Introduction
Inflammation is involved with diverse pathological
processes, including infection, diabetes, atherosclerosis,
neurodegenerative disease, and cancer (1). In particular, inflammation plays a
critical role in either inhibiting or promoting tumor progression,
depending on the context, such as the stage and origin of the
cancer. Inflammatory cells were initially thought to actively
target tumor cells (2); however,
recent research suggests that cancer could be provoked by the
signaling crosstalk that occurs between inflammatory cells and
tumor cells (3,4). For example, tumor-associated
macrophages that express cathepsins B and S are known to promote
cancer growth and invasion in a pancreatic tumor model (5). Therefore, further investigation of
the connection between inflammation and cancer is important for a
better understanding of tumor pathology and developing improved
tumor-directed therapeutics.
Bacterial infection is a major cause of inflammation
and is mediated by a variety of bacterial cell compartments
(6). LPS in the outer membrane is
a principal pathogenic molecule in the case of gram-negative
bacteria, whereas plasma membrane lipoprotein plays a similar role
in mycoplasma, which lacks a cell wall and outer membrane (7). Both LPS and lipoprotein contain lipid
moieties, lipid A and a lipoylated amino-terminal cysteinyl
residue, respectively, that are responsible for stimulating the
host immune response (7,8). Ag 243-5 lipoprotein, originally
isolated from Mycoplasma arginini, but also described as the
P47 lipoprotein of M. hyorhinis, is reported to have a
metastasis-promoting activity (9,10).
Interestingly, Ag 243-5 shows significant sequence homology with
macrophage-activating lipoprotein-404 (MALP-404) from M.
fermentans, which is known to increase cytokine production in
human monocytes (11).
Several LPS binding molecules facilitate the
biological effect of LPS on host cells. For instance, LPS-binding
protein (LBP) binds aggregated LPS and then delivers monomeric LPS
to CD14 (12). Membrane anchored
protein CD14 functions as a critical toll-like receptor 4 (TLR4)
co-receptor, and transfers LPS to the myeloid differentiation
factor 2 (MD2)-TLR4 complex (13).
Finally, LPS triggers the dimerization of TLR4, which subsequently
initiates intracellular signaling cascades (14,15).
Besides, several other proteins are reported to bind LPS, including
high mobility group box 1 protein (HMGB1) (16), CXCR4 (17), and β2-glycoprotein I
(18). These proteins are actively
studied as inflammatory regulators and therapeutic candidates in
inflammatory diseases, but the current data are not sufficient to
address any meaningful applications.
Ninjurin1 was originally identified as an
upregulated protein in injured nerves and is comprised of two
transmembrane domains (aa 72–100 and aa 118–139), N-terminal (aa
1–71) and C-terminal (aa 140–152) extracellular domains, and
cytosolic region (aa 101–117) (19). Our group and others have reported
on the various roles of Ninjurin1 in macrophages, such as
increasing adhesion to endothelial cells and motility during early
ocular development and experimental autoimmune encephalomyelitis
(20,21). Moreover, it was recently revealed
that Ninjurin1 modulates the TLR4-dependent inflammatory response
triggered by LPS via p38 phosphorylation and activator protein-1
(AP-1) activation (22); however,
the precise mechanism of its roles in the inflammatory response is
enigmatic. In this study, we report that Ninjurin1 mediates
LPS-induced inflammation by directly binding to LPS. This
identification of Ninjurin1 and LPS binding likely provides an
important insight into the regulation of macrophage-mediated
inflammatory response and diseases.
Materials and methods
Cell culture
HEK293T and Raw264.7 cells were obtained from the
American Type Culture Collection (ATCC, Manassas, VA, USA) and the
Korean Cell Line Bank (KCLB, Seoul, Korea), respectively. Cells
were cultured in Dulbecco's modified Eagle's medium (DMEM,
GenDEPOT, Barker, TX, USA) supplemented with 10% fetal bovine serum
(FBS, GenDEPOT), and 100 U/ml penicillin and 100 μg/ml streptomycin
(GenDEPOT) at 37°C in a humidified 5% CO2
atmosphere.
Construction of expression plasmids and
transfection
Expression plasmids for human (NM_004148) and mouse
(NM_013610) Ninjurin1 were constructed as described previously
(21). To construct expression
plasmids for N-terminal MYC-tagged human and mouse Ninjurin1, cDNA
were amplified by PCR and subcloned into pCS2+-Myc.
Truncated forms of mouse Ninjurin1, MYC-mNINJ1 (1–71), MYC-mNINJ1
(72–152), MYC-mNINJ1 (1–100), MYC-mNINJ1 (1–90), MYC-mNINJ1 (1–80),
MYC-mNINJ1 (81–152), MYC-mNINJ1 (91–152), MYC-mNINJ1 (101–152), and
MYC-mNINJ1 (71–110) plasmids were also constructed using
pCS2+-Myc as backbone. Designing and cloning of
expression plasmid of non-tagged Ninjurin1 was described previously
(23). Briefly, mouse Ninjurin1
cDNA was subcloned into pcDNA3.1+ myc/his backbone
without removing a stop codon.
HEK293T cells at 3×106 cells/dish were
cultured in 100-mm culture dishes for 24 h and then expression
plasmids were transfected using polyethylenimine (PEI,
Sigma-Aldrich, St. Louis, MO, USA). After 48 h of culture, cells
were washed with PBS and collected.
RNA interference
Ninjurin1 downregulation was performed with RNA
interference. siNinj1 targeted to mouse Ninjurin1 was purchased
from Life Technologies (Grand Island, NY, USA). Negative control of
RNA interference, siControl with the scrambled sequence was
purchased from Bioneer (Daejeon, Korea). The following sequences
were used: siControl: 5′-CCTACGCCACCAAUUUCGUdTdT-3′; siNinj1:
5′-ACCGGCCCAUCAAUGUAAACCAUUA-3′. Raw264.7 cells at 2×105
cells/dish were cultured in 60-mm culture dishes for 12 h. siRNAs
(20 nM) were transfected using Lipofectamine RNAiMAX transfection
reagent (Life Technologies). After 24 h of transfection, media were
changed with presence or absence of 1 μg/ml LPS (Sigma-Aldrich).
After another 24 h, cultured supernatant and cells were
collected.
Immunoblot analysis
Proteins were extracted in cell lysis buffer
containing 50 mM Tris-Cl (pH 7.4), 300 mM NaCl, 5 mM EDTA, 0.02%
(w/v) sodium azide, 1% (w/v) Triton X-100, 10 mM iodoacetamide, 1
mM phenylmethane-sulfonyl fluoride, 2 μg/ml leupeptin, and protease
inhibitor cocktail (Calbiochem, Billerica, MA, USA). Lysates were
separated with SDS-PAGE and transferred to nitrocellulose membrane
(GE Healthcare Life Sciences, Pittsburgh, PA, USA). The transferred
membrane was probed with the specific antibodies. Antibodies to
NOS2 purchased from BD Bioscience (San Diego, CA, USA), MYC and
GAPDH from Santa Cruz Biotechnology (Dallas, TX, USA). Endogenous
Ninjurin1 was detected by custom-made antibody that was raised in
rabbit with aa 139–152 of mouse Ninjurin1 (Ninj1
Ab139–152) (23). Image
was acquired using LAS3000 machine (GE Healthcare Life
Sciences).
Immunoprecipitation and silver
staining
Protein lysates (1,000 μg) and 1 μg of MYC antibody
were incubated overnight at 4°C with gentle rotation. Protein A
agarose (10 μl) (EMD Millipore, Billerica, MA, USA) was added to
each sample and the mixture was incubated at 4°C for 4 h with
gentle rotation. After washing 5 times with washing buffer
containing 50 mM Tris-Cl (pH 7.4), 300 mM NaCl, 5 mM EDTA, 0.02%
(w/v) sodium azide, and 0.1% (w/v) Triton X-100, precipitated
proteins were eluted by heating with SDS sample buffer at 95°C for
10 min. Eluted sample was separated with SDS-PAGE followed by
silver staining procedure using PlusOne Silver Staining kit (GE
Healthcare Life Sciences) as recommended by the manufacturer.
Binding assay of Ninjurin1 with
MALP-2
Macrophage-activating lipopeptide-2 (2 μg) (MALP-2,
Enzo Life Sciences, Farmingdale, NY, USA) was conjugated to
NHS-activated agarose beads (Life Technologies) as recommended by
the manufacturer. Protein lysates (500 μg) and MALP-2 conjugated
beads were incubated overnight at 4°C with gentle rotation. After
washing 5 times with washing buffer, binding proteins were eluted
by heating with SDS sample buffer at 95°C for 10 min. Eluted
samples were further analyzed by immunoblot analysis.
Binding assay of Ninjurin1 with LPS
Binding assays were performed by pull-down with
streptavidin sepharose beads (GE Healthcare Life Science) or
immunoprecipitation with MYC antibody. For pull-down with
streptavidin sepharose beads, 500 μg of protein lysates and 4 μg of
LPS-biotin (InvivoGen, San Diego, CA, USA) were incubated overnight
at 4°C with gentle rotation. Streptavidin sepharose beads (10 μl)
were added to each sample and incubated at 4°C for 2 h with gentle
rotation. After washing 5 times with washing buffer, the bound
proteins were eluted by heating with SDS sample buffer at 95°C for
10 min. The eluted samples were further analyzed by immunoblot
analysis. In case of immunoprecipitation with MYC antibody, 1,000
μg of protein lysates, 1 μg of MYC antibody, and 10 μl of protein A
agarose were incubated overnight at 4°C with gentle rotation, after
which the unbound molecules were removed using 5 washes with
washing buffer. Washed protein A agarose was eluted for silver
staining or incubated with 10 μg of LPS-biotin at 4°C for 4 h.
After washing 5 times with washing buffer, the bound molecules were
eluted. Eluted samples were detected using streptavidin-HRP (Thermo
Fisher Scientific, Waltham, MA, USA).
Mass spectrometry
Candidate protein bands excised from silver stained
gels were destained with destaining solution consisted of 30 mM
potassium ferricyanide and 100 mM sodium thiosulfate for 5 min and
then incubated with 200 mM ammonium bicarbonate for 20 min. The
gels were dehydrated with acetonitrile and dried in a vacuum
centrifuge. The dried gels were rehydrated with 50 mM ammonium
bicarbonate containing 200 ng trypsin (Promega, Madison, WI, USA)
for 45 min, replaced solution to 50 mM ammonium bicarbonate, and
incubated overnight at 37°C. Digested peptide was purified using a
desalting column (GE loader tip, Eppendorf, Hamburg, Germany). Each
peptide sample was applied to ESI-Q-TOF MS/MS spectrometer
(ABSciex, Framingham, MA, USA). The deduced peptide sequence after
MS/MS were analyzed by MASCOT search engine (http://www.matrixscience.com) against Swiss-Prot and
NCBI databases.
Nitric oxide (NO) assay
Raw264.7 cells were plated into 60-mm culture dishes
at 2×105 cells/dish for 12 h and then transfected with
Ninj1 or negative control siRNA. After 24 h of culture,
media were changed with or without LPS (1 μg/ml). After incubation
for 24 h, the culture supernatant was collected and cells were
removed by centrifugation at 500 g for 3 min. Culture supernatant
(100 μl) was mixed with 100 μl of Griess reagent (1:1 mixture of 1%
sulfanilamide in 30% acetate and 0.1% N-1-naphthylethylenediamine
dihydrochloride in 60% acetate) at room temperature for 10 min. The
absorbance of the incubated samples was measured by using
microplate reader at 540 nm. A standard curve drawn with known
concentrations of sodium nitrite was applied to calculate the
concentration of nitrite, the stable end product of NO.
Measurement of TNFα secretion
Raw264.7 cells were cultured in 96-well culture
plates at 1×104 cells/well for 12 h. The Raw264.7 cells
were transfected with Ninj1 or negative control siRNA for 24
h and then media was removed and replaced with 200 μl of fresh
media with or without LPS (1μg/ml). After 24 h of incubation, the
culture supernatant was collected. Amount of secreted TNFα was
measured using Mouse TNFα ELISA MAX kit (BioLegend, San Diego, CA,
USA) following the manufacturer's instructions.
Statistical analysis
The data are expressed as the mean ± SD. Differences
between groups were analyzed by the unpaired two-tailed Student's
t-test. P<0.05 denoted the presence of statistically
significance.
Results
Ag 243-5 protein binds human and mouse
Ninjurin1
At the start of this study, immunoprecipitation, gel
separation, and mass spectrometry analysis were performed to
identify novel Ninjurin1 binding partners. For this, HEK293T cells
were transfected with MYC-tagged human Ninjurin1 (MYC-hNINJ1) or
empty control (mock) plasmid. Total lysates from transfected
HEK293T cells were immunoprecipitated with MYC antibody, separated
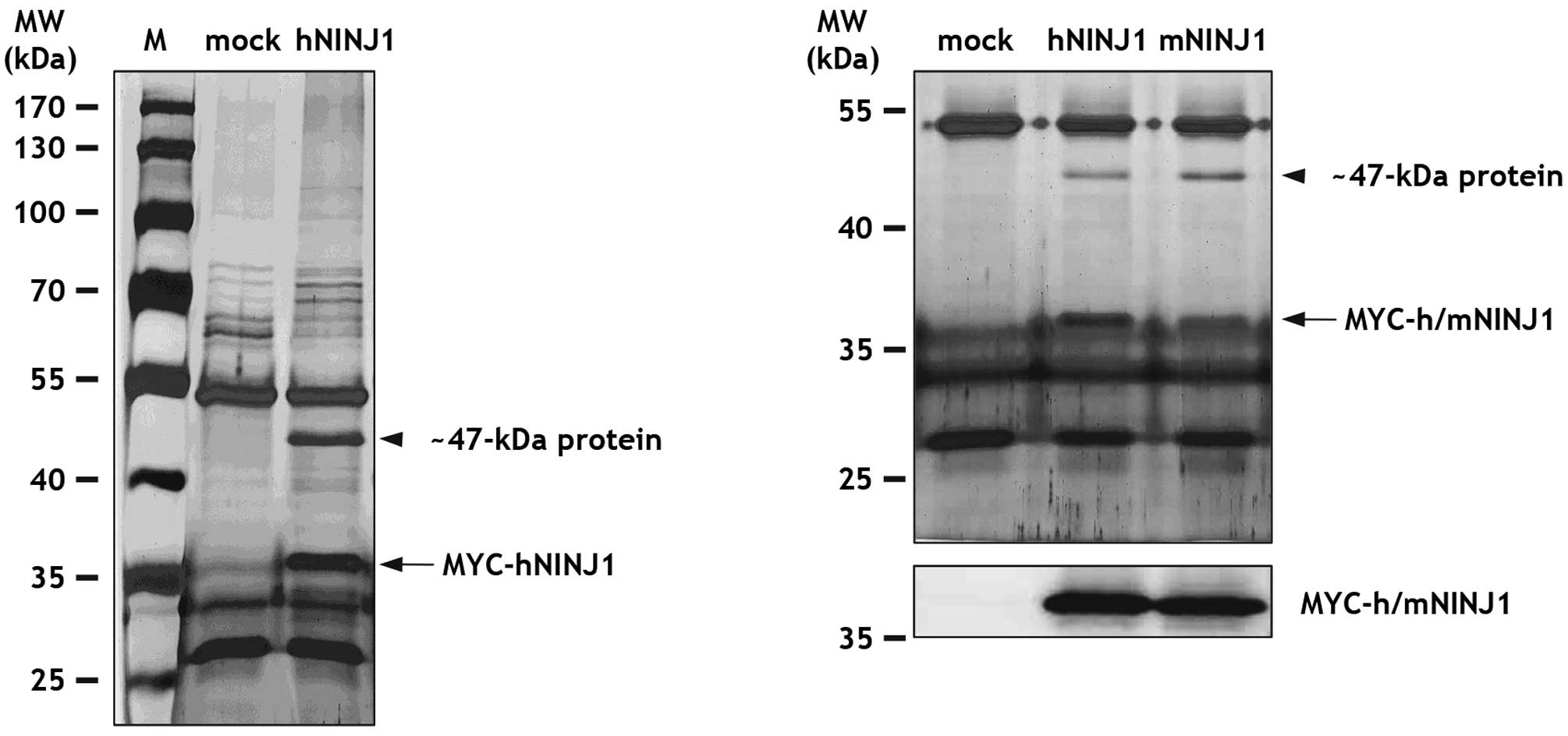
by SDS-PAGE, and then stained with silver nitrate. Notably, we
found a ~47-kDa protein that co-immunoprecipitated with ~36 kDa
MYC-hNINJ1 protein, but was not observed in the control sample
(Fig. 1). To examine whether this
~47-kDa protein bound mouse Ninjurin1, immunoprecipitations were
repeated with lysates from HEK293T cells transfected with
MYC-tagged mouse Ninjurin1 (MYC-mNINJ1). Significantly, both hNINJ1
and mNINJ1 were capable of pulling-down the ~47-kDa protein
(Fig. 1). As a control,
MYC-h/mNINJ1 expression was confirmed by immunoblot analysis with
MYC antibody (Fig. 1).
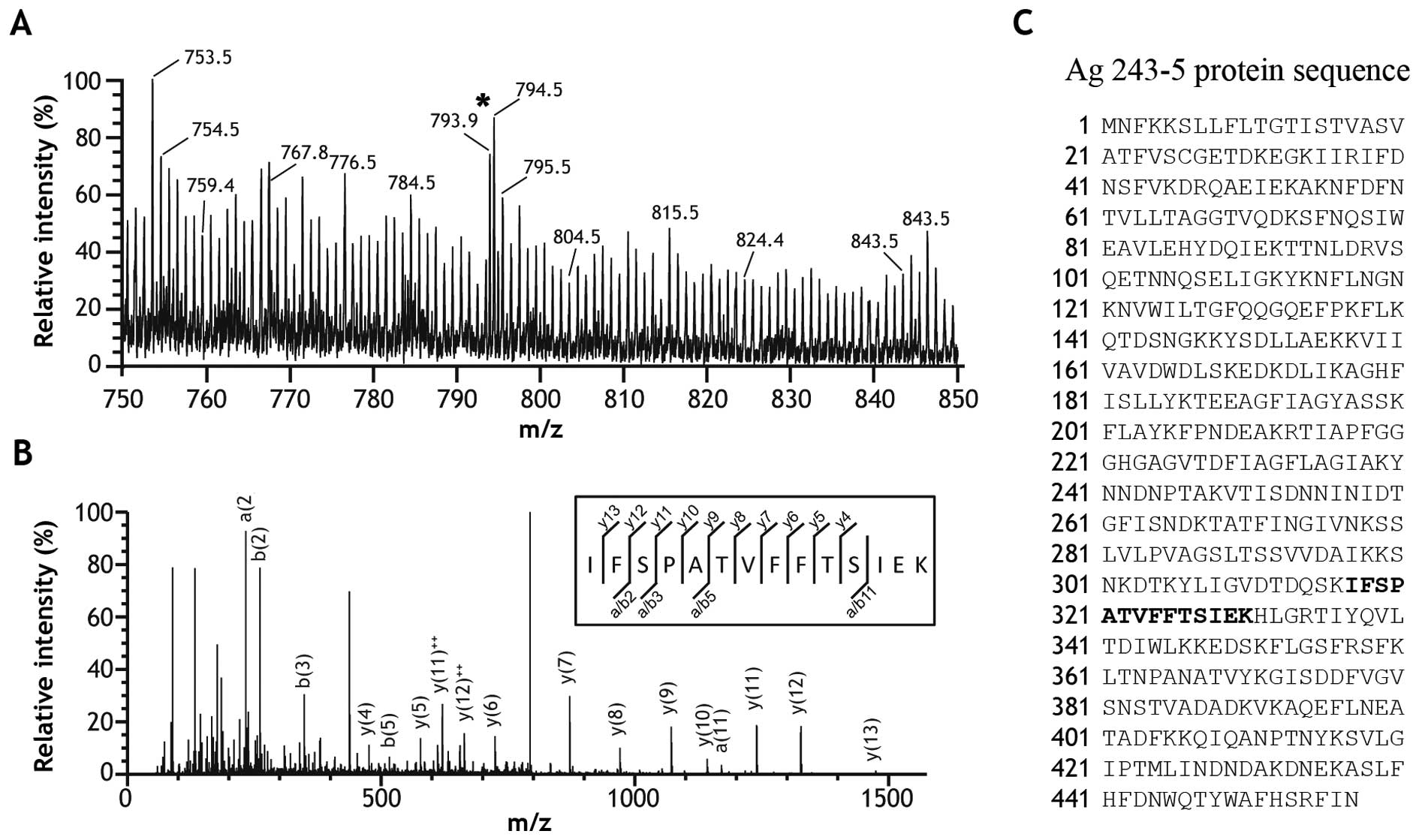
The ~47-kDa band was excised and analyzed by mass
spectrometry in order to identify the protein of interest. The
resulting ESI-MS spectrum presented with m/z peaks ranging from 750
to 850 (Fig. 2A). The peak at
793.9 m/z was sequenced and identified as a 14-amino acid peptide
(IFSPATVFFTSIEK) in further MS/MS analysis (Fig. 2B). Unexpectedly, queries in the
Swiss-Prot and NCBI databases using the MASCOT search engine
revealed that the peptide sequence was identical to aa 317–330 of
Ag 243-5 (BAA04082) (Fig. 2C). To
determine the reason for mycoplasma protein existence in our
HEK293T cell lysates, we tested our cultures for mycoplasma and
found that the HEK293T cells used in the analysis were positive for
contamination. Subsequent analyses revealed that non-contaminated
HEK293T cell lysates did not contain this ~47-kDa protein band
(data not shown). Although unexpected, these observations were
intriguing since several previous reports demonstrated a role for
Ninjurin1 in the inflammatory response, but the precise mechanism
was unknown. Thus, we hypothesized that Ninjurin1 could recognize
microbial pathogens conjugated with lipid moieties, such as
mycoplasma lipopeptide MALP-2, the MALP-404 N-terminus, and
LPS.
MALP-2 and LPS bind human and mouse
Ninjurin1
Both MALP-2 and LPS are bacterial endotoxins that
induce inflammatory macrophage activation in a manner dependent on
their lipid moieties. Therefore, we tested whether MALP-2 and LPS
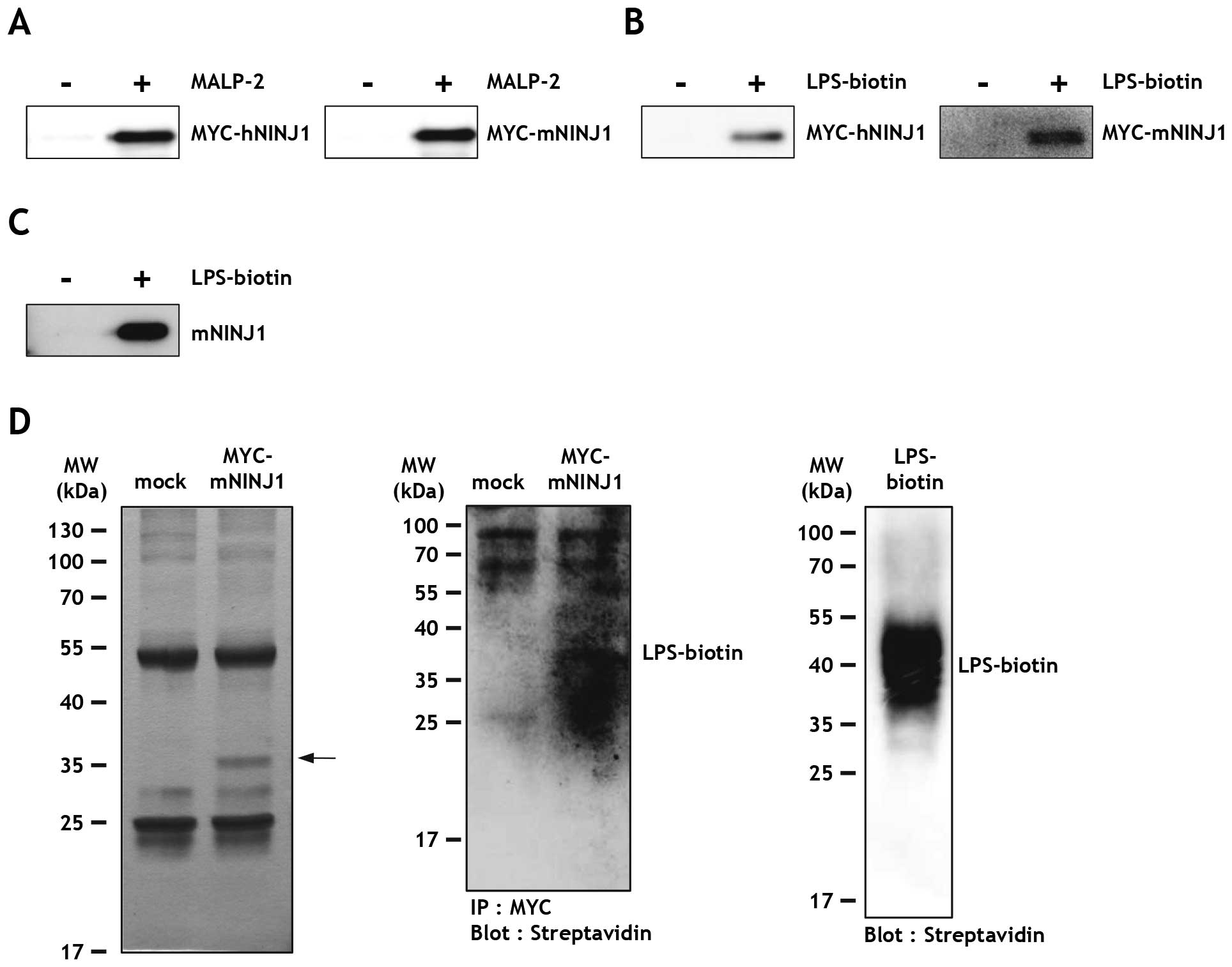
were also able to bind Ninjurin1 as observed with Ag 243-5. For
this, MYC-h/mNINJ1 HEK293T cell lysates were incubated with
MALP-2-conjugated beads, and the bound proteins were eluted and
examined by immunoblot analysis. The same process was used for
control samples with unconjugated beads. Results showed that human
and mouse Ninjurin1 was efficiently pulled down with MALP-2 beads
(Fig. 3A), but not control
samples. In the case of LPS-biotin, Ninjurin1-expressing cell
lysates were incubated with or without LPS-biotin and streptavidin
sepharose beads. Similar to that observed with MALP-2, human and
mouse Ninjurin1 were both able to bind LPS-biotin (Fig. 3B). To rule out the possibility of
an interaction between the MYC peptide tag and LPS, LPS binding was
assessed using lysates from HEK293T cells transfected with
non-tagged mouse Ninjurin1 (mNINJ1) plasmid. As expected,
non-tagged mouse Ninjurin1 also bound LPS-biotin (Fig. 3C). To exclude indirect binding
through potential adaptor proteins, the unbound proteins were
washed away from the immunoprecipitated MYC-mNINJ1 cell lysates,
followed by addition of LPS-biotin. MYC-mNINJ1 protein was detected
in the silver nitrate stained gel (Fig. 3D, arrow) and MYC-mNINJ1-bound
LPS-biotin was detected by streptavidin-HRP, thus confirming a
direct interaction between Ninjurin1 and LPS (Fig. 3D, right).
The aa 81–100 region of Ninjurin1 is
responsible for LPS binding
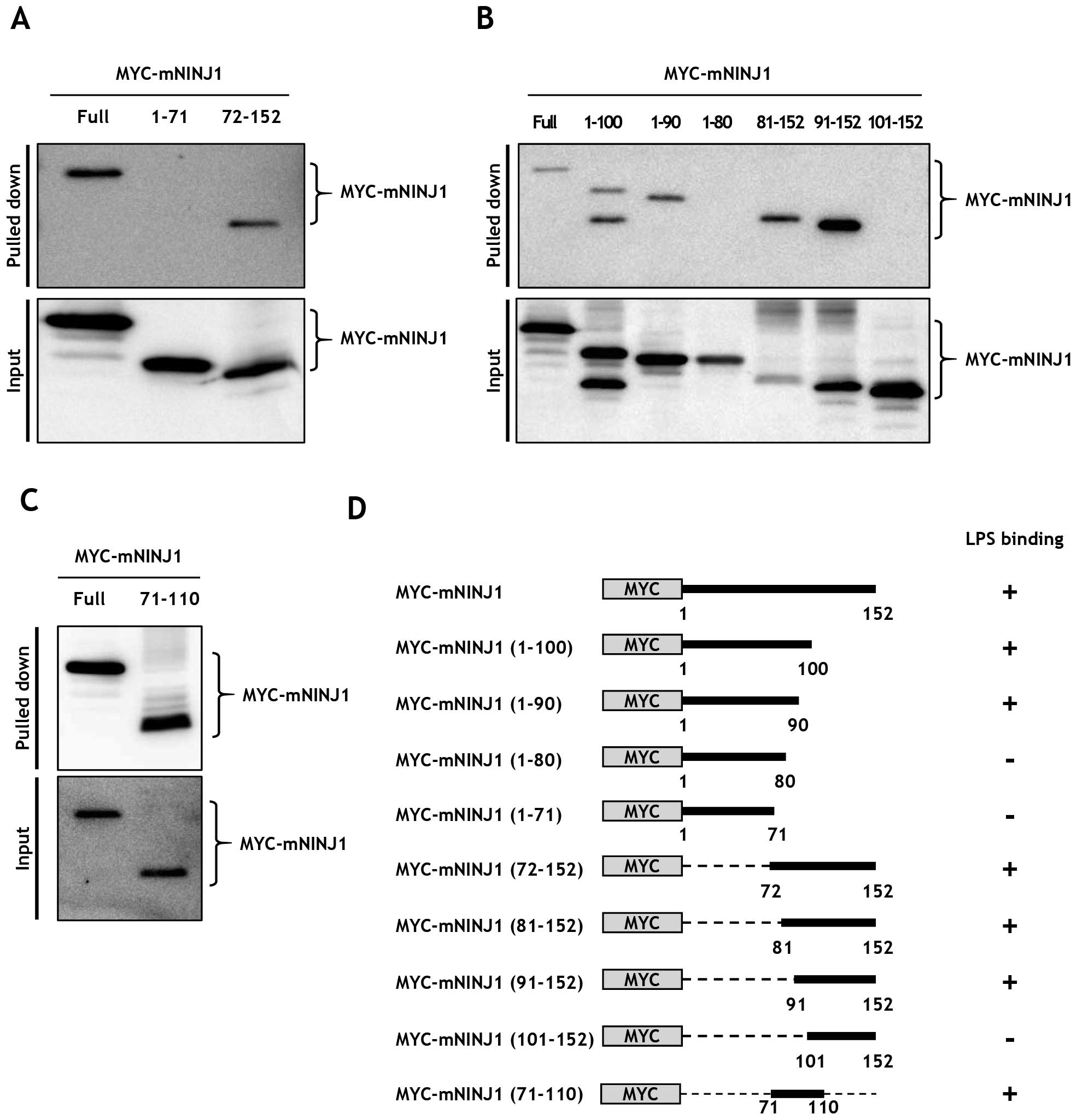
To specify the region of Ninjurin1 responsible for
LPS binding, truncated mouse Ninjurin1 expression plasmids were
constructed. MYC-mNINJ1 (1–71) and MYC-mNINJ1 (72–152), encoding
the extracellular N-terminal region and the two transmembrane,
cytosolic, and extracellular C-terminus domains, respectively, were
cloned into pCS2+-Myc backbone plasmid. Full length
MYC-mNINJ1, MYC-mNINJ1 (1–71), and MYC-mNINJ1 (72–152) were
transfected to HEK293T cells and expression was confirmed by
immunoblot analysis (Fig. 4A,
lower). LPS binding assays were then performed using equal amounts
of cell lysates and LPS-biotin (Fig.
4A, upper). Results showed that full-length MYC-mNINJ1 and
MYC-mNINJ1 (72–152) bound LPS, whereas MYC-mNINJ1 (1–71) did not.
To further delineate the binding region within Ninjurin1,
additional expression plasmids of the truncated N- and C-terminals
of mouse Ninjurin1 were constructed as follows: MYC-mNINJ1 (1–100),
MYC-mNINJ1 (1–90), MYC-mNINJ1 (1–80), MYC-mNINJ1 (81–152),
MYC-mNINJ1 (91–152), MYC-mNINJ1 (101–152), and MYC mNINJ1 (71–100).
Ninjurin1 mutant expression was then examined by immunoblot
analysis (Fig. 4B and 4C, lower).
Notably, binding assays demonstrated that MYC-mNINJ1 (1–100),
MYC-mNINJ1 (1–90), MYC-mNINJ1 (81–152), MYC-mNINJ1 (91–152), and
MYC-mNINJ1 (71–110) bound LPS, whereas MYC-mNINJ1 (1–80) and
MYC-mNINJ1 (101–152) did not (Fig. 4B
and C, upper). The binding abilities of these recombinant
Ninjurin1 mutant proteins are summarized in Fig. 4D, and indicate that the aa 72–152
region of Ninjurin1 is required for LPS binding. Probably an aa
81–100 region of Ninjurin1 is essential for LPS binding, but it
requires further experiments to define the essential region.
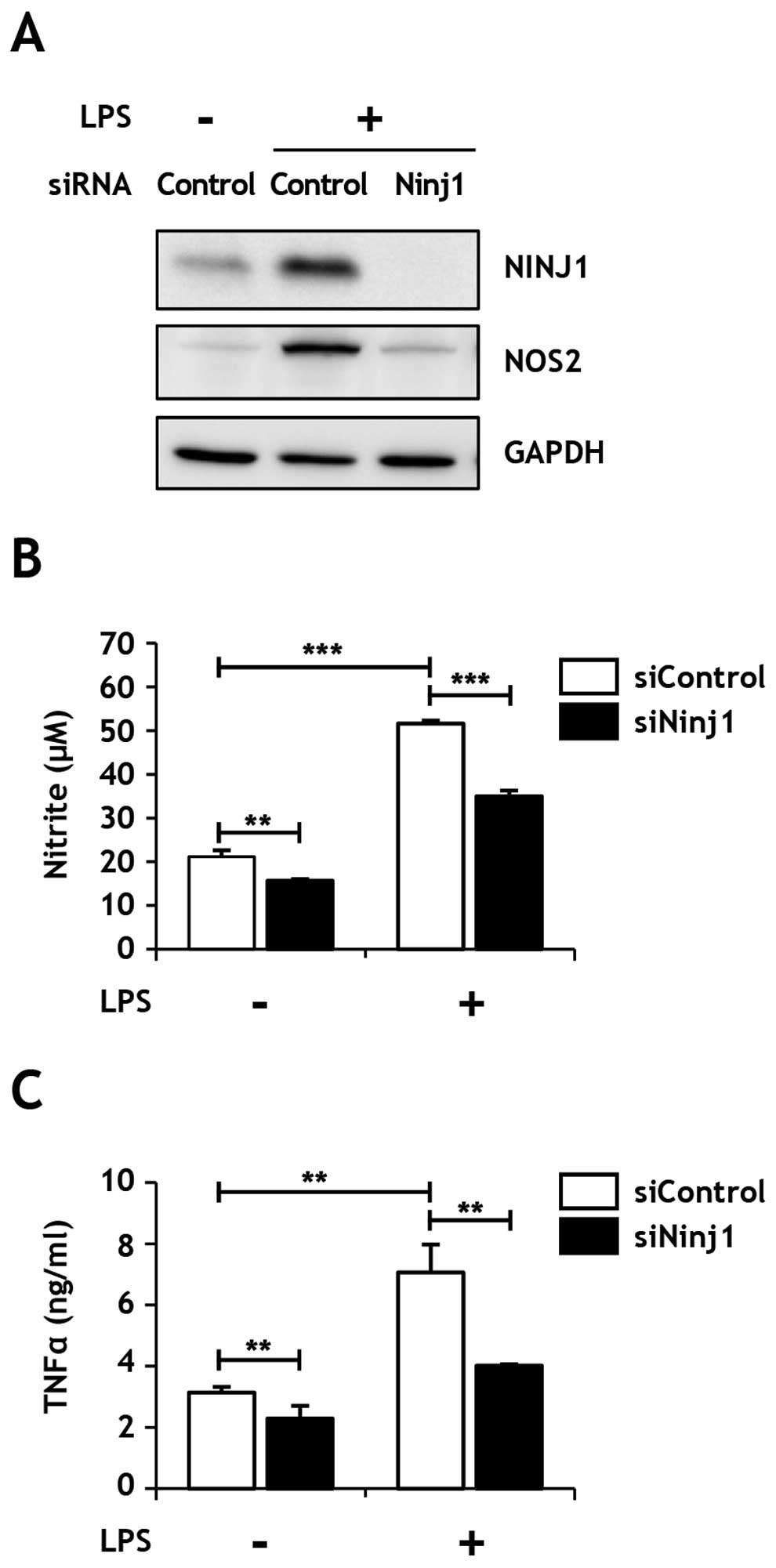
Ninjurin1 downregulation inhibits
LPS-induced NO and TNFα secretion in Raw264.7 macrophages
Next, we investigated the physiological basis of
Ninjurin1 and LPS binding in the macrophage-mediated inflammatory
response. Notably, LPS induced an increase of Ninjurin1 expression
in Raw264.7 macrophages, consistent with a previous report
(22). To elucidate the role of
Ninjurin1 in the LPS-mediated inflammatory response, we silenced
Ninjurin1 expression with specific or negative control siRNAs
(siNinj1 and siControl, respectively) in Raw264.7 cells, and
subsequently, analyzed the production of two well-known macrophage
activation markers, NOS2 and TNFα. Interestingly, the induction of
NOS2 protein expression by LPS treatment was inhibited in cells
transfected siNinj1 (Fig. 5A), as
was NO release (Fig. 5B).
Similarly, TNFα secretion induced by LPS treatment was markedly
inhibited in Ninjurin1-knockdown Raw264.7 cells (Fig. 5C). These results suggest that the
LPS-induced inflammatory response was significantly inhibited by
Ninjurin1-knockdown, likely due to the decreased direct interaction
of Ninjurin1 and LPS.
Discussion
In this study, we identified Ninjurin1 as a novel
LPS binding partner (Fig. 3). To
determine the region of Ninjurin1 that conveyed its ability to bind
LPS, binding assays were performed with Ninjurin1 mutant proteins.
These results showed that LPS bound to Ninjurin1 aa 81–100, which
belongs to first transmembrane domain (Fig. 4). Ninjurin1 has two transmembrane
domains (aa 72–100 and 118–139), and both the regions are highly
hydrophobic. In the binding assay using MYC-mNINJ1 (101–152),
containing second transmembrane domain, LPS-biotin failed to bind
to Ninjurin1 (Fig. 4B). Based on
this observation, we proposed that LPS binds specifically to aa
81–100 of Ninjurin1. To address whether Ninjurin1-LPS binding
affects cellular function, we repressed Ninjurin1 expression with
siRNA in Raw264.7 cells. Notably, Ninjurin1 downregulation
inhibited LPS-induced NOS2 enzyme induction, NO release, and TNFα
secretion in Raw264.7 macrophages (Fig. 5), suggesting that the direct
binding of LPS to Ninjurin1 was required for the inflammatory
activation of macrophages by LPS.
The binding properties of Ninjurin1 were previously
investigated mainly focussing on its homophilic binding domain
(20,24). In addition, a heterophilic
interaction with unknown molecules has also been suggested based on
results that the basal adhesion of wild-type Jurkat cells was
inhibited by treatment with peptides containing Ninjurin1 adhesion
motif (25). Moreover, Ninjurin1
overexpression led to enhanced macrophage adhesion to umbilical
vein endothelial cells and extracellular matrix proteins, such as
fibronectin, type I collagen, vitronectin, and type IV collagen
(21,26). In this study, we identified LPS as
a novel heterophilic binding partner of Ninjurin1. Since the lipid
moiety of LPS is crucial for its binding with Ninjurin1, it would
be worthwhile to investigate the interaction between Ninjurin1 and
other lipid-containing molecules.
Binding region of LPS-interacting partners would
likely be a potent therapeutic target for inflammatory diseases.
For example, synthetic peptides of the HMGB1 LPS-binding region, aa
3–15 or aa 80–96, inhibits the interaction between LPS to HMGB1
in vitro, and also decreases TNFα production in a
subclinical endotoxemia mouse model (27). Thus, we also sought to determine
the region of Ninjurin1 responsible for LPS binding. It is already
known that Ninjurin1 contains a homophilic binding domain (aa
26–37) important for its role in immune cell aggregation and
macrophage-endothelial cell adhesion. Moreover, the aggregation of
Ninjurin1-expressing Jurkat cells is completely abolished by
treatment with Ninjurin1 aa 26–37 peptide (25), and treatment with antibody directed
towards this protein fragment blocks macrophage adhesion and
transmigration to endothelial cells (20). Furthermore, LPS-induced IL-6
and TNFα transcription is inhibited by treatment with this
aa 26–37 peptide (22); however,
according to our result, LPS specifically bound aa 81–100 of
Ninjurin1, but not the N-terminus containing the homophilic binding
domain. This result indicates that Ninjurin1 harbors an LPS-binding
motif separated from its homophilic binding domain. Therefore,
identification of the specific LPS binding region in Ninjurin1
could be valuable for the precise regulation of LPS-induced
inflammation, as it would presumably not affect the protein's role
in cell adhesion.
Besides macrophages, Ninjurin1 is expressed in
various cell types, such as endothelial cells, pericytes,
fibroblasts, and epithelial cells (26,28,29).
Moreover, Ninjurin1 is implicated in several human diseases,
including carcinogenesis of non-muscle-invasive urothelial bladder
cancer (30) and hepatocellular
carcinoma (31). Likewise, LPS
also affects various cell types and pathological conditions, which
are not only restricted to immune cells. For example, human
endothelial cells treated with LPS produce neutrophil chemotactic
factor (32), whereas LPS
stimulation in mouse CT26 colon cancer cells triggers NF-κB-DNA
binding (33).
Collectively, the Ninjurin1-LPS interaction would
likely affect various cellular functions beyond macrophage
inflammation. Thus, the Ninjurin1 LPS-binding domain would be an
attractive therapeutic target for inflammatory diseases.
Acknowledgements
This study was supported by the Global Research
Laboratory Program (2011-0021874), the Global Core Research Center
(GCRC) Program (2011-0030001), the International Cooperation
Program (2014K2A1C2074279), the Basic Science Research Program
(2013R1A1A2058956) through the National Research Foundation (NRF)
funded by the Korean Ministry of Science, ICT and Future Planning
(MSIP).
References
|
1
|
Medzhitov R: Inflammation 2010: New
adventures of an old flame. Cell. 140:771–776. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Urban JL, Shepard HM, Rothstein JL,
Sugarman BJ and Schreiber H: Tumor necrosis factor: A potent
effector molecule for tumor cell killing by activated macrophages.
Proc Natl Acad Sci USA. 83:5233–5237. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Grivennikov SI, Greten FR and Karin M:
Immunity, inflammation, and cancer. Cell. 140:883–899. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Coussens LM and Werb Z: Inflammation and
cancer. Nature. 420:860–867. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gocheva V, Wang HW, Gadea BB, Shree T,
Hunter KE, Garfall AL, Berman T and Joyce JA: IL-4 induces
cathepsin protease activity in tumor-associated macrophages to
promote cancer growth and invasion. Genes Dev. 24:241–255. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Akira S: Pathogen recognition by innate
immunity and its signaling. Proc Jpn Acad, Ser B, Phys Biol Sci.
85:143–156. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chambaud I, Wróblewski H and Blanchard A:
Interactions between mycoplasma lipoproteins and the host immune
system. Trends Microbiol. 7:493–499. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Beutler B and Rietschel ET: Innate immune
sensing and its roots: The story of endotoxin. Nat Rev Immunol.
3:169–176. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Calcutt MJ, Kim MF, Karpas AB, Mühlradt PF
and Wise KS: Differential posttranslational processing confers
intraspecies variation of a major surface lipoprotein and a
macrophage-activating lipopeptide of Mycoplasma fermentans. Infect
Immun. 67:760–771. 1999.PubMed/NCBI
|
|
10
|
Ushio S, Iwaki K, Taniai M, Ohta T, Fukuda
S, Sugimura K and Kurimoto M: Metastasis-promoting activity of a
novel molecule, Ag 243–5, derived from mycoplasma, and the complete
nucleotide sequence. Microbiol Immunol. 39:393–400. 1995.
View Article : Google Scholar
|
|
11
|
Rosati S, Pozzi S, Robino P, Montinaro B,
Conti A, Fadda M and Pittau M: P48 major surface antigen of
Mycoplasma agalactiae is homologous to a malp product of Mycoplasma
fermentans and belongs to a selected family of bacterial
lipoproteins. Infect Immun. 67:6213–6216. 1999.PubMed/NCBI
|
|
12
|
Hailman E, Lichenstein HS, Wurfel MM,
Miller DS, Johnson DA, Kelley M, Busse LA, Zukowski MM and Wright
SD: Lipopolysaccharide (LPS)-binding protein accelerates the
binding of LPS to CD14. J Exp Med. 179:269–277. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wright SD, Ramos RA, Tobias PS, Ulevitch
RJ and Mathison JC: CD14, a receptor for complexes of
lipopolysaccharide (LPS) and LPS binding protein. Science.
249:1431–1433. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jerala R: Structural biology of the LPS
recognition. Int J Med Microbiol. 297:353–363. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Miyake K: Innate recognition of
lipopolysaccharide by Toll-like receptor 4-MD-2. Trends Microbiol.
12:186–192. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Youn JH, Oh YJ, Kim ES, Choi JE and Shin
JS: High mobility group box 1 protein binding to lipopolysaccharide
facilitates transfer of lipopolysaccharide to CD14 and enhances
lipopolysaccharide-mediated TNF-alpha production in human
monocytes. J Immunol. 180:5067–5074. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Triantafilou K, Triantafilou M and Dedrick
RL: A CD14-independent LPS receptor cluster. Nat Immunol.
2:338–345. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Agar C, de Groot PG, Mörgelin M, Monk SD,
van Os G, Levels JH, de Laat B, Urbanus RT, Herwald H, van der Poll
T, et al: β(2)-glycoprotein I: A novel component of innate
immunity. Blood. 117:6939–6947. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Araki T and Milbrandt J: Ninjurin, a novel
adhesion molecule, is induced by nerve injury and promotes axonal
growth. Neuron. 17:353–361. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ahn BJ, Le H, Shin MW, Bae SJ, Lee EJ, Wee
HJ, Cha JH, Lee HJ, Lee HS, Kim JH, et al: Ninjurin1 deficiency
attenuates susceptibility of experimental autoimmune
encephalomyelitis in mice. J Biol Chem. 289:3328–3338. 2014.
View Article : Google Scholar :
|
|
21
|
Lee HJ, Ahn BJ, Shin MW, Jeong JW, Kim JH
and Kim KW: Ninjurin1 mediates macrophage-induced programmed cell
death during early ocular development. Cell Death Differ.
16:1395–1407. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jennewein C, Sowa R, Faber AC, Dildey M,
von Knethen A, Meybohm P, Scheller B, Dröse S and Zacharowski K:
Contribution of ninjurin1 to toll-like receptor 4 signaling and
systemic inflammation. Am J Respir Cell Mol Biol. 53:656–663. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ahn BJ, Le H, Shin MW, Bae SJ, Lee EJ, Wee
HJ, Cha JH, Park JH, Lee HS, Lee HJ, et al: The N-terminal
ectodomain of Ninjurin1 liberated by MMP9 has chemotactic activity.
Biochem Biophys Res Commun. 428:438–444. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ifergan I, Kebir H, Terouz S, Alvarez JI,
Lécuyer MA, Gendron S, Bourbonnière L, Dunay IR, Bouthillier A,
Moumdjian R, et al: Role of Ninjurin-1 in the migration of myeloid
cells to central nervous system inflammatory lesions. Ann Neurol.
70:751–763. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Araki T, Zimonjic DB, Popescu NC and
Milbrandt J: Mechanism of homophilic binding mediated by ninjurin,
a novel widely expressed adhesion molecule. J Biol Chem.
272:21373–21380. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee HJ, Ahn BJ, Shin MW, Choi JH and Kim
KW: Ninjurin1: A potential adhesion molecule and its role in
inflammation and tissue remodeling. Mol Cells. 29:223–227. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Youn JH, Kwak MS, Wu J, Kim ES, Ji Y, Min
HJ, Yoo JH, Choi JE, Cho HS and Shin JS: Identification of
lipopolysaccharide-binding peptide regions within HMGB1 and their
effects on subclinical endotoxemia in a mouse model. Eur J Immunol.
41:2753–2762. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Matsuki M, Kabara M, Saito Y, Shimamura K,
Minoshima A, Nishimura M, Aonuma T, Takehara N, Hasebe N and Kawabe
J: Ninjurin1 is a novel factor to regulate angiogenesis through the
function of pericytes. Circ J. 79:1363–1371. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cho SJ, Rossi A, Jung YS, Yan W, Liu G,
Zhang J, Zhang M and Chen X: Ninjurin1, a target of p53, regulates
p53 expression and p53-dependent cell survival, senescence, and
radiation-induced mortality. Proc Natl Acad Sci USA. 110:9362–9367.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mhawech-Fauceglia P, Ali L, Cheney RT,
Groth J and Herrmann FR: Prognostic significance of
neuron-associated protein expression in non-muscle-invasive
urothelial bladder cancer. J Clin Pathol. 62:710–714. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim JW, Moon AR, Kim JH, Yoon SY, Oh GT,
Choe YK and Choe IS: Up-regulation of ninjurin expression in human
hepatocellular carcinoma associated with cirrhosis and chronic
viral hepatitis. Mol Cells. 11:151–157. 2001.PubMed/NCBI
|
|
32
|
Strieter RM, Kunkel SL, Showell HJ, Remick
DG, Phan SH, Ward PA and Marks RM: Endothelial cell gene expression
of a neutrophil chemotactic factor by TNF-alpha, LPS, and IL-1
beta. Science. 243:1467–1469. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Luo JL, Maeda S, Hsu LC, Yagita H and
Karin M: Inhibition of NF-kappaB in cancer cells converts
inflammation- induced tumor growth mediated by TNFalpha to
TRAIL-mediated tumor regression. Cancer Cell. 6:297–305. 2004.
View Article : Google Scholar : PubMed/NCBI
|