Introduction
As one of the most common pathogens in human
bacteria-related chronic infection and carcinomatous diseases,
Helicobacter pylori infects almost 50% of the world
population (1). In 1994, the World
Health Organization (WHO) and International Agency for Research on
Cancer (IARC) classified it as a type I carcinogen (2). It has been reported that H.
pylori plays a crucial role in the occurrence and development
of chronic active gastritis, peptic ulcers, mucosa-associated
lymphoid tissue lymphoma (MALT) and even gastric adenocarcinoma
(3). Moreover, H. pylori is
also closely related to some hematological systemic disorders, for
example, hypoferric anemia and idiopathic thrombocytopenic purpura.
Recently, it was demonstrated that it is involved in extragas-tric
diseases such as atherosclerosis, ischemic heart disease,
immunologic dysfunction, migraines and pediatric growth retardation
(4–7). According to abundant clinical studies
which had demonstrated morphological and molecular biological
evidence, the infection rate of H. pylori is higher in
patients with chronic hepatic diseases such as chronic hepatitis,
liver cirrhosis and liver cancer than the rate of healthy persons
(8–14). Xuan et al (14) even positively cultured H.
pylori in 3 (10.7%) of their 28 primary hepatic carcinoma
patients.
Primary hepatic cancer is an epidemic malignant
tumor worldwide, and ~665,000 new cases are diagnosed every year
(15). With an extremely high
mortality rate, it ranks second as a cancer-related cause of death
and is responsible for 746,000 deaths worldwide in 2012 (16). Its 5-year survival rate in
developed countries, such as the United States, is 9% (15), whereas <5% worldwide (17), due to its high postoperative
recurrence rate and poor prognosis (18,19).
Thus, interference from the source is the most effective measure
that might etiologically prevent the occurrence and development of
primary hepatic cancer. Hepatitis B virus, hepatitis C virus,
aflatoxin, chemical carcinogens and parasitic infections have been
identified as pathogenic factors in primary hepatic cancer
(20–22). However, basing on the discovery
that H. pylori was found in the liver of patients who
suffered from chronic hepatic diseases, researchers have proposed
that it plays a role in hepatitis and hepatic cancer. This novel
hypothesis prompted studies concerning the correlation between
H. pylori and hepatic cancer (23,24).
The findings may also be helpful in prophylaxis and treatment of
hepatic cancer.
Ito et al (25), Zhang et al (26) and Chen et al (27) co-cultured human hepatoma cells with
H. pylori in vitro and determined the interaction of H.
pylori with hepatocyte surface molecular receptors. H.
pylori adhered to and invaded hepatoma cells. As a result, it
caused a cytotoxic effect that upregulated tumor-related cyclin D1
and PCNA (proliferating cell nuclear antigen). However, they mainly
performed studies in vitro that could hardly simulate the
microenvironment of liver in vivo. Thus, the function of
H. pylori in liver cancer remains unclear. In the present
study, BALB/cAnSlac mice were orally inoculated with an infective
dose of H. pylori. Then, we developed a tumor-bearing mouse
model. Finally, we evaluated the infection status and explored the
role of H. pylori in the development of hepatic cancer.
Materials and methods
Animals
Six to eight weeks old male and female
specific-pathogen-free (SPF) BALB/cAnSlac mice whose fecal DNA were
determined by C97/C98 Helicobacter spp. primers were
obtained from Slaccas Laboratory Animal Co., Ltd., (Shanghai,
China; License no. SCXK 2002–0003, Certificate no. 2007000551273)
and housed under SPF conditions in Fujian Medical University
Laboratory Animal Center (Fuzhou, China; License no. SYXK
2012–0001). All the following pathogens were excluded from SPF
BALB/cAnSlac mice [SPF Laboratory Animal Standard of the People's
Republic of China (GB 14922.2–2011)]. Salmonella spp.;
Yersinia pseudotuberculosis; Yersinia enterocolitica;
Pathogenic dermal fungi; Streptobacillus
moniliformis; Bordetella bronchiseptica;
Mycoplasma spp.; Corynebacterium kutscheri;
Tyzzer's organism; Escherichia coli O115 a, C,
K (B); Pasteurella pneumotropica; Klebsiella
pneumonia; Staphylococcus aureus; Streptococcus
pnemoniae; β-hemolytic streptococcus; Pseudomonas
aeruginosa; lymphocytic choriomenigitis virus (LCMV);
hantavirus (HV); ectromelia virus (Ect.); mouse hepatitis virus
(MHV); sendai virus (SV); pneumonia virus of mice (PVM); reovirus
type III (Reo-3); minute virus of mice (MVM); Theiler's mouse
encephalomyelitis virus (TMEV); mouse adenovirus (Mad);
polyomavirus (POLY); rat parvovirus (KRV); rat parvovirus (H-1);
rat coronavirus (RCV)/sialodacryoadenitis virus (SDAV). All mice
were fed a sterilized commercial diet, given water ad
libitum, and allowed to acclimatize for at least 1 week before
the experiments. All animal protocols met the approval of the
Institutional Animal Care Committee.
Bacterial strains and culture
conditions
H. pylori type strain NCTC 11637 (National
Institute For Communicable Disease Control And Prevention, Chinese
Center For Disease Control And Prevention, Beijing, China) was used
in the present study. Columbia agar base was used (Oxoid Ltd.,
Hampshire, UK) that was supplemented with 8% sheep blood (Bio-Kont
Technology Co., Ltd., Xiamen, China) and H. pylori selective
supplements (Oxoid) containing vancomycin (0.01 mg/ml), cefsulodin
(0.5 mg/ml), trimethoprim (0.5 mg/ml) and amphotericin B (0.5
mg/ml). The bacteria were incubated in microaerophilic conditions
(5% O2, 10% CO2, 85% N2, 37°C,
humidity >90%; LEEC Touch 190S; LEEC Ltd., Nottingham, UK) and
harvested in sterile phosphate-buffered saline (PBS) after 48 h of
growth, centrifuged at 4000 × g for 2 min, and adjusted in sterile
PBS to a final concentration of 5×109 colony-forming
units per milliliter (CFU/ml). Additionally, H. pylori was
not only assessed by Gram staining and phase microscopy for purity,
morphology and motility, but also tested for urease, catalase and
oxidase activity before any animal experiments.
Cell line and cultivation
H22 murine hepatic hepatoma cells were obtained from
China Center for Type Culture Collection (Wuhan, China) and
syngeneic to BALB/cAnSlac mice. The cells were initially grown in a
complete RPMI-1640 medium (HyClone Laboratories Inc., Logan, UT,
USA) containing 10% fetal bovine serum (FBS) at 37°C and 5%
CO2 in vitro for 2–3 days. Subsequently, the cell
suspension was tested for Helicobacter and other
microorganisms prior to mouse injection or implantation. The cell
suspension was spread evenly over Columbia agar base and kept in
microaerophilic condition for microaerophilic bacteria detection.
So did the Luria-Bertani base in usual condition (37°C) for aerobic
bacteria detection. Additionally, after centrifugation the DNA was
extracted by Genomic DNA Mini extraction kit (Beyotime Institute of
Biotechnology, Haimen, China) for Helicobacter
genus-specific 16S rRNA (C97/C98) test. Under the circumstance that
all detection methods above were negative for microorganisms, we
conducted the further protocols. The cell suspension was collected
and centrifuged at 2000 × g for 5 min and adjusted with a
serum-free RPMI-1640 medium to a final concentration of
1×107/ml. Every BALB/cAnSlac mouse was intraperitoneally
injected with a 0.2-ml cell suspension for the first in vivo
subcultivation for a 7-day period. Finally, the mouse was
euthanized by cervical dislocation, and its ascites was collected
and centrifuged. Ascitic precipitate cells were washed and adjusted
to the same concentration. The cell suspension was
intraperitoneally injected into another BALB/cAnSlac mouse for the
second in vivo subcultivation. Following similar, previously
discussed protocols, a third in vivo subcultivation was
performed. Then, the centrifuged cells were adjusted to
4×107/ml and were used in an orthotopic
implantation.
Drug pretreatment
Lactobacilli which inhabit the stomachs of
the mice would interfere with the growth of H. pylori even
eradicate it (28). All 70
experimental mice were treated on the 1st to 3rd day with the drugs
listed in Table I, according to
the methods described by Thalmaier et al (29). A mixed solution of ciprofloxacin
(Zhejiang Jingxin Pharmaceutical, Co., Ltd., Zhejiang, China),
amikacin (Shanghai Harvest Pharmaceutical Co., Ltd., Shanghai,
China), imipenem (Merck Sharp & Dohme Corp., Kenilworth, NJ,
USA), vancomycin (Zhejiang Hisun Pharmaceutical Co., Ltd.,
Zhejiang, China), and fluconazole (Yangtze River Pharmaceutical
Group, Taizhou, China) were administered once daily.
 | Table IDrugs and dose scheme (μg/mouse/day). |
Table I
Drugs and dose scheme (μg/mouse/day).
| Antibiotics | Oral dose | Intraperitoneal
dose |
|---|
| Ciprofloxacin | 500 | 0 |
| Amikacin | 375 | 375 |
| Imipenem | 1250 | 1250 |
| Vancomycin | 1000 | 1000 |
| Fluconazole | 150 | 0 |
Infection protocols
On day 3 (after the last drug treatment), 70 mice
were randomly divided into group A (n=40, H. pylori infected
mice) and group B (n=30, uninfected mice). From 3rd to 9th day, all
mice were fasted for 15–18 h overnight before the treatment on the
following day. The next day, every mouse was orally inoculated with
a 0.2-ml sterilized alkalescent buffer (300 mM NaHCO3)
to neutralize gastric acidity. Then, 15–30 min later, each mouse in
group A was administered an oral H. pylori suspension
(1×109 CFU; in 0.2 ml of sterilized PBS) once daily for
a continuous 7-day period (4th to 10th day; defined as the first
week); group B was administered a 0.2-ml sham dose of sterilized
PBS in a same manner and schedule (30–32).
All mice underwent hepatic surgery during the 9th week.
Orthotopic hepatic carcinoma model
At the beginning of the 9th week, the mice in group
A were randomly divided into group A1 (n=20; H. pylori
infected and tumor positive mice) and group A2 (n=20; H.
pylori infected and tumor negative mice), and group B was
divided into group B1 (n=20, uninfected and tumor positive mice)
and group B2 (n=10, uninfected and tumor negative mice). The left
liver lobe of the mice was implanted with 50 μl of H22 cell
suspension (2×106 cells; mice in group A1 and B1) or
just 50 μl serum-free RPMI-1640 medium (mice in group A2 and B2)
with a subcapsular intrahepatic injection according to Yao et
al (33) and Aprahamian et
al (34). Each mouse was
anesthetized with 80 mg/kg ketamine (Sunkind Co., Ltd., Shanxi,
China) by intraperitoneal injection. The mouse was placed in a
supine position on the operating table when it was fully
anesthetized. A small median longitudinal incision was made below
the xiphoid to expose the left lobe of the liver, and sterile gauze
was placed under it to prevent peritoneal sowing. H22 cells in a
serum-free RPMI-1640 medium were slowly injected into the
parenchyma of the left hepatic lobe at a 30-degree angle with a
100-μl microinjector (Shanghai Bolige Industry & Trade Co.,
Ltd., Shanghai, China), thus, a transparent bleb of medium could be
seen within the hepatic capsule. A sterile cotton swab was gently
compressed on the injection site for hemostasis, and the abdomen
was closed separately in a two layer method (peritoneum and skin)
with a 4-0 silk suture. The mice were kept in SPF warm incubator
for observation and finally returned to the SPF animal room when
they had fully recovered from the anesthesia.
Tissue collection and bacterial
isolation
The mice were fasted for 24 h overnight before
necropsy. At the end of the 13th week, all mice were euthanized by
cervical dislocation and necropsied for bacterial isolation and
histopathology. The hemorrhagic ascites was firstly collected from
bulge abdomen of tumor positive mouse by a 2-ml aseptically
injector. The ascites was fully mixed and piped (200 μl) onto the
Columbia agar base for H. pylori culture in microaerophilic
environment. The remainder was centrifuged at 4000 × g for 5 min.
The supernatant was tested for AFP (α-fetoprotein) by ELISA kit
(Wuhan Boster Biological Technology, Co., Ltd., Wuhan, China) and
compared with that in serum of both tumor positive and negative
mouse. The DNA of cellular precipitate was extracted by Genomic DNA
Mini extraction kit (Beyotime Institute of Biotechnology) and
tested for Helicobacter genus-specific 16S rRNA (C97/C98).
Samples of blood, stomach and liver were aseptically collected and
cultured during necropsy. A total of 150 μl of blood samples were
collected from inferior vena cava puncture. One hundend microliters
were spread evenly over a Columbia agar plate for H. pylori
cultivation and 50 μl were stored at −80°C until for DNA and AFP
analysis. The liver, as a whole, was removed and weighed. Then, the
left liver lobe (group A2 and B2) was directly divided into three
sections. The first section (50–100 mg) was ground in sterile
grinders with 100 μl of sterile PBS and spread evenly over a
Columbia agar plate for H. pylori cultivation. The second
section (15×15×3 mm) used for histological analysis was placed into
10% normal buffered formalin for 24 h, and embedded in paraffin.
Sections (3-μm) were cut. The slides were stained with hematoxylin
and eosin (H&E; conducted by the Pathology Department of
Affiliated Union Hospital of Fujian Medical University, Fuzhou,
China) for assessment of histopathological changes and
immunohistochemistry (IHC) staining for H. pylori, PCNA,
B-cell lymphoma 2 (Bcl-2) and Bcl-2-associated X protein (Bax). The
third section was stored at −80°C for DNA and protein analysis.
Equally, the tumor in the left liver lobe (A1 and B1 group) was
also divided, processed and analyzed as the protocols described
above. Finally, the stomach was removed, opened along its greater
curvature, and rinsed gently in sterile cold PBS. When the contents
had been totally removed, the stomach was placed (mucosal side up)
on sterile gauze and longitudinally dissected along the greater
curvature into three fragments, so that each one contained the
gastric cardia, body and antrum. The three gastric fragments were
processed and analyzed. All Columbia agar plates were incubated in
a microaerophilic condition (5% O2, 10% CO2,
85% N2, humidity >90%) at 37°C. The plates were
evaluated for H. pylori growth from 3rd to 10th day after
necropsy. The presence of colonies were confirmed using morphology,
Gram staining, biochemical tests (urease, catalase and oxidase
reactions) and polymerase chain reaction (PCR). The plates were
discarded and defined as negative if no positive colony was
detected until the 10th day.
DNA extraction and PCR amplification
DNA of samples (blood, stomachs, livers and tumors)
were isolated with Genomic DNA Mini extraction kit (Beyotime
Institute of Biotechnology). According to the manufacturer's
instruction, ~25 mg of tissue was completely triturated with an
electric grinder (Tiangen Biotech, Co., Ltd., Beijing, China), and
then, the homogenate (or 50 μl of blood) was mixed in a vortex with
180 μl of sample lysis buffer A and 20 μl of Proteinase K. The
mixture was incubated overnight at 55°C, thus, the tissues could be
fully cleaved. The next day, 200 μl of sample lysis buffer B was
added to and incubated with them for 10 min at 70°C. The sample was
fully mixed with 200 μl of dehydrated ethanol and then added to a
clean spun column. The mixture was washed and centrifuged with a
series of washing buffer. Finally, 50 μl of elution buffer was
pipetted into the spun column. The DNA was collected with the last
high-speed centrifugation, and its concentration was adjusted to 80
ng/μl for the following analysis. The extracted DNA was amplified
with Helicobacter genus-specific 16S rRNA primers (35): sense primer, 5′-GCT ATG ACG GGT ATC
C-3′ (C97F) and antisense primer, 5′-GAT TTT ACC CCT ACA CCA-3′
(C98R). The sense and antisense primers amplified a 400-bp product.
We mixed 2 μl of 80 ng/μl extracted DNA, 1 μl of 20 pmol from each
primer, 12.5 μl of 2X Power Taq PCR MasterMix (Bioteke Corp.,
Beijing, China), and 8.5 μl of sterilized ultrapure water. The
mixture was amplified after a PCR cycle: initial denaturation with
a Taq polymerase at 95°C for 10 min, denaturation at 95°C for 30
sec, annealing at 55°C for 30 sec, extension at 75°C for 30 sec (35
cycles), and a final extension step at 75°C for 5 min. The
amplified products were loaded onto 1.5% (weight/volume) agarose
(Solarbio Technology Co., Ltd., Beijing, China) gels containing
0.04% (volume/volume) GoodView (Beijing SBS Genetech Co., Ltd.,
Beijing, China), and the production size was compared with a 100-bp
DNA marker ladder (Pregene Biotechnology Co., Ltd., Beijing,
China). H. pylori type strain NCTC 11637 was used as a
positive control, while sterilized ultrapure water was used as a
negative control for each PCR cycle. Bands were visualized and
photographed under UV light. The 16S rRNA positive samples were
sequenced by Biosune Biotechnology Co., Ltd., (Beijing, China) and
blasted with H. pylori type strain NCTC 11637.
Immunohistochemical staining and
quantitative analysis
Tissue sections were deparaffinized in xylene and
rehydrated through graded alcohols to water. Slides were antigen
unmasked by maintaining in a citrate buffer (10 mmol/l; pH 6.0) at
a sub-boiling temperature for 20 min. The sections were cooled on a
bench top for 30 min and washed with PBS (pH 7.4) three times for 5
min each. Hydrogen peroxide (3%) was incubated with the slides for
10 min and washed away, so endogenous peroxidase activity would be
ceased. Sections were then incubated with polyclonal rabbit
anti-H. pylori antibody (1:50 diluted in PBS, Product no.
B047101; Dako, Glostrup, Denmark), monoclonal mouse anti-PCNA
antibody (1:200 diluted in PBS, Product no. sc-56; Santa Cruz
Biotechnology, Dallas, TX, USA), mono-clonal mouse anti-Bcl-2
antibody (1:50 diluted in PBS, Product no. sc-7382; Santa Cruz
Biotechnology), or polyclonal rabbit anti-Bax antibody (1:50
diluted in PBS, Product no. sc-526; Santa Cruz Biotechnology)
overnight at 4°C. The next day, we removed the primary antibody and
washed the sections. A prepared rabbit or mouse SP kit (ZSGB-BIO,
Beijing, China) was used according to the manufacturer's
instructions, and we incubated the sections with solution A for 30
min and then with solution B for 45 min at 37°C. We removed the
reagents and washed them. DAB (ZSGB-BIO) was added to each section
and the staining closely monitored. As soon as the sections had
developed, they were immersed in PBS and counterstained in
hematoxylin. Finally, the sections were dehydrated with graded
alcohols to form xylene before mounting. The tissue sections must
not dry out during the whole immunocytochemical staining procedure.
Brown granules which were observed in the gastric lumens, hepatic
sinusoid, cytoplasm, or nucleus were regarded as positive staining.
The slides were observed under a light microscope (Olympus Corp.,
Tokyo, Japan), and five photographs of each section were randomly
obtained at ×40 magnification using the same conditions (light
source, color saturation, brightness, gain and contrast).
Photograph quantification was performed using Image-Pro Plus
software, version 6.0 (Media Cybernetics, Inc., Silver Spring, MD,
USA). In each field, the integrated optical density (IOD) for all
positive staining was measured.
Protein extraction and western
blotting
Twenty milligrams of the liver or tumor was
completely triturated with an electric grinder, and the homogenate
was mixed with 250 μl of cold RIPA lysis buffer (Beyotime Institute
of Biotechnology, Shanghai, China) containing 1 mM of
phenylmethylsulfonyl fluoride (PMSF). The mixture was centrifuged
(14,000 × g, 4°C for 5 min), and the supernatant was collected. The
protein concentrations were detected using a BCA kit (Beyotime
Institute of Biotechnology). The proteins (50–100 μg/lane) were
electrophoresed in a 12% SDS-polyacrylamide gel and then
transferred onto a PVDF membrane (Millipore Corp., Bedford, MA,
USA). The membranes were blocked with 5% skim milk in TBST at room
temperature for 1 h and subsequently incubated overnight at 4°C
with a primary antibody (PCNA at 1:500, Product no. sc-56; Bax at
1:500, Product no. sc-526; and Bcl-2 at 1:500, Product no.
sc-7382). The next day, the membranes were incubated for 1 h at
room temperature with a horseradish peroxidase-conjugated goat
anti-rabbit or goat anti-mouse IgG secondary antibody (Zhongshan
Golden Bridge Biotechnology Co., Ltd., Beijing, China) at 1:5,000.
Finally, the protein bands on the membranes were detected using an
ECL kit (Beyotime Institute of Biotechnology) and scanned with the
ImageQuant LAS 4000 digital imaging system (GE Healthcare,
Buckinghamshire, UK) or exposed to a medical radiography film
(Kodak, Tokyo, Japan). The bands were quantified by Image-Pro Plus
software. The IOD of each band was measured and expressed as mean ±
standard deviation (mean ± SD).
Statistical analysis
Measurement data are presented as mean ± SD for
normal data and median with ranges for non-normal data, while
enumeration data are presented as ratios. The data were analyzed
using one-way ANOVA if they corresponded with both a normal
distribution and homoscedasticity. The SPSS program package
(version 11.0; SPSS, Inc., Chicago, IL, USA) was used for our
statistical analysis. Differences were considered significant at
P-value <0.05.
Results
Morphological, serologic and ascitic
features of mice
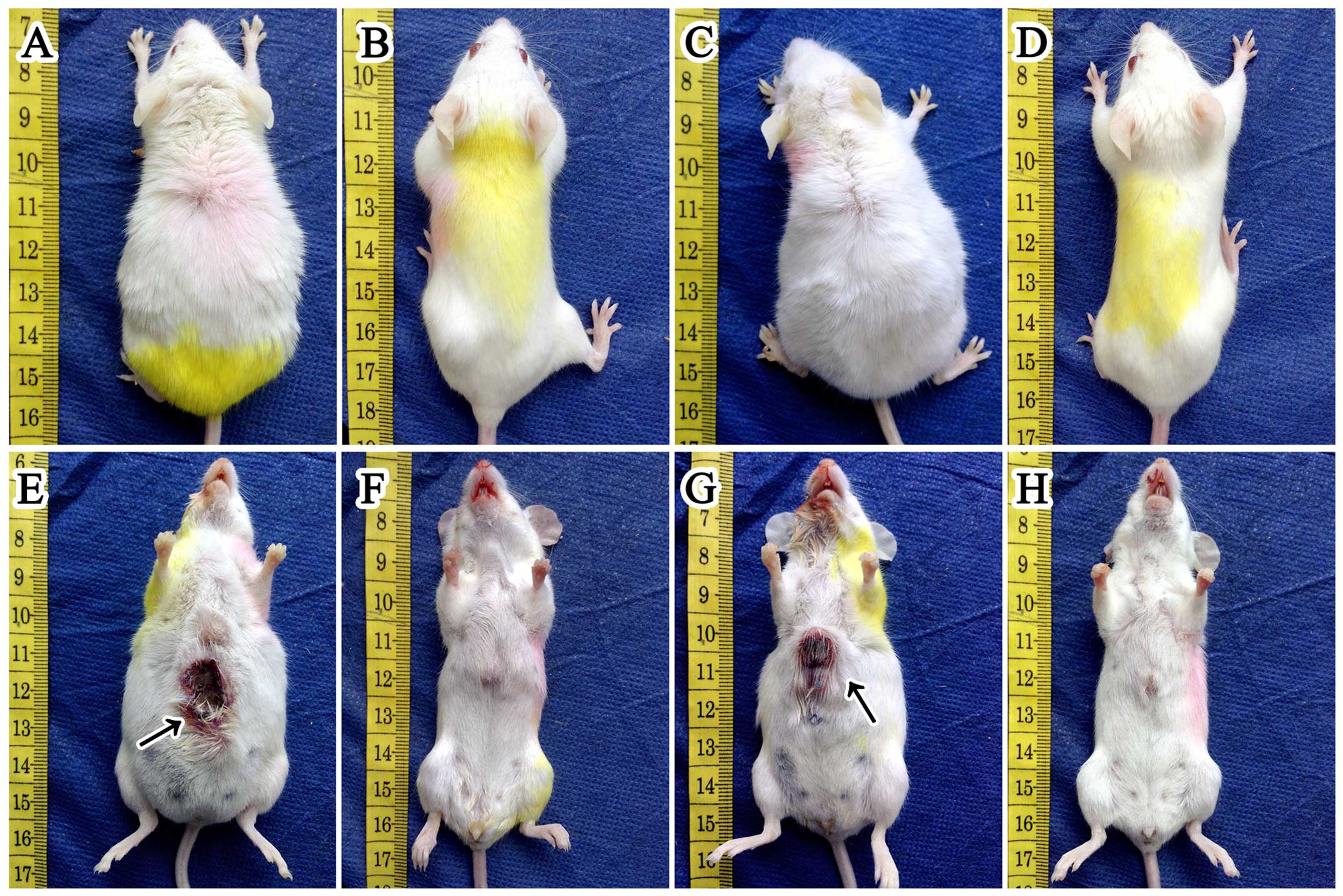
The skin ulceration (Fig. 1) was observed in 27.8% (5/18) and
30% (6/20) of mice in group A1 and B1, respectively. However, there
was no significant (P=0.583) difference between the two groups. We
were unable to collect the skin ulceration for morphological
detection. But we analyzed the ‘link tissue’ which connected
hepatic tumor to abdominal wall instead of ulceration by H&E
stain. Consequently, we were not surprised at finding that it was
full of tumor cells which were characteristic of cellular
pleomorphism. Additionally, 50% (9/18) and 45% (9/20) of mice in
group A1 and B1 had developed ascites before they were necropsied.
Moreover, H. pylori could not be cultured from any blood and
ascites samples. AFP level (mean ± SD, ng/ml) of each group was
11.11±0.69 (ascites of A1 group), 10.89±0.70 (serum of A1 group),
10.99±0.67 (serum of A2 group), 10.98±0.72 (ascites of B1 group),
10.88±0.54 (serum of B1 group) and 11.08±0.71 (serum of B2 group),
respectively. However, no significant difference (P=0.962) could be
found between any of the two groups above. Moreover,
Helicobacter genus-specific 16S rRNA was not detected in
blood or ascites samples.
Gastric H. pylori determination and
histopathological assessment
H. pylori was cultured from gastric
homogenate from 90% (18 of 20) of the mice in group A1 (H.
pylori infected and tumor positive mice) and 80% (16 of 20) of
the mice in group A2 (H. pylori infected and tumor negative
mice). Helicobacter genus-specific 16S rRNA (Fig. 2) was detected using DNA extracted
from the stomach, and a 95% (19 of 20) positive rate was found in
both group A1 and A2. Three to five positive PCR products in each
group were randomly selected and sequenced. All the sequenced
samples were completely homologous to H. pylori type strain
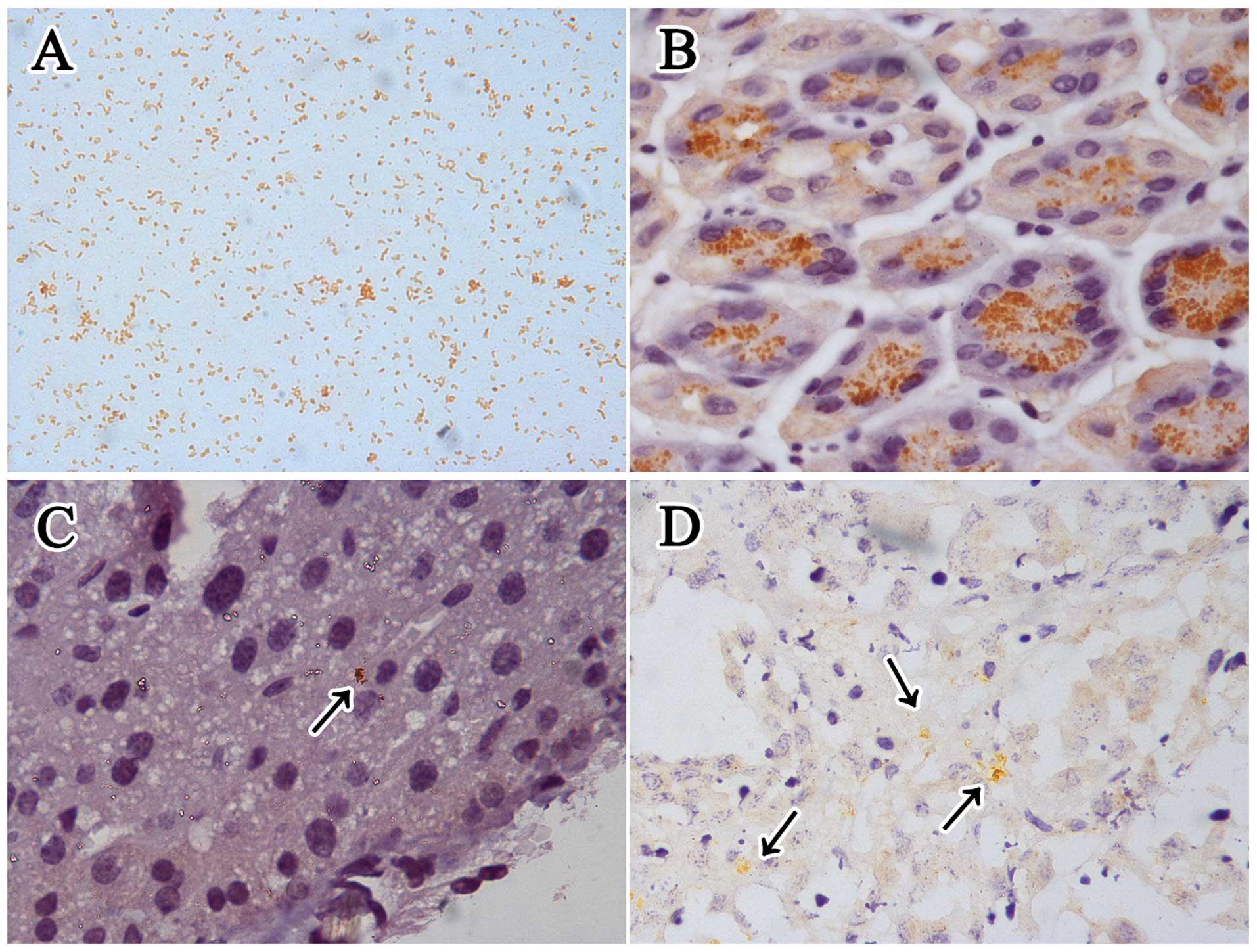
NCTC 11637. IHC staining for H. pylori in
Helicobacter genus-specific 16S rRNA positive gastric slides
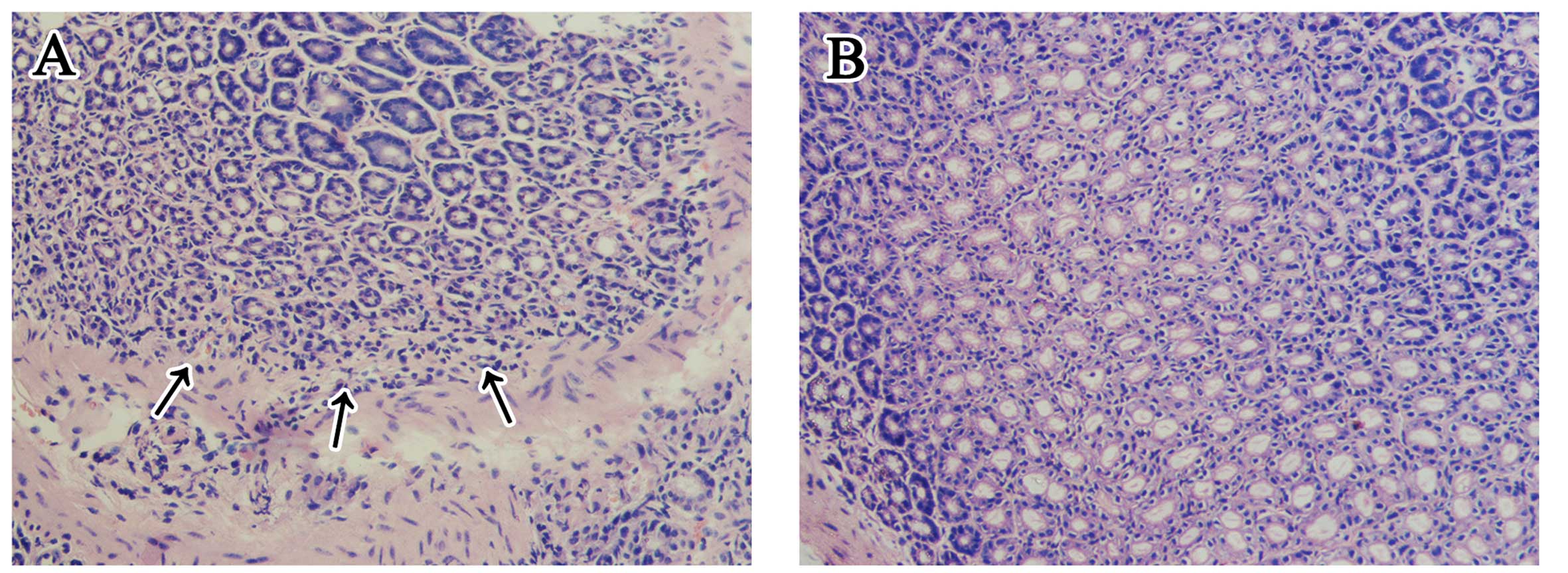
showed that it mainly colonized the gastric glands (Fig. 3). Although there was no apparent
ulceration in any H. pylori-infected stomach (group A1 and
A2), the histopathological changes (Fig. 4) indicated that chronic
inflammatory cells infiltrated the pyloric antrum area, and the
inflammatory changes were mild to moderate. All the pathogen
detections above were negative for H. pylori in the
uninfected stomachs (group B1 and B2), and chronic inflammatory
cells were not apparent.
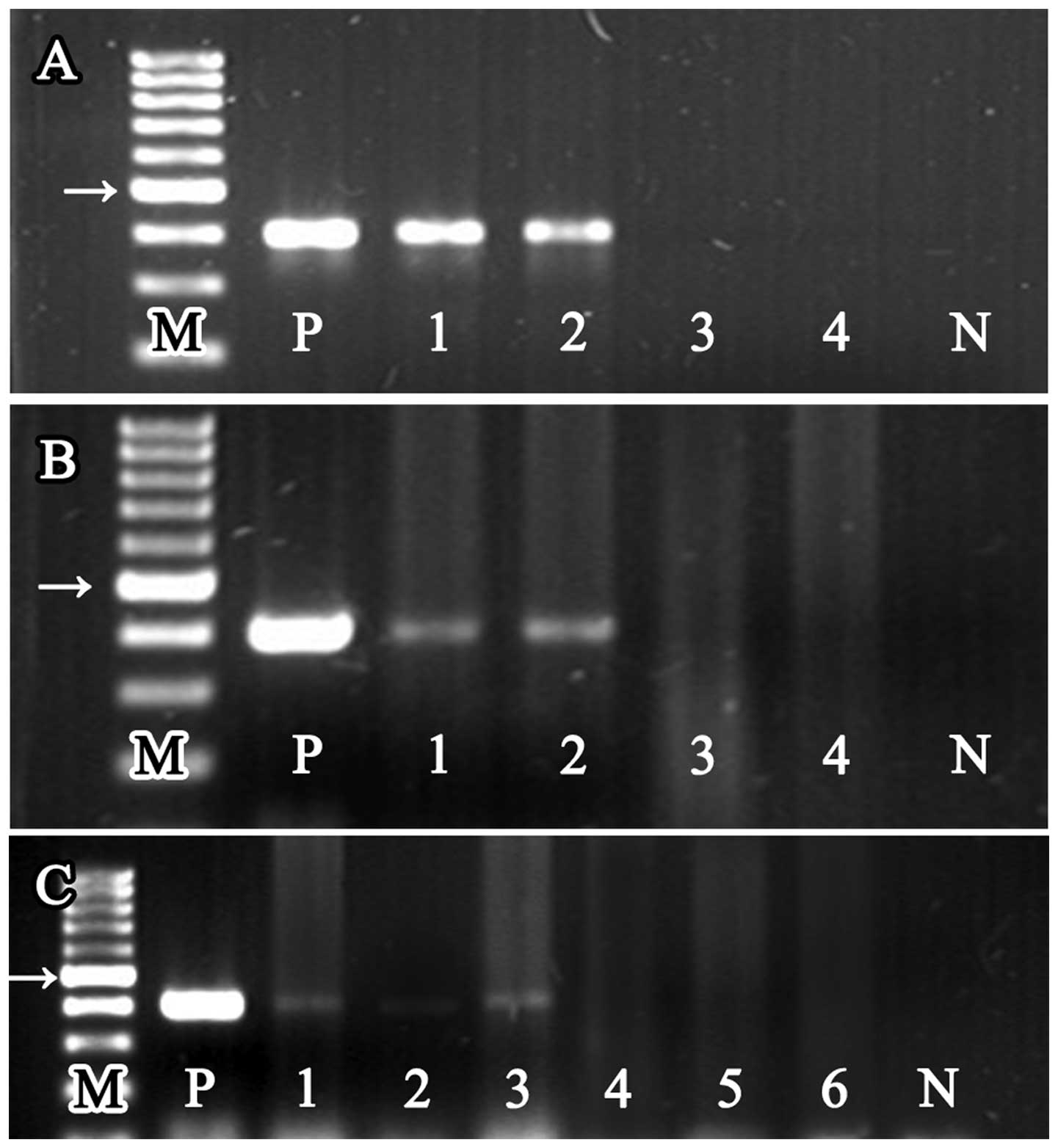 | Figure 2The PCR products of the
Helicobacter genus-specific 16S rRNA from DNA of different
mouse tissue samples, the size of all product was 400 bp. Lane ‘M’
represents 100 bp DNA marker ladder, and lanes ‘P’ and ‘N’
represent positive control and negative control, respectively. The
arrows, 500 bp location of the marker. (A) 16S rRNA PCR products
detected from bacterial colonies which were cultured from gastric
suspensions. Lanes 1 and 2 represent the DNA samples extracted from
bacterial colonies cultured from H. pylori-infected stomach
(group A1 and A2), and lanes 3 and 4 represent the DNA samples
extracted from bacterial colonies cultured from uninfected stomach
(group B1 and B2). (B) 16S rRNA PCR products detected from gastric
samples. Lanes 1 and 2, H. pylori-infected gastric DNA
samples (group A1 and A2), and lanes 3 and 4, the uninfected
gastric DNA samples (group B1 and B2). (C) 16S rRNA PCR products
detected from liver and tumor samples. Lanes 1 and 2, liver and
tumor DNA samples extracted from H. pylori-infected and
tumor positive mice (group A1). Lane 3, hepatic DNA samples
extracted from H. pylori-infected and tumor negative mice
(group A2). Lanes 4 and 5, liver and tumor DNA samples extracted
from uninfected and tumor positive mice (group B1). Lane 6, hepatic
DNA samples extracted from uninfected and tumor negative mice
(group B2). |
Detection of H. pylori in livers
The mice in group A1 (n=18) and A2 (n=16) whose
gastric homogenates were positive for H. pylori by culture
method as well as all individuals in group B1 (n=20) and B2 (n=10)
were taken into the following detection and analysis procedures.
H. pylori could not be cultured in any liver or tumor
sample. The positive rate of Helicobacter genus-specific 16S
rRNA (Fig. 2) for the liver and
tumor samples in group A1 was 83.3% (15 of 18) and 66.7% (12 of
18), respectively. The rate for the liver samples in group A2 was
87.5% (14 of 16). Three to five positive PCR products were also
sequenced as previously described. All sequenced products were also
completely homologous to H. pylori type strain NCTC 11637.
H. pylori was mainly observed in the hepatic sinusoid and
necrotic area of the tumors in the infected mice using IHC staining
detection (Fig. 3), while none
could be found in the tumor parenchyma. All the H. pylori
detection methods were negative for the uninfected livers and
tumors (group B1 and B2).
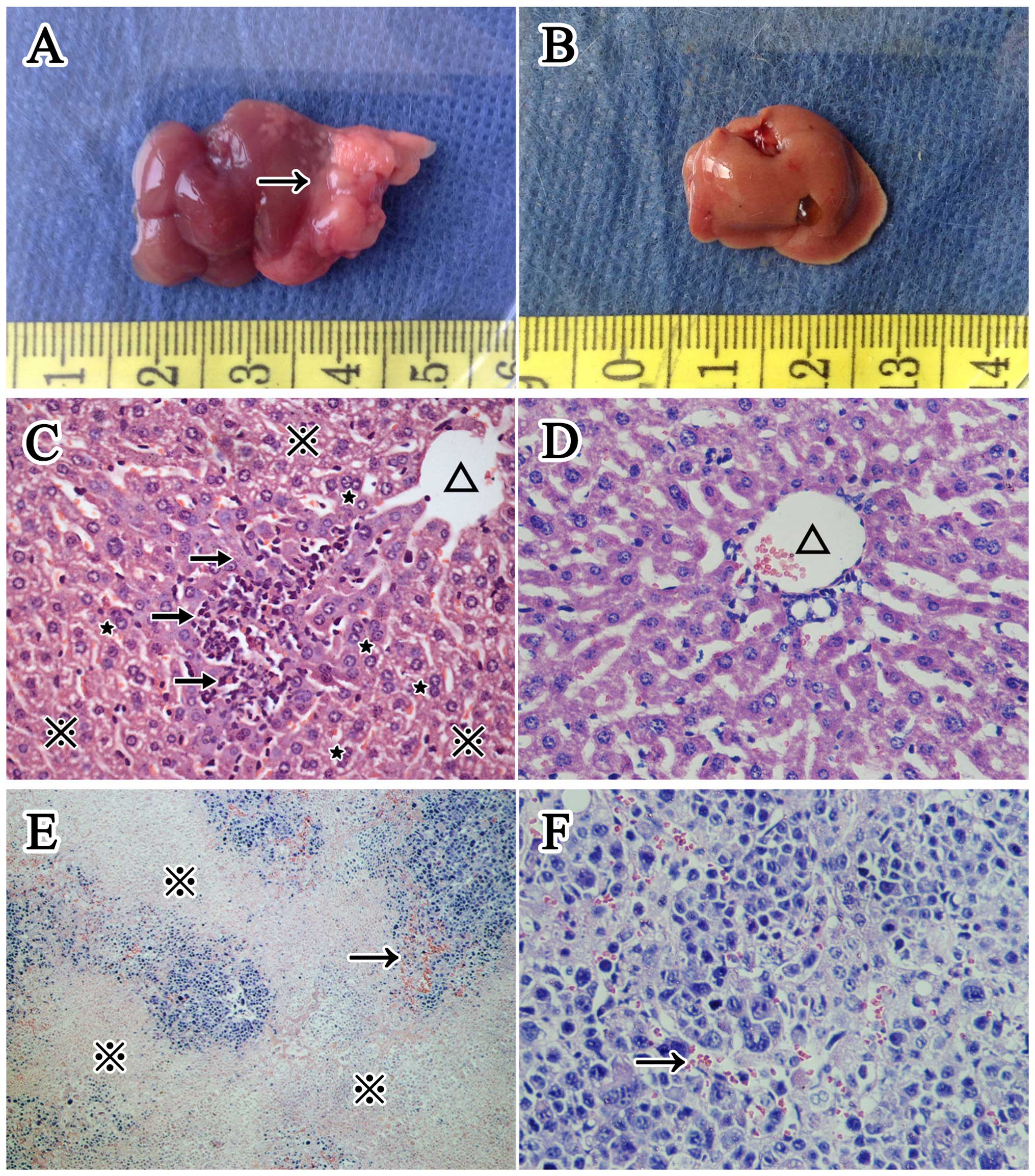
Morphological changes in the livers
We determined liver to body weight ratio for each
group and show the average ratio (mean ± SD) of the mice in groups
A1, A2, B1 and B2 was
0.072261±0.024696,0.045331±0.003885,0.062900±0.020324, and
0.042018±0.004516, respectively. The ratio of tumor positive mice
(group A1 and B1) was significantly higher (P<0.001) than that
of tumor negative mice (group A2 and B2), while there was no
significant difference between H. pylori infected and
uninfected mice, namely group A1 and B1 (P=0.178) or group A2 and
B2 (P=0.635). Cytoplasmic vacuolation (ballooning degeneration) and
hepatic binucleate cells could be observed in the H. pylori
infected livers, and the sinusoids were compressed by swollen
hepatic cells. In addition, the infiltrated inflammatory cells were
thinly distributed in the hepatic lobules and also observed in the
H. pylori infected livers. However, no visible microscopic
difference existed between group A1 and B1 tumors. Both had
characterized structural disorders and cellular pleomorphism such
as atypia, increased nuclear-cytoplasmic ratio, hyperchromatic
nuclei and pathological mitosis. Additionally, inflammatory cells
and vasodilation could be observed among the tumor cells. The
necrotic areas, which were homogeneous and lightly stained, were
distributed around the tumor parenchyma (Figs. 1 and 5).
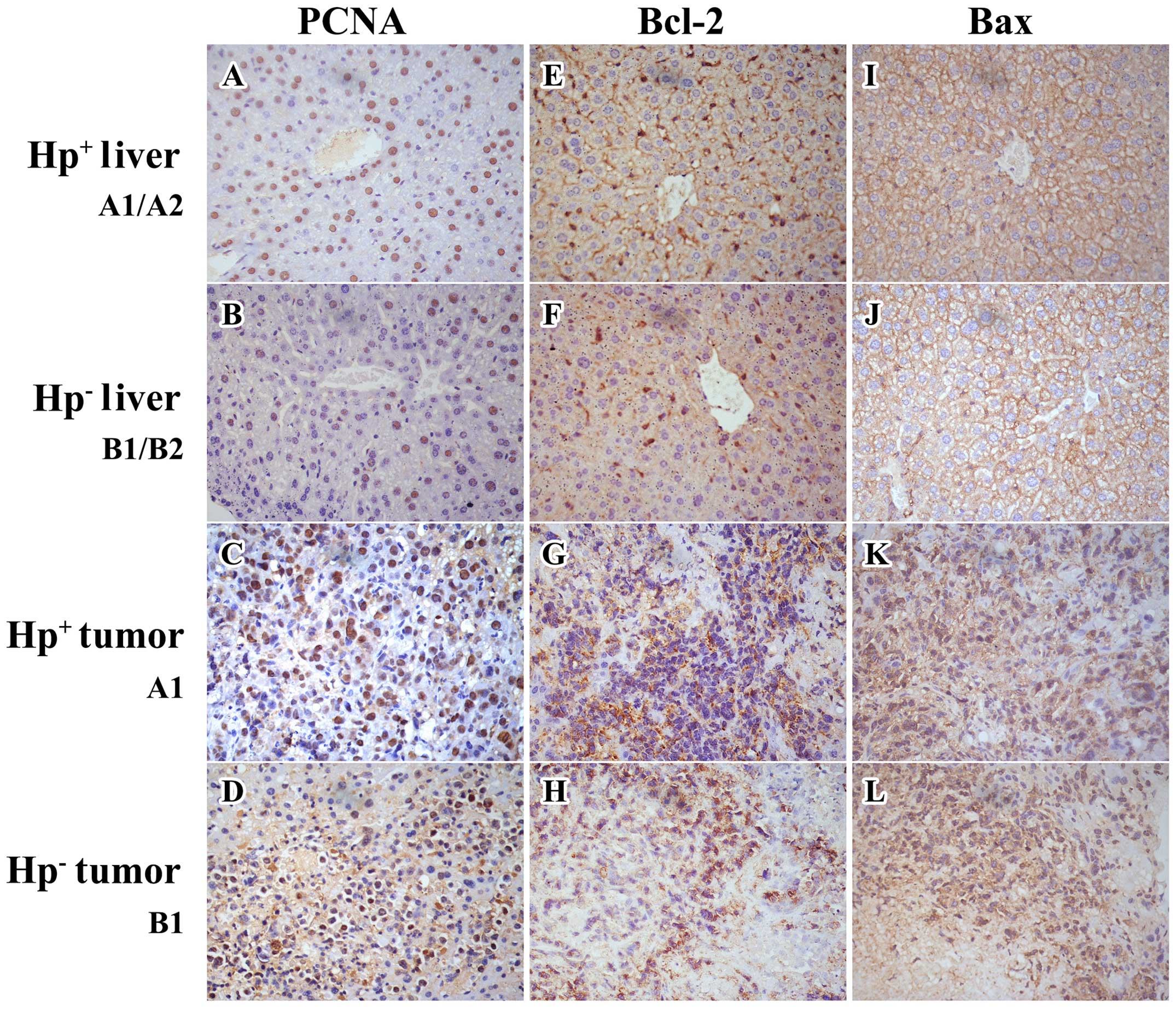
Protein expression in livers and
tumors
We detected the expression of specific proteins in
the livers and tumors by performing IHC staining and western blot
analysis. Similar trends with IHC staining and western blot
analysis were noted regarding PCNA and Bcl-2 expression. The PCNA
expression (Figs. 6 and 7 and Table
II) was significantly (P≤0.01) increased in the H.
pylori infected tumors (group A1) as being compared with the
uninfected tumors (group B1). Similarly, it was also significantly
(P<0.05) increased in the H. pylori infected livers
(group A1 and A2) as being compared with the uninfected livers
(group B1 and B2). Additionally, the tumor PCNA expression (group
A1 and B1) was dramatically increased (P<0.001) as compared with
the liver (group A1, A2, B1 and B2). There was no difference
(P>0.1) in the Bcl-2 expression (Figs. 6 and 7 and Table
II) in any tumor or liver. With western blot analysis, a
significant reduction (P=0.035) in the Bax expression (Fig. 7 and Table II) of the H.
pylori-infected tumors was demonstrated (group A1) as compared
with the uninfected tumors (group B1); there was no difference
(P=0.176) in Bax expression (Fig.
6 and Table II) between these
two groups with IHC staining. However, both western blot analysis
and IHC staining demonstrated a significant upregulation
(P<0.05) of Bax expression in the H. pylori-infected
livers (group A1 and A2), as compared with the uninfected livers
(group B1 and B2). Moreover, the Bax expression in the tumors
(group A1 and B1) was significantly decreased (P<0.001), as
compared with the livers (group A1, A2, B1 and B2).
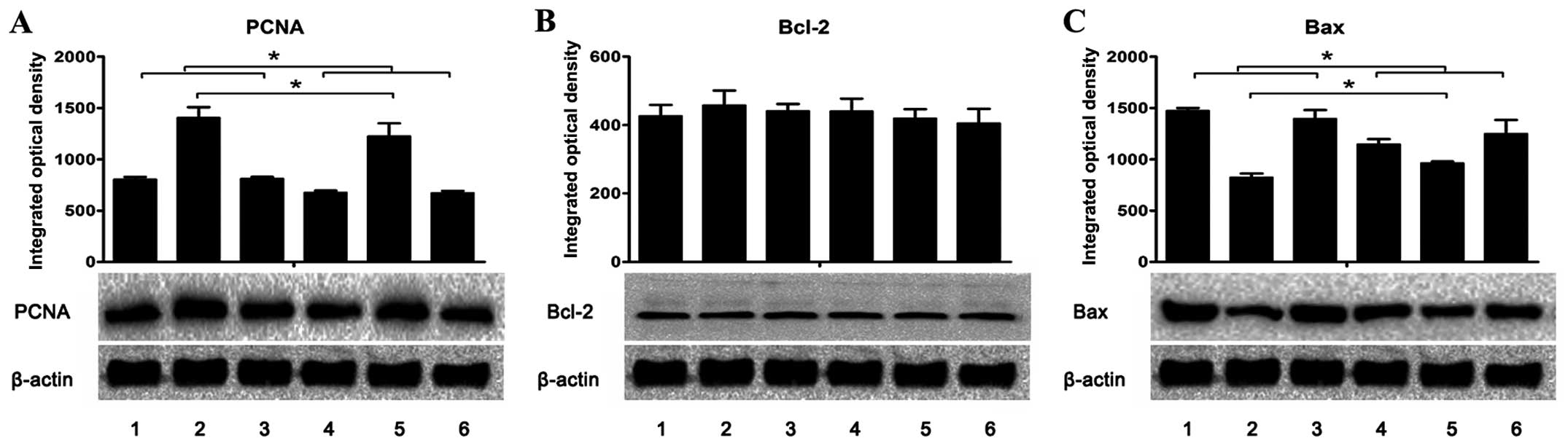 | Figure 7Expression of (A) PCNA, (B) Bcl-2 and
(C) Bax in the livers and tumors was assessed using western blot
analysis. The integrated optical density (IOD) of each antigen band
was measured. The results are expressed as mean ± SD in the
histogram. Lanes 1 and 2, the protein expression in the livers and
tumors, respectively, of the H. pylori-infected and tumor
positive mice (group A1). Lane 3, the protein expression in the
livers of H. pylori-infected and tumor negative mice (group
A2). Lanes 4 and 5, the protein expression in the livers and
tumors, respectively, of the uninfected and tumor positive mice
(group B1). Lane 6, the protein expression in the livers of
uninfected and tumor negative mice (group B2).
*P<0.05 as lane 1 compared with lanes 4 or 6, lane 3
compared with lanes 4 or 6, and lane 2 compared with lane 5. |
| Table IIIntegrated optical density of the
proteins expressed in representative liver or tumor samples
detected by immunohisto-chemical stain and western blot analysis
(mean ± SD). |
Table II
Integrated optical density of the
proteins expressed in representative liver or tumor samples
detected by immunohisto-chemical stain and western blot analysis
(mean ± SD).
| | PCNA | Bcl-2 | Bax |
|---|
| |
|
|
|
|---|
| Group | Sample | IHC | WB | IHC | WB | IHC | WB |
|---|
| A1 | Liver |
660.384±24.196a,b |
804.811±21.937a,b | 257.009±25.241 | 426.883±31.775 |
568.872±35.024a,b |
1473.967±25.356a,b |
| A1 | Tumor |
769.849±16.381c |
1406.576±100.647c | 284.988±12.188 | 458.303±43.060 | 309.306±20.036 |
822.853±38.193c |
| A2 | Liver |
657.051±13.166a,b |
812.288±15.339a,b | 243.345±25.150 | 441.563±20.182 |
533.919±15.386a,b |
1395.167±85.053a,b |
| B1 | Liver | 452.964±15.665 | 677.803±17.419 | 283.233±38.032 | 440.803±36.415 | 449.545±30.011 |
1145.367±50.639 |
| B1 | Tumor | 725.417±17.978 |
1226.722±124.089 | 275.016±21.806 | 419.970±26.492 | 336.763±15.308 | 961.160±18.509 |
| B2 | Liver | 442.964±18.235 | 672.141±18.418 | 262.746±40.646 | 405.830±41.563 | 481.272±16.762 |
1248.333±134.814 |
Discussion
H. pylori has been classified as a type I
carcinogen (2), that is a major
causative factor in many gastric diseases. Furthermore, some
extragastric diseases are also correlated with its pre-infection
during childhood (4–7). Recent studies on the correlation
between H. pylori infection and chronic hepatic disease have
found that the anti-H. pylori antibody level in the serum of
patients with chronic liver disease is significantly higher than
that of healthy patients (8–12,36–40).
In addition, H. pylori was also detected and confirmed in
many liver samples from chronic hepatic disease patients using
morphological and genetic detection (8,13,14,24,41–43).
Moreover, researchers have successfully cultured H. pylori
from the liver samples of some clinical hepatic disease cases
(14,44). All the above mentioned details
suggest that H. pylori could infect the human liver and may
play a role in the development or progression of hepatic
disease.
However, when talking about the role of H.
pylori in the progression of hepatic diseases, there is no
consistent opinion among researchers. Some investigator, such as
Fox et al (35), Matsukura
et al (45) and García
et al (46) claimed that it
could not colonize livers and was barely able to promote hepatic
diseases. They rather regard it as contaminant instead of
infection. However, others, such as Goo et al (23), Ito et al (24) and Rocha et al (47), reported that H. pylori not
only colonize the livers, but also contribute to the hepatic
disease evolution. Nowadays, investigators are gradually
discovering a growing kinds of Helicobacter spp., such as
H. bilis, H. pullorum and H. pylori, which has
been found in the liver of hepatic disease patients. However,
attention is mainly focused on H. bilis or H.
pullorum (35,45,46)
rather than H. pylori for the following reasons. It has been
confirmed by in vitro study that H. pylori was unable
to survive in bile products (48).
Moreover, cholecystectomy would increase the gastric
Helicobacter infection risk (49). Relying on the crucial findings that
H. pylori was unable to survive in hepatobiliary condition,
the presence of H. pylori was considered a result of
contamination rather than colonization, even though it had ever
been found in gallbladder and bile of patients (50). In humans, bile is produced
continuously by the hepatocytes (liver bile), but stored and
concentrated in the gallbladder (gallbladder bile) (51). Ito et al (25), Zhang et al (26) and Chen et al (27) found H. pylori exhibited
hepatotoxicity while it was co-cultured with hepatocytes which were
capable of producing bile in vitro. Hence, they insisted
H. pylori might play a potential role in
hepatocarcinogenesis. Additionally, Goo et al (23), Ito et al (24) and Rocha et al (47) found all liver samples were negative
for other gut organisms, such as E. coli, which mainly
inhabited gut and more likely present in liver than H.
pylori as a contaminant. Moreover, the successful H.
pylori-cultivation from livers of patients (14,44)
also suggest its colonization rather than contamination. Finally,
basing on the finding that H. pylori could be recovered from
feces, Kelly et al (52)
and Thomas et al (53)
agreed with the opinion that it would survived even after exposure
to bile. We came to the same conclusion as that summarized by Ito
et al (25), Zhang et
al (26) and Chen et al
(27). However, bile products
might transform H. pylori from virulent helical form to
insufficient coccoid form (54).
The colonization and pathology ability of H. pylori in liver
may depend on its virulence factors, such as CagA or VacA (25,55–57)
and forms. Therefore, we thought the reasons for low H.
pylori frequency in the study of Fox et al (35) may due to the insufficient strain or
form of H. pylori which the researchers had adopted.
By using morphological (IHC staining) and genetic
detection (PCR), the present study confirmed that H. pylori
or only its structural components were both detected in stomachs
and livers. It was mainly detected in the hepatic sinusoids and
necrotic areas of the tumors. Currently, the following possible
perspectives or hypotheses are widely accepted regarding the
presence of H. pylori in the liver. Firstly, because its
genetic components had been detected in cholecystic samples or the
bile of gallbladder disease patients, some researchers consider the
findings related to duodenal reflux (58–62).
Gastrointestinal motility will transport H. pylori from the
stomach to the descending duodenum. It would be gradually
transmitted in a retrograde manner through the major duodenal
papilla, sphincter of Oddi, common bile duct, common hepatic duct,
left and right hepatic ducts, and cholangiole to its final
destination in the liver or gallbladder. Secondly, the genetic
components of H. pylori were detectable in the endarterium
samples of atherosclerotic patients using PCR detection by Danesh
et al (7) and others who
indicated that H. pylori might enter into the circulatory
system (5,6,63). A
long-term chronic H. pylori infection would result in
chronic gastritis and a chronic gastric ulcer. Consequently, a
small amount of H. pylori would constantly leak into the
blood through impaired vessels that are in or around gastric ulcers
and then wander aimlessly along the portal or lymph circulation.
Tian et al (66) found that
inflammatory cells were mostly thinly distributed along the central
veins, hepatic sinusoids, arteriolar, and venules in the periportal
area of H. pylori-infected livers. Although similar
morphological changes were found in H. pylori-infected mouse
livers of the present study, we could not detect H. pylori
genus-specific 16S rRNA in any of the blood samples. The low
bacteria concentration (<103 CFU/μl) in the blood may
be responsible for the negative results (60). Thus, even though it was
undetectable in the blood of hepatic positive individuals, we
cannot deny the abovementioned secondary hypothesis. Thirdly,
inflammatory cells would phagocytize H. pylori and arrive at
liver when the hepatic homeostatic equilibrium had been broken, for
example, an increasing of bile pH value.
With a growing concern regarding the correlativity
of H. pylori infection and hepatic disease, the positivity
rate of H. pylori genetic components detected in hepatic
samples is increasing. Although it is not uncommon for H.
pylori to be cultured from the gastric samples of infected
patients and animals, a positive culture from hepatic samples is
still rarely reported. It has been reported that H. pylori
could be cultured from clinical hepatic samples (14,44).
However, no positive culture result could be found in hepatic or
blood samples of H. pylori-infected animal models which were
developed by various researchers, including ourselves. By referring
to the findings of researchers in the field, we analyzed the
results and summarized the possible reasons as follows. First, by
analyzing the results of H. pylori-specific IHC staining, we
found that the densities of a positive reaction in H.
pylori-infected livers were significantly lower than those of
the stomachs. Silva et al (60) indicated the reasons why they were
unable to visualize the H. pylori colonies stating that the
bacteria were cultured in the conventional conditions and methods.
Hence, a low detection sensitivity of bacterial culture may produce
a false-negative result. H. pylori prefers gaseous
conditions (2–5% of oxygen and 5–10% of carbon dioxide), certain
temperatures (34 to 40°C; 37°C is its optimum), humidity (>90%),
and acidity (pH 5.5 to 8.0; neutral is the optimum). H.
pylori transforms from its normal form to a coccoid form if its
growing environment changes (54),
such as cholestasis in which hepatic cells excrete abnormal acidic
bile. The coccoid form of H. pylori is widely known as a
degenerative dead form or a nonculturable form (64,65).
In addition to its alkalinity, bile may also contain some other
unfavorable ingredients that are unsuitable for the growth of H.
pylori and would play a crucial role in restraining or killing
it. Because the genetic components or structural proteins would not
be eliminated completely by inflammatory or hepatic cells. Thus, we
were able to realize the existence of H. pylori in samples
by a more sensitive method, such as morphological IHC stain and
PCR. Narikawa et al (54)
also hypothesized that the coccoid transformation of H.
pylori would contribute to the inability of H. pylori
culture in hepatic samples despite a positive genetic
detection.
Although, the liver to body weight ratio in H.
pylori-infected livers was not significant as compared with
uninfected livers, cytoplasmic vacuolation and hepatic binucleate
cells were observed by Goo et al (23), and the present study. In addition,
infiltrated inflammatory cells that were thinly distributed in the
H. pylori-infected hepatic lobules were also found by Tian
et al (66). No macroscopic
or microscopic differences existed in the H. pylori-infected
and uninfected tumors, except for the pathogenic detection of H.
pylori. In the present study, the skin ulceration bearing mouse
usually bore an extremely gross exophytic growing hepatic tumor
which stuck to the abdominal wall. We did not have the resources
for skin ulceration detection, we analyzed the ‘link tissue’
instead and found that it was full of pleomorphic tumor cells. On
that account, we insist that the skin ulceration was a part of the
necrotic tumor, which had invaded into the abdominal wall. In order
to explore the impact of H. pylori in the development and
progression of liver and hepatic orthotopic graft tumors, we
introduced three tumor markers, namely PCNA, Bax and Bcl-2, which
are cellular growth, apoptosis-promotion and apoptosis-inhibition
proteins, respectively. In the present study, H. pylori may
have upregulated the expression of PCNA both in tumor and liver,
whereas may have downregulated the Bax expression in the tumor and
upregulated it in the liver. No significant impact of H.
pylori on the Bcl-2 expression could be seen in the tumor or
the liver.
PCNA was a 36-kDa antigen which was associated with
cellular growth activity (67–70).
It plays an important role in cellular proliferation and DNA
synthesis, and its expression is usually proportional to DNA
synthesis. Hence, it is widely thought to be an indicator and is
used to estimate the occurrence, development, metastasis,
progression, classification, neoplasm staging, treatment evaluation
and prognosis of many tumors (71–74).
In actively proliferative cells, such as tumor cells, the
expression of PCNA is significantly upregulated (75–80).
In the present study, besides the significantly unregulated
expression in tumors, PCNA in H. pylori-infected livers and
tumors was also markedly higher than that in the uninfected ones.
Our results corresponded to the findings of Goo et al
(23) and Tian et al
(81). We noted that the swollen
cells in the H. pylori-infected livers compressed the
hepatic sinusoids, and subsequently, might lead to the blood and
oxygen deficits. The impaired hepatic cells might induce the
expression of PCNA indirectly as the deficits in nutrition, oxygen
and blood supply. Moreover, inflammatory cells and their infection
factors might contribute to PCNA upregulation in hepatic cells.
This is our hypothesis for the reasons for PCNA upregulation that
was detected in H. pylori-infected livers. Moreover, Tian
et al (81) performed an
in vitro experiment involving the human liver HepG2 cells
and H. pylori co-culture indicating an increasing PCNA
expression. However, the liver cells in their study were in an
abundant nutritious, oxygen and blood condition (82). Thus, we infer from the results that
H. pylori may promote cellular proliferation in some direct
but unknown pathways.
The Bcl-2 family is divided into two categories.
One is a type of regulator protein that induces cell apoptosis,
such as Bax. The other, especially Bcl-2, is considered an
important anti-apoptosis protein. The Bcl-2 family antigen would
combine with each other and become a protein dimer which act as a
molecular switch in the progression of cell death (83–88).
The occurrence of carcinoma is an imbalance between cellular growth
or proliferation and cellular death or apoptosis. The upregulation
of proto-oncogenes or downregulation of anti-oncogenes would
finally promote tumor progression. When the Bax expression were
higher than that of Bcl-2, they would combine as Bax/Bax homologous
dimers and promote cellular apoptosis. Alternatively, the Bcl-2 and
Bax antigens would combine as Bcl-2/Bax heterodimers and
Bcl-2/Bcl-2 homodimers and play a role in inhibiting cellular
apoptosis (86,89–92).
Deficits in nutrients, oxygen, and blood supply might be a crucial
reason for cellular apoptosis in the present study (93). Additionally, inflammatory factors
or mediators excreted by infiltrating inflammatory cells were also
responsible for cellular apoptosis. All the above mentioned reasons
enabled the cellular apoptosis in H. pylori-infected livers
and led them to have a higher Bax expression. Hemangiectasis was
noted in the tumors, thus, their blood and oxygen supply were
abundant. Moreover, the percentage of death and apoptosis was
significantly lower in the tumor than the normal liver tissue.
Thus, we were able to explain the downregulation of the Bax
expression demonstrated in the present study. We conclude from the
results that inflammation might be the primary cause of apoptosis
in the H. pylori-infected livers, but H. pylori might
act as a crucial factor to inhibit or reduce tumorous
apoptosis.
The present study has some shortcomings. We could
not establish the H. pylori forms and the exact reasons for
hepatic inflammation. Additionally, it is unknown whether the
difference among groups will be more significantly visible when the
model-build period is prolonged. Therefore, further research is
essential for the worldwide unsolved issues that were found in
exploring the relationship between H. pylori and hepatic
diseases.
Acknowledgements
Our research was supported by the Provincial
Natural Science Foundation of Fujian (code: 2012J01358) and by the
Construction Project of National Key Clinical Subject of General
Surgery.
Abbreviations:
|
H. pylori
|
Helicobacter pylori
|
|
PCNA
|
proliferating cell nuclear
antigen
|
|
Bcl-2
|
B-cell lymphoma 2
|
|
Bax
|
Bcl-2-associated X protein
|
References
|
1
|
Rybicka M, Nakonieczna J, Stalke P and
Bielawski KP: Host response to the presence of Helicobacter spp.
DNA in the liver of patients with chronic liver diseases. Pol J
Microbiol. 60:175–178. 2011.PubMed/NCBI
|
|
2
|
Schistosomes, liver flukes and
Helicobacter pylori. In: IARC Working Group on the Evaluation of
Carcinogenic Risks to Humans; Lyon. 7–14 June 1994; IARC monographs
on the evaluation of carcinogenic risks to humans. World Health
Organization, International Agency for Research on Cancer; 61. pp.
1–241. 1994
|
|
3
|
Wedi B and Kapp A: Helicobacter pylori
infection in skin diseases: a critical appraisal. Am J Clin
Dermatol. 3:273–282. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pellicano R, Menard A, Rizzetto M and
Megraud F: Helicobacter species and liver diseases: association or
causation? Lancet Infect Dis. 8:254–260. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Farsak B, Yildirir A, Akyön Y, Pinar A, Oç
M, Böke E, Kes S and Tokgözoğlu L: Detection of Chlamydia
pneumoniae and Helicobacter pylori DNA in human atherosclerotic
plaques by PCR. J Clin Microbiol. 38:4408–4411. 2000.PubMed/NCBI
|
|
6
|
Ameriso SF, Fridman EA, Leiguarda RC and
Sevlever GE: Detection of Helicobacter pylori in human carotid
atheroscle-rotic plaques. Stroke. 32:385–391. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Danesh J, Koreth J, Youngman L, Collins R,
Arnold JR, Balarajan Y, McGee J and Roskell D: Is Helicobacter
pylori a factor in coronary atherosclerosis? J Clin Microbiol.
37:16511999.PubMed/NCBI
|
|
8
|
Nilsson I, Lindgren S, Eriksson S and
Wadstrom T: Serum antibodies to Helicobacter hepaticus and
Helicobacter pylori in patients with chronic liver disease. Gut.
46:410–414. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pellicano R, Leone N, Berrutti M, Cutufia
MA, Fiorentino M, Rizzetto M and Ponzetto A: Helicobacter pylori
seroprevalence in hepatitis C virus positive patients with
cirrhosis. J Hepatol. 33:648–650. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ponzetto A, Pellicano R, Leone N, Berrutti
M, Turrini F and Rizzetto M: Helicobacter pylori seroprevalence in
cirrhotic patients with hepatitis B virus infection. Neth J Med.
56:206–210. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ponzetto A, Pellicano R, Leone N, Cutufia
MA, Turrini F, Grigioni WF, D'Errico A, Mortimer P, Rizzetto M and
Silengo L: Helicobacter infection and cirrhosis in hepatitis C
virus carriage: is it an innocent bystander or a troublemaker? Med
Hypotheses. 54:275–277. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Queiroz DM, Rocha AM, Rocha GA, Cinque SM,
Oliveira AG, Godoy A and Tanno H: Association between Helicobacter
pylori infection and cirrhosis in patients with chronic hepatitis C
virus. Dig Dis Sci. 51:370–373. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Avenaud P, Marais A, Monteiro L, Le Bail
B, Bioulac Sage P, Balabaud C and Mégraud F: Detection of
Helicobacter species in the liver of patients with and without
primary liver carcinoma. Cancer. 89:1431–1439. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xuan SY, Li N, Qiang X, Zhou RR, Shi YX
and Jiang WJ: Helicobacter infection in hepatocellular carcinoma
tissue. World J Gastroenterol. 12:2335–2340. 2006.PubMed/NCBI
|
|
15
|
Farazi PA and DePinho RA: Hepatocellular
carcinoma pathogenesis: from genes to environment. Nat Rev Cancer.
6:674–687. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
17
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Iwatsuki S, Starzl TE, Sheahan DG,
Yokoyama I, Demetris AJ, Todo S, Tzakis AG, Van Thiel DH, Carr B,
Selby R, et al: Hepatic resection versus transplantation for
hepatocellular carcinoma. Ann Surg. 214:221–228; discussion
228–229. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
McPeake JR, O'Grady JG, Zaman S, Portmann
B, Wight DG, Tan KC, Calne RY and Williams R: Liver transplantation
for primary hepatocellular carcinoma: tumor size and number
determine outcome. J Hepatol. 18:226–234. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kensler TW, Roebuck BD, Wogan GN and
Groopman JD: Aflatoxin: a 50-year odyssey of mechanistic and
translational toxicology. Toxicol Sci. 120(Suppl 1): S28–S48. 2011.
View Article : Google Scholar :
|
|
21
|
Chuang SC, La Vecchia C and Boffetta P:
Liver cancer: Descriptive epidemiology and risk factors other than
HBV and HCV infection. Cancer Lett. 286:9–14. 2009. View Article : Google Scholar
|
|
22
|
Arzumanyan A, Reis HM and Feitelson MA:
Pathogenic mechanisms in HBV- and HCV-associated hepatocellular
carcinoma. Nat Rev Cancer. 13:123–135. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Goo MJ, Ki MR, Lee HR, Yang HJ, Yuan DW,
Hong IH, Park JK, Hong KS, Han JY, Hwang OK, et al: Helicobacter
pylori promotes hepatic fibrosis in the animal model. Lab Invest.
89:1291–1303. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ito K, Nakamura M, Toda G, Negishi M,
Torii A and Ohno T: Potential role of Helicobacter pylori in
hepatocarcinogenesis. Int J Mol Med. 13:221–227. 2004.PubMed/NCBI
|
|
25
|
Ito K, Yamaoka Y, Ota H, El-Zimaity H and
Graham DY: Adherence, internalization, and persistence of
Helicobacter pylori in hepatocytes. Dig Dis Sci. 53:2541–2549.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang Y, Fan XG, Huang YK, Chen R and Dai
H: Helicobacter pylori enhances cyclin D1, PCNA expression in HepG2
cell line. Zhonghua Gan Zang Bing Za Zhi. 12:695–696. 2004.(In
Chinese). PubMed/NCBI
|
|
27
|
Chen R, Fan XG, Huang Y, Li N and Chen CH:
In vitro cytotoxicity of Helicobacter pylori on hepatocarcinoma
HepG2 cells. Ai Zheng. 23:44–49. 2004.(In Chinese). PubMed/NCBI
|
|
28
|
Karita M, Li Q, Cantero D and Okita K:
Establishment of a small animal model for human Helicobacter pylori
infection using germ-free mouse. Am J Gastroenterol. 89:208–213.
1994.PubMed/NCBI
|
|
29
|
Thalmaier U, Lehn N, Pfeffer K, Stolte M,
Vieth M and Schneider-Brachert W: Role of tumor necrosis factor
alpha in Helicobacter pylori gastritis in tumor necrosis factor
receptor 1-deficient mice. Infect Immun. 70:3149–3155. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Algood HM, Allen SS, Washington MK, Peek
RM Jr, Miller GG and Cover TL: Regulation of gastric B cell
recruitment is dependent on IL-17 receptor A signaling in a model
of chronic bacterial infection. J Immunol. 183:5837–5846. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Maurer KJ, Ihrig MM, Rogers AB, Ng V,
Bouchard G, Leonard MR, Carey MC and Fox JG: Identification of
cholelithogenic enterohepatic Helicobacter species and their role
in murine cholesterol gallstone formation. Gastroenterology.
128:1023–1033. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee CW, Rickman B, Rogers AB, Muthupalani
S, Takaishi S, Yang P, Wang TC and Fox JG: Combination of sulindac
and antimicrobial eradication of Helicobacter pylori prevents
progression of gastric cancer in hypergastrinemic INS-GAS mice.
Cancer Res. 69:8166–8174. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yao X, Hu JF, Daniels M, Yien H, Lu H,
Sharan H, Zhou X, Zeng Z, Li T, Yang Y, et al: A novel orthotopic
tumor model to study growth factors and oncogenes in
hepatocarcinogenesis. Clin Cancer Res. 9:2719–2726. 2003.PubMed/NCBI
|
|
34
|
Aprahamian M, Bour G, Akladios CY,
Fylaktakidou K, Greferath R, Soler L, Marescaux J, Egly JM, Lehn JM
and Nicolau C: Myo-InositolTrisPyroPhosphate treatment leads to
HIF-1α suppression and eradication of early hepatoma tumors in
rats. Chem Biochem. 12:777–783. 2011.
|
|
35
|
Fox JG, Dewhirst FE, Shen Z, Feng Y,
Taylor NS, Paster BJ, Ericson RL, Lau CN, Correa P, Araya JC, et
al: Hepatic Helicobacter species identified in bile and gallbladder
tissue from Chileans with chronic cholecystitis. Gastroenterology.
114:755–763. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Leone N, Pellicano R, Brunello F, Cutufia
MA, Berrutti M, Fagoonee S, Rizzetto M and Ponzetto A: Helicobacter
pylori seroprevalence in patients with cirrhosis of the liver and
hepatocellular carcinoma. Cancer Detect Prev. 27:494–497. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ponzetto A, Pellicano R, Redaelli A,
Rizzetto M and Roffi L: Helicobacter pylori infection in patients
with hepatitis C Virus positive chronic liver diseases. New
Microbiol. 26:321–328. 2003.PubMed/NCBI
|
|
38
|
Spinzi G, Pellicano R, Minoli G, Terreni
N, Cutufia MA, Fagoonee S, Rizzetto M and Ponzetto A: Helicobacter
pylori seroprevalence in hepatitis C virus positive patients with
cirrhosis. The Como cross-sectional study. Panminerva Med.
43:85–87. 2001.PubMed/NCBI
|
|
39
|
Fan XG, Zou YY, Wu AH, Li TG, Hu GL and
Zhang Z: Seroprevalence of Helicobacter pylori infection in
patients with hepatitis B. Br J Biomed Sci. 55:176–178. 1998.
|
|
40
|
Zhang S, Bao Y and Zu MH: Research of
relationship between Hp infection and HCC. Chin J Clin Oncol.
45–48. 2004.
|
|
41
|
Huang Y, Fan XG, Wang ZM, Zhou JH, Tian XF
and Li N: Identification of helicobacter species in human liver
samples from patients with primary hepatocellular carcinoma. J Clin
Pathol. 57:1273–1277. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pellicano R, Mazzaferro V, Grigioni WF,
Cutufia MA, Fagoonee S, Silengo L, Rizzetto M and Ponzetto A:
Helicobacter species sequences in liver samples from patients with
and without hepatocellular carcinoma. World J Gastroenterol.
10:598–601. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Leelawat K, Suksumek N, Leelawat S and
Lek-Uthai U: Detection of VacA gene specific for Helicobactor
pylori in hepatocellular carcinoma and cholangiocarcinoma specimens
of Thai patients. Southeast Asian J Trop Med Public Health.
38:881–885. 2007.PubMed/NCBI
|
|
44
|
de Magalhaes Queiroz DM and Santos A:
Isolation of a Helicobacter strain from the human liver.
Gastroenterology. 121:1023–1024. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Matsukura N, Yokomuro S, Yamada S, Tajiri
T, Sundo T, Hadama T, Kamiya S, Naito Z and Fox JG: Association
between Helicobacter bilis in bile and biliary tract malignancies:
H. bilis in bile from Japanese and Thai patients with benign and
malignant diseases in the biliary tract. Jpn J Cancer Res.
93:842–847. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
García A, Feng Y, Parry NM, McCabe A,
Mobley MW, Lertpiriyapong K, Whary MT and Fox JG: Helicobacter
pylori infection does not promote hepatocellular cancer in a
transgenic mouse model of hepatitis C virus pathogenesis. Gut
Microbes. 4:577–590. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Rocha M, Avenaud P, Menard A, Le Bail B,
Balabaud C, Bioulac-Sage P, de Magalhães Queiroz DM and Mégraud F:
Association of Helicobacter species with hepatitis C cirrhosis with
or without hepatocellular carcinoma. Gut. 54:396–401. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hanninen ML: Sensitivity of Helicobacter
pylori to different bile salts. Eur J Clin Microbiol Infect Disy.
10:515–518. 1991. View Article : Google Scholar
|
|
49
|
Caldwell MT, McDermott M, Jazrawi S,
O'Dowd G, Byrne PJ, Walsh TN, Hourihane DO and Hennessy TP:
Helicobacter pylori infection increases following cholecystectomy.
Ir J Med Sci. 164:52–55. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kawaguchi M, Saito T, Ohno H, Midorikawa
S, Sanji T, Handa Y, Morita S, Yoshida H, Tsurui M, Misaka R, et
al: Bacteria closely resembling Helicobacter pylori detected
immunohistologically and genetically in resected gallbladder
mucosa. J Gastroenterol. 31:294–298. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Guyton JE and Hall AC: Textbook of Medical
Physiology. Saunders Elsevier; Philadelphia, PA: 2011
|
|
52
|
Kelly SM, Pitcher MC, Farmery SM and
Gibson GR: Isolation of Helicobacter pylori from feces of patients
with dyspepsia in the United Kingdom. Gastroenterology.
107:1671–1674. 1994.PubMed/NCBI
|
|
53
|
Thomas JE, Gibson GR, Darboe MK, Dale A
and Weaver LT: Isolation of Helicobacter pylori from human faeces.
Lancet. 340:1194–1195. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Narikawa S, Kawai S, Aoshima H, Kawamata
O, Kawaguchi R, Hikiji K, Kato M, Iino S and Mizushima Y:
Comparison of the nucleic acids of helical and coccoid forms of
Helicobacter pylori. Clin Diagn Lab Immunol. 4:285–290.
1997.PubMed/NCBI
|
|
55
|
Dore MP, Realdi G, Mura D, Graham DY and
Sepulveda AR: Helicobacter infection in patients with HCV-related
chronic hepatitis, cirrhosis, and hepatocellular carcinoma. Dig Dis
Sci. 47:1638–1643. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Silva LD, Rocha AM, Rocha GA, de Moura SB,
Rocha MM, Dani R, de Melo FF, Guerra JB, de Castro LP, Mendes GS,
et al: The presence of Helicobacter pylori in the liver depends on
the Th1, Th17 and Treg cytokine profile of the patient. Mem Inst
Oswaldo Cruz. 106:748–754. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Boonyanugomol W, Chomvarin C, Sripa B,
Bhudhisawasdi V, Khuntikeo N, Hahnvajanawong C and Chamsuwan A:
Helicobacter pylori in Thai patients with cholangiocarcinoma and
its association with biliary inflammation and proliferation. HPB
(Oxford). 14:177–184. 2012. View Article : Google Scholar
|
|
58
|
Myung SJ, Kim MH, Shim KN, Kim YS, Kim EO,
Kim HJ, Park ET, Yoo KS, Lim BC, Seo DW, et al: Detection of
Helicobacter pylori DNA in human biliary tree and its association
with hepatolithiasis. Dig Dis Sci. 45:1405–1412. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Chen W, Li D, Cannan RJ and Stubbs RS:
Common presence of Helicobacter DNA in the gallbladder of patients
with gallstone diseases and controls. Dig Liver Dis. 35:237–243.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Silva CP, Pereira-Lima JC, Oliveira AG,
Guerra JB, Marques DL, Sarmanho L, Cabral MM and Queiroz DM:
Association of the presence of Helicobacter in gallbladder tissue
with cholelithiasis and cholecystitis. J Clin Microbiol.
41:5615–5618. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Maurer KJ, Rogers AB, Ge Z, Wiese AJ,
Carey MC and Fox JG: Helicobacter pylori and cholesterol gallstone
formation in C57L/J mice: A prospective study. Am J Physiol
Gastrointest Liver Physiol. 290:G175–G182. 2006. View Article : Google Scholar
|
|
62
|
Abayli B, Colakoglu S, Serin M, Erdogan S,
Isiksal YF, Tuncer I, Koksal F and Demiryurek H: Helicobacter
pylori in the etiology of cholesterol gallstones. J Clin
Gastroenterol. 39:134–137. 2005.PubMed/NCBI
|
|
63
|
Kowalski M, Rees W, Konturek PC, Grove R,
Scheffold T, Meixner H, Brunec M, Franz N, Konturek JW, Pieniazek
P, et al: Detection of Helicobacter pylori specific DNA in human
atheromatous coronary arteries and its association to prior
myocardial infarction and unstable angina. Dig Liver Dis.
34:398–402. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Kusters JG, Gerrits MM, Van Strijp JA and
Vandenbroucke-Grauls CM: Coccoid forms of Helicobacter pylori are
the morphologic manifestation of cell death. Infect Immun.
65:3672–3679. 1997.PubMed/NCBI
|
|
65
|
Andersen LP, Dorland A, Karacan H, Colding
H, Nilsson HO, Wadström T and Blom J: Possible clinical importance
of the transformation of Helicobacter pylori into coccoid forms.
Scand J Gastroenterol. 35:897–903. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Tian XF, Fan XG, Fu CY, Huang Y and Zhu C:
Experimental study on the pathological effect of Helicobacter
pylori on liver tissues. Zhonghua Gan Zang Bing Za Zhi. 13:780–783.
2005.(In Chinese). PubMed/NCBI
|
|
67
|
Egelkrout EM, Mariconti L, Settlage SB,
Cella R, Robertson D and Hanley-Bowdoin L: Two E2F elements
regulate the proliferating cell nuclear antigen promoter
differently during leaf development. Plant Cell. 14:3225–3236.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Essers J, Theil AF, Baldeyron C, van
Cappellen WA, Houtsmuller AB, Kanaar R and Vermeulen W: Nuclear
dynamics of PCNA in DNA replication and repair. Mol Cell Biol.
25:9350–9359. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Shivji KK, Kenny MK and Wood RD:
Proliferating cell nuclear antigen is required for DNA excision
repair. Cell. 69:367–374. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Leonardi E, Girlando S, Serio G, Mauri FA,
Perrone G, Scampini S, Dalla Palma P and Barbareschi M: PCNA and
Ki67 expression in breast carcinoma: correlations with clinical and
biological variables. J Clin Pathol. 45:416–419. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Kobayashi I, Matsuo K, Ishibashi Y, Kanda
S and Sakai H: The proliferative activity in dysplasia and
carcinoma in situ of the uterine cervix analyzed by proliferating
cell nuclear antigen immunostaining and silver-binding argyrophilic
nucleolar organizer region staining. Hum Pathol. 25:198–202. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Allegranza A, Girlando S, Arrigoni GL,
Veronese S, Mauri FA, Gambacorta M, Pollo B, Dalla Palma P and
Barbareschi M: Proliferating cell nuclear antigen expression in
central nervous system neoplasms. Virchows Arch A Pathol Anat
Histopathol. 419:417–423. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Tsai ST and Jin YT: Proliferating cell
nuclear antigen (PCNA) expression in oral squamous cell carcinomas.
J Oral Pathol Med. 24:313–315. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Willett CG1, Warland G, Hagan MP, Daly WJ,
Coen J, Shellito PC and Compton CC: Tumor proliferation in rectal
cancer following preoperative irradiation. J Clin Oncol.
13:1417–1424. 1995.PubMed/NCBI
|
|
75
|
Prosperi E: Multiple roles of the
proliferating cell nuclear antigen: DNA replication, repair and
cell cycle control. Prog Cell Cycle Res. 3:193–210. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Moldovan GL, Pfander B and Jentsch S:
PCNA, the maestro of the replication fork. Cell. 129:665–679. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Wang SC, Nakajima Y, Yu YL, Xia W, Chen
CT, Yang CC, McIntush EW, Li LY, Hawke DH, Kobayashi R and Hung MC:
Tyrosine phosphorylation controls PCNA function through protein
stability. Nat Cell Biol. 8:1359–1368. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Pal HC1, Sharma S, Strickland LR, Agarwal
J, Athar M, Elmets CA and Afaq F: Delphinidin reduces cell
proliferation and induces apoptosis of non-small-cell lung cancer
cells by targeting EGFR/VEGFR2 signaling pathways. PLoS One.
8:e772702013. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Aziz MH, Wheeler DL, Bhamb B and Verma AK:
Protein kinase C delta overexpressing transgenic mice are resistant
to chemically but not to UV radiation-induced development of
squamous cell carcinomas: A possible link to specific cytokines and
cyclo-oxygenase-2. Cancer Res. 66:713–722. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Ojeh N, Hiilesvuo K, Warri A, Salmivirta
M, Henttinen T and Maatta A: Ectopic expression of syndecan-1 in
basal epidermis affects keratinocyte proliferation and wound
re-epithelialization. J Invest Dermatol. 128:26–34. 2008.
View Article : Google Scholar
|
|
81
|
Tian XF, Fan XG, Huang Y, Zhang Y and Zhu
C: Expression of cyclin D1 proliferating cell nuclear antigen in
liver of C57BL/6 mice infected with Helicobacter pylori. World Chin
J Digestol. 14:1341–1345. 2006.
|
|
82
|
Zhang Y, Fan XG, Chen R, Xiao ZQ, Feng XP,
Tian XF and Chen ZH: Comparative proteome analysis of untreated and
Helicobacter pylori-treated HepG2. World J Gastroenterol.
11:3485–3489. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Komatsu K1, Miyashita T, Hang H, Hopkins
KM, Zheng W, Cuddeback S, Yamada M, Lieberman HB and Wang HG: Human
homologue of S. pombe Rad9 interacts with BCL-2/BCL-xL and promotes
apoptosis. Nat Cell Biol. 2:1–6. 2000.PubMed/NCBI
|
|
84
|
Lin B, Kolluri SK, Lin F, Liu W, Han YH,
Cao X, Dawson MI, Reed JC and Zhang XK: Conversion of Bcl-2 from
protector to killer by interaction with nuclear orphan receptor
Nur77/TR3. Cell. 116:527–540. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Hoetelmans RW: Nuclear partners of Bcl-2:
Bax and PML. DNA Cell Biol. 23:351–354. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Oltvai ZN, Milliman CL and Korsmeyer SJ:
Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that
accelerates programmed cell death. Cell. 74:609–619. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Tsujimoto Y, Finger LR, Yunis J, Nowell PC
and Croce CM: Cloning of the chromosome breakpoint of neoplastic B
cells with the t(14;18) chromosome translocation. Science.
226:1097–1099. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Cleary ML, Smith SD and Sklar J: Cloning
and structural analysis of cDNAs for bcl-2 and a hybrid
bcl-2/immunoglobulin transcript resulting from the t(14;18)
translocation. Cell. 47:19–28. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Yang LJ, Cao XT and Yu LZ: Bcl-2 and Bax
in apoptosis of tumor cells. Chin J Cancer Biother. 10:232–234.
2003.
|
|
90
|
Yin XM, Oltvai ZN and Korsmeyer SJ: BH1
and BH2 domains of Bcl-2 are required for inhibition of apoptosis
and heterodimerization with Bax. Nature. 369:321–323. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Yang E and Korsmeyer SJ: Molecular
thanatopsis: a discourse on the BCL2 family and cell death. Blood.
88:386–401. 1996.PubMed/NCBI
|
|
92
|
Yang E, Zha J, Jockel J, Boise LH,
Thompson CB and Korsmeyer SJ: Bad, a heterodimeric partner for
Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell.
80:285–291. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Cotran RS, Kumar V and Collins T: Robbins
Pathologic Basis of Disease. 6th edition. W.B Saunders Company;
Philadelphia, PA: 1998
|