Introduction
Chemotherapy is one of the most common strategies in
the treatment of cancer, although it is frequently unsuccessful due
to the development of chemoresistance, and especially multi-drug
resistance (MDR). A number of mechanisms involved in the occurrence
of MDR have been described, including the overexpression of one or
more ATP binding cassette (ABC) transporter proteins that mediate
the efflux of many clinically relevant drugs. More than 40 ABC
transporters have been identified so far, but just a few play a
role in MDR, including the breast cancer resistance protein
(BCRP/ABCG2). BCRP expression was detected in several different
solid tumours (1–5), as well as in malignant hematopoietic
and lymphoid cells (6). Moreover,
BCRP is very often upregulated in cells undergoing mitoxantrone
(MTX) treatment (7).
Several compounds have been shown to inhibit the
efflux activity of ABC transporters, thereby increasing
intracellular drug accumulation and sensitising cancer cells to
therapy. It was previously reported that proadifen (SKF-525A), a
well-known cytochrome P450 monooxygenase inhibitor, not only has
anti-proliferative potential in some cancer cell lines, but it is
also able to inhibit BCRP and MRP1 transporter proteins (8). The exact molecular mechanism of
proadifen antitumour action is not yet fully understood. However,
it seems not to be dependent on the inhibitory activity against
cytochrome P450 enzymes. In our previous study, we demonstrated the
anti-proliferative and pro-apoptotic activity of proadifen in the
HT-29 cancer cell line (9).
Moreover, we revealed the ability of proadifen to increase the
intracellular hypericin content in HT-29 cells most likely via the
inhibition of BCRP and MRP1 transporters (8). Based on these results, we
investigated the effect of proadifen pre-treatment on MTX toxicity.
We examined not just the effect of proadifen and MTX on the
expression of BCRP, but we were also interested in other molecular
mechanisms involved in the possible antitumour activity of
proadifen alone and in combination with MTX, such as the expression
of anti-apoptotic proteins, proteins involved in the regulation of
BCRP and proteins involved in the reparation of chemotherapeutic
drug-induced DNA damage. Because MTX is a preferential and strong
substrate of BCRP, MTX-sensitive HL-60 cell line and an
MTX-resistant ABCG2-overexpressing subclone (here referred to as
cBCRP) were chosen as the experimental models.
Materials and methods
Cell culture
Human promyelocytic leukemia cell line HL-60 was
purchased from the American Type Culture Collection (ATCC,
Rockville, MD, USA) and its ABCG2-overexpressing subclone (here
referred to as cBCRP) was kindly provided by Dr Balazs Sarkadi
(Membrane Research Group, Hungarian Academy of Sciences, Budapest,
Hungary) (10). Both cell lines
were grown in complete RPMI-1640 medium (Gibco, Grand Island, NY,
USA) supplemented with 10% fetal bovine serum (FBS; Gibco), 7.5%
NaHCO3 and antibiotics (penicillin 100 U/ml,
streptomycin 100 μg/ml and amphotericin 25 μg/ml; Invitrogen,
Carlsbad, CA, USA) at 37°C, 95% humidity and in atmosphere of 5%
CO2.
Reagents
Proadifen (SKF-525A; α-phenyl-α-propylbenzene-acetic
acid 2-(diethylamino)ethylester; CAS no. 62-68-0; Sigma-Aldrich,
St. Louis, MO, USA) and mitoxantrone dihydrochloride (MTX;
1,4-dihydroxy-5,8-bis[[2-[(2-hydroxyethyl)
amino]-9,10-anthracenedione dihydrochloride; CAS no. 70476-82-3;
HPLC grade, Sigma-Aldrich) stock solutions were prepared in
distilled water and stored at −20°C). The concentrations of
prepared stock solutions were 10 and 1 mM for proadifen and MTX,
respectively. Working solutions of both reagents were always
freshly prepared before being added to the cultures. The final
concentrations of distilled water did not influence the cytokinetic
parameters. Because no significant differences in the response to
distilled water were observed, these data are referred to as a
control.
Experimental design
The cells were seeded in 96-well plates (MTT assay),
in 6-well plates (flow cytometry analyses) or in 60-mm Petri dishes
(western blotting, membrane-bound BCRP expression, RNA isolation)
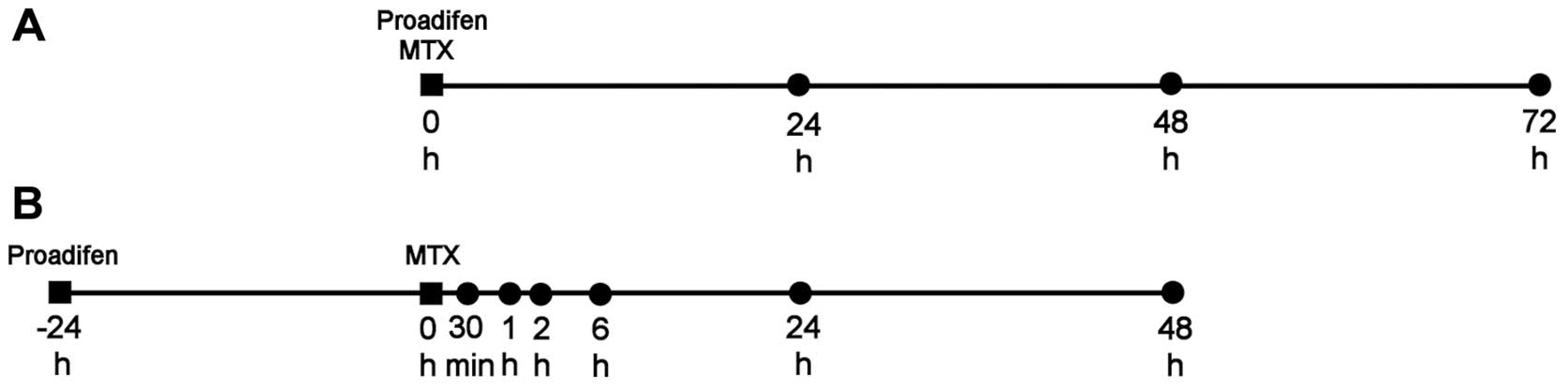
(all TPP, Trasadingen, Switzerland). Both cell lines were treated
immediately after seeding (Fig.
1).
Experimental scheme A
For the determination of the IC20 values
(20% inhibitory concentration) of proadifen and MTX, MTT assays
were performed 24, 48 and 72 h after proadifen and MTX addition (0
in the time schedule).
Experimental scheme B
Cells were pre-treated with proadifen for 24 h (−24
h in the time schedule) prior to MTX addition (0 in the time
schedule). Changes in metabolic activity, mitochondrial membrane
potential, cell death and cell cycle distribution were analysed 24
and 48 h after MTX treatment. Changes in the phosphorylation of the
H2AX histone were analysed 2 and 24 h after MTX addition.
Membrane-bound BCRP expression was examined 30 min, 1 h and 2 h
after MTX treatment. BCRP mRNA level and the expression of selected
proteins were detected 30 min, 1, 2 and 6 h after MTX addition.
MTT assay
To analyse the changes in the cell metabolic
activity that occurred as the consequence of single and combined
drug treatment, MTT assays were carried out as previously described
(11). The results were evaluated
as the percentage of the absorbance (λ = 584 nm) of the untreated
control. Proadifen and MTX IC20 values were extrapolated
from an exponential fit to the metabolic activity data using
OriginPro 8.5.0 SR1 software (OriginLab Corp., Northampton, MA,
USA). To test the synergistic effect of proadifen pre-treatment on
MTX action, MTT results were analysed using the Chou and Talalay
(12) median-effect method with
CalcuSyn software (Biosoft, Ferguson, MO, USA). The extent of
interaction between proadifen and MTX was expressed through
combination index (CI) values.
Detection of mitochondrial membrane
depolarisation
Cells were treated according to the experimental
design (Fig. 1B), harvested,
centrifuged, washed with HBSS and stained with 0.1 μM TMRE
(tetramethylrhodamine ethyl ester perchlorate; Sigma-Aldrich) in
Hank's balanced salt solution (HBSS) for 20 min at RT in the dark.
Thereafter, the cells were analysed (1×104 cells per
sample) using a BD FACSCalibur flow cytometer (Becton-Dickinson,
San Jose, CA, USA) with a 488-nm argon-ion excitation laser.
Fluorescence was detected via a 585/42 band-pass filter (FL-2). The
obtained results were analysed using FlowJo software (TreeStar
Inc., Ashland, OR, USA) and are presented as the percentage of
cells with dissipated MMP (mitochondrial membrane potential).
Analysis of viability and
phosphatidylserine externalisation
Analysis of cell viability and phosphatidylserine
externalisation was performed using the Apoptest™-FITC (Dako
Denmark A/S, Glostrup, Denmark) according to the manufacturer's
instructions. The cells were harvested at scheduled times (Fig. 1B), centrifuged, washed with HBSS
and stained with Annexin V-FITC (0.75 μg/ml) in 1X binding buffer
for 20 min at RT in the dark. Subsequently, the cells were stained
with propidium iodide (PI; 1 μg/ml) for 5 min and were analysed
(1×104 cells per sample) using a BD FACSCalibur flow
cytometer. Fluorescence was detected via a 530/30 nm band-pass
filter (FL-1; Annexin V/FITC) and a 670 nm long-pass filter (FL-3;
PI). The results were evaluated using FlowJo software.
Cell cycle analysis
For the flow cytometric analysis of cell cycle
distribution, cells were harvested at scheduled times (Fig. 1B), washed in cold
phosphate-buffered saline (PBS), fixed in cold 70% ethanol and kept
at −20°C overnight. Prior to analysis, the cells were washed twice
in PBS, resuspended in staining buffer (0.1% Triton X-100, 0.137
mg/ml ribonuclease A and 0.02 mg/ml PI), incubated for 30 min at RT
in the dark and analysed using a BD FACSCalibur flow cytometer
(2×104 cells per sample). Fluorescence was detected via
a 585/42 nm band-pass filter (FL-2). ModFit 3.0 software (Verity
Software House, Topsham, ME, USA) was used to generate DNA content
frequency histograms and quantify the number of cells in the
individual cell cycle phases (G0/G1, S and G2/M).
Intracellular accumulation of MTX
The cells were pre-treated with proadifen for 24 h.
Immediately after incubation, the cells were harvested and
resuspended in HBSS. Afterwards, MTX was added to the cells and its
intracellular accumulation was detected continuously for the
duration of 45 min. The measurement was performed by BD FACSAria II
SORP (Becton-Dickinson) using excitation with a 640 nm laser and
emission acquisition in an Alexa Fluor 700 (DM685LP-710/50)
channel.
Cell surface expression of BCRP
For the flow cytometry assay that was intended to
detect cell surface expression of BCRP, cells (3×105)
were harvested at scheduled time-points (Fig. 1B), washed in staining buffer (HBSS
with 5% FBS) and incubated with FITC-conjugated anti-BCRP primary
antibody [ABCG2 (5D3) FITC, 1:100, sc-18841 FITC, Santa Cruz
Biotechnology Inc.] or the FITC-conjugated mouse IgG2b as an
isotype control (normal mouse IgG2b-FITC, 1:100, sc-2857, Santa
Cruz Biotechnology Inc.) for 30 min at RT in the dark. The cells
were then washed in HBSS and analysed on a BD FACSCalibur flow
cytometer (2×104 cells per sample). The obtained data
were analysed using FlowJo software. Cell surface BCRP was
expressed as a ratio of the median fluorescence of ABCG2 (5D3) and
of the IgG2b isotype control.
Histone H2AX phosphorylation
Flow cytometric analysis of the histone H2AX
phosphorylation on Ser139 (γH2AX) was performed to examine possible
changes in MTX-mediated DNA double strand breaks (DSBs), as
previously described (13).
Fluorescence intensity was detected using a BD FACSCalibur flow
cytometer via a 530/30 nm band-pass filter (FL-1) and the results
were analysed using FlowJo software. The expression of γH2AX was
expressed as a ratio of the median fluorescence of Alexa
Fluor® 488 Mouse anti-H2AX (pS139) (cat. no. 560445, BD
Biosciences) and of Alexa Fluor 488 Mouse IgG1 κ Isotype Control
(cat. no. 557782, BD Biosciences). The results are presented as the
fold of untreated control.
Western blot analysis
For detection of the expression of selected
proteins, cells were treated in line with the experimental design
(Fig. 1B). Western blot analysis
was performed, as previously described (13). The blots were incubated overnight
at 4°C with the specific primary antibodies: polyclonal rabbit
anti-human survivin (#2803, 1:500), monoclonal rabbit anti-human
Bcl-xL (#2764, 1:1,000), monoclonal rabbit anti-human p-Akt
(Ser473) (#4060, 1:1,000), polyclonal rabbit anti-human Akt (#9272,
1:1,000) (all from Cell Signaling Technology, Danvers, MA, USA),
monoclonal mouse anti-human Bcl-2 (sc-7382, 1:250), monoclonal
mouse anti-human caspase-3 (sc-7272, 1:500), mouse monoclonal
anti-human B23 (sc-32256, 1:500), mouse monoclonal anti-human BCRP
(sc-58222, 1:400), monoclonal mouse anti-human Ku-86 (sc-5280,
1:500), monoclonal mouse anti-human E2F1 (sc-251, 1:250) (all from
Santa Cruz Biotechnology, Santa Cruz, CA, USA), polyclonal rabbit
anti-human Mcl-1 (M8484, 1:1,000) (Sigma-Aldrich). After 30 min of
washing in TBS, the membranes were incubated with appropriate
horseradish peroxidase-conjugated (HRP) secondary antibodies for 1
h at RT (goat anti-rabbit IgG-HRP, 1:5,000, #31461; goat anti-mouse
IgG-HRP, 1:5,000, #31436, Pierce Biotechnology, Rockford, IL, USA).
Antibody reactivity was detected with Pierce ECL Western Blotting
Substrate (Pierce Biotechnology) and was visualised on medical
X-ray films (Agfa HealthCare NV, Mortsel, Belgium). Equal sample
loading was verified by immunodetection of β-actin (A5441,
monoclonal, mouse, Sigma-Aldrich). The densitometry of proteins was
evaluated using ImageJ software (NIH, Bethesda, MD, USA). Relative
protein levels were normalised to the expression of β-actin.
RNA extraction and semiquantitative
RT-PCR
In order to evaluate ABCG2 expression in the
HL-60 and cBCRP cell lines, cells were seeded and treated according
to the experimental schedule (Fig.
1B). Total RNA isolation, the evaluation of RNA concentration,
purity, integrity and the whole RT-PCR procedure was performed, as
previously described (13). The
samples (50 ng cDNA) were subjected to PCR amplifications with
primers specific for ABCG2 (F, 5′-GGCCATAGCAGC
AGGTCAGAGTG-3′; R, 5′-TGCAAAGCCGTAAATCCATA TCGTG-3′). As an
internal loading control, GAPDH (F, 5′-ATG
GGGAAGGTGAAGGTCGGAGTC-3′; R, 5′-CTCGCTCCT GGAAGATGGTGATGG-3′) and
also ACTB genes (F, 5′-GAT CCGCCGCCCGTCCACAC-3′; R,
5′-TTGCACATGCCGG AGCCGTTG-3′) were used. Each PCR mixture contained
cDNA; deoxynucleoside triphosphates (dNTP Mix, Fermentas, Thermo
Fisher Scientific, Vilnius, Lithuania) at a concentration of 200
μM; 0.2 μM of each primer; and 1 unit of BioThermAB™ Hot Start Taq
DNA Polymerase (GeneCraft Köln, Germany) in a total volume of 25
μl. The PCR was performed on a Mastercycler pro S (Eppendorf,
Hamburg, Germany), and the reaction conditions were as follows:
initial denaturation at 94°C for 2 min, followed by numerous cycles
(20 cycles for ABCG2 and GAPDH, 25 cycles for
ACTB) of denaturation at 94°C for 30 sec, annealing at the
annealing temperature for 30 sec and elongation at 72°C for 45 sec,
with a final extension at 72°C for 10 min. The number of cycles was
adjusted to allow detection in the linear range. The densitometry
analysis of the PCR products was evaluated using ImageJ software
and the ABCG2 levels were normalised to GAPDH.
Statistical analysis
The results were analysed using a one-way ANOVA with
Tukey's post-test or t-test and are expressed as the mean ±
standard deviation (SD) of at least three independent experiments.
Significance levels are indicated in the legend for each
figure.
Results
Effects of proadifen and MTX on cell
metabolic activity
The metabolic activity was assessed after exposure
of HL-60 and cBCRP cells to 0–50 μM proadifen and 0–20 μM MTX.
Proadifen and MTX showed a time- and dose-dependent inhibitory
effect on both cell lines, with a more pronounced effect on
sensitive HL-60 cells (data not shown). IC20 values for
proadifen, as well as for MTX (Table
I), were evaluated for further combined drug treatment
experiments.
 | Table IThe IC20 of proadifen and
MTX in HL-60 and cBCRP cells.a |
Table I
The IC20 of proadifen and
MTX in HL-60 and cBCRP cells.a
| | HL-60 | | cBCRP |
|---|
| |
| |
|
|---|
| 24 h | 48 h | 72 h | 24 h | 48 h | 72 h |
|---|
| Proadifen |
| IC20
(μM) | 11.28 | 6.95 | 5.51 | 40.49 | 37.19 | 30.74 |
| MTX |
| IC20
(μM) | 0.029 | 0.011 | 0.002 | 3.68 | 1.53 | 0.36 |
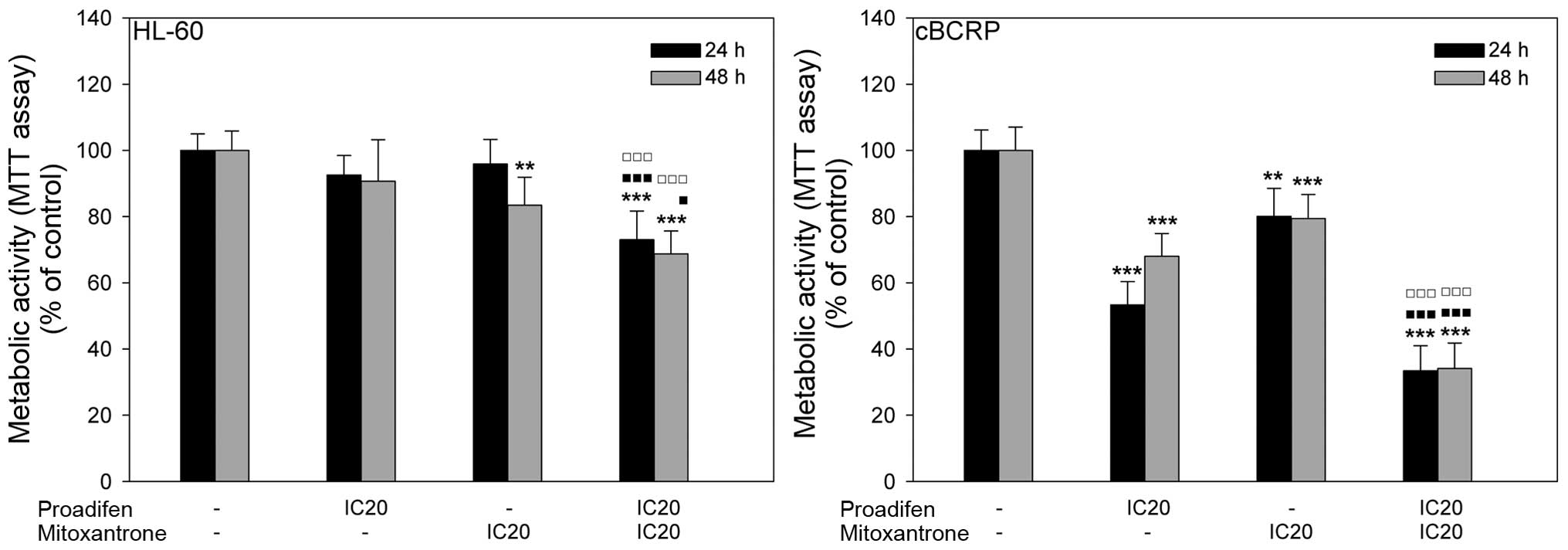
To determine the impact of proadifen on MTX action,
cells were pre-treated with proadifen for 24 h. The inhibitory
effect of MTX on cell metabolic activity was potentiated by
proadifen in both cell lines. A much stronger effect was observed
in resistant cBCRP cells, where the metabolic activity
significantly decreased in the experimental group administered the
combined treatment compared to the group treated with MTX alone
(Fig. 2).
Based on these results, proadifen-MTX combination
index (CI) values were evaluated using CalcuSyn software (Table II) and synergism was revealed in
both cell lines.
| Table IICombination index (CI) values of
proadifen-MTX mutual combinations.a |
Table II
Combination index (CI) values of
proadifen-MTX mutual combinations.a
| Proadifen (μM) | Mitoxantrone
(μM) | CI | CalcuSyn
effect |
|---|
| HL-60 |
| 24 h | 5 | 0.05 | 0.630 | Synergism |
| 5 | 0.01 | 0.965 | Nearly
additive |
| 48 h | 5 | 0.01 | 0.744 | Moderate
synergism |
| BCRP |
| 24 h | 35 | 5 | 0.780 | Moderate
synergism |
| 40 | 5 | 0.855 | Slight
synergism |
| 48 h | 30 | 1 | 0.886 | Slight
synergism |
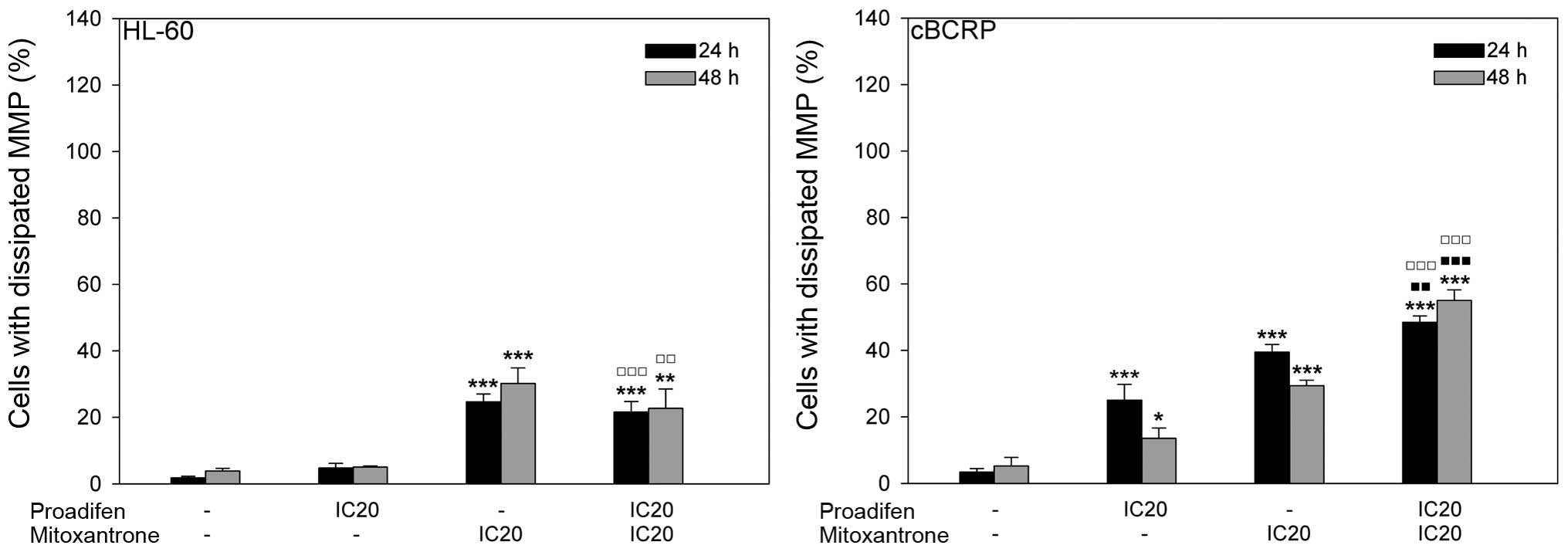
Proadifen effect on MTX cytotoxicity
To determine whether proadifen-MTX synergism arising
from the MTT assay may reflect the impact of proadifen on MTX
cytotoxicity, various cell death parameters were analysed. Changes
in mitochondrial membrane depolarisation revealed different effects
in sensitive HL-60 and resistant cBCRP cells. In the resistant cell
line proadifen alone and MTX alone significantly increased the
percentage of cells with dissipated mitochondrial membrane
potential (MMP) compared to the untreated control. Additionally,
the cytotoxic effect of MTX was more pronounced after proadifen
pre-treatment, particularly at 48 h after MTX addition (Fig. 3). In contrast, proadifen did not
influence MMP in HL-60 cells either alone or combined with MTX. The
cytotoxic action of MTX was slightly reduced by proadifen in the
HL-60 cell line (Fig. 3).
Consistent with the results of the MMP analysis,
Annexin V-FITC/PI double staining revealed no effect of proadifen
on MTX cytotoxicity in HL-60 cells (Table III). In the case of the cBCRP
cell line, proadifen markedly potentiated MTX-induced cell death.
Proadifen significantly increased the percentage of cells in the
later stages of apoptosis (Annexin V+/PI+) 24
and 48 h after MTX treatment. Moreover, 48 h after MTX addition,
proadifen even caused massive accumulation of the cells in the
early stages of programmed cell death (Annexin
V+/PI−).
| Table IIIAnalysis of cell death.a |
Table III
Analysis of cell death.a
| Annexin
V+/PI− | Annexin
V−/PI+ | Annexin
V+/PI+ |
|---|
| HL-60 |
| 24 h |
| Control | 0.47±0.16 | 1.54±0.49 | 2.04±0.44 |
| Proadifen | 0.64±0.21 | 1.15±0.27 | 2.96±0.53 |
| MTX | 1.21±0.59 | 1.98±0.99 |
6.02±2.12b |
| Proadifen +
MTX | 0.89±0.35 | 1.43±0.69 |
6.31±1.52b,e |
| 48 h |
| Control | 0.44±0.17 | 1.61±0.78 | 3.42±1.16 |
| Proadifen | 0.57±0.33 | 1.04±0.57 | 3.12±0.85 |
| MTX | 0.56±0.31 | 4.95±2.08 |
14.36±6.09b |
| Proadifen +
MTX | 0.42±0.22 | 4.70±2.52 | 12.94±5.57 |
| cBCRP |
| 24 h |
| Control | 0.36±0.04 | 0.84±0.13 | 2.17±0.14 |
| Proadifen | 0.53±0.26 | 2.86±0.52 | 7.00±3.62 |
| MTX |
7.89±0.76d |
7.11±0.48d |
14.27±2.85c |
| Proadifen +
MTX |
1.79±0.13b,i |
4.82±1.41c,h |
22.68±2.42d,f,h |
| 48 h |
| Control | 0.74±0.18 | 0.72±0.08 | 3.11±0.71 |
| Proadifen | 0.79±0.05 | 1.15±0.05 | 5.37±1.70 |
| MTX | 0.71±0.20 |
6.30±0.35c |
12.97±3.63b |
| Proadifen +
MTX |
14.26±2.23d,g,i |
6.41±1.99c,f |
31.39±3.87d,g,i |
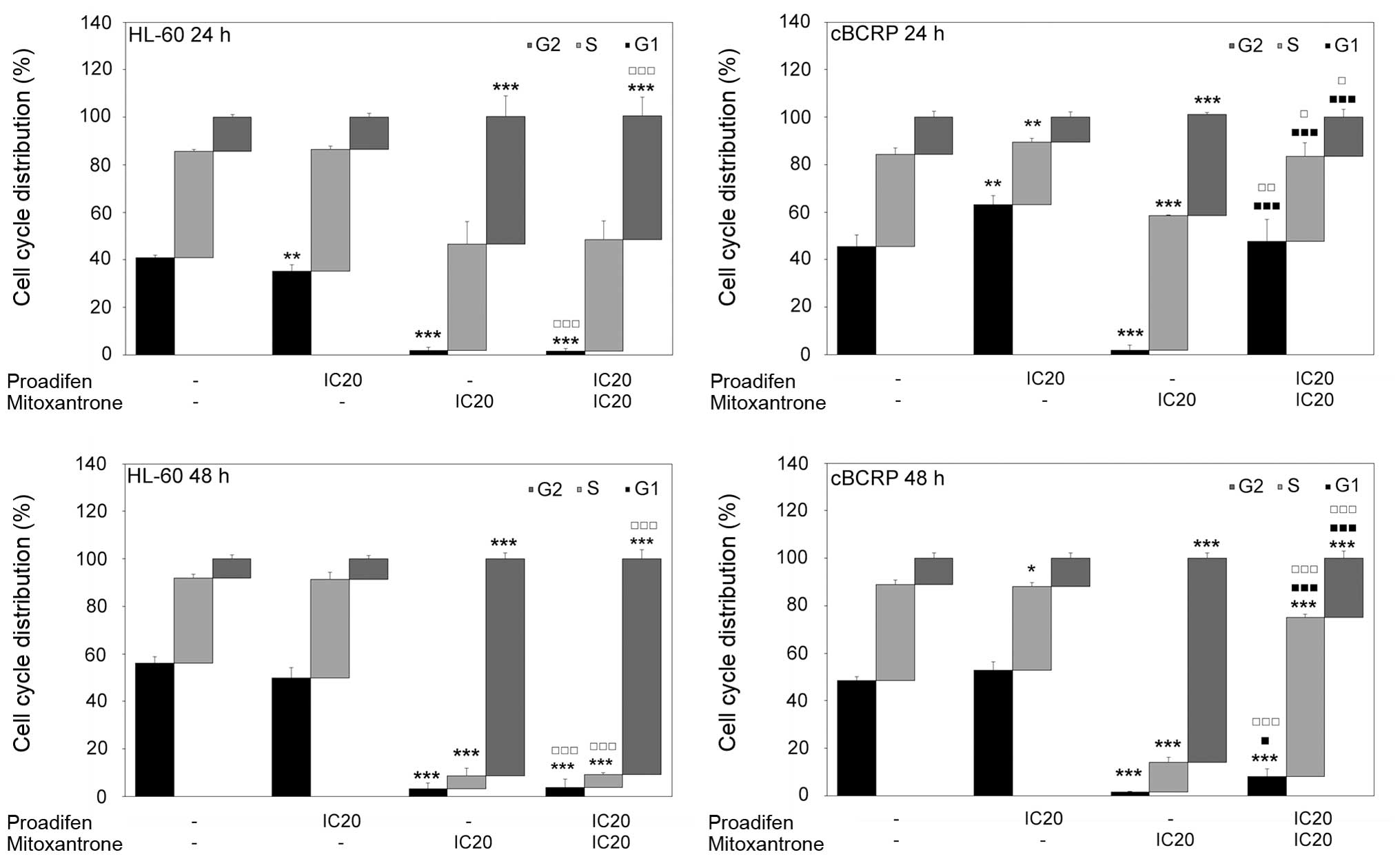
Effects of proadifen, MTX and combined
treatment on cell cycle distribution
Cell cycle distribution was affected by both drugs
(Fig. 4). Proadifen alone caused a
significant accumulation of cBCRP cells in the G0/G1 phase 24 h
after treatment. Simultaneously, a reduction of the cells in the
S-phase was observed. In the case of the HL-60 cell line, proadifen
alone caused the opposite effect. On the other hand, MTX alone
markedly affected cell cycle progression in both cell lines,
leading to a massive G2/M arrest and reduction of cells in the
G0/G1 phase. The MTX-induced accumulation of HL-60 and cBCRP cells
in the G2/M phase was even stronger 48 h after treatment. The
effect of proadifen pre-treatment on MTX cytostatic action was
observed only in resistant cBCRP cells. Proadifen caused cell cycle
redistribution by the significant attenuation of MTX-mediated G2/M
arrest and increased accumulation of cBCRP cells in the G0/G1 phase
(Fig. 4).
Effects of proadifen, MTX and combined
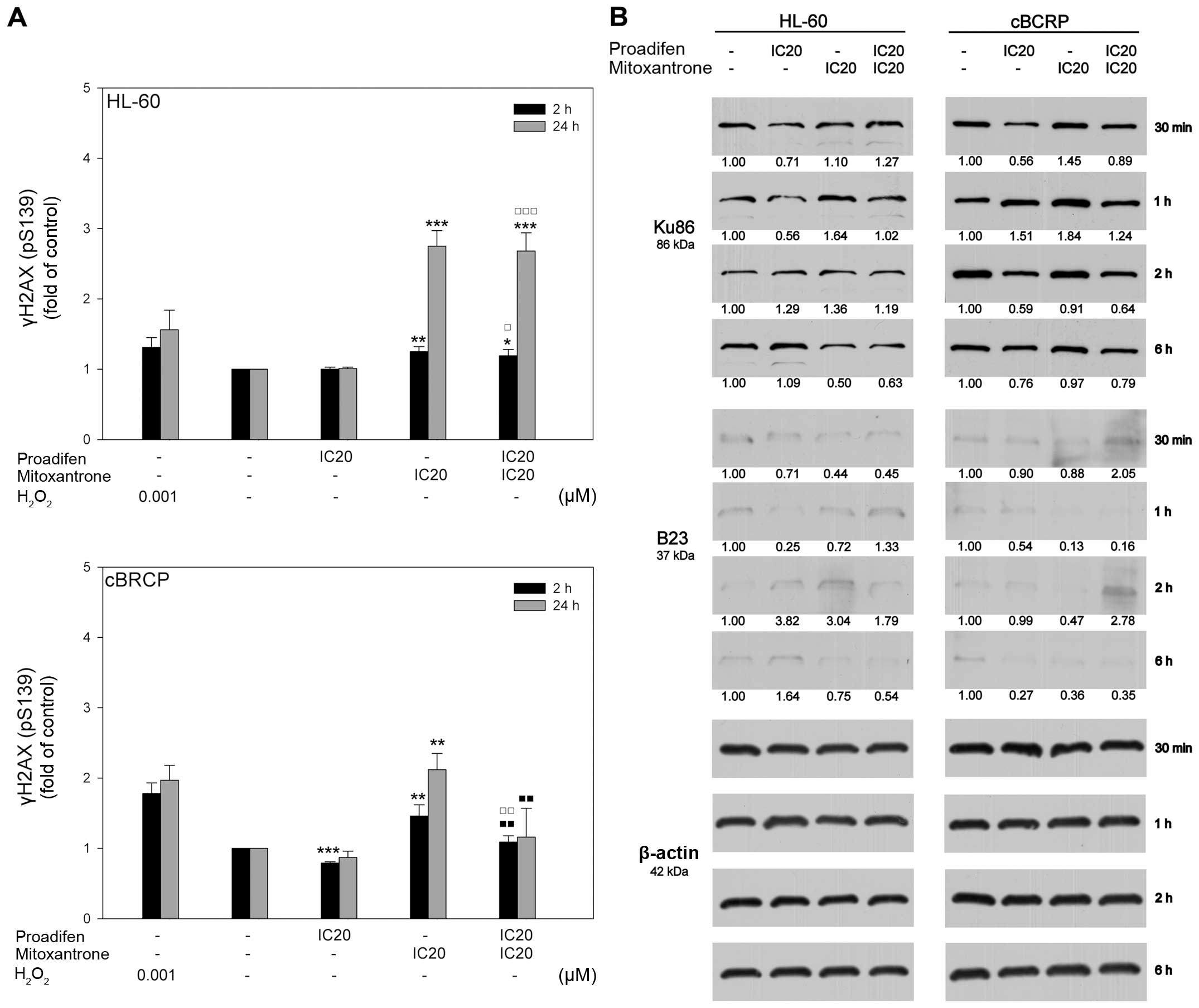
treatment on histone H2AX phosphorylation
To determine whether the cytotoxic effect of MTX was
correlated with its ability to cause DSBs, the phosphorylation of
histone H2AX on Ser139 (γH2AX) was analysed. MTX alone elevated the
expression of γH2AX, especially in the sensitive cell line. The
effect of proadifen on the γH2AX level was cell line-dependent,
leading to a slight decrease in γH2AX expression in cBCRP cells.
However, no changes in the phosphorylation of H2AX in the HL-60
cells were detected. Interestingly, pre-treatment of cBCRP cells
with proadifen markedly decreased the MTX-induced γH2AX expression,
with a more significant effect 24 h after MTX addition (Fig. 5A).
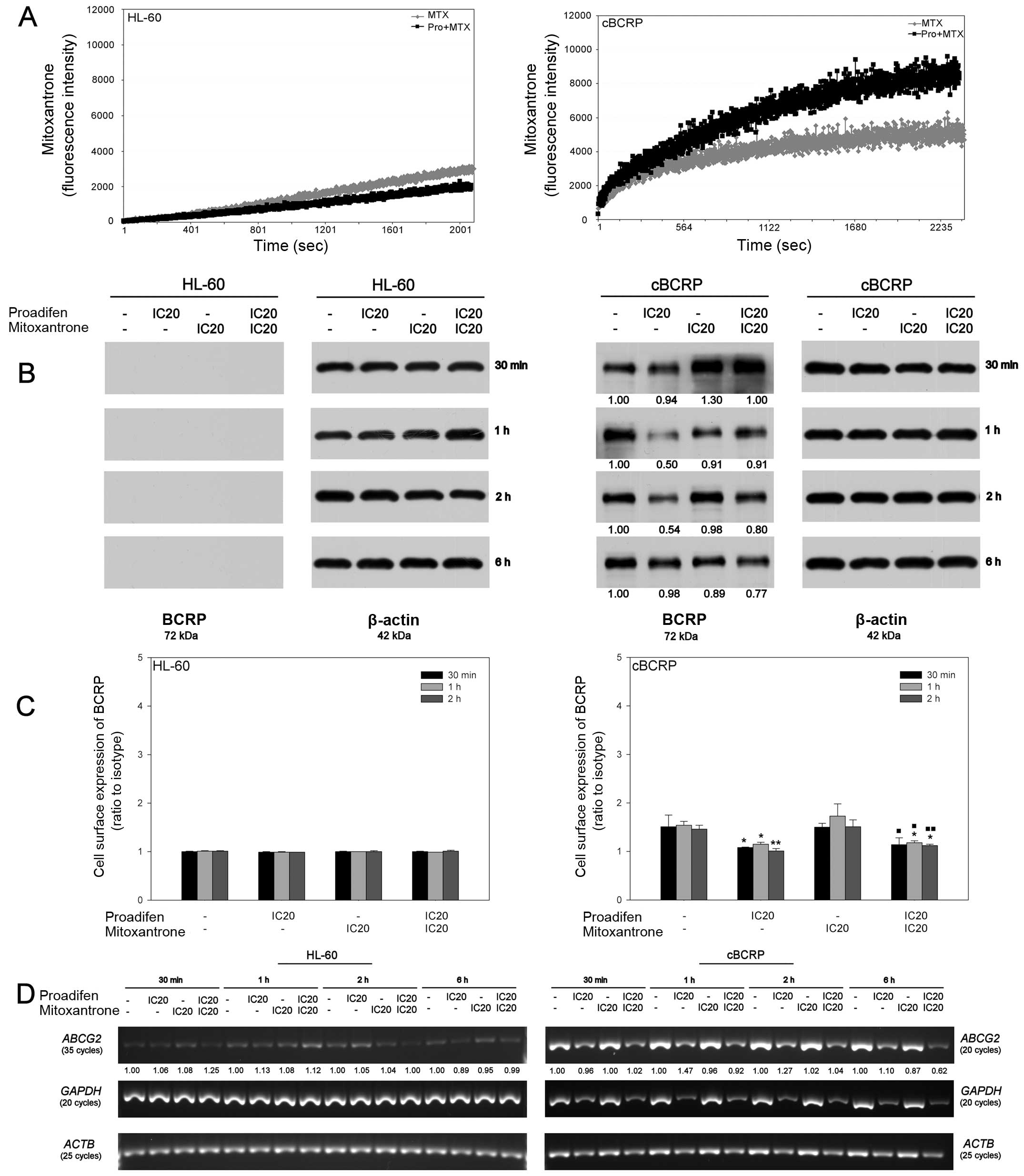
Proadifen effect on BCRP expression and
activity
The analysis of MTX accumulation in HL-60 and cBCRP
cells was performed to verify the possible inhibitory effect of
proadifen on the BCRP transporter (Fig. 6A). Changes in MTX fluorescence were
measured continuously over time (duration of 45 min). In the case
of the ABCG2-overexpressing cBCRP cell line, 24 h proadifen
pre-treatment led to an increase in the intracellular MTX level. On
the other hand, proadifen slightly decreased the intracellular
accumulation of MTX in the HL-60 cells (Fig. 6A).
To determine whether the changes in the increased
intracellular accumulation of MTX after combined drug treatment
were associated with decreased expression of BCRP, whole cell
protein levels, as well as cell surface expression of BCRP, were
detected. In the case of the BCRP transporter, no protein levels
were detected in the HL-60 cells (Fig.
6B). Simultaneously, BCRP overexpression was confirmed in the
cBCRP cell line. The level of this transporter protein was
decreased by proadifen and by the combined drug treatment,
particularly 6 h after MTX addition (Fig. 6B). The 5D3 antibody recognises an
extracellular epitope of BCRP transporter protein. In HL-60 cells,
no changes in membrane-bound BCRP were detected. However, proadifen
alone, as well as in combined treatment, led to a slight reduction
in the expression of cell surface BCRP in resistant cBCRP cells
(Fig. 6C).
To verify whether proadifen-mediated reduction of
the BCRP protein level correlated with its mRNA content,
semi-quantitative RT-PCR was performed (Fig. 6D). Marked differences in BCRP
(ABCG2) expression between the cell lines were confirmed.
Although BCRP mRNA was detectable in HL-60 cells, its content was
very low compared to cBCRP cells. Considering the impact of
proadifen on the ABCG2 mRNA level, no downregulation was
observed, except for the later time-point (6 h) in the case of the
cBCRP experimental group given the combined treatment (Fig. 6D). Interestingly, proadifen
treatment decreased the mRNA content for internal loading control
GAPDH in cBCRP cells, despite the fact that equal amounts of
mRNA were used for cDNA synthesis. This effect was confirmed in
ACTB, another housekeeping gene, even though no changes in
β-actin protein levels were detected.
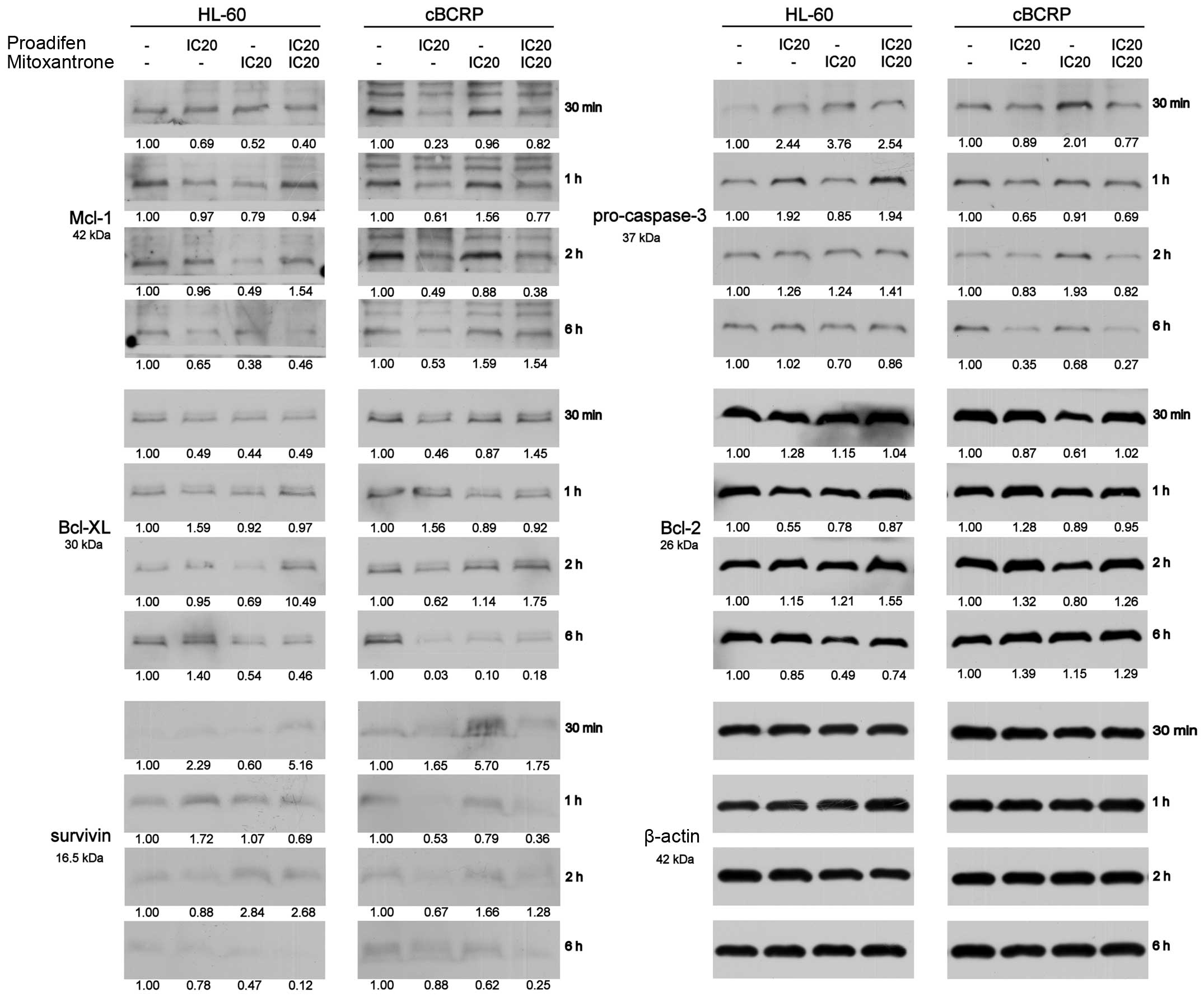
Proadifen, MTX and combined
treatment-mediated changes in protein levels
To determine whether the changes in cytotoxicity of
the combined drug treatment were associated with altered expression
of anti-apoptotic proteins, proteins engaged in DNA damage repair
or proteins regulating the expression of BCRP transporter, western
blot analysis was performed at early time-points after the MTX
addition (30 min, 1, 2 and 6 h) (Fig.
7).
In cBCRP cells, proadifen alone and combined with
MTX markedly downregulated several anti-apoptotic proteins,
especially Mcl-1, Bcl-xL and survivin (Fig. 7). On the other hand, proadifen, MTX
and their combination increased the level of anti-apoptotic protein
Bcl-2 above that of the untreated control (Fig. 7). This effect was most pronounced 6
h after the MTX addition. Moreover, single and combined drug
treatments led to the activation of procaspase-3 (Fig. 7).
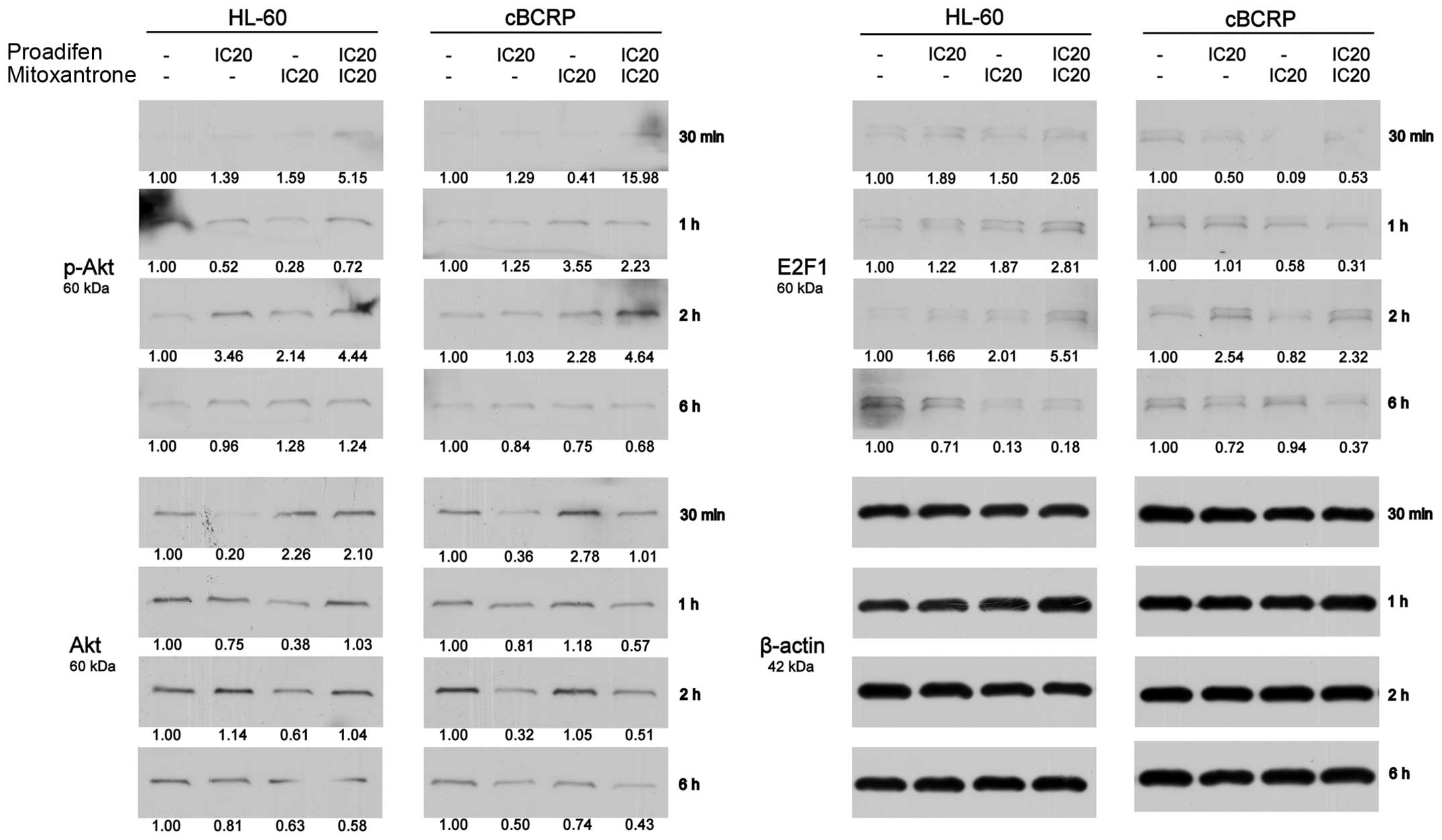
The reduction of transcription factor E2F1 that is
potentially involved in the regulation of BCRP expression was
observed, except for one time-point (2 h) (Fig. 8). However, protein levels of pAkt,
which may also be engaged in the regulation of BCRP activity and
expression, were increased after single and combined drug
treatment, except for the latest time-point (Fig. 8). On the other hand, we detected
downregulated protein levels of Akt (Fig. 8).
Proteins involved in DSB repair were also
investigated (Fig. 5B). We
observed slightly elevated levels of Ku86 (XRCC5) in cBCRP cells
compared to HL-60 cells. Proadifen alone and in combination with
MTX downregulated the expression of both Ku86 and B23 (NPM1). The
strongest effect of single and combined drug treatment was observed
at the third time-point (2 h) in the case of Ku86. On the other
hand, the levels of B23 were markedly decreased 1 h and 6 h after
MTX treatment.
Discussion
Cancer cell chemoresistance remains a major obstacle
to successful therapy. Great effort has been made to identify
various agents that could sensitise cancer cells to treatment,
thereby improving chemotherapy outcome. Proadifen (SKF-525A), an
inhibitor of cytochrome P450 monooxygenases (P450), is a drug
approved by the US Food and Drug Administration (14) and is used as a local anaesthetic
(15). Additionally,
anti-proliferative properties of proadifen on various cancer cell
lines have also been demonstrated (9). Moreover, proadifen has been shown to
potentiate the efficiency of hypericin-mediated photodynamic
therapy (HY-PDT), most likely via the inhibition of ABC transporter
activity, primarily BCRP (8).
However, the exact mechanism or mechanisms engaged in the
anti-proliferative and chemosensitising potential of proadifen
remain unclear. Therefore, the main purpose of this study was to
verify the proadifen-mediated enhancement of MTX cytotoxicity
through potential BCRP inhibition using MTX-sensitive HL-60 cells
and MTX-resistant ABCG2-overexpressing cBCRP subclone. Another aim
was to explore the mechanisms that could be involved in the
anti-proliferative properties of proadifen in the cBCRP cell
line.
Proadifen showed a time- and dose-dependent
inhibitory effect on HL-60 and cBCRP cells, with markedly lower
concentrations inhibiting the sensitive HL-60 cells. On the
contrary, such a low proadifen concentration stimulated metabolic
activity in resistant cBCRP cells and much higher concentrations
were required for significant decreases in this parameter (data not
shown). A similar effect of different proadifen concentrations in
various cancer cell models was previously reported by other authors
(9,11,16).
Consistent with previous studies (16,17),
we also detected dose-dependent variations in the cell cycle
distribution. Whereas higher concentrations of proadifen (30.74 and
37.19 μM) caused an elevated accumulation of cBCRP cells in G0/G1
phase, lower concentrations (5.51 and 6.95 μM) increased the
accumulation of HL-60 cells in the S-phase. These results indicate
the cytostatic action of proadifen in both cell lines. However,
through the determination of multiple cell death markers, we
revealed that proadifen IC20 was cytotoxic only in the
case of resistant cBCRP cell lines. Furthermore, in cBCRP cells, we
revealed decreased mRNA levels of two commonly used reference
housekeeping genes, GAPDH and ACTB, after proadifen
treatment. No such effect was observed in HL-60 cells. Since there
was no change in β-actin protein levels, we assume that proadifen
cytotoxicity in cBCRP cells could be related to the impairment of
overall transcriptional activity or to the increase in mRNA
degradation. Previously, it was found that proadifen is able to
inhibit viral RNA amplification (18). On the contrary, Fernandez et
al (19) indicated an
inhibitory effect of proadifen on RNA degradative processes in rat
liver.
Proadifen as an inhibitor of cytochrome P450
monooxygenases, along with cyclooxygenase and lipoxygenase
inhibitors, could suppress the conversion of free arachidonic acid.
Extensive research has focused on the role of cyclo-oxygenase-2
(COX-2) inhibitors in sensitising cancer cells to chemotherapy,
mainly through non-specific inhibition of one or more ABC
transporter proteins. Our previous study indicates that proadifen
represents a potential agent capable of sensitising cancer cells to
HY-PDT, most likely via inhibition of more than one transporter
protein (8). In the present study,
we showed for the first time that proadifen enhanced the
sensitivity of cancer cells towards MTX treatment. Pre-treatment of
MTX-resistant ABCG2-overexpressing cBCRP cells with proadifen
markedly increased the cytotoxic properties of this
chemotherapeutic agent. Since proadifen was able to suppress the
expression of total, as well as membrane-bound BCRP protein in
cBCRP cells, we suggest proadifen-mediated downregulation of the
BCRP level as one of the most probable causes of its
chemosensitising effect. Furthermore, 6 h after combined drug
treatment, decrease of ABCG2 mRNA level was revealed.
Moreover, downregulation of BCRP by proadifen was also demonstrated
by increased intracellular MTX accumulation in cBCRP cells within
45 min after MTX addition. Taken together, these results show that
proadifen is not only able to inhibit BCRP expression, but also
BCRP activity in the resistant cBCRP cell line. These findings are
in line with our previous study (8) where proadifen suppressed
hypericin-mediated induction of BCRP expression at protein level,
and also increased the intracellular content of hypericin in HT-29
cells. Similarly, Elahian et al (20) showed that dexamethasone, a COX-2
inhibitor, was able to enhance MTX cytotoxicity in the MTX
resistant breast cancer cell line, MCF-7, through BCRP
inhibition.
The underlying mechanism by which proadifen may
affect BCRP expression and function is not yet fully understood.
Several transcription factors and signalling pathways have been
shown to regulate BCRP expression or activity (6). It was revealed that the
downregulation of Akt might lead to decreased BCRP activity, but
not to the changes in BCRP protein levels (2,21).
However, Nakanishi et al (22) demonstrated that inhibition of the
Akt signalling pathway in leukemia cells affected not only the
plasma membrane localisation of BCRP, but also downregulated the
transporter protein levels. These studies are in contrast with our
results, where we detected elevated protein levels of
phosphorylated Akt, except for the latest time-point (6 h). We
suppose that the expression and activity of BCRP in cBCRP cells is
not mediated through Akt signalling. A recent study identified the
transcription factor E2F1, implicated in various cellular
functions, as another possible activator of BCRP expression in
various human lung cancer cell lines (23). Moreover, Rosenfeld et al
(23) revealed that the E2F-BCRP
axis suppresses chemotherapy-induced cell death. Another important
fact corresponding with our results is the attenuation of this
resistance via BCRP inhibition. We found that proadifen was able to
decrease the protein levels of the transcription factor E2F in
cBCRP cells. The above mentioned results indicate that
downregulation of E2F1 might play a role in the regulation of BCRP
expression in the resistant cBCRP leukemic cells and that
proadifen-mediated potentiation of MTX cytotoxicity could be
related to these changes. However, further investigations will be
necessary to elucidate the exact mechanisms.
Subsequently, we focused on the expression of
proteins involved in the induction of apoptosis, including Bcl-2,
Bcl-xL, Mcl-1 and the inhibitor of apoptosis, survivin, as well as
caspase-3, a protease engaged in the execution phase of apoptosis.
Interestingly, we detected slightly elevated Bcl-2 protein levels
after treatment with both drugs alone and in combination,
indicating Bcl-2 independent induction of apoptosis in cBCRP cells.
However, proadifen alone and in combined treatment downregulated
the expression of survivin, Mcl-1, and Bcl-xL and led to the
activation of procaspase-3. These results are consistent with our
previous study (9), where
proadifen decreased the expression level of Mcl-1 and led to the
activation of caspase-3 in HT-29 colorectal adenocarcinoma cells.
However, it has also been determined that no effect on Bcl-xL
expression existed (24,25). Mcl-1 was reported to prevent
apoptosis and confer drug resistance in acute myeloid leukemia.
Overexpression of Mcl-1 in HL-60 also led to cross-resistance to
MTX and ABT-737 (26).
Downregulation of these important pro-survival factors by proadifen
may contribute to the increased anti-proliferative impact of
combined therapy in cBCRP cells. Several other studies showed that
downregulation of survivin (27,28),
Mcl-1 (29), Bcl-xL (30) and activation of caspase-3 (31) sensitised cancer cells to
chemotherapy.
Since proadifen sensitised cBCRP cells to
MTX-induced cell death and even affected the cytostatic action of
MTX by cell cycle redistribution, we investigated the impact of
proadifen on certain proteins involved in DNA damage repair. MTX, a
topoisomerase II inhibitor and DNA intercalator, induces DSBs due
to the collision of the progressing RNA polymerase with the
stabilised topoisomerase II-DNA complex. Therefore, we focused on
several proteins engaged in the DSBs repair. The generation of DSBs
causes rapid phosphorylation of histone H2AX on Ser139 (γH2AX).
γH2AX represents an important factor in DNA damage signalling
pathways (32). While proadifen
had no effect on H2AX phosphorylation in HL-60 cells, we observed
decreased γH2AX expression below the untreated control level in
cBCRP cells after proadifen treatment. Moreover, proadifen reverted
the MTX induced expression of γH2AX. It is well-known that
nonhomologous end-joining (NHEJ) and homologous repair (HR) are the
major pathways of DSBs repair. The NHEJ DSBs repair is mediated by
several repair proteins, including Ku86 (XRCC5), which is the key
protein involved in NHEJ (33).
Recent studies demonstrated the role of nucleolar protein B23
(NPM1) in the HR pathway (33,34).
However, Koike et al (34)
showed that only B23 phosphorylated on residue Thr199 is recruited
to DSBs and plays a role in DNA repair. Our results showed that
proadifen reduced the levels of repair proteins Ku86 and B23.
Moreover, we determined proadifen-mediated decreases in the
expression of γH2AX, which is involved in the recruitment of the
proteins necessary for the reparation process (32). These findings indicate a possible
role of proadifen in DNA repair blockage, thus suppressing the rate
of MTX-induced DSB reparation. However, more analyses of
Thr199-phosphorylated B23 will be necessary to elucidate the role
of this protein in cBCRP cells.
In conclusion, our results revealed
anti-proliferative properties of proadifen in MTX-resistant human
promyelocytic leukemia cells. Moreover, we demonstrated the ability
of proadifen to increase the intracellular accumulation of MTX,
thereby enhancing its cytotoxicity in the MTX-resistant cBCRP cell
line. This effect was mediated through proadifen-mediated
inhibition of BCRP activity and expression. Furthermore, our study
indicates that proadifen also affects the expression levels of
various anti-apoptotic proteins, as well as proteins engaged in the
DNA damage repair pathways, thus enhancing chemotherapy efficacy.
Taken together, based on our recent findings, as well as on
previous results, we suggest that proadifen may be beneficial in
combination with chemotherapy for overcoming multidrug resitance,
enhancing the cytotoxicity of these drugs.
Acknowledgements
This study was supported by the Slovak Research and
Development Agency under the contract no. APVV-0040-10, the
Scientific Grant Agency of the Ministry of Education of the Slovak
Republic under the contract no. VEGA 1/0626/11 and the Cancer
Research Foundation under the contract no. 0-12-102/0001-00. The
authors are very grateful to Viera Balážová for the assistance with
technical procedures.
References
|
1
|
Nakanishi T, Chumsri S, Khakpour N, Brodie
AH, Leyland-Jones B, Hamburger AW, Ross DD and Burger AM:
Side-population cells in luminal-type breast cancer have
tumour-initiating cell properties, and are regulated by HER2
expression and signalling. Br J Cancer. 102:815–826. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hu C, Li H, Li J, Zhu Z, Yin S, Hao X, Yao
M, Zheng S and Gu J: Analysis of ABCG2 expression and side
population identifies intrinsic drug efflux in the HCC cell line
MHCC-97L and its modulation by Akt signaling. Carcinogenesis.
29:2289–2297. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hu L, McArthur C and Jaffe RB: Ovarian
cancer stem-like side-population cells are tumourigenic and
chemoresistant. Br J Cancer. 102:1276–1283. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Salcido CD, Larochelle A, Taylor BJ,
Dunbar CE and Varticovski L: Molecular characterisation of side
population cells with cancer stem cell-like characteristics in
small-cell lung cancer. Br J Cancer. 102:1636–1644. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu T, Xu F, Du X, Lai D, Liu T, Zhao Y,
Huang Q, Jiang L, Huang W, Cheng W, et al: Establishment and
characterization of multi-drug resistant, prostate
carcinoma-initiating stem-like cells from human prostate cancer
cell lines 22RV1. Mol Cell Biochem. 340:265–273. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Natarajan K, Xie Y, Baer MR and Ross DD:
Role of breast cancer resistance protein (BCRP/ABCG2) in cancer
drug resistance. Biochem Pharmacol. 83:1084–1103. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Diah SK, Smitherman PK, Aldridge J, Volk
EL, Schneider E, Townsend AJ and Morrow CS: Resistance to
mitoxantrone in multidrug-resistant MCF7 breast cancer cells:
Evaluation of mitoxantrone transport and the role of multidrug
resistance protein family proteins. Cancer Res. 61:5461–5467.
2001.PubMed/NCBI
|
|
8
|
Jendzelovský R, Mikes J, Koval' J, Soucek
K, Procházková J, Kello M, Sacková V, Hofmanová J, Kozubík A and
Fedorocko P: Drug efflux transporters, MRP1 and BCRP, affect the
outcome of hypericin-mediated photodynamic therapy in HT-29
adenocarcinoma cells. Photochem Photobiol Sci. 8:1716–1723. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jendželovský R, Koval J, Mikeš J, Papčová
Z, Plšíková J and Fedoročko P: Inhibition of GSK-3β reverses the
pro-apoptotic effect of proadifen (SKF-525A) in HT-29 colon
adenocarcinoma cells. Toxicol In Vitro. 26:775–782. 2012.
View Article : Google Scholar
|
|
10
|
Ozvegy-Laczka C, Hegedus T, Várady G,
Ujhelly O, Schuetz JD, Váradi A, Kéri G, Orfi L, Német K and
Sarkadi B: High-affinity interaction of tyrosine kinase inhibitors
with the ABCG2 multidrug transporter. Mol Pharmacol. 65:1485–1495.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kleban J, Mikes J, Szilárdiová B, Koval J,
Sacková V, Solár P, Horváth V, Hofmanová J, Kozubík A and Fedorocko
P: Modulation of hypericin photodynamic therapy by pretreatment
with 12 various inhibitors of arachidonic acid metabolism in colon
adenocarcinoma HT-29 cells. Photochem Photobiol. 83:1174–1185.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: The combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jendželovská Z, Jendželovský R, Hiľovská
L, Kovaľ J, Mikeš J and Fedoročko P: Single pre-treatment with
hypericin, a St. John's wort secondary metabolite, attenuates
cisplatin- and mitoxantrone-induced cell death in A2780, A2780cis
and HL-60 cells. Toxicol In Vitro. 28:1259–1273. 2014. View Article : Google Scholar
|
|
14
|
Kretschy N, Teichmann M, Kopf S, Atanasov
AG, Saiko P, Vonach C, Viola K, Giessrigl B, Huttary N, Raab I, et
al: In vitro inhibition of breast cancer spheroid-induced
lymphendothelial defects resembling intravasation into the
lymphatic vasculature by acetohexamide, isoxsuprine, nifedipin and
proadifen. Br J Cancer. 108:570–578. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Spitzmaul G, Gumilar F, Dilger JP and
Bouzat C: The local anaesthetics proadifen and adiphenine inhibit
nicotinic receptors by different molecular mechanisms. Br J
Pharmacol. 157:804–817. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hoferová Z, Soucek K, Hofmanová J, Hofer
M, Chramostová K, Fedorocko P and Kozubik A: In vitro proliferation
of fibrosarcoma cells depends on intact functions of lipoxygenases
and cytochrome P-450-monooxygenase. Cancer Invest. 22:234–247.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hofmanová J, Soucek K, Dusek L, Netíková J
and Kozubík A: Inhibition of the cytochrome P-450 modulates
all-trans-retinoic acid-induced differentiation and apoptosis of
HL-60 cells. Cancer Detect Prev. 24:325–342. 2000.PubMed/NCBI
|
|
18
|
Huang YC and Han YS: Determining
anti-betanodavirus compounds through a GF-1 cell-based screening
platform. Antiviral Res. 105:47–53. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fernández G, Villarruel MC, Bernacchi A,
de Castro CR and Castro JA: Effects of repeated administration of
rat 2-diethyl-aminoethyl-2-2-diphenylvalerate-HCI (SKF 525 A) on
liver. Toxicology. 20:185–193. 1981. View Article : Google Scholar
|
|
20
|
Elahian F, Kalalinia F and Behravan J:
Evaluation of indo-methacin and dexamethasone effects on
BCRP-mediated drug resistance in MCF-7 parental and resistant cell
lines. Drug Chem Toxicol. 33:113–119. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chu TS, Chen JS, Lopez JP, Pardo FS,
Aguilera J and Ongkeko WM: Imatinib-mediated inactivation of Akt
regulates ABCG2 function in head and neck squamous cell carcinoma.
Arch Otolaryngol Head Neck Surg. 134:979–984. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nakanishi T, Shiozawa K, Hassel BA and
Ross DD: Complex interaction of BCRP/ABCG2 and imatinib in
BCR-ABL-expressing cells: BCRP-mediated resistance to imatinib is
attenuated by imatinib-induced reduction of BCRP expression. Blood.
108:678–684. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rosenfeldt MT, Bell LA, Long JS, O'Prey J,
Nixon C, Roberts F, Dufès C and Ryan KM: E2F1 drives
chemotherapeutic drug resistance via ABCG2. Oncogene. 33:4164–4172.
2014. View Article : Google Scholar
|
|
24
|
Breitenbuecher F, Markova B, Kasper S,
Carius B, Stauder T, Böhmer FD, Masson K, Rönnstrand L, Huber C,
Kindler T, et al: A novel molecular mechanism of primary resistance
to FLT3-kinase inhibitors in AML. Blood. 113:4063–4073. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Glaser SP, Lee EF, Trounson E, Bouillet P,
Wei A, Fairlie WD, Izon DJ, Zuber J, Rappaport AR, Herold MJ, et
al: Anti-apoptotic Mcl-1 is essential for the development and
sustained growth of acute myeloid leukemia. Genes Dev. 26:120–125.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hermanson DL, Das SG, Li Y and Xing C:
Overexpression of Mcl-1 confers multidrug resistance, whereas
topoisomerase IIβ downregulation introduces mitoxantrone-specific
drug resistance in acute myeloid leukemia. Mol Pharmacol.
84:236–243. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fuessel S, Herrmann J, Ning S, Kotzsch M,
Kraemer K, Schmidt U, Hakenberg OW, Wirth MP and Meye A:
Chemosensitization of bladder cancer cells by survivin-directed
antisense oligodeoxy-nucleotides and siRNA. Cancer Lett.
232:243–254. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dong H, Liu G, Jiang B, Guo J, Tao G, Yiu
W, Zhou J and Li G: The effects of aspirin plus cisplatin on
SGC7901/CDDP cells in vitro. Biomed Rep. 2:344–348. 2014.PubMed/NCBI
|
|
29
|
Li G, Zhang S, Fang H, Yan B, Zhao Y, Feng
L, Ma X and Ye X: Aspirin overcomes Navitoclax-resistance in
hepatocellular carcinoma cells through suppression of Mcl-1.
Biochem Biophys Res Commun. 434:809–814. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bank A, Yu J and Zhang L: NSAIDs
downregulate Bcl-X(L) and dissociate BAX and Bcl-X(L) to induce
apoptosis in colon cancer cells. Nutr Cancer. 60(Suppl 1):
S98–S103. 2008. View Article : Google Scholar
|
|
31
|
Zhang DQ, Guo Q, Zhu JH and Chen WC:
Increase of cyclo-oxygenase-2 inhibition with celecoxib combined
with 5-FU enhances tumor cell apoptosis and antitumor efficacy in a
subcutaneous implantation tumor model of human colon cancer. World
J Surg Oncol. 11:162013. View Article : Google Scholar
|
|
32
|
Paull TT, Rogakou EP, Yamazaki V,
Kirchgessner CU, Gellert M and Bonner WM: A critical role for
histone H2AX in recruitment of repair factors to nuclear foci after
DNA damage. Curr Biol. 10:886–895. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kalra RS and Bapat SA: Enhanced levels of
double-strand DNA break repair proteins protect ovarian cancer
cells against genotoxic stress-induced apoptosis. J Ovarian Res.
6:662013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Koike A, Nishikawa H, Wu W, Okada Y,
Venkitaraman AR and Ohta T: Recruitment of phosphorylated NPM1 to
sites of DNA damage through RNF8-dependent ubiquitin conjugates.
Cancer Res. 70:6746–6756. 2010. View Article : Google Scholar : PubMed/NCBI
|