Introduction
Prostate cancer (PCa) is the second most frequently
diagnosed cancer and bone metastasis is the principal issue,
accounting for as many as 90% of patients with advanced PCa
(1). The main therapeutic option
for bone metastasis in hormone-responsive PCa is androgen
deprivation therapy. Despite initial response rates of 80–90%,
virtually all treated patients progress to androgen-insensitive
disease, a state referred to as metastatic castration-resistant PCa
(mCRPC). Although these agents effectively palliate symptoms and
prolong life, mCRPC remains incurable (2,3).
Therefore, an increased understanding of the mechanisms of PCa bone
metastasis and metastatic castration-resistance is needed to
develop novel therapeutic approaches.
Epithelial to mesenchymal transition (EMT), as a
transient phenomenon involving in the process of metastasis of
cancers, plays a key role in tumor cells invasion and metastasis
(4). Cancer stem cells (CSCs) are
a rare subpopulation of cells with stem cell-like properties which
have been found in solid malignancies (5–7), and
also are thought to be responsible for cancer relapse and
metastasis (8,9). Recent evidence has showed that EMT
can generate cancer cells with stemness properties (10). The study of Ribeiro and Paredes
revealed that P-cadherin expression, which has been already
identified as a breast cancer stem cell marker and invasive
promoter, was probably able to identify an intermediate EMT state
associated with a metastatic phenotype (11). This important finding implies a
direct link between EMT and properties of CSCs. Therefore,
unveiling the molecular mechanisms responsible for EMT and CSCs
would be helpful to develop new promising therapies for PCa
patients (12).
N-cadherin, as a marker of ongoing EMT, is not
expressed in normal epithelial cells, but its expression has been
demonstrated in several types of carcinomas (13–15).
Recent study showed that FGFR signaling was responsible for the
initiation of N-cadherin-driven EMT and stemness properties in
breast cancer cells (16). In PCa,
simultaneous upregulation of N-cadherin and downregulation of
E-cadherin have been found in more aggressive PCa lines, primary
and metastatic PCa. Importantly, aberrant N-cadherin expression has
been reported as crucial in PCa progression not only to metastasis,
but also to castration resistance (13,17).
Furthermore, in xenografts of castration-resistant PCa, a
monoclonal antibody that targeted the ectodomain of N-cadherin
inhibited androgen-independent growth, local invasion and
metastasis. However, the underlying mechanism of N-cadherin in
promoting PCa progression is not fully understood. As a switch from
E-cadherin to N-cadherin plays a critical role in EMT and
progression of PCa and high mortality (18), we hypothesize that N-cadherin
positively regulates metastatic abilities of PCa cells by
modulating EMT and stemness properties of PCa cells.
The ErbB family tyrosine kinases consists of four
members, ErbB1-4, and ErbB1 and ErbB2 also known as EGFR and HER2,
respectively. EGFR was the first receptor evidenced with a
relationship between receptor overexpression and epidermoid
carcinoma (19). Amplification and
mutation of EGFR has been proved to be associated with poor
prognosis in cancers, such as glioma (20–22),
lung cancer (23–26) and breast cancer (27,28).
HER2 has been also reported to be amplified and been widely studied
in breast cancer. HER2 serves as an important prognosis maker and
therapy target for breast cancer (29–31).
Furthermore, it has been reported that HER2 could induce EMT in
both mammary epithelial cells (32) and breast cancer cells (33,34).
Furthermore, activation of ERRB2 and ERRB3 was able to mediate
glioblastoma cancer stem-like cell resistance to EGFR-targeted
inhibition (35) and activation of
EGFR was reported to promote acquisition of stem cell-like
properties in head and neck squamous cell carcinoma (36). Currently, the underlying mechanism
between ErbB signaling and EMT and stemness of cancer cells is
poorly understood, and the role of ErbB signaling in PCa is largely
unknown.
In the present study, we reported that N-cadherin
positively regulated invasion, migration, EMT and stemness
properties of PCa cells. Importantly, through microarray analysis
and further test, we found that overexpression of N-cadherin
activated ErbB signaling, but not FGF signaling. Furthermore, our
results demonstrated that N-cadherin regulated EMT and stem
cell-like property linked with ErbB signaling in PCa cells. Taken
together, N-cadherin might serve as a novel potential therapeutic
target in PCa.
Materials and methods
Cells and cell culture
The brain metastatic cell line DU145 and the bone
metastatic PCa cell line PC-3 were purchased from the American Type
Culture Collection (ATCC) and grown in DMEM culture medium
(Hyclone) and Ham's F-12 culture medium (Hyclone) respectively,
supplemented with 10% fetal bovine serum (Hyclone). Cells were
grown at a humidified atmosphere of 5% CO2 at 37°C.
Labatinib was purchased and prepared as a 10 mM concentrated stock
solution in dimethyl sulphoxide (Fisher Scientific).
Vectors and retroviral infection
N-cadherin gene was amplified from cDNA by RT-PCR
and cloned into the pMSCV-EF2 lentiviral vector. Two human
N-cadherin-targeting shRNA sequences were cloned into
pSuper-retro-puro to generate pSuper-retro-N-cadherin RNAi (s) and
the sequences of RNAi#1 and RNAi#2 are GCTGAAAGAACTGAAGCATTT and
AAATGCTTAGTTCTTTCAGC, respectively. Retroviral production and
infection were performed as previously described (37). Stable cell lines expressing
N-cadherin or N-cadherin shRNAs were selected for 10 days with 0.5
mg/ml puromycin.
Microarray analysis
Total RNA from PC-3/vector, PC-3/N-cadherin-RNAi and
N-cadherin-overexpression were extracted. Total RNA from each
sample was quantified by the NanoDrop ND-1000 and RNA integrity was
assessed by standard denaturing agarose gel electrophoresis
(38). Microarray analysis was
performed commercially by the Shanghai Biochip Corp. according to
standard Agilent protocol. Briefly, integrity and concentration of
RNA was assessed after RNA extraction and prior to sample labeling.
Total RNA of each sample was used for labeling and array
hybridization with the following steps: i) reverse transcription
with Invitrogen Superscript ds-cDNA synthesis kit; ii) ds-cDNA
labeling with NimbleGen one-color DNA labeling kit; iii) array
hybridization using the NimbleGen Hybridization system and followed
by washing with the NimbleGen wash buffer kit; iv) array scanning
using the Axon GenePix 4000B microarray scanner (Molecular Devices
Corp.). Data were extracted and normalized using NimbleScan v2.5
software. Results are provided in the NimbleScan Generated Data
Folder. Further data analysis was performed using Agilent
GeneSpring GX v11.5.1 software. Bioinformatics analysis and
visualization of microarray data were performed with the MeV v4.4
program (http://www.tm4.org/mev/) (39).
Quantitative reverse
transcription-PCR
The procedure was performed according to the
instrcution of All-in-One™ miRNA qRT-PCR Detection kit
(GeneCopoeia, USA), as described previously (40). The relative expression levels from
three independent experiments were counted following the
2−ΔΔCt method of Livak and Schmittgen (41). The qRT-PCR primers for N-cadherin,
c-Myc, OCT-4, SOX2, Klf4 and glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) were designed by the Primer Express version
2.0 software (Applied Biosystems). N-cadherin forward,
5′-GGCATACACCATG CCATCTT-3′; reverse, 5′-GTGCATGAAGGACAGCCTCT-3′;
c-Myc forward, 5′-CACCGA GTCGTAGTCGAGGT-3′; reverse,
5′-GCTGCTTAGACGCTGGATTT-3′; OCT-4 forward,
5′-TCTCCAGGTTGCCCTCACT-3′; reverse, 5′-GTGGAG GAAGCTGACAACAA-3′;
SOX2 forward, 5′-GTCATTTG CTGTGGGTGATG-3′; reverse,
5′-AGAAAAACGAGGGA AATGGG-3′; Klf4 forward, 5′-CCCCGTGTGTTTACG
GTAGT-3′; reverse, 5′-GAGTTCCCATCTCAAGGCAC-3′; GAPDH forward,
5′-ACATCCCCTCACCAATAACAAC-3′; reverse,
5′-TAGCCAAATCATACTGCTCGTC-3′.
Western blotting
For the analysis of expression of related proteins,
western blot assay was performed according to a standard method, as
described previously (42). The
following primary antibodies were used: mouse anti-vimentin, mouse
anti-E-cadherin (CST, cell signal technique); mouse
antifibronectin, mouse anti-N-cadherin (BD Biosciences); mouse
anti-Grb2, anti-pShc and anti-pERK1/2 (Abcam). Blotting membranes
were stripped and re-probed with anti-tubulin antibody (Sigma) as a
loading control. Nuclear extracts were prepared using the Nuclear
Extraction kit (Active Motif), according to the manufacturer's
instructions.
Wound healing assay
PCa cells were cultured on 6-well plates with DMEM
containing 10% FBS to 90% conflucency and scratched with a sterile
10 μl pipette tip to create artificial wounds. After scratching,
the detached and damaged cells were carefully washed with
phosphate-buffered solution (PBS) and maintained in 10% fetal
bovine serum media. Progression of migration was observed and
photographed at 24 h after wounding. Images of the cells migrating
into the wound were taken at the time points of 0, 6 and 12 h by an
inverted microscope (x40).
Invasion assays
Cell invasion assays were performed using Transwell
chambers consisting of 8-mm membrane filter inserts (Corning;
Corning Inc.) coated with Matrigel (BD Biosciences). Briefly, the
trypsinized PCa cells were resuspended in serum-free medium and
seeded in the upper chamber. Then, the lower chamber of the
Transwell was filled with 1 ml Ham's F-12 medium or T-medium
supplemented with 10% FBS. After incubation for 24–48 h at 37°C in
5% CO2, cells passing through the coated membrane to the
bottom side of the inserts were fixed with 4% paraformaldehyde and
stained with hematoxylin. The non-migratory cells on the upper
chamber were removed with cotton swabs, and the migratory cells
were stained, photographed, and quantified by counting them in 5
random high-power fields.
Colony formation assay
The cells were trypsinized as single cells and
suspended in the media with 10% FBS. Indicated cells (300 cells per
well) were seeded into of 6-well plate for ~10–14 days. Colonies
were stained with 1% crystal violet for 10 min after fixation with
10% formaldehyde for 5 min. Plating efficiency = number of colonies
(≥50 cells per colony)/per input cells × 100%. Different colony
morphologies were captured under a light microscope (Olympus).
Self-renewing spheroid formation
assay
Indicated cells (500 cells/well) were seeded into
6-well Ultra Low Cluster plate (Corning) and were cultured in
suspension in serum-free DMEM/F12 (BioWhittaker), supplemented with
2% B27 (Invitrogen), 20 ng/ml EGF (BD Biosciences), 20 ng/ml bFGF
(PeproTech), 5 μg/ml insulin (Sigma) and 0.4% bovine serum albumin
(Sigma). After 10–12 days, the number of cell spheres (tight,
spherical, non-adherent masses >50 μm in diameter) were counted,
and image of the spheres were captured under inverse microscope.
Sphere formation efficiency = colonies/input cells ×100%.
Luciferase reporter assay
Luciferase assays were carried out in 293FT cells
that were co-transfected with miRNAs and luciferase reporter
plasmids in 24-well plates and cultured for 48 h before the cells
were harvested and lysed for luminescence detection. Subsequent
processing and detection were performed by using the luciferase
assay kit (Promega) according to the manufacturer's protocols.
Renilla luciferase was activated to emit primary luminescence, and
firefly luminescence was used for normalization. Each test was
repeated in triplicate.
Statistical analyses
All statistical analyses were carried out using SPSS
17.0 statistical software package. Means ± SD was calculated and
two-tailed Student's t-test or one-way ANOVA was performed using
data analysis tools provided in the software package. In all cases,
P<0.05 was considered statistically significant.
Results
N-cadherin promotes invasion, migration
and EMT of PCa cells
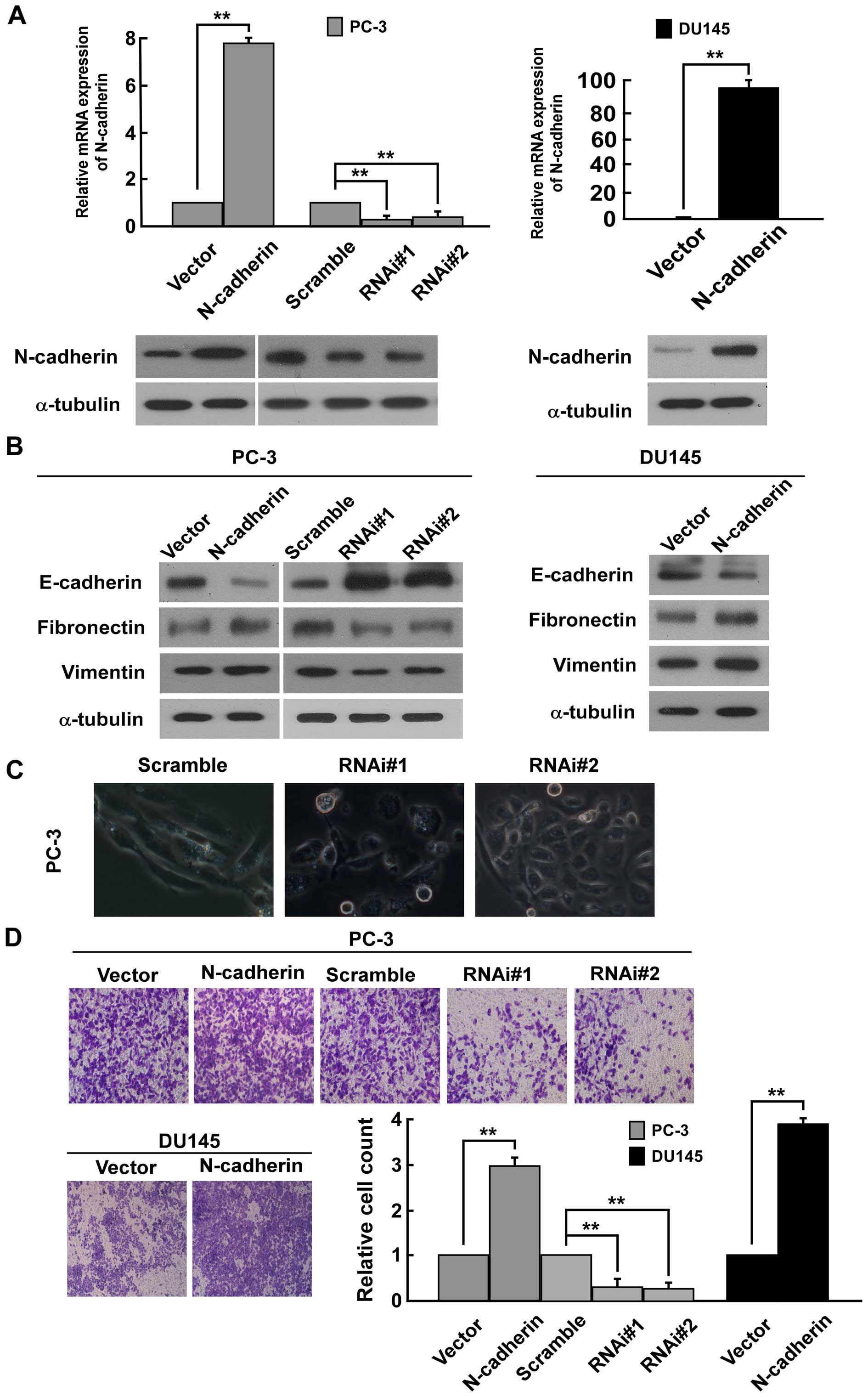
To investigate the biological function of N-cadherin
in PCa cells, we transducted PC-3 and DU145 PCa cells with
constructed pMSCV/N-cadherin-derived retrovirus to establish
N-cadherin-overexpressing stable cell lines. As N-cadherin was not
highly expressed in DU145 human prostate cell lines (43,44),
N-cadherin-specific RNA interference (RNAi) oligonucleotides were
cloned into a retroviral transfer vector pSuper-retro-puro to
establish N-cadherin-low expressing stable cell lines only in PC-3
(Fig. 1A). Western blot analysis
suggested that N-cadherin expression is positively related with the
expression of mesenchymal makers fibronectin and vimentin, but
negatively related with the expression of epithelial marker
E-cadherin in PC-3 and DU145 cells (Fig. 1B). On the contrary, the low
expression of N-cadherin significantly reduced the expression of
fibronectin and vimentin and increased the expression of E-cadherin
in PC3 cells (Fig. 1B). The result
suggested that N-cadherin expression might be involved in EMT of
PCa cells. Firstly, upon knockdown of N-cadherin in PC-3 cells, we
noted that PC-3 cells underwent a marked change in morphology, from
spindle like morphology to epithelial transition (Fig. 1C). Moreover, Transwell matrix
penetration assay revealed that overexpression of N-cadherin
promoted, while silencing N-cadherin strongly repressed the
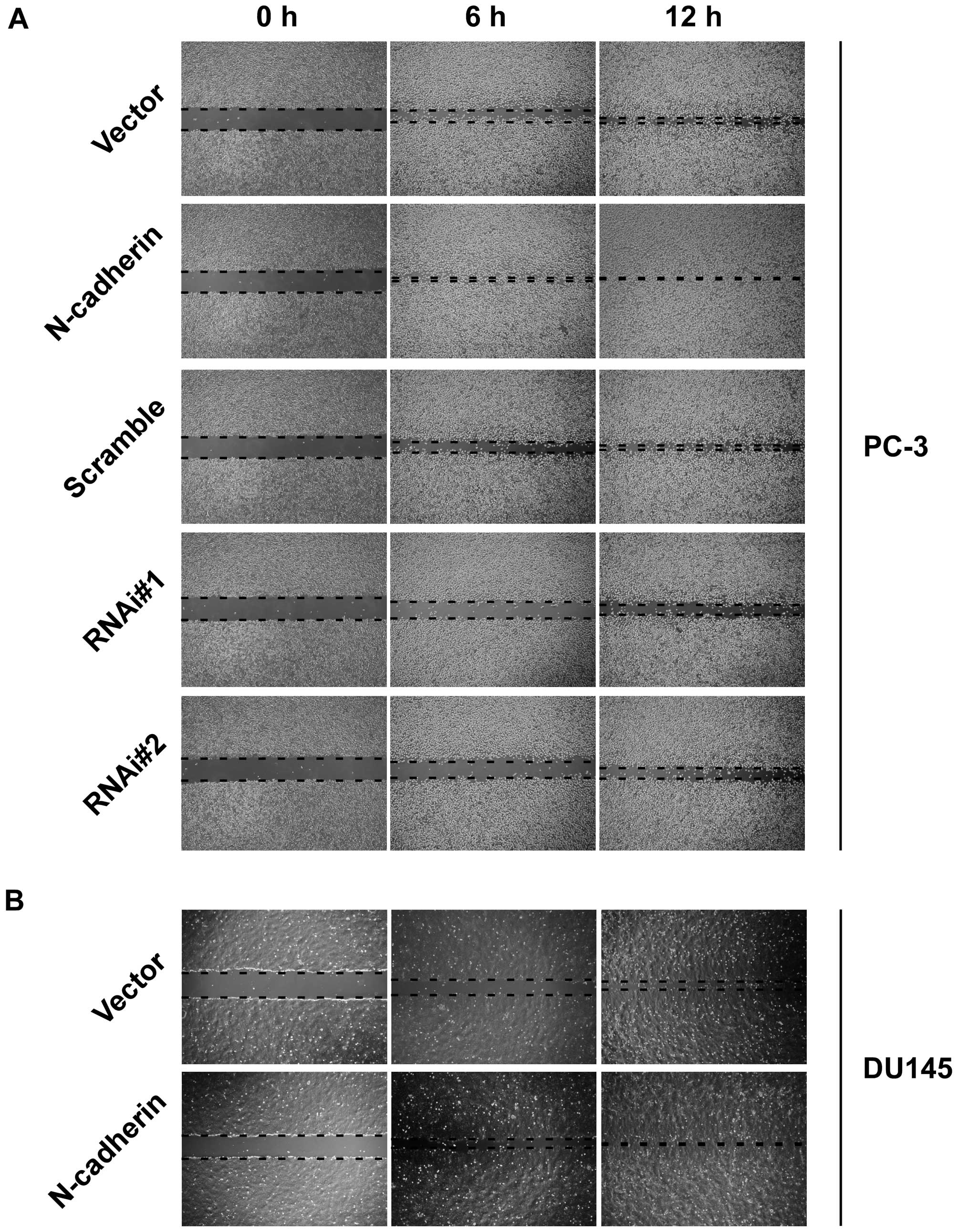
invasive ability compared to vector cells (Fig. 1D). In addition, wound healing assay
showed that upregulation of N-cadherin increased, while knockdown
of N-cadherin decreased healing speed of the scratch in transducted
cells (Fig. 2). These data
demonstrated that N-cadherin was able to modulate the invasion and
migration properties of PCa cells.
N-cadherin promotes colony, spheroid
formation and Sox2, c-Myc, Oct4 and Klf4 expression in PCa
cells
Numerous studies suggested that cancer stem cells
are involved in tumor metastasis (10,45).
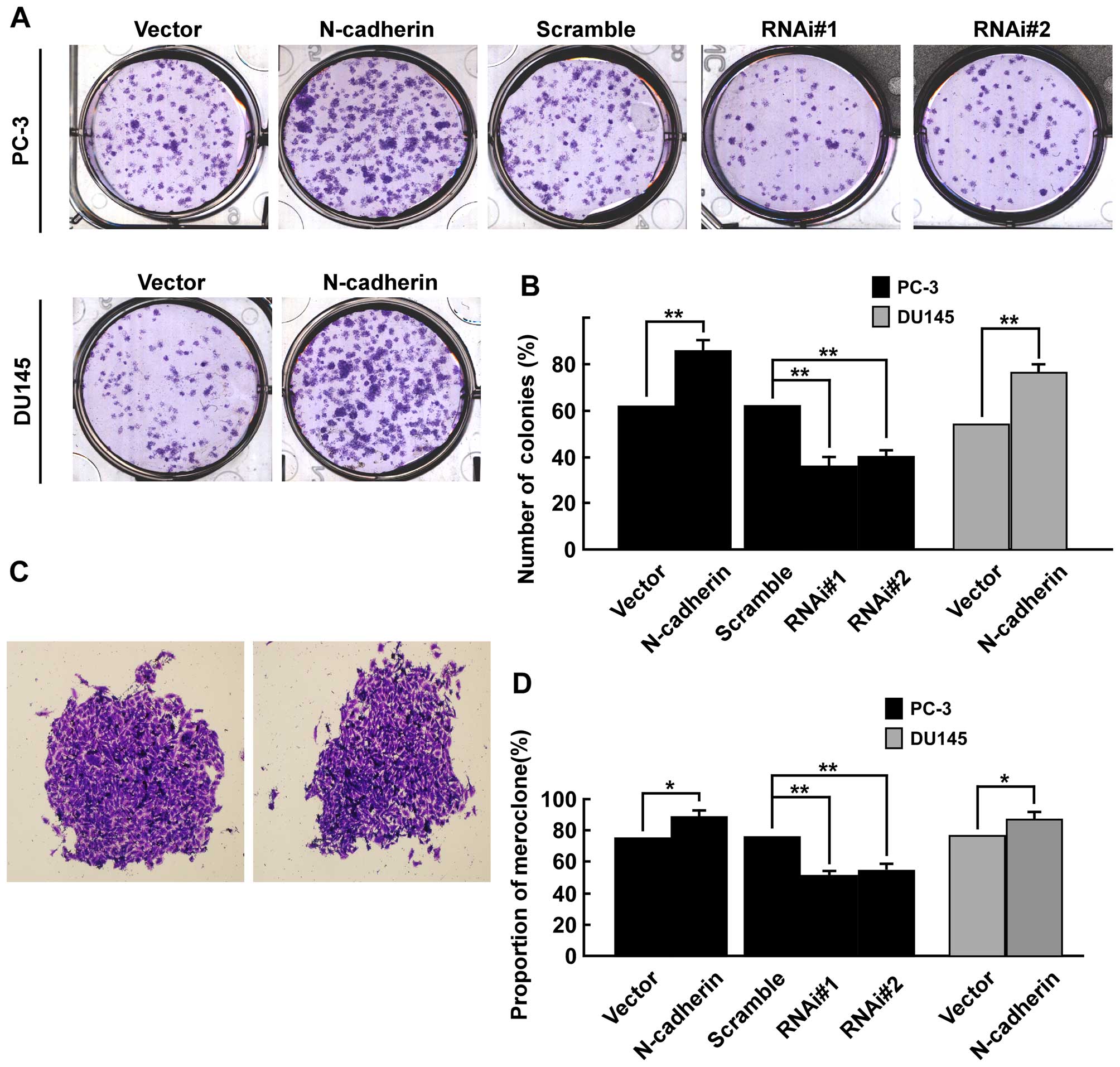
Firstly, colony formation assays was used to explore whether
N-cadherin had an effect on viability of PCa cells in vitro.
Results showed overexpression of N-cadherin significantly enhanced
colony formation efficiency in both PC-3 and DU145 cells, while
downregulation of N-cadherin markedly inhibited colony formation
efficiency in PC-3 cells (Fig.
3A). As shown (Fig. 3B), the
number of colonies (% plating efficiency) was 85.7% in
PC-3/N-cadherin versus 62.8% in PC-3/vector, 63.3% in PC-3/scramble
versus 38.1% in PC-3/N-cadherin-RNAi#1 and 40.5% in
PC-3/N-cadherin-RNAi#2 cells, and 77.6% in DU145 cells transfected
with N-cadherin versus 55.9% in DU145/vector. Colonies with
different morphologies in vitro are classified as
holoclones, meroclones and paraclones (46). Holoclones are generally more round
and tightly packed; paraclones are irregular in composition and
often contain more elongated or flattened cells; and meroclones are
an intermediate phenotype (Fig.
3C). We did not find typical holoclones in PC-3 and DU145
cells. The proportion of meroclones was 87.6% in PC-3/N-cadherin,
77.2% in PC-3/vector and 78.1% in PC-3/scramble, 52.2% in
PC-3/N-cadherin-RNAi#1 and 55.4% in PC-3/N-cadherin-RNAi#2 cells,
and 84.7% in DU145 cells of N-cadherin overexpression versus 75.3%
in DU145/vector. Overexpression of N-cadherin significantly
increased the proportion of meroclones of PCa cells (p<0.01,
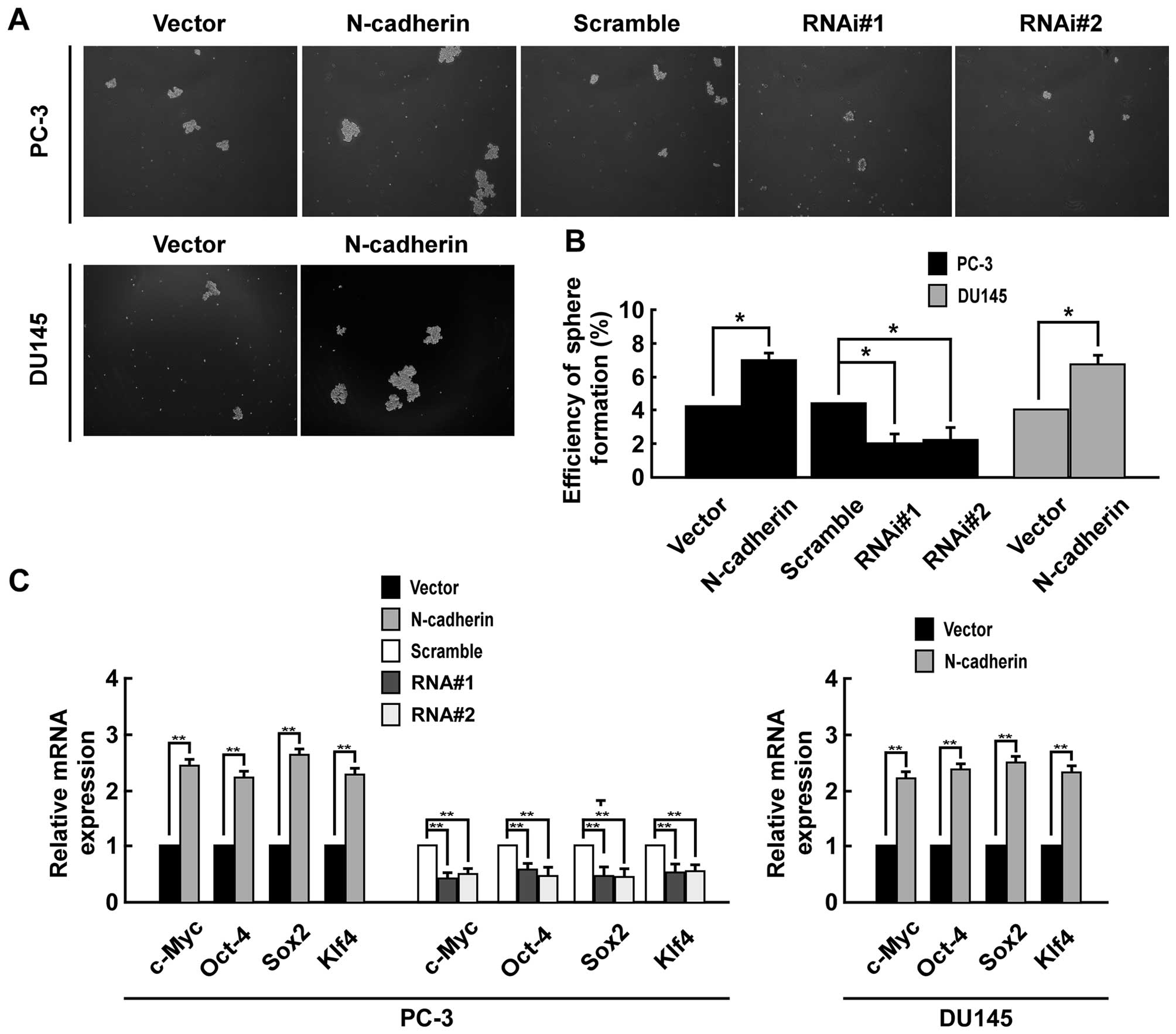
Fig. 3D). Moreover, sphere
formation assays suggested that upregulating N-cadherin enhanced
the number and size of tumor spheroids in both PC-3 and DU145
cells, while downregulating N-cadherin inhibited the number and
size of tumor spheroids in PC-3 cells (Fig. 4A). The spheroid formation
efficiency was 7.1% in PC-3/N-cadherin versus 4.2% in PC-3/vector,
4.4% in PC-3/scramble versus 2.0% in PC-3/N-cadherin-RNAi#1 cells
and 2.2% PC-3/N-cadherin-RNAi#2, and 6.7% in DU145 cells
overexpressing N-cadherin versus 4.0% in DU145/vector (Fig. 4B). Real-time PCR was performed to
examine the mRNA level of pluripotency-associated markers including
Sox2, c-Myc, Oct4 and Klf4. The result suggested that CSC markers
were significantly higher expressed in N-cadherin-transduced cells
than control group and significantly lower expressed in
N-cadherin-RNAi cells (Fig. 4C).
Thus, our results suggested that overexpression of N-cadherin
promotes prostate CSC-like traits.
N-cadherin promotes EMT and CSC-like
traits of PCa cells via ErbB signaling pathway
To understanding the underlying mechanism of
N-cadherin in regulation of EMT and stemness of PCa cells, we
performed microarray analysis on PC-3/vector and PC-3/N-cadherin
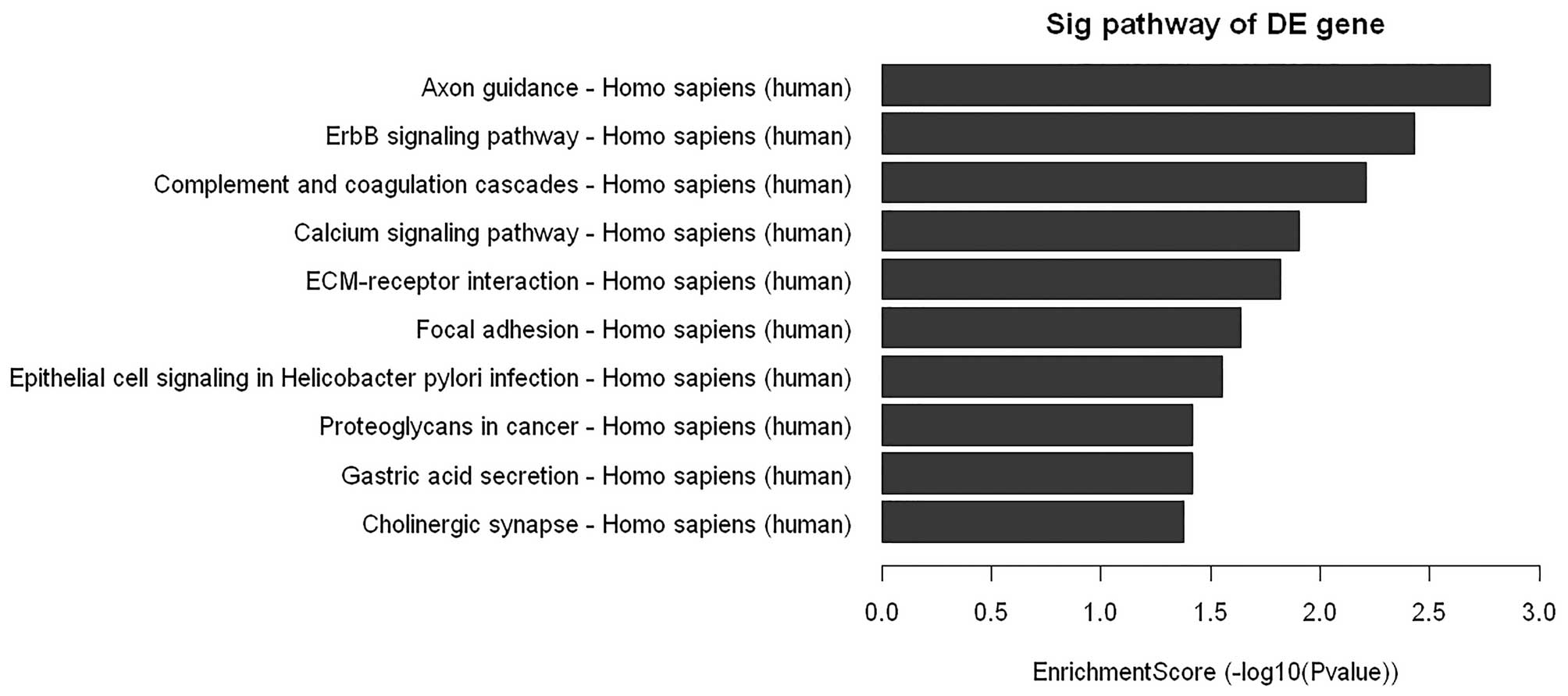
cells. As shown in Fig. 5, the
microarray data showed a comprehensive activation of ErbB in
PC-3/N-cadherin cells compared to PC-3/vector cells, which
indicated that N-cadherin might achieve its function via ErbB
signaling. To further confirm the data, downstream ERBB signaling
component Grb2 and phosphorylation of components Shc and ERK were
examined by western blot analysis. We found that overexpression of
N-cadherin enhanced the expression of Grb2, pShc and pERK1/2
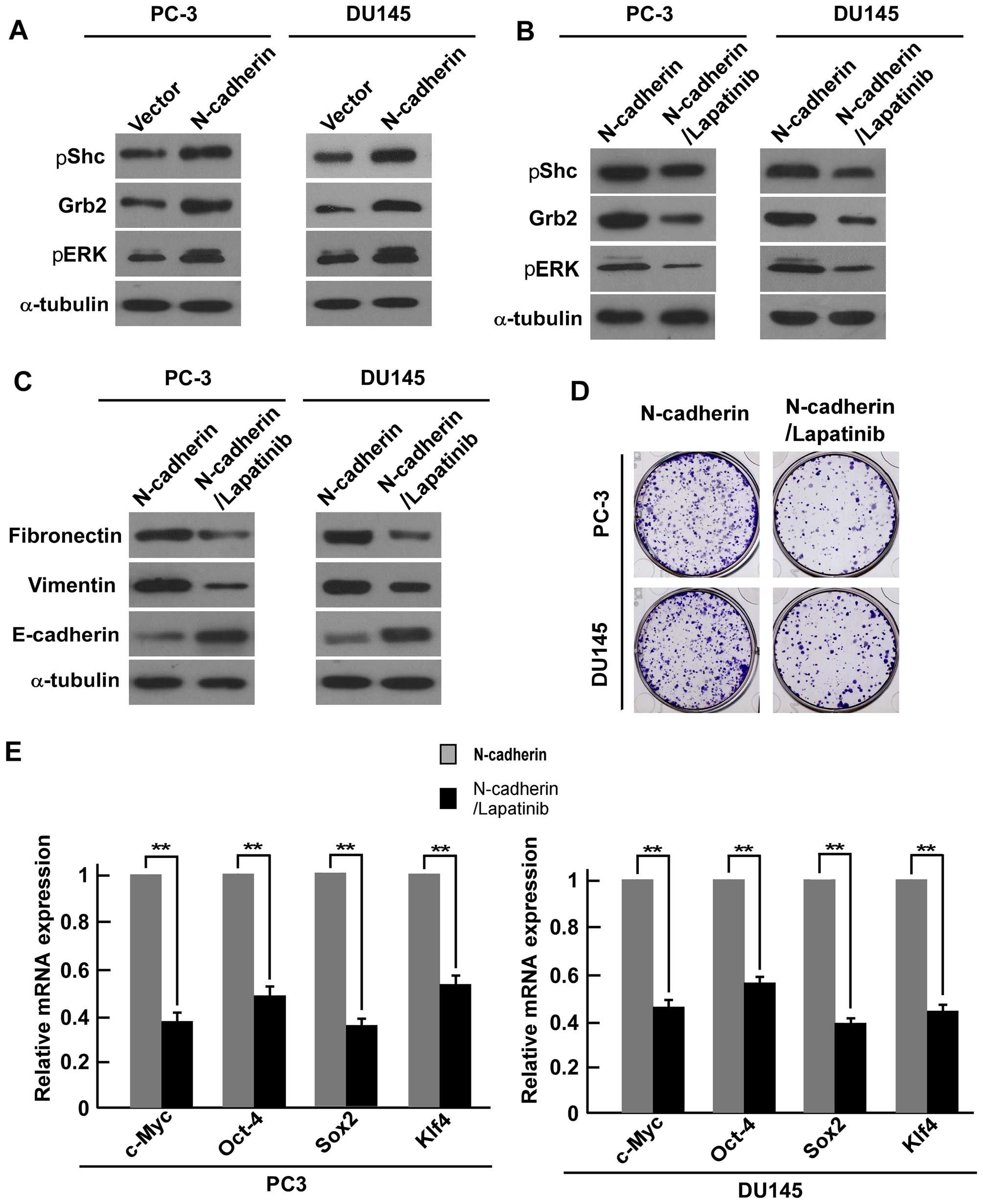
(Fig. 6A).
In order to explore whether the stem cell-like
phenotype in PCa cells is associated with ErbB signaling
activation, we also analyzed the impact of blocking ErbB signaling
on the stem cell population capability of N-cadherin transduced
PC-3 and DU145 cell lines. We used lapatinib, a potent
ATP-competitive inhibitor, to inhibit both EGFR and HER2 (100 nM).
Western blot analysis showed that lapatinib successfully reduced
downstream Grb2, pShc and pERK1/2 in PC-3 and DU145 cells (Fig. 6B). Furthermore, western blot
analysis showed that fibronectin and vimentin expression were
suppressed in the lapatinib treated PCa cells, while expression of
E-cadherin was increased (Fig.
6C). Colony formation assays suggested that the number of
colonies were inhibited in the treatment cells compared with
N-cadherin-transduced cells (Fig.
6D). Real-time PCR analysis showed that the mRNA expression
level of ‘stemness’ factors c-Myc, Oct4, Klf4, and Sox2 were
downregulated in the lapatinib-treated cells compared with
N-cadherin-transduced cells (Fig.
6E). All the above data revealed that N-cadherin regulates EMT
and CSC-like traits of PCa cells via ErbB signaling pathway.
N-cadherin does not mediate the function
of miR-145 in PC-3 cells
Analysis using the publicly available algorithms
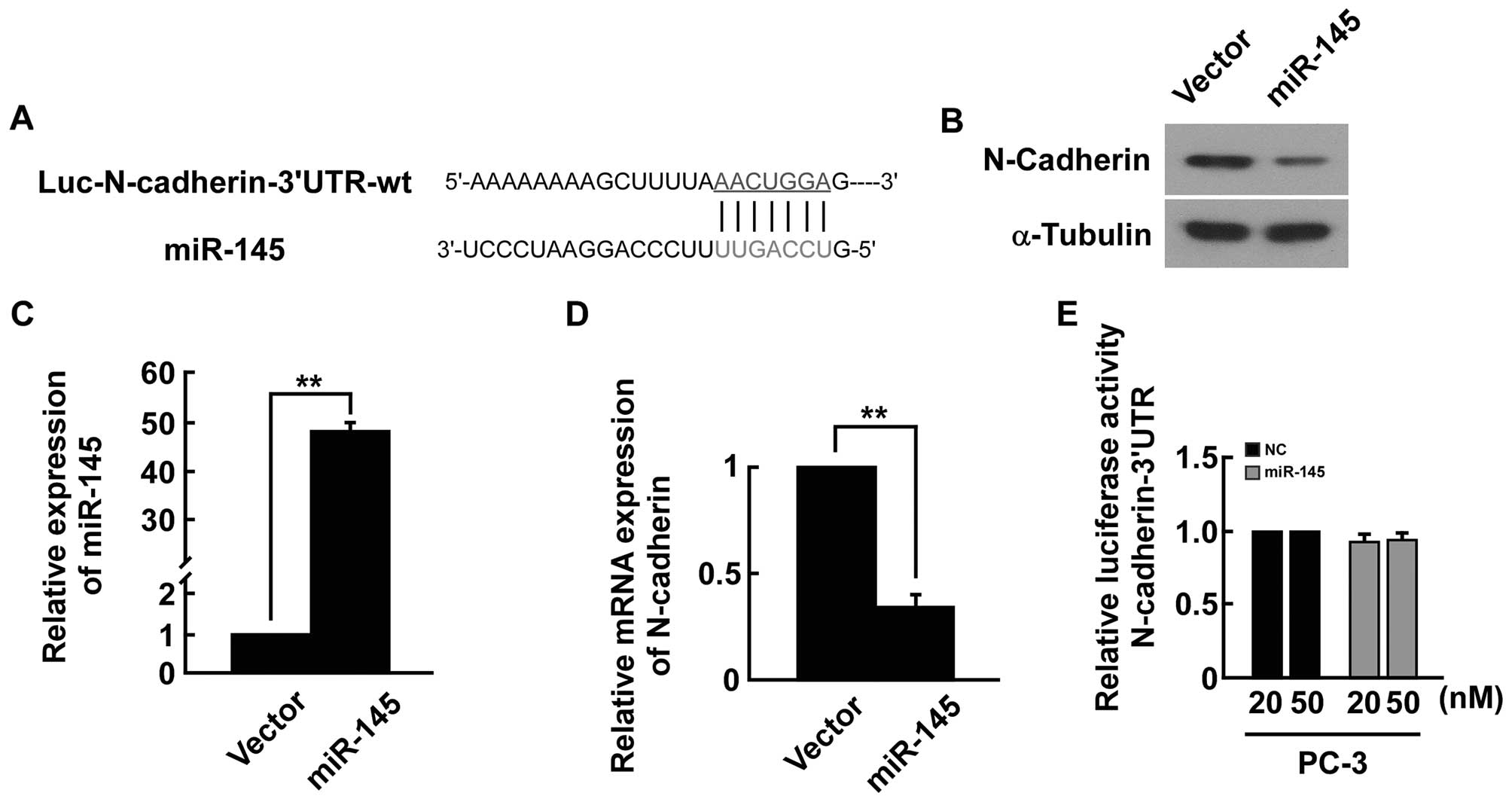
(e.g., TargetScan, miRANDA) indicated that the N-cadherin-3′-UTR
region is the theoretical conserved target of miR-145 (Fig. 7A). The expression level of the
N-cadherin mRNA and protein was significantly decreased in
miRNA-145-transfected cells (Fig.
7B–D). However, upon transfection with miR-145, we did not find
strong reduction in luciferase activity from pGL3-N-cadherin-3′-UTR
(Fig. 7E). Our results indicated
that miR-145 did not directly target N-cadherin in PC-3 cells.
Discussion
In this study, we present a pivotal finding that
overexpression of N-cadherin promoted, while knockdown of
N-cadherin repressed EMT and stem cell-like property in PCa.
Microarray analysis and further test mechanistically demonstrated
that N-cadherin achieve EMT and stemness promoting function by
activating the ErbB signaling. These findings provide strong
evidence that, for the first time, elevated N-cadherin promotes
EMT, stemness and metastatic ability via ErbB signaling pathway in
PCa cells, which further supported that N-cadherin could be a
therapeutic target in PCa.
N-cadherin, a mesenchymal cadherin associated with
epithelial-to-mesenchymal transition, has been widely studied in
various tumor types. Hazan and colleagues reported that N-cadherin
promotes adhesion between invasive breast cancer cells and the
stroma (47) and overexpression of
N-cadherin correlates with invasiveness in breast carcinoma
(48). In PCa, N-cadherin has also
been demonstrated to be elevated and associated with poor prognosis
(13,49). An important finding by Tanaka and
colleagues demonstrated that monoclonal antibody targeting of
N-cadherin could inhibit PCa growth, metastasis and castration
resistance (17). Here, we found
that increasing N-cadherin enhanced, while silencing N-cadherin
impaired the invasion and migration of PCa cells. Western blot
analysis showed that overexpression of N-cadhein enhanced the
expression of mesenchymal cell makers, fibronectin and vimentin,
and decreased the expression of epithelial cell markers,
E-cadherin, and downexpression of N-cadherin inhibited the
expression of fibronectin and vimentin, and increased the
expression of E-cadherin. Moreover, upregulation of N-cadherin also
enhanced the stem cell-like property of PCa cells as indicated by
higher tumor spheroids and colony formation efficiency and
increased the expression of stem cell property-associated factors,
including Sox2, c-Myc, Oct4 and Klf4, and vice versa. All together,
the above presented the pivotal role of N-cadherin in promoting
metastasis in PCa.
Although a recent study showed that
N-cadherin-driven EMT and stemness properties depend on FGFR
activation, ERK activity, matrix metalloproteinase 9 production and
on selective inhibition of the AKT3 isoform in many solid tumors
(14,16,50),
our results showed that the mechanism via which N-cadherin
regulates invasion, migration, EMT and stem cell-like property in
PCa is different from that in breast cancer. The present findings
indicated that N-cadherin-driven EMT and stemness properties at
least partially depend on ErbB signaling in PCa cells. Furthermore,
accumulating evidence also shows that ErbB signaling pathway is
associated with poor prognosis in various cancers. In glioma,
ErbB1, known as EGFR, extracellular missense mutations as a novel
mechanism for oncogenic EGFR activation may help identify patients
who can benefit from EGFR kinase inhibitors for treatment of
glioblastoma (22); in lung
cancer, especially non-small cell lung carcinoma (NSCLC), it has
been reported that EGFR amplication is universal and EGFR mutations
in lung carcinomas make the disease more responsive to treatment
with tyrosine kinase inhibitors (23–25);
in breast cancer, EGFR was regarded as a predictor of early
recurrence and death (27), and
ErbB2, also known as HER2, serves as an important prognosis maker
and therapy target for breast cancer (29). Further studies have demonstrated
that HER2 could induce EMT in both mammary epithelial cells
(32) and breast cancer cells
(33,34). Furthermore, some evidence reported
that glioblastoma cancer stem-like cell resistance to EGFR-targeted
inhibition was mediated by activation of multiple ERBB family
receptors (35). In head and neck
squamous cell carcinoma, EGFR kinase promotes acquisition of stem
cell-like properties (36).
Therefore, all the above-mentioned evidence indicated that ErbB
signaling pathway plays an important role in metastasis of
cancer.
For most PCa patients who were identified in the
early stages the initial therapies mostly result in significant
long-term remission (51).
However, for advanced metastatic cases these treatments, such as
prostatectomy, radiation and cryotherapy, show little benefit and
without effective control the patients eventually die of the
disease. Although androgen deprivation and chemotherapy are
currently effective treatments for these patients, development of
hormone ablation resistance is inevitable (52), which is termed castration-resistant
PCa. Consequently, improved understanding of the mechanisms
underlying mCRPC progression has contributed to the recognition of
multiple molecular targets and advances in the therapeutic
landscape. Recent study indicated that N-cadherin is crucial in PCa
progression not only to metastasis, but also to castration
resistance (17). Our results
showed that N-cadherin promoted EMT and stemness of CSCs of PCa
cells by upregulating ErbB signaling. Because EMT and CSCs play
crucial roles during the development of castration-resistance in
PCa (12), one of the important
mechanism by which N-cadherin increased castration resistance of
metastatic PCa cells may promote EMT and stemness of CSCs of PCa
cells by upregulating ErbB signaling. Therefore, understanding the
inhibition of this signaling pathway may be useful to develop new
therapies for metastatic PCa.
A wide range of studies have demonstrated that many
microRNAs (miRNAs), as crucial post-transcriptional regulators
repressing the expression of their target genes, play a pivotal
role in solid tumor metastasis via regulating migration, invasion
and EMT of cancer cells and the properties of CSCs (53–58).
Our previous studies found that miR-145 played an important role in
inhibiting migration, invasion, EMT and stemness properties of PCa
cells via different targets (40,59,60).
Furthermore, recent study showed that N-cadherin was a direct
target of miR-145 and promoted the invasion-metastasis cascade in
gastric cancer (61). Moreover,
our present results showed that N-cadherin promoted EMT and
stemness properties of PCa cells. Therefore, we can suppose that
N-cadherin mediates the function of miR-145 in regulating EMT and
stemness properties in PCa cells. However, we used dual-luciferase
reporter gene assay to check the relationship between miR-145 and
N-cadherin, and found miR-145 did not targets N-cadherin in PC-3
cell. This difference may be due to the using of different tumor
models. It will be of great interest to investigate the reasons why
miR-145 is off-target for N-cadherin in PCa cells.
In conclusion, this study demonstrated that
N-cadherin promotes invasion, migration, EMT and stemness of PCa
cells, which suggest a pivotal role of N-cadherin in metastasis and
castration resistance of PCa cells. Importantly, these findings
exploit interesting and realistic avenues for cancer therapies
using N-cadherin antagonists.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (nos. 81272938 and
81472505).
Abbreviations:
|
PCa
|
prostate cancer
|
|
miRNAs
|
microRNAs
|
|
EMT
|
epithelial-mesenchymal transition
|
|
CSCs
|
cancer stem cells
|
|
mCRPC
|
metastatic castration-resistant
prostate cancer
|
References
|
1
|
Carlin BI and Andriole GL: The natural
history, skeletal complications, and management of bone metastases
in patients with prostate carcinoma. Cancer. 88(Suppl): 2989–2994.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Karantanos T, Corn PG and Thompson TC:
Prostate cancer progression after androgen deprivation therapy:
Mechanisms of castrate resistance and novel therapeutic approaches.
Oncogene. 32:5501–5511. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Friedlander TW, Ngo VT, Dong H,
Premasekharan G, Weinberg V, Doty S, Zhao Q, Gilbert EG, Ryan CJ,
Chen WT, et al: Detection and characterization of invasive
circulating tumor cells derived from men with metastatic
castration-resistant prostate cancer. Int J Cancer. 134:2284–2293.
2014. View Article : Google Scholar
|
|
4
|
Berx G, Raspé E, Christofori G, Thiery JP
and Sleeman JP: Pre-EMTing metastasis? Recapitulation of
morphogenetic processes in cancer. Clin Exp Metastasis. 24:587–597.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Al-Hajj M, Wicha MS, Benito-Hernandez A,
Morrison SJ and Clarke MF: Prospective identification of
tumorigenic breast cancer cells. Proc Natl Acad Sci USA.
100:3983–3988. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Singh SK, Hawkins C, Clarke ID, Squire JA,
Bayani J, Hide T, Henkelman RM, Cusimano MD and Dirks PB:
Identification of human brain tumour initiating cells. Nature.
432:396–401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ricci-Vitiani L, Lombardi DG, Pilozzi E,
Biffoni M, Todaro M, Peschle C and De Maria R: Identification and
expansion of human colon-cancer-initiating cells. Nature.
445:111–115. 2007. View Article : Google Scholar
|
|
8
|
Nagata T, Sakakura C, Komiyama S,
Miyashita A, Nishio M, Murayama Y, Komatsu S, Shiozaki A, Kuriu Y,
Ikoma H, et al: Expression of cancer stem cell markers CD133 and
CD44 in locoregional recurrence of rectal cancer. Anticancer Res.
31:495–500. 2011.PubMed/NCBI
|
|
9
|
Merlos-Suárez A, Barriga FM, Jung P,
Iglesias M, Céspedes MV, Rossell D, Sevillano M, Hernando-Momblona
X, da Silva-Diz V, Muñoz P, et al: The intestinal stem cell
signature identifies colorectal cancer stem cells and predicts
disease relapse. Cell Stem Cell. 8:511–524. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pirozzi G, Tirino V, Camerlingo R, Franco
R, La Rocca A, Liguori E, Martucci N, Paino F, Normanno N and Rocco
G: Epithelial to mesenchymal transition by TGFβ-1 induction
increases stemness characteristics in primary non small cell lung
cancer cell line. PLoS One. 6:e215482011. View Article : Google Scholar
|
|
11
|
Ribeiro AS and Paredes J: P-cadherin
linking breast cancer stem cells and invasion: A promising marker
to identify an ‘intermediate/metastable’ EMT state. Front Oncol.
4:3712014.
|
|
12
|
Li P, Yang R and Gao WQ: Contributions of
epithelial-mesenchymal transition and cancer stem cells to the
development of castration resistance of prostate cancer. Mol
Cancer. 13:552014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jennbacken K, Tesan T, Wang W, Gustavsson
H, Damber JE and Welén K: N-cadherin increases after androgen
deprivation and is associated with metastasis in prostate cancer.
Endocr Relat Cancer. 17:469–479. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hulit J, Suyama K, Chung S, Keren R,
Agiostratidou G, Shan W, Dong X, Williams TM, Lisanti MP, Knudsen
K, et al: N-cadherin signaling potentiates mammary tumor metastasis
via enhanced extracellular signal-regulated kinase activation.
Cancer Res. 67:3106–3116. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hui L, Zhang S, Dong X, Tian D, Cui Z and
Qiu X: Prognostic significance of twist and N-cadherin expression
in NSCLC. PLoS One. 8:e621712013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Qian X, Anzovino A, Kim S, Suyama K, Yao
J, Hulit J, Agiostratidou G, Chandiramani N, McDaid HM, Nagi C, et
al: N-cadherin/FGFR promotes metastasis through
epithelial-to-mesenchymal transition and stem/progenitor cell-like
properties. Oncogene. 33:3411–3421. 2014. View Article : Google Scholar :
|
|
17
|
Tanaka H, Kono E, Tran CP, Miyazaki H,
Yamashiro J, Shimomura T, Fazli L, Wada R, Huang J, Vessella RL, et
al: Monoclonal antibody targeting of N-cadherin inhibits prostate
cancer growth, metastasis and castration resistance. Nat Med.
16:1414–1420. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gravdal K, Halvorsen OJ, Haukaas SA and
Akslen LA: A switch from E-cadherin to N-cadherin expression
indicates epithelial to mesenchymal transition and is of strong and
independent importance for the progress of prostate cancer. Clin
Cancer Res. 13:7003–7011. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Thompson DM and Gill GN: The EGF receptor:
Structure, regulation and potential role in malignancy. Cancer
Surv. 4:767–788. 1985.PubMed/NCBI
|
|
20
|
Heimberger AB, Suki D, Yang D, Shi W and
Aldape K: The natural history of EGFR and EGFRvIII in glioblastoma
patients. J Transl Med. 3:382005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Puputti M, Tynninen O, Sihto H, Blom T,
Mäenpää H, Isola J, Paetau A, Joensuu H and Nupponen NN:
Amplification of KIT, PDGFRA, VEGFR2, and EGFR in gliomas. Mol
Cancer Res. 4:927–934. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee JC, Vivanco I, Beroukhim R, Huang JH,
Feng WL, DeBiasi RM, Yoshimoto K, King JC, Nghiemphu P, Yuza Y, et
al: Epidermal growth factor receptor activation in glioblastoma
through novel missense mutations in the extracellular domain. PLoS
Med. 3:e4852006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sharma SV, Bell DW, Settleman J and Haber
DA: Epidermal growth factor receptor mutations in lung cancer. Nat
Rev Cancer. 7:169–181. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Marchetti A, Martella C, Felicioni L,
Barassi F, Salvatore S, Chella A, Camplese PP, Iarussi T, Mucilli
F, Mezzetti A, et al: EGFR mutations in non-small-cell lung cancer:
Analysis of a large series of cases and development of a rapid and
sensitive method for diagnostic screening with potential
implications on pharmacologic treatment. J Clin Oncol. 23:857–865.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sholl LM, Yeap BY, Iafrate AJ,
Holmes-Tisch AJ, Chou YP, Wu MT, Goan YG, Su L, Benedettini E, Yu
J, et al: Lung adenocarcinoma with EGFR amplification has distinct
clinicopathologic and molecular features in never-smokers. Cancer
Res. 69:8341–8348. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dacic S, Flanagan M, Cieply K, Ramalingam
S, Luketich J, Belani C and Yousem SA: Significance of EGFR protein
expression and gene amplification in non-small cell lung carcinoma.
Am J Clin Pathol. 125:860–865. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sainsbury JR, Farndon JR, Needham GK,
Malcolm AJ and Harris AL: Epidermal-growth-factor receptor status
as predictor of early recurrence of and death from breast cancer.
Lancet. 1:1398–1402. 1987.PubMed/NCBI
|
|
28
|
Bhargava R, Gerald WL, Li AR, Pan Q, Lal
P, Ladanyi M and Chen B: EGFR gene amplification in breast cancer:
Correlation with epidermal growth factor receptor mRNA and protein
expression and HER-2 status and absence of EGFR-activating
mutations. Mod Pathol. 18:1027–1033. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kallioniemi OP, Kallioniemi A, Kurisu W,
Thor A, Chen LC, Smith HS, Waldman FM, Pinkel D and Gray JW: ERBB2
amplification in breast cancer analyzed by fluorescence in situ
hybridization. Proc Natl Acad Sci USA. 89:5321–5325. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dybdal N, Leiberman G, Anderson S, McCune
B, Bajamonde A, Cohen RL, Mass RD, Sanders C and Press MF:
Determination of HER2 gene amplification by fluorescence in situ
hybridization and concordance with the clinical trials
immunohistochemical assay in women with metastatic breast cancer
evaluated for treatment with trastuzumab. Breast Cancer Res Treat.
93:3–11. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhao J, Wu R, Au A, Marquez A, Yu Y and
Shi Z: Determination of HER2 gene amplification by chromogenic in
situ hybridization (CISH) in archival breast carcinoma. Mod Pathol.
15:657–665. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jenndahl LE, Isakson P and Baeckström D:
c-erbB2-induced epithelial-mesenchymal transition in mammary
epithelial cells is suppressed by cell-cell contact and initiated
prior to E-cadherin downregulation. Int J Oncol. 27:439–448.
2005.PubMed/NCBI
|
|
33
|
Lu J, Guo H, Treekitkarnmongkol W, Li P,
Zhang J, Shi B, Ling C, Zhou X, Chen T, Chiao PJ, et al: 14-3-3zeta
cooperates with ErbB2 to promote ductal carcinoma in situ
progression to invasive breast cancer by inducing
epithelial-mesenchymal transition. Cancer Cell. 16:195–207. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ai M, Liang K, Lu Y, Qiu S and Fan Z:
Brk/PTK6 cooperates with HER2 and Src in regulating breast cancer
cell survival and epithelial-to-mesenchymal transition. Cancer Biol
Ther. 14:237–245. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Clark PA, Iida M, Treisman DM, Kalluri H,
Ezhilan S, Zorniak M, Wheeler DL and Kuo JS: Activation of multiple
ERBB family receptors mediates glioblastoma cancer stem-like cell
resistance to EGFR-targeted inhibition. Neoplasia. 14:420–428.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Abhold EL, Kiang A, Rahimy E, Kuo SZ,
Wang-Rodriguez J, Lopez JP, Blair KJ, Yu MA, Haas M, Brumund KT, et
al: EGFR kinase promotes acquisition of stem cell-like properties:
A potential therapeutic target in head and neck squamous cell
carcinoma stem cells. PLoS One. 7:e324592012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li J, Gong L-Y, Song L-B, Jiang L-L, Liu
L-P, Wu J, Yuan J, Cai J-C, He M, Wang L, et al: Oncoprotein Bmi-1
renders apoptotic resistance to glioma cells through activation of
the IKK-nuclear factor-κB pathway. Am J Pathol. 176:699–709. 2010.
View Article : Google Scholar :
|
|
38
|
Chen T, Xu C, Chen J, Ding C, Xu Z, Li C
and Zhao J: MicroRNA-203 inhibits cellular proliferation and
invasion by targeting Bmi1 in non-small cell lung cancer. Oncol
Lett. 9:2639–2646. 2015.PubMed/NCBI
|
|
39
|
Saeed AI, Sharov V, White J, Li J, Liang
W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, et
al: TM4: A free, open-source system for microarray data management
and analysis. Biotechniques. 34:374–378. 2003.PubMed/NCBI
|
|
40
|
Guo W, Ren D, Chen X, Tu X, Huang S, Wang
M, Song L, Zou X and Peng X: HEF1 promotes epithelial mesenchymal
transition and bone invasion in prostate cancer under the
regulation of microRNA-145. J Cell Biochem. 114:1606–1615. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
42
|
Guan H, Song L, Cai J, Huang Y, Wu J, Yuan
J, Li J and Li M: Sphingosine kinase 1 regulates the Akt/FOXO3a/Bim
pathway and contributes to apoptosis resistance in glioma cells.
PLoS One. 6:e199462011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nalla AK, Estes N, Patel J and Rao JS:
N-cadherin mediates angiogenesis by regulating monocyte
chemoattractant protein-1 expression via PI3K/Akt signaling in
prostate cancer cells. Exp Cell Res. 317:2512–2521. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Rosenberg EE, Prudnikova TY, Zabarovsky
ER, Kashuba VI and Grigorieva EV: D-glucuronyl C5-epimerase cell
type specifically affects angiogenesis pathway in different
prostate cancer cells. Tumour Biol. 35:3237–3245. 2014. View Article : Google Scholar
|
|
45
|
Allegra A, Alonci A, Penna G, Innao V,
Gerace D, Rotondo F and Musolino C: The cancer stem cell
hypothesis: A guide to potential molecular targets. Cancer Invest.
32:470–495. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pfeiffer MJ and Schalken JA: Stem cell
characteristics in prostate cancer cell lines. Eur Urol.
57:246–254. 2010. View Article : Google Scholar
|
|
47
|
Hazan RB, Kang L, Whooley BP and Borgen
PI: N-cadherin promotes adhesion between invasive breast cancer
cells and the stroma. Cell Adhes Commun. 4:399–411. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hazan RB, Phillips GR, Qiao RF, Norton L
and Aaronson SA: Exogenous expression of N-cadherin in breast
cancer cells induces cell migration, invasion, and metastasis. J
Cell Biol. 148:779–790. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jaggi M, Nazemi T, Abrahams NA, Baker JJ,
Galich A, Smith LM and Balaji KC: N-cadherin switching occurs in
high Gleason grade prostate cancer. Prostate. 66:193–199. 2006.
View Article : Google Scholar
|
|
50
|
Caramel J, Papadogeorgakis E, Hill L,
Browne GJ, Richard G, Wierinckx A, Saldanha G, Osborne J,
Hutchinson P, Tse G, et al: A switch in the expression of embryonic
EMT-inducers drives the development of malignant melanoma. Cancer
Cell. 24:466–480. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Siegel R, DeSantis C, Virgo K, Stein K,
Mariotto A, Smith T, Cooper D, Gansler T, Lerro C, Fedewa S, et al:
Cancer treatment and survivorship statistics, 2012. CA Cancer J
Clin. 62:220–241. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Pagliarulo V, Bracarda S, Eisenberger MA,
Mottet N, Schröder FH, Sternberg CN and Studer UE: Contemporary
role of androgen deprivation therapy for prostate cancer. Eur Urol.
61:11–25. 2012. View Article : Google Scholar :
|
|
53
|
Brabletz T: EMT and MET in metastasis:
Where are the cancer stem cells? Cancer Cell. 22:699–701. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Bracken CP, Gregory PA, Kolesnikoff N,
Bert AG, Wang J, Shannon MF and Goodall GJ: A double-negative
feedback loop between ZEB1-SIP1 and the microRNA-200 family
regulates epithelial-mesenchymal transition. Cancer Res.
68:7846–7854. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Korpal M, Lee ES, Hu G and Kang Y: The
miR-200 family inhibits epithelial-mesenchymal transition and
cancer cell migration by direct targeting of E-cadherin
transcriptional repressors ZEB1 and ZEB2. J Biol Chem.
283:14910–14914. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Moes M, Le Béchec A, Crespo I, Laurini C,
Halavatyi A, Vetter G, Del Sol A and Friederich E: A novel network
integrating a miRNA-203/SNAI1 feedback loop which regulates
epithelial to mesenchymal transition. PLoS One. 7:e354402012.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Nicoloso MS, Spizzo R, Shimizu M, Rossi S
and Calin GA: MicroRNAs - the micro steering wheel of tumour
metastases. Nat Rev Cancer. 9:293–302. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Siemens H, Jackstadt R, Hünten S, Kaller
M, Menssen A, Götz U and Hermeking H: miR-34 and SNAIL form a
double-negative feedback loop to regulate epithelial-mesenchymal
transitions. Cell Cycle. 10:4256–4271. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ren D, Wang M, Guo W, Huang S, Wang Z,
Zhao X, Du H, Song L and Peng X: Double-negative feedback loop
between ZEB2 and miR-145 regulates epithelial-mesenchymal
transition and stem cell properties in prostate cancer cells. Cell
Tissue Res. 358:763–778. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Huang S, Guo W, Tang Y, Ren D, Zou X and
Peng X: miR-143 and miR-145 inhibit stem cell characteristics of
PC-3 prostate cancer cells. Oncol Rep. 28:1831–1837.
2012.PubMed/NCBI
|
|
61
|
Gao P, Xing AY, Zhou GY, Zhang TG, Zhang
JP, Gao C, Li H and Shi DB: The molecular mechanism of microRNA-145
to suppress invasion-metastasis cascade in gastric cancer.
Oncogene. 32:491–501. 2013. View Article : Google Scholar
|