factors (15). During mid G1 phase, cdk4 and cdk6
interact with D type cyclins to form heterodimer kinase complex.
This event follows the interaction of cyclin E with cdk2 to
phosphorylate Rb in the late G1 phase (16–18).
The cdk inhibitors, including p21Cip1, p15
and p19, have been shown to inhibit activity of cyclin-cdk
complex to decrease phosphorylation of Rb. Phosphorylation of Rb
has been shown to be critical for the stabilization of active E2F1
to translocate into the nucleus and transcribe various genes
required for the entry of the cell from G1 to S phase (19).
TGF-β/Smads signaling pathway is involved in a broad
spectrum of biological responses throughout embryonic development
and adult life, including cell proliferation, differentiation,
epithelial-to-mesenchymal transition, apoptosis and angiogenesis
(20,21). Transforming growth factor-β (TGF-β)
is a member of the TGF-β superfamily. The cytokine signals carried
by TGF-β were transmitted through a heterogenic complex of type I
and type II serine/threonine kinase receptors (TβR-I and TβR-II).
Activation of the receptor complex through ligand binding results
in the phosphorylation of the TβR-I by the TβR-II. Subsequently,
active TβR-I phosphorylate receptor-regulated Smads (R-Smads),
which are intracellular transducers of TGF-β signals, including
Smad2 and Smad3. Phosphorylated R-Smads then associate with Smad4,
the common Smad (Co-Smad) and shuttle to the nucleus. The complexes
interact with a large repertoire of transcription factors and
result in corresponding biological function subsequently (22–25).
TGF-β has been described as a potent tumor suppressor for promoting
cell growth inhibition, apoptosis and differentiation (26,27).
Mutations in the components of the TGF-β signaling cascade have
been identified in a number of human cancers, including hereditary
non-polyposis colon cancer, hepatocellular carcinoma, and
pancreatic and ovarian cancers (28).
TGF-β1 is a potent inhibitor of cell proliferation.
TGF-β1-induced arrest occurs during G1 and is mediated by Smad
proteins, which regulate transcriptional targets, including c-myc
(29–31). Downregulation of c-myc allows
induction of p15Ink4b, which inhibits Cdk4-cyclin
D (32,33). The p27Kip1
inhibitor is also utilized by TGF-β1 to inhibit Cdk2-cyclin E
(34). Cdk suppression prevents
hyperphosphorylation of Rb (18),
causing Rb to remain in a hypophosphorylated, growth-suppressive
form (35).
TGF-β1 inhibits the growth of cells of epithelial
origin by downregulating components of the cell cycle and
upregulating cell cycle inhibitors (36). In most epithelial cell types TGF-β1
acts in late G1 phase and prevents further progression to the G1/S
phase transition (37). Cdks and
cyclin complexes phosphorylate specific target molecules, such as
the retinoblastoma proteins pRb, p107 and p130 (38). The Cdk inhibitors mediate cell
cycle arrest at different points of G1 (39). TGF-β1 inhibitory actions in late G1
phase are mediated in part by the inhibition of cyclin D1 and
cyclin E expression which prevents Cdk kinase activity resulting in
RB hypophosphorylation (40,41)
and/or by the upregulation the expression of various Cdk inhibitors
such as p21 and p27 (42). The
transcription factor E2F regulates the expression of S and G2 phase
cyclins such as cyclins E, A and B (43). Hypophosphorylated Rb binds to and
represses E2F by recruiting histone deacetylases and forming a
repressor complex at E2F-responsive promoters to block
transcription of cyclins necessary for cell cycle progression
(44,45).
In the present study, we found that EA inhibits the
proliferation of MCF-7 breast cancer cells mainly mediated by
arresting cell cycle in the G0/G1 phase. TGF-β/Smads signaling
pathway was further found as the potential molecular mechanism of
EA to regulate cell cycle arrest in vitro. Therefore, the
regulation of TGF-β/Smads pathway in breast cancer cells could be a
novel therapeutic approach for treatment of patients with breast
cancer. Further studies with in vivo models, as well as an
analysis of additional human samples are still needed to confirm
the molecular mechanisms of EA in inhibition or prevention of
breast cancer growth.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (nos. 81372612 and 81302059),
the Outstanding Youth Science Foundation of Heilongjiang Province
(no. JC201203), the Study Abroad Returnees Science Foundation of
Heilongjiang (no. LC201009), and the Natural Science Foundation of
Heilongjiang province (no. H201425).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dai Z, Nair V, Khan M and Ciolino HP:
Pomegranate extract inhibits the proliferation and viability of
MMTV-Wnt-1 mouse mammary cancer stem cells in vitro. Oncol Rep.
24:1087–1091. 2010.PubMed/NCBI
|
|
3
|
Larrosa M, Tomas-Barberan FA and Espin JC:
The dietary hydrolysable tannin punicalagin releases ellagic acid
that induces apoptosis in human colon adenocarcinoma Caco-2 cells
by using the mitochondrial pathway. J Nutr Biochem. 17:611–625.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Seeram NP, Adams LS, Henning SM, et al: In
vitro antiproliferative, apoptotic and antioxidant activities of
punicalagin, ellagic acid and a total pomegranate tannin extract
are enhanced in combination with other polyphenols as found in
pomegranate juice. J Nutr Biochem. 16:360–367. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Herrera MC and Luque de Castro MD:
Ultrasound-assisted extraction for the analysis of phenolic
compounds in strawberries. Anal Bioanal Chem. 379:1106–1112.
2004.PubMed/NCBI
|
|
6
|
Tsao DA, Chang HJ, Lin CY, et al: Gene
expression profiles for predicting the efficacy of the anticancer
drug 5-fluorouracil in breast cancer. DNA Cell Biol. 29:285–293.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Berger E, Rome S, Vega N, Ciancia C and
Vidal H: Transcriptome profiling in response to adiponectin in
human cancer-derived cells. Physiol Genomics. 42A:61–70. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Poulogiannis G, Luo F and Arends MJ: RAS
signalling in the colorectum in health and disease. Cell Commun
Adhes. 19:1–9. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Areshkov PO, Avdieiev SS, Balynska OV,
Leroith D and Kavsan VM: Two closely related human members of
chitinase-like family, CHI3L1 and CHI3L2, activate ERK1/2 in 293
and U373 cells but have the different influence on cell
proliferation. Int J Biol Sci. 8:39–48. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huang KF, Zhang GD, Huang YQ and Diao Y:
Wogonin induces apoptosis and down-regulates survivin in human
breast cancer MCF-7 cells by modulating PI3K-AKT pathway. Int
Immunopharmacol. 12:334–341. 2012. View Article : Google Scholar
|
|
11
|
Rodriguez-Berriguete G, Fraile B,
Martinez-Onsurbe P, Olmedilla G, Paniagua R and Royuela M: MAP
kinases and prostate cancer. J Signal Transduct. 2012:1691702012.
View Article : Google Scholar
|
|
12
|
Wang CY, Tsai AC, Peng CY, et al:
Dehydrocostuslactone suppresses angiogenesis in vitro and in vivo
through inhibition of Akt/GSK-3beta and mTOR signaling pathways.
PLoS One. 7:e311952012. View Article : Google Scholar
|
|
13
|
Cirera-Salinas D, Pauta M, Allen RM, et
al: Mir-33 regulates cell proliferation and cell cycle progression.
Cell Cycle. 11:922–933. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dong Y, Ganther HE, Stewart C and Ip C:
Identification of molecular targets associated with
selenium-induced growth inhibition in human breast cells using cDNA
microarrays. Cancer Res. 62:708–714. 2002.PubMed/NCBI
|
|
15
|
Uhlmann F, Bouchoux C and Lopez-Aviles S:
A quantitative model for cyclin-dependent kinase control of the
cell cycle: revisited. Philos Trans R Soc Lond B Biol Sci.
366:3572–3583. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu B, Lane ME and Wadler S: SU9516, a
cyclin-dependent kinase 2 inhibitor, promotes accumulation of high
molecular weight E2F complexes in human colon carcinoma cells.
Biochem Pharmacol. 64:1091–1100. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Maiti B, Li J, de Bruin A, et al: Cloning
and characterization of mouse E2F8, a novel mammalian E2F family
member capable of blocking cellular proliferation. J Biol Chem.
280:18211–18220. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cheng L, Rossi F, Fang W, Mori T and
Cobrinik D: Cdk2-dependent phosphorylation and functional
inactivation of the pRB-related p130 protein in pRB(−),
p16INK4A(+) tumor cells. J Biol Chem. 275:30317–30325.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Muller H, Moroni MC, Vigo E, Petersen BO,
Bartek J and Helin K: Induction of S-phase entry by E2F
transcription factors depends on their nuclear localization. Mol
Cell Biol. 17:5508–5520. 1997.PubMed/NCBI
|
|
20
|
Gui T, Sun Y, Shimokado A and Muragaki Y:
The roles of mitogen-activated protein kinase pathways in
TGF-beta-induced epithelial-mesenchymal transition. J Signal
Transduct. 2012:2892432012. View Article : Google Scholar
|
|
21
|
Pardali E and Ten Dijke P: TGFbeta
signaling and cardiovascular diseases. Int J Biol Sci. 8:195–213.
2012. View Article : Google Scholar
|
|
22
|
Attisano L and Wrana JL: Signal
transduction by the TGF-beta superfamily. Science. 296:1646–1647.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Feng XH and Derynck R: Specificity and
versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev
Biol. 21:659–693. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Siegel PM and Massague J: Cytostatic and
apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev
Cancer. 3:807–821. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Moustakas A, Souchelnytskyi S and Heldin
CH: Smad regulation in TGF-beta signal transduction. J Cell Sci.
114:4359–4369. 2001.
|
|
26
|
de Caestecker MP, Piek E and Roberts AB:
Role of transforming growth factor-beta signaling in cancer. J Natl
Cancer Inst. 92:1388–1402. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Derynck R, Akhurst RJ and Balmain A:
TGF-beta signaling in tumor suppression and cancer progression. Nat
Genet. 29:117–129. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Levy L and Hill CS: Alterations in
components of the TGF-beta superfamily signaling pathways in human
cancer. Cytokine Growth Factor Rev. 17:41–58. 2006. View Article : Google Scholar
|
|
29
|
Chen CR, Kang Y, Siegel PM and Massague J:
E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to
c-myc repression. Cell. 110:19–32. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Coffey RJ Jr, Bascom CC, Sipes NJ,
Graves-Deal R, Weissman BE and Moses HL: Selective inhibition of
growth-related gene expression in murine keratinocytes by
transforming growth factor beta. Mol Cell Biol. 8:3088–3093.
1988.PubMed/NCBI
|
|
31
|
Pietenpol JA, Holt JT, Stein RW and Moses
HL: Transforming growth factor beta 1 suppression of c-myc gene
transcription: role in inhibition of keratinocyte proliferation.
Proc Natl Acad Sci USA. 87:3758–3762. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hannon GJ and Beach D: p15INK4B is a
potential effector of TGF-beta-induced cell cycle arrest. Nature.
371:257–261. 1994. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Warner BJ, Blain SW, Seoane J and Massague
J: Myc downregulation by transforming growth factor beta required
for activation of the p15Ink4b G1 arrest
pathway. Mol Cell Biol. 19:5913–5922. 1999.PubMed/NCBI
|
|
34
|
Polyak K, Kato JY, Solomon MJ, et al:
p27Kip1, a cyclin-Cdk inhibitor, links transforming growth
factor-beta and contact inhibition to cell cycle arrest. Genes Dev.
8:9–22. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Laiho M, DeCaprio JA, Ludlow JW,
Livingston DM and Massague J: Growth inhibition by TGF-beta linked
to suppression of retinoblastoma protein phosphorylation. Cell.
62:175–185. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ramos C, Becerril C, Montano M, et al:
FGF-1 reverts epithelial-mesenchymal transition induced by
TGF-{beta}1 through MAPK/ERK kinase pathway. Am J Physiol Lung Cell
Mol Physiol. 299:L222–L231. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Derynck R: TGF-beta-receptor-mediated
signaling. Trends Biochem Sci. 19:548–553. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Claudio PP, Tonini T and Giordano A: The
retinoblastoma family: twins or distant cousins? Genome Biol.
3:reviews3012. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Coqueret O: New roles for p21 and p27
cell-cycle inhibitors: a function for each cell compartment? Trends
Cell Biol. 13:65–70. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ko TC, Sheng HM, Reisman D, Thompson EA
and Beauchamp RD: Transforming growth factor-beta 1 inhibits cyclin
D1 expression in intestinal epithelial cells. Oncogene. 10:177–184.
1995.PubMed/NCBI
|
|
41
|
Massague J and Polyak K: Mammalian
antiproliferative signals and their targets. Curr Opin Genet Dev.
5:91–96. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Robson CN, Gnanapragasam V, Byrne RL,
Collins AT and Neal DE: Transforming growth factor-beta1
up-regulates p15, p21 and p27 and blocks cell cycling in G1 in
human prostate epithelium. J Endocrinol. 160:257–266. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cam H and Dynlacht BD: Emerging roles for
E2F: beyond the G1/S transition and DNA replication. Cancer Cell.
3:311–316. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Brehm A, Miska EA, McCance DJ, Reid JL,
Bannister AJ and Kouzarides T: Retinoblastoma protein recruits
histone deacetylase to repress transcription. Nature. 391:597–601.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Magnaghi-Jaulin L, Groisman R, Naguibneva
I, et al: Retinoblastoma protein represses transcription by
recruiting a histone deacetylase. Nature. 391:601–605. 1998.
View Article : Google Scholar : PubMed/NCBI
|