Introduction
Hepatocellular carcinoma (HCC) is one of the most
common and generally incurable malignancies, which represent the
third-leading cause of cancer-related deaths worldwide (1,2). The
high mortality is due to late stage detection of this cancer when
most of the therapies available are not effective (3). The disease is progressive and death
usually occurs within 10 months of initial diagnosis (3). Most HCC related deaths are due to
advanced metastatic disease, resulting from lymphatic, blood, or
contiguous local spread, highlighting the need for a better
understanding of this disease pathogenesis (4).
Characterized by a RING B-box-coiled-coil protein
domain architecture, the tripartite motif (TRIM) families of
proteins are involved in the pathogenic mechanism of various
cancers, acting as either oncogenes or tumor suppressors (5). TRIM16 is highly expressed in basal
keratinocytes and increases differentiation markers in
keratinocytes, but is downregulated during the proliferative phase
of wound healing (6). In a
previous study, it was found that the expression of TRIM16 was
significantly reduced in vivo during the progression from
normal skin to squamous cell carcinoma (SCC). Moreover, TRIM16
inhibited SCC cell migration in vitro (7). In addition, TRIM16 downregulates
protein-binding partners, cytoplasmic vimentin and nuclear E2F1 in
neuroblastoma cells (8).
Collectively, these data suggest that TRIM16 plays a role in
repressing cancer cell replication and migration. However, the role
of TRIM16 in HCC is unknown.
In this study, excised human HCC samples and human
HCC cell lines were examined to show that the expression of TRIM16
was significantly downregulated in HCC lesions, compared with
paired normal liver tissues of clinical cancer samples. The
knockdown of TRIM16 promoted EMT in a manner correlated with HCC
metastasis in vitro and in vivo. Mechanistically,
TRIM16 inhibited the expression of ZEB2, which in turn inhibited
transcription of the pivotal ZEB2 target gene E-cadherin. RNA
interference-mediated silencing of ZEB2 attenuated
shTRIM16-enhanced cell migration and invasion. In conclusion, our
findings define TRIM16 as an inhibitor of EMT and metastasis in HCC
that predicts poor clinical outcomes, collectively indicating that
TRIM16 represents a novel therapeutic target in HCC.
Materials and methods
Chemicals and antibodies
Lipofectamine transfection and TRIzol reagents were
purchased from Invitrogen (Grand Island, NY, USA). MG132 was
purchased from Selleckchem (Houston, TX, USA). Antibodies against
TRIM16, E-cadherin, N-cadherin, vimentin, HA, Myc, snail, slug,
ZEB1, ZEB2 and β-actin antibodies were from Cell Signaling
Technology (Danvers, MA, USA). Anti-α-catenin antibody and Matrigel
were from BD (Franklin Lakes, NJ, USA). Unless otherwise noted, all
other chemicals were from Sigma (St. Louis, MO, USA).
Cell lines and cell culture
Liver cancer cell lines HepG2, SMMC-7721, HCCLM3,
MHCC97H, and HEK 293 Phoenix ampho-packaging cells were purchased
from Cell Bank of Type Culture Collection of Chinese Academy of
Sciences, Chinese Academy of Sciences; liver cancer cell lines were
routinely cultured as previously described (9). Cell lines were maintained at 37°C in
an atmosphere containing 5% CO2 in Dulbecco's modified
Eagle's medium or RPMI-1640 supplemented with 10% fetal bovine
serum.
Patients and specimens
Sixty-one tumor and para-cancerous tissues which,
were used for qRT-PCR and western blot analysis, were randomly
collected from HCC patients who underwent curative resection with
informed consent between 2011 and 2014 at the Department of
Laparoscopic Surgery, First Affiliated Hospital of Dalian Medical
University. Study protocols were approved by the Hospital Ethics
Committee of Dalian Medical University, and written informed
consent was obtained from patients based on the Declaration of
Helsinki.
Establishment of TRIM16 stable expression
and TRIM16, ZEB2 knockdown cell lines
pBabe retroviral construct containing human TRIM16
cDNA and pSuper with shRNA against human TRIM16 were prepared as
described previously (10). The
generation of retrovirus supernatants and transfection of cancer
cells were conducted as described previously. Infected cells were
selected by adding 2 μg/ml puromycin to the culture medium for 48 h
and then maintained in complete medium with 0.5 μg/ml puromycin.
Empty retroviral-infected stable cell lines were also produced by
the above protocols. siRNA against ZEB2 expressed in pSuper
vector were prepared as described previously (11). The generation of retrovirus
supernatants and transfection of cancer cells were conducted as
described above except infected cells were selected by adding 400
μg/ml of G418. The expression of TRIM16 and ZEB2 was confirmed by
qRT-PCR and western blot analysis.
Cell invasion and motility assay
Invasion assay was performed using Matrigel
(BD)-coated Transwell inserts (Costar, Manassas, VA, USA)
containing polycarbonate filters with 8-μm pores as detailed
previously (12). According to the
manufacturer's instructions, the inserts were coated with 50 μl of
1 mg/ml Matrigel matrix. Cells (2×105) in 200 μl of
serum-free medium were plated in the upper chamber, whereas 600 μl
of medium with 10% fetal bovine serum were added to lower chamber.
After 24-h incubation, cells that migrated to the lower surface of
the membrane were fixed and stained. For each membrane, five random
fields were counted at ×10 magnification. Motility assays were
similar to Matrigel invasion assay except that the Transwell insert
was not coated with Matrigel.
Confocal immunofluorescence
microscopy
Cell lines were plated on culture slides for 24 h.
In addition, the cells were rinsed with phosphate-buffered saline
fixed with 4% paraformaldehyde in PBS. Next, cell membrane was
permeabilized using 0.5% Triton X-100. These cells were then
blocked for 30 min in 10% BSA in PBS and incubated with primary
antibodies in 10% BSA overnight at 4°C. After three washes in PBS,
the slides were incubated for 1 h in the dark with FITC-conjugated
secondary antibodies. Slides were stained with DAPI for 5 min to
visualize the nuclei, and examined using a confocal imaging system
(LSM 780) (Carl Zeiss, Jena, Germany).
Western blot analysis
Standard methods were used for western blot
analysis. Cell lysates were prepared by extraction with lysis
buffer. Proteins (10 μg) were separated by SDS-PAGE under reducing
conditions and blotted onto a polyvinylidene difluoride membrane.
Membranes were probed with specific antibodies. Blots were washed
and probed with respective secondary peroxidase-conjugated
antibodies, and the bands visualized by chemiluminescence.
qRT-PCR
Total RNA was extracted using TRIzol reagent and
cDNA was synthesized using SuperScript II reverse transcriptase.
qRT-PCR and data collection were performed with an ABI PRISM 7900HT
sequence detection system. The primers used in this study are
listed in Table I.
 | Table IPrimer sequences used for qRT-PCR. |
Table I
Primer sequences used for qRT-PCR.
| Primer | Sequence (5′-3′) |
|---|
| hGAPDH-S370 | GCT GGC GCT GAG TAC
GTC GT |
| hGAPDH-AS821 | ACG TTG GCA GTG GGG
ACA CG |
| hslug-S84 | CAG CGG CTC TGA TTA
CAG ACC TCG |
| hslug-AS285 | GTC TTC ACA GGC CTG
ACG CAG T |
| hTRIM16-S358 | ATT GAG CTG TTG CCG
CTG TTG CTG |
| hTRIM16-AS614 | GCC CTT CCT TTC CTG
TGT CAT CCT C |
|
hE-cadherin-S1117 | TGG GCT GGA CCG AGA
GAG TTT C |
|
hE-cadherin-AS1562 | ATC CAG CAC ATC CAC
GGT GAC G |
|
hN-cadherin-S1152 | CCG GTT TCA TTT GAG
GGC ACA TGC |
|
hN-cadherin-AS1562 | GCC GTG GCT GTG TTT
GAA AGG C |
|
hFibronectin-S5527 | TCC AAG TTG ATG CCG
TTC CA |
|
hFibronectin-AS5986 | GAG AGA GCT TCT TGT
CCT GTC T |
| hVimentin-S83 | AAC TTA GGG GCG CTC
TTG TC |
| hVimentin-AS518 | GGT GGA CGT AGT CAC
GTA GC |
| hα-catenin-S961 | TCA TTG TGG ACC CCT
TGA GC |
|
hα-catenin-AS1168 | TTA CGT CCA GCA TTG
CCC AT |
| hsnail-S1964 | GCA CAT GTC CTG ATT
TGT TCT TGA |
| hsnail-AS1821 | CCC CTA ACG CTG AAC
AGA CA |
| hZEB1-S1539 | GGC CAT AGG CAC TGT
AGC AA |
| hZEB1-AS1322 | GGT TTC ACA AGT GAT
AAT TCT GAG C |
| hZEB2-S453 | GGC AAA GTG GAG TGG
GAA AGT A |
| hZEB2-AS226 | AGT GCG GAA AGA AGC
AAC AG |
In vivo tumor metastasis
Different cells were resuspended in PBS at a
concentration of 1×107 cells/ml in metastasis assays.
Cell suspension (0.1 ml) was injected into tail veins of nude mice.
The mice were sacrificed by CO2 60 days after
inoculation.
Statistical analysis
Data are presented as means ± standard deviation
(SD). Data were analyzed by SPSS/Win11.0 software (SPSS, Inc.,
Chicago, IL, USA) in t-test (unpaired, two-tailed), and results
were considered significant at P<0.05.
Results
TRIM16 is downregulated and correlated
with distant metastasis in HCCs
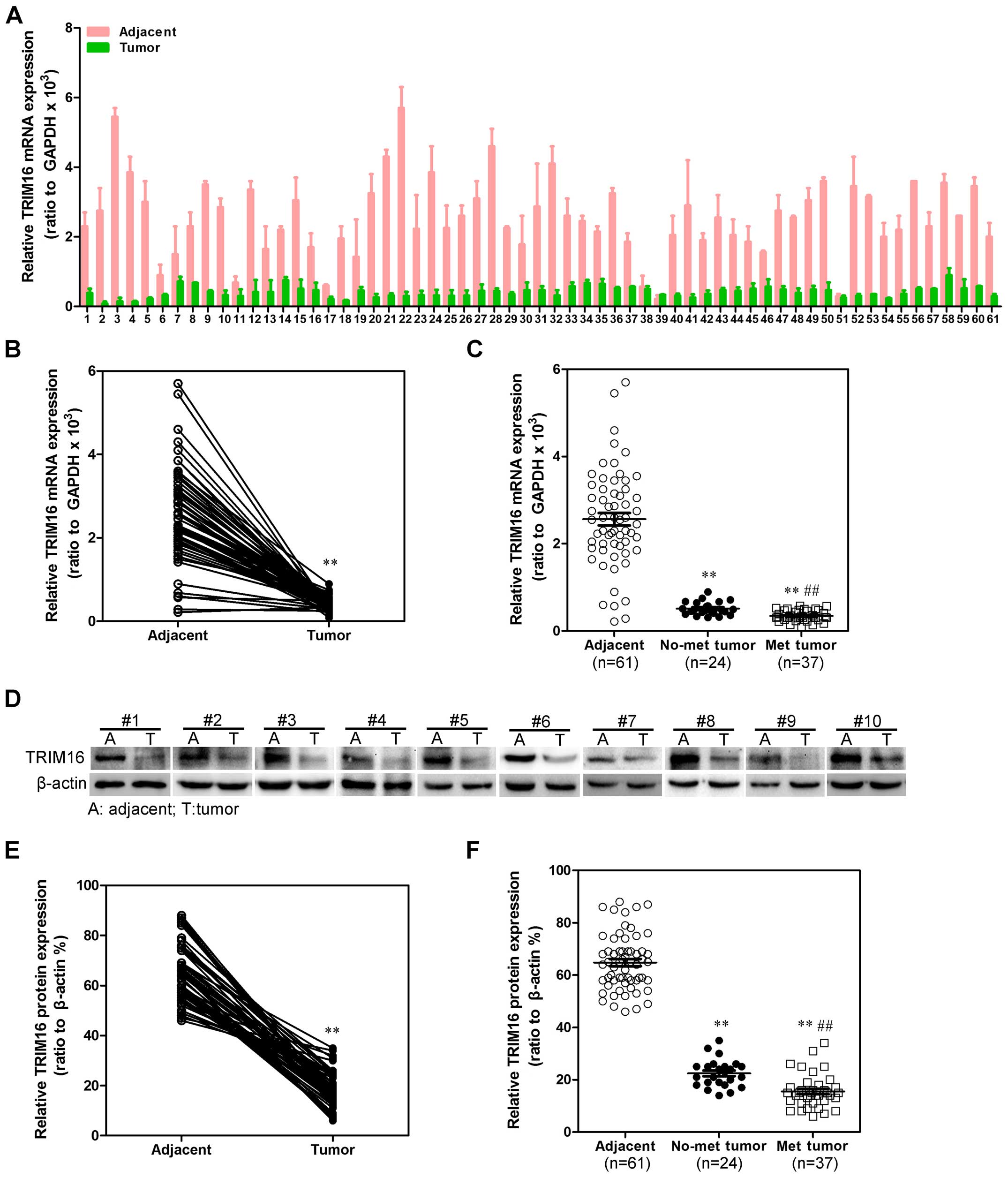
To investigate whether TRIM16 might be involved in
HCC, the mRNA expression level of TRIM16 in HCC tissues and their
matched normal adjacent tissues was determined by qRT-PCR in 61
samples (Fig. 1A). As compared
with normal tissues, HCC specimens showed lower expression of
TRIM16 (Fig. 1B). We then analyzed
TRIM16 expression in HCCs without or with metastasis property; we
found that TRIM16 mRNA lower expression was significantly
correlated with metastasis property in HCC tissues (Fig. 1C). We examined TRIM16 protein
expression in the same HCC samples by western blot analysis
(Fig. 1D). We observed that the
level of TRIM16-positive cells was markedly lower in HCC tissues
than the level in the normal liver tissues (Fig. 1E). Most importantly, TRIM16 lower
expression was consistently significantly correlated to distant
metastasis in these HCC samples (Fig.
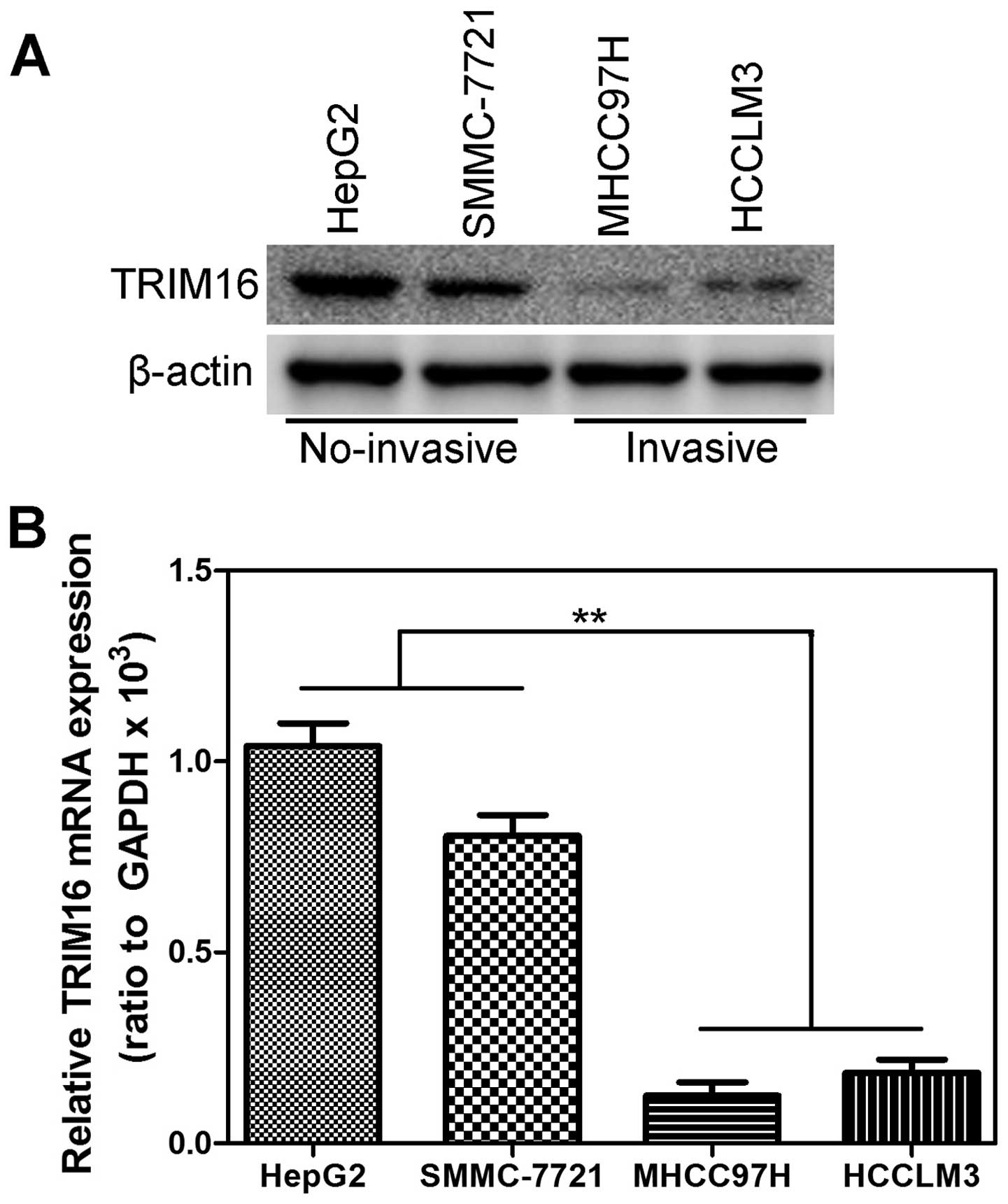
1F). As showed in Fig. 2,
expression level of TRIM16 protein (Fig. 2A) and mRNA (Fig. 2B) in invasive HCC cell lines was
lower than that in the no-invasive HCC cell lines. These data
demonstrated that the downregulation of TRIM16 might be relevant to
invasive property of HCC.
Suppression of TRIM16 promotes HCC cell
migration and invasion in vitro
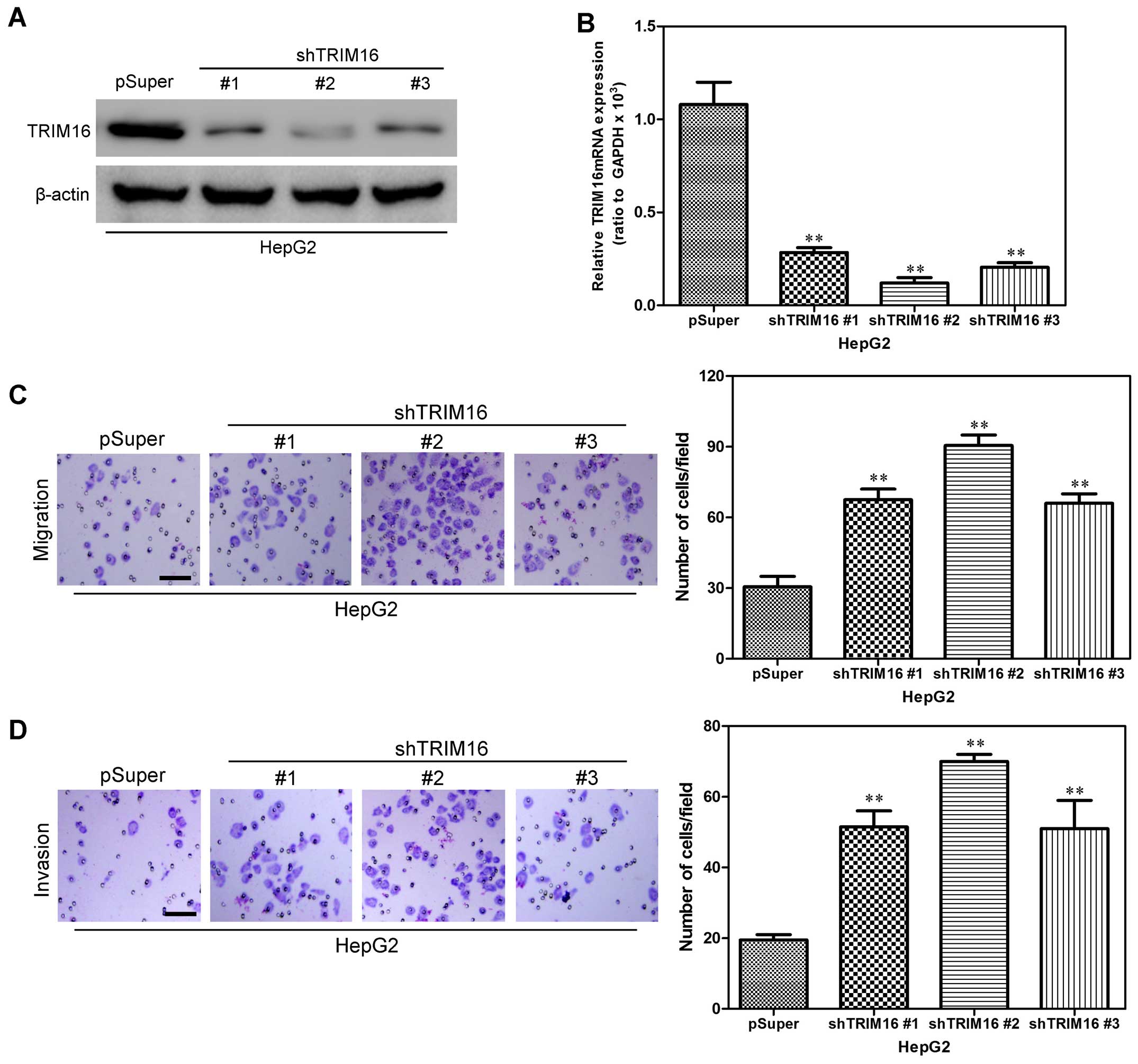
To investigate the effect of TRIM16 knockdown on
hepatoma carcinoma cell migration and invasion, HepG2 human
hepatoma cell line, was first infected with shTRIM16 or the
control. We used western blot and qRT-PCR analyses to detect
transfection efficiency. Compared with the control cells, the HepG2
cells, transfected with the TRIM16 shRNA plasmid, displayed
significantly decreased TRIM16 expression at the protein and mRNA
levels (Fig. 3A and B). Boyden
chamber assay was first used to assess the influence of TRIM16 on
HCC cell migration to detect the effect of TRIM16 on HCC cells. As
indicated by the Transwell assay and Matrigel assay, knockdown of
TRIM16 expression significantly inhibited HCC cell migration and
invasion (Fig. 3C and D). These
results further demonstrated that silencing TRIM16 promotes HCC
cell migration and invasion in vitro.
Ectopic TRIM16 expression inhibits HCC
cell migration and invasion in vitro
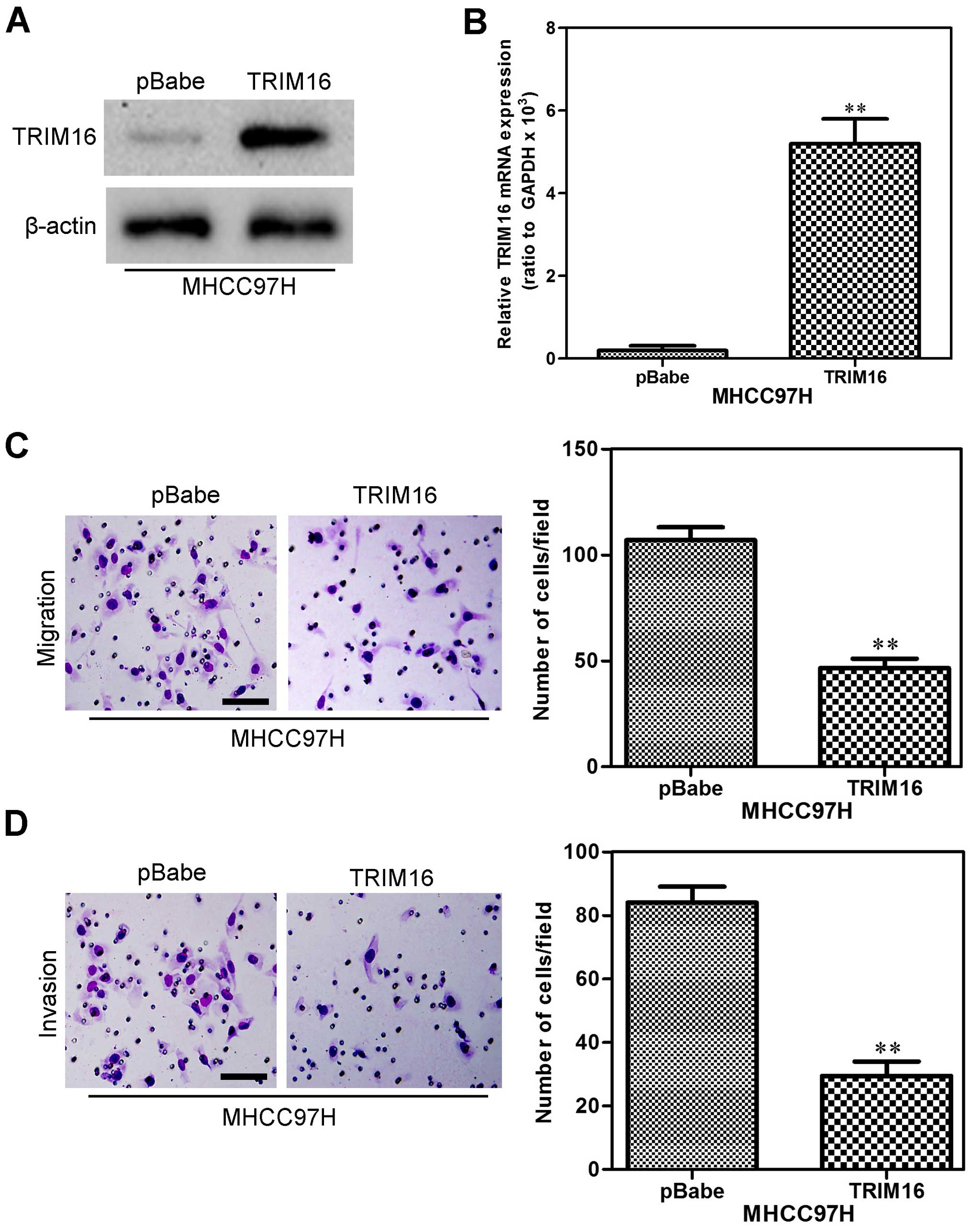
We also used MHCC97H cells to establish a stable
cell line that constitutively overexpressed TRIM16. The
transfection efficiency was confirmed using western blot and
qRT-PCR analyses. As shown in Fig. 4A
and B, the MHCC97H cells that had been transfected with the
TRIM16 expression plasmid displayed significantly increased TRIM16
expression both at the protein and mRNA levels compared with the
vector cells. In order to measure the effect of TRIM16 on HCC cell
migration and invasion, the effect of TRIM16 on HCC cell migration
was assessed by the Boyden chamber assay. The overexpression TRIM16
markedly reduced the migratory (Fig.
4C) and invasive (Fig. 4D)
capacity of MHCC97H cells. These results indicate that ectopic
TRIM16 expression inhibits HCC cell migration and invasion in
vitro.
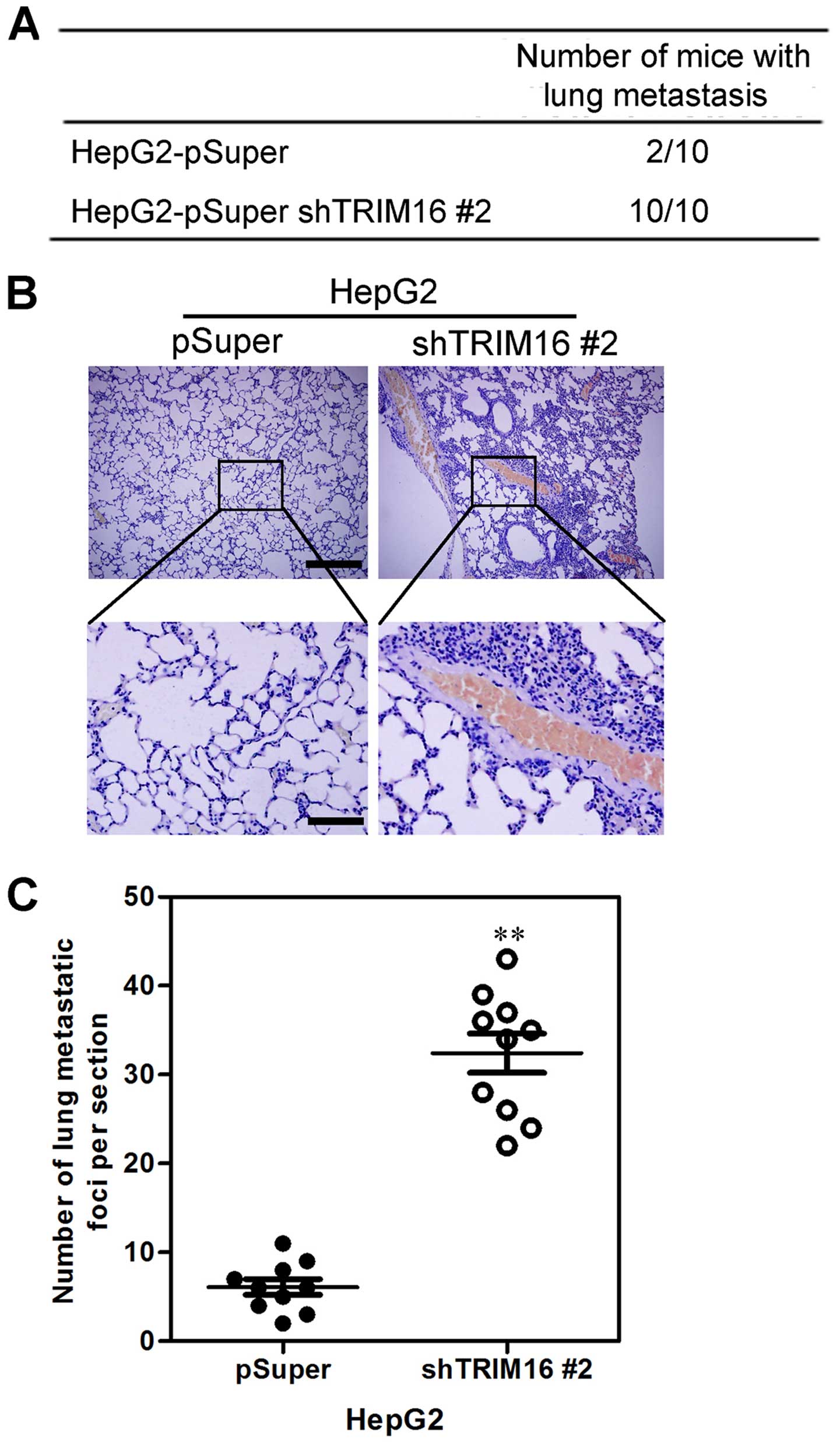
Silencing TRIM16 promotes HCC cell
distant metastasis in vivo
We then investigated the functional relevance of
TRIM16 for metastasis in vivo. HepG-2-shTRIM16 #2 and its
control cells were injected into nude mice through the tail vein.
We observed that silencing TRIM16 not only significantly increased
the number of mice with distant metastasis (Fig. 5A), but also markedly increased the
number of metastatic tumors in the lungs of each mouse (Fig. 5B and C). Therefore, the in
vivo results further demonstrate the critical role of TRIM16 in
HCC metastasis.
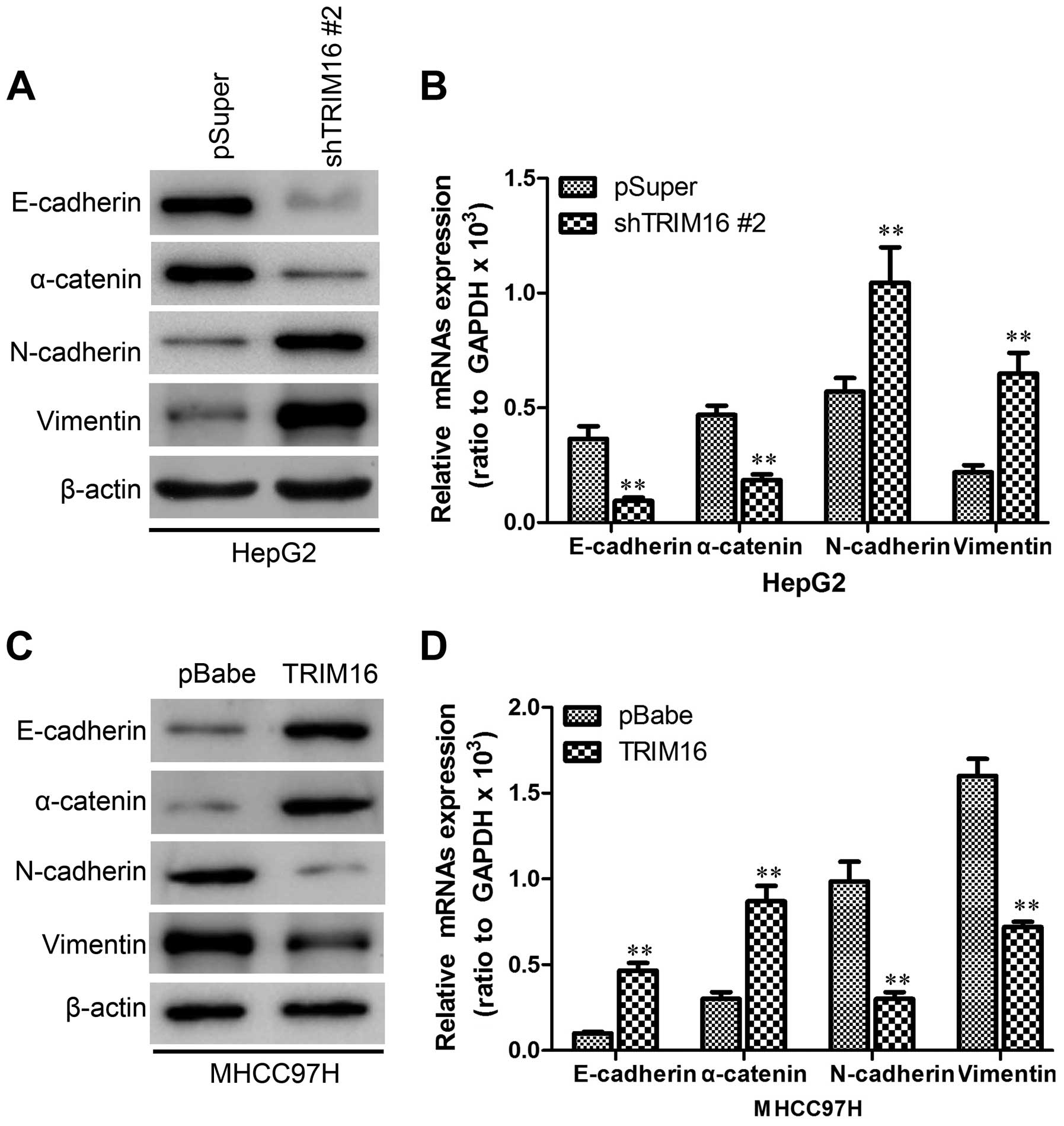
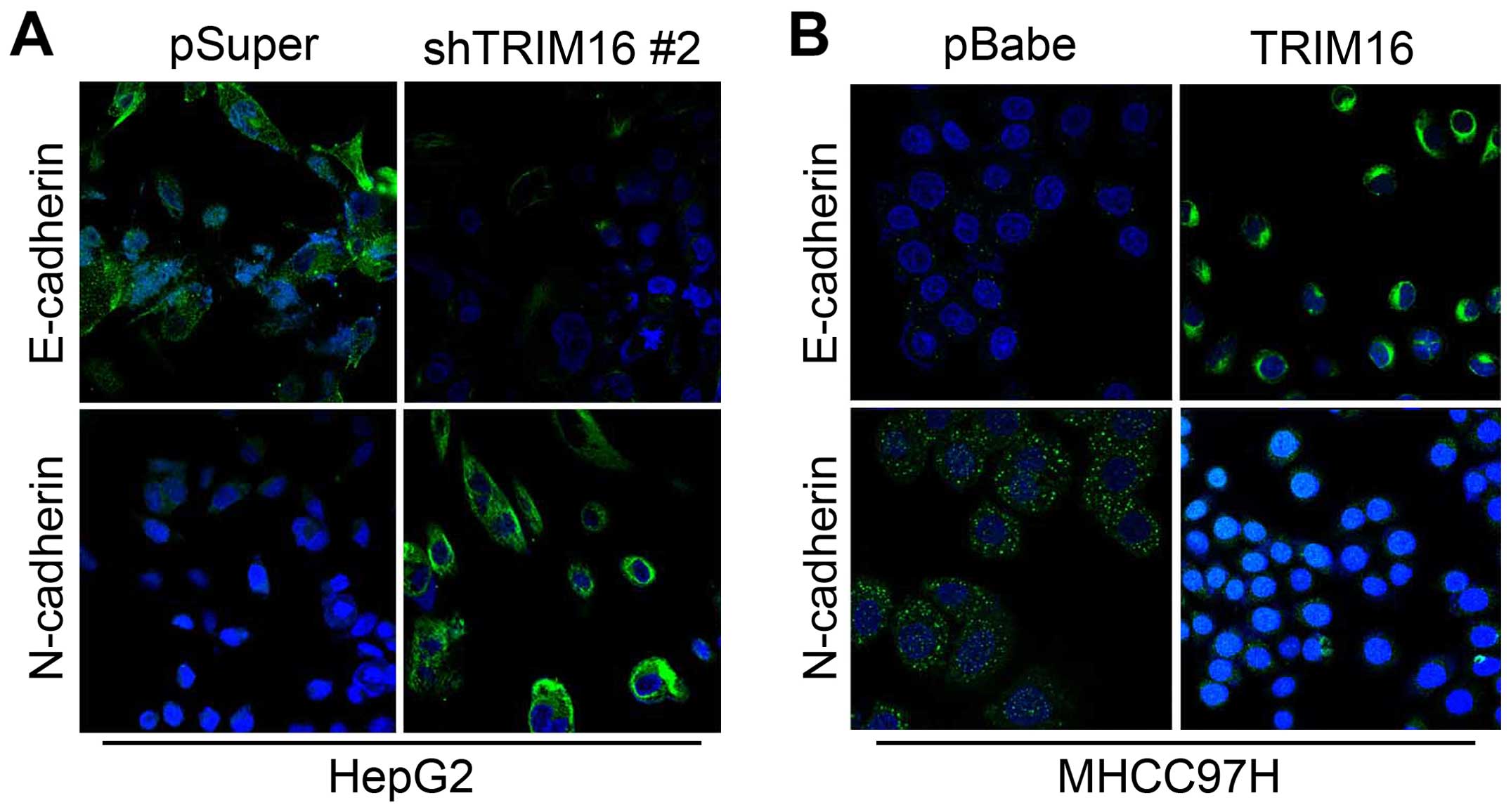
TRIM16 inhibits EMT behavior in HCC
cells
During the establishment of these cell lines, we
observed that HepG-2-shTRIM16 cells exhibited fibroblastic
morphology, compared to their respective control cells (data not
shown). This observation was further confirmed by western blot and
immunofluorescence analyses of epithelial and mesenchymal molecular
marker expression. We showed that TRIM16 knockdown decreased the
levels of epithelial markers (E-cadherin and α-catenin) and
increased the levels of mesenchymal markers (N-cadherin and
vimentin) (Figs. 6A and 7A). Moreover, expression levels of mRNA
correlated with the corresponding protein levels (Fig. 6B), suggesting that TRIM16 affected
the expression of epithelial and mesenchymal markers at the
transcript level. Conversely, MHCC97H-TRIM16 cells reverted to a
mesenchymal phenotype as compared to their respective control cells
(data not shown). Consistent with this, ectopic TRIM16 expression
resulted in an increase in expression of epithelial markers, and a
decrease in expression of mesenchymal markers (Figs. 6C and D and 7B).
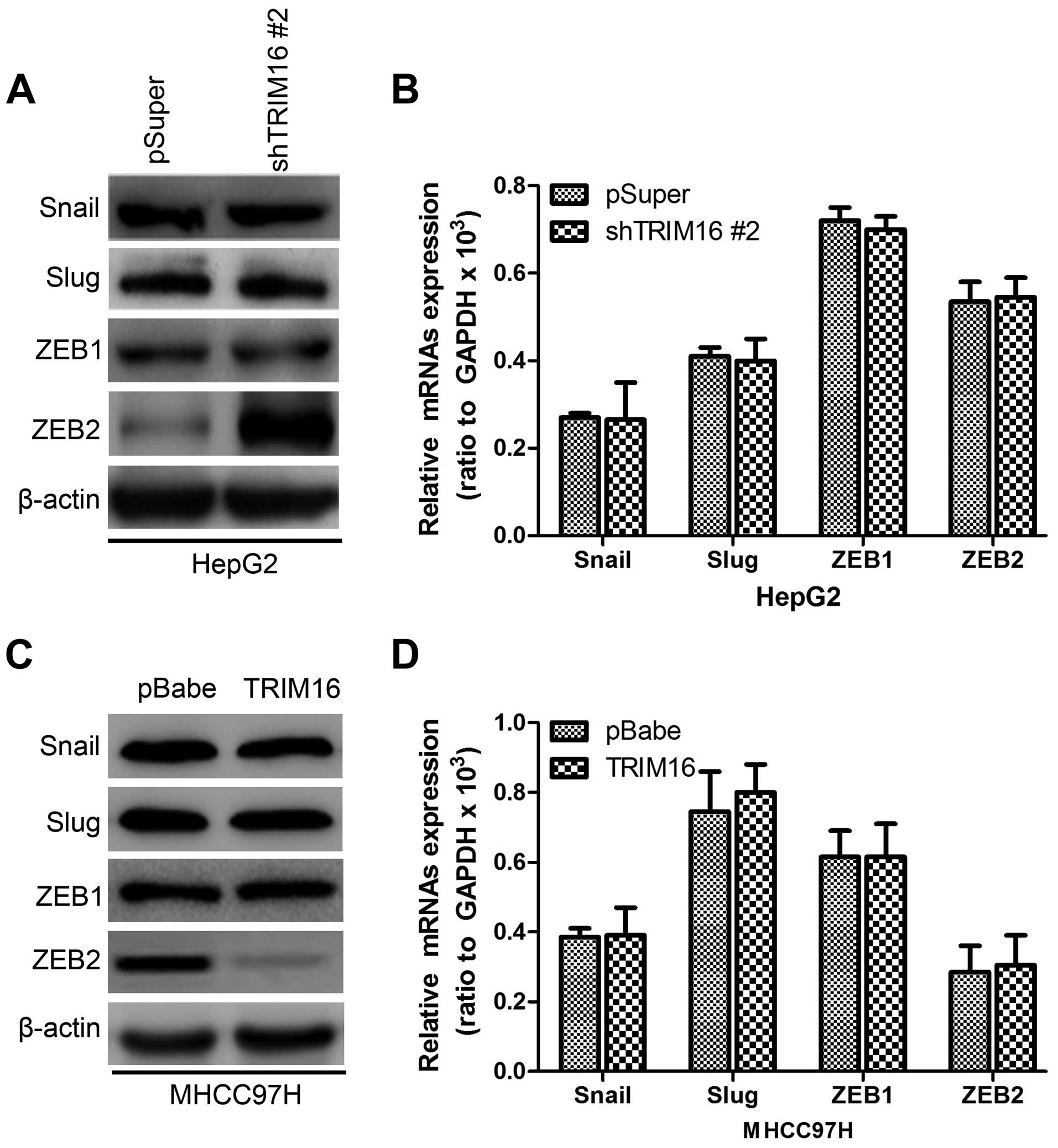
TRIM16 downregulates ZEB2 expression
through interaction
To better understand the mechanisms by which TRIM16
engaged in the EMT program, we assessed the expression of the EMT
regulating genes, such as Snail, Slug, ZEB1 and ZEB2, on
HepG2-shTRIM16, MHCC97H-TRIM16 and their control cells. The results
shown that HepG2-shTRIM16 cells exhibited greatly increased ZEB2
expression at protein level (Fig.
8A), whereas, ectopic TRIM16 in MHCC97H cells markedly
decreased ZEB2 expression at protein levels (Fig. 8C). While ZEB2 mRNA level had no
significant change in either TRIM16 knockdown or ectopic expression
(Fig. 8B and D), indicating that
the regulation function of TRIM16 on ZEB2 expression only took
place on the post-transcriptional level.
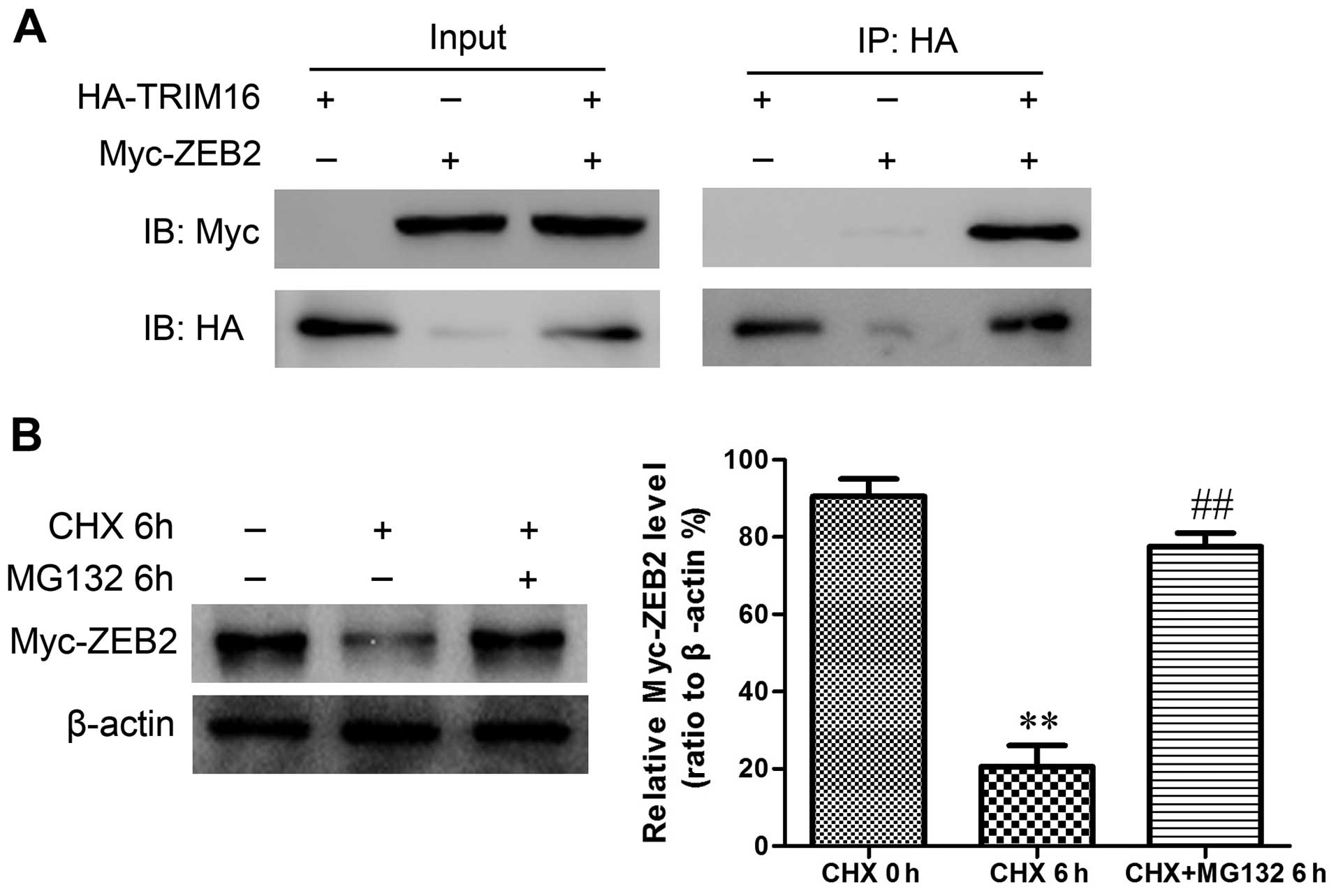
To understand how TRIM16 exerts its effects on ZEB2,
293T cells were co-transfected with HA-tagged TRIM16 and Myc-tagged
ZEB2 constructs. Immunoprecipitation and subsequent immunoblot
analyses revealed that Myc-tagged ZEB2 co-immunoprecipitated with
HA-tagged TRIM16 (Fig. 9A),
confirming a physical interaction between the two proteins. The
physical interaction between TRIM16 and ZEB2 suggested that TRIM16
maybe target ZEB2 for ubiquitination and subsequent degradation.
Next, in order to investigate if the regulation of TRIM16 on ZEB2
protein is dependent on the ubiquin-proteasome pathway, we combined
the treatment of proteasome inhibitor MG132 with ectopic TRIM16
treatment. We found that ZEB2 degradation caused by TRIM16 ectopic
expression was completely abrogated by proteasome inhibition
(Fig. 9B), demonstrating that the
regulation of ZEB2 expression by TRIM16 is indeed mediated via
proteasome-dependent pathway.
ZEB2 is a mediator for shTRIM16-induced
migration and invasion behavior in HCC cells
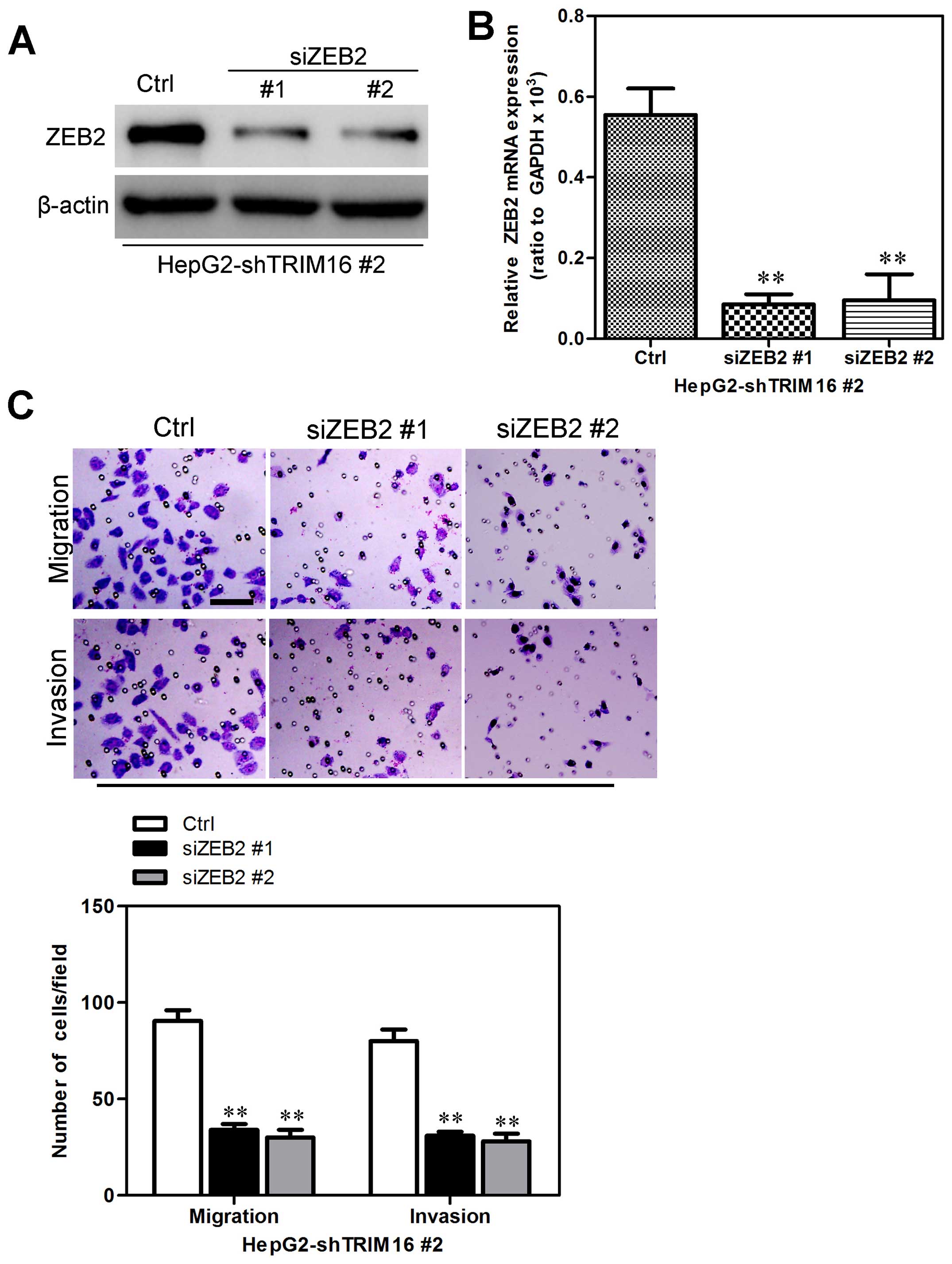
We transfected HepG2-shTRIM16 cells with ZEB2 siRNAs
to silence ZEB2 gene expression to investigate whether
TRIM16-induced metastatic capacity was mediated by ZEB2 expression
(Fig. 10A and B). It was found
that knockdown of ZEB2 in HepG2-shTRIM16 cells were accompanied by
the reduction of migratory and invasive capacities (Fig. 10C). Collectively, these results
show that ZEB2 mediates shTRIM16-induced migration and invasion in
HCC cells.
Discussion
Hepatocellular carcinoma (HCC) is a highly lethal
malignancy with increasing incidence globally (1). Recent studies have indicated that
early frequent metastasis has critical roles in the development and
progression of HCC (13).
Therefore, identifying novel molecules that regulate HCC metastasis
will promote the development of anti-metastasis strategies.
Mounting evidence shows that in epithelial cancers, including HCC,
induction of EMT is a major event that provides mobility to cancer
cells in order to generate metastases (14).
EMT is a process where cells undergo a morphological
switch from the epithelial phenotype to esenchymal phenotype. In
this process, epithelial cells not only lose defined
cell-cell/cell-substratum contacts and their structural polarity,
but also they become spindle shaped (15). On the molecular level, EMT is
defined as the loss of cell-cell adhesion molecules, downregulation
of epithelial differentiation markers and transcriptional induction
of mesenchymal markers (13). It
has been shown that EMT is associated with cancer progression and
cancer-related death. Moreover, EMT has been recognized to play
pivotal roles in several diverse processes during embryonic
development, chronic inflammation and fibrosis, as well as tumor
progression. Numerous observations support the concept that the EMT
process plays a role in the progression of tumors, including HCCs
(16).
In this study, the clinical significance of TRIM16
in HCC and the mechanistic role of TRIM16 in inhibiting HCC cell
metastasis were first delineated to our knowledge. We found that
TRIM16 down-expression in HCC cells induced EMT, migration and
invasion in vitro and enhanced metastatic capacity in
vivo. In contrast, ectopic TRIM16 expression reversed these
events in other aggressive and invasive HCC cells. It was also
shown that a mechanistic link exists between TRIM16 and migration
through TRIM16-mediated downregulation of ZEB2, which may
subsequently lead to transcriptional downregulation of E-cadherin
expression. Moreover, knockdown of ZEB2 attenuated shTRIM16 induced
migration and invasion. Considering all these results, we propose a
model for TRIM16 regulation of EMT and metastasis through
downregulation of ZEB2 in HCC cells.
The tripartite motif or TRIM family of proteins were
originally described by its multi-domain design of three
structurally distinct motifs, the RING finger zinc-binding domain,
a B-box zinc-binding domain and the coiled-coil domain (17). TRIM16 is also known as the
estrogen-responsive B-box protein due to its original discovery as
an estrogen responsive protein in human mammary epithelial cells
(17). TRIM16 has been shown to
suppress tumor progression through regulatory pathways involved in
growth inhibition, migration, differentiation and apoptosis.
Recently research has demonstrated that TRIM16 can heterodimerize
with other TRIM proteins and has E3 ubiquitin ligase activity
(6). These data strongly support a
role for TRIM16 as a tumor suppressor gene. However, the exact
mechanisms of TRIM16 involvement in HCC remain unclear. Our study
points to a novel function of TRIM16 in HCC metastasis through
inhibiting essential characteristics of metastatic disease in HCC:
EMT. First, HCC cells expressing low level of TRIM16 displayed an
EMT phenotype, including the associated stimulatory effects on
in vitro migration and invasion. Interestingly, our results
indicate that TRIM16 not only inhibits EMT, but ectopic expression
of TRIM16 also leads to MET. All of these characteristics that are
induced by shTRIM16 in vitro culminated to increased number
of distant metastases in vivo. These empirical findings
provide a mechanistic framework to explain the clinical
observations that HCC patients with low levels of TRIM16 in tissue
samples have a greater chance of distant metastasis.
The roles of several transcription factors as EMT
regulators have been extensively reported. In our effort to
elucidate the mechanism how TRIM16 inhibits metastasis in HCC
cells, we identified ZEB2 as an effective mediator of shTRIM16
inducing these phenomena. The mechanistic connection between TRIM16
and ZEB2 was previously unknown. In this study, we founded that
modulation of TRIM16 expression altered ZEB2 expression. Thus, in
conclusion, TRIM16 inhibits migration and invasion in vitro
and metastasis in vivo by downregulating ZEB2
expression.
Metastasis and EMT are essential for HCC cells to
disseminate from adjacent tissues and seed new tumors in distant
sites (18). Our results
demonstrated that TRIM16 regulated these two essential
characteristics of metastatic disease and TRIM16-induced processes
are reversible with the suppression of TRIM16 expression, providing
us an optimal therapeutic option to manipulate TRIM16 levels in
clinical HCC practice.
Acknowledgements
This study was supported by the Liaoning Province
Natural Science Foundation of China (grant no. 2014B007).
References
|
1
|
DeSantis CE, Lin CC, Mariotto AB, Siegel
RL, Stein KD, Kramer JL, Alteri R, Robbins AS and Jemal A: Cancer
treatment and survivorship statistics, 2014. CA Cancer J Clin.
64:252–271. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hato T, Goyal L, Greten TF, Duda DG and
Zhu AX: Immune checkpoint blockade in hepatocellular carcinoma:
Current progress and future directions. Hepatology. 60:1776–1782.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
El-Serag HB and Kanwal F: Epidemiology of
hepatocellular carcinoma in the United States: Where are we? Where
do we go? Hepatology. 60:1767–1775. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Marshall GM, Bell JL, Koach J, Tan O, Kim
P, Malyukova A, Thomas W, Sekyere EO, Liu T, Cunningham AM, et al:
TRIM16 acts as a tumour suppressor by inhibitory effects on
cytoplasmic vimentin and nuclear E2F1 in neuroblastoma cells.
Oncogene. 29:6172–6183. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bell JL, Malyukova A, Holien JK, Koach J,
Parker MW, Kavallaris M, Marshall GM and Cheung BB: TRIM16 acts as
an E3 ubiquitin ligase and can heterodimerize with other TRIM
family members. PLoS One. 7:e374702012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sutton SK, Koach J, Tan O, Liu B, Carter
DR, Wilmott JS, Yosufi B, Haydu LE, Mann GJ, Thompson JF, et al:
TRIM16 inhibits proliferation and migration through regulation of
interferon beta 1 in melanoma cells. Oncotarget. 5:10127–10139.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cheung BB, Koach J, Tan O, Kim P, Bell JL,
D'andreti C, Sutton S, Malyukova A, Sekyere E, Norris M, et al: The
retinoid signalling molecule, TRIM16, is repressed during squamous
cell carcinoma skin carcinogenesis in vivo and reduces skin cancer
cell migration in vitro. J Pathol. 226:451–462. 2012. View Article : Google Scholar :
|
|
9
|
You H, Ding W and Rountree CB: Epigenetic
regulation of cancer stem cell marker CD133 by transforming growth
factor-beta. Hepatology. 51:1635–1644. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang Y, Wen M, Kwon Y, Xu Y, Liu Y, Zhang
P, He X, Wang Q, Huang Y, Jen KY, et al: CUL4A induces
epithelial-mesenchymal transition and promotes cancer metastasis by
regulating ZEB1 expression. Cancer Res. 74:520–531. 2014.
View Article : Google Scholar :
|
|
11
|
Wang Y, Zhang P, Liu Z, Wang Q, Wen M,
Wang Y, Yuan H, Mao JH and Wei G: CUL4A overexpression enhances
lung tumor growth and sensitizes lung cancer cells to erlotinib via
transcriptional regulation of EGFR. Mol Cancer. 13:2522014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sun Y, Wang Y, Fan C, Gao P, Wang X, Wei G
and Wei J: Estrogen promotes stemness and invasiveness of
ER-positive breast cancer cells through Gli1 activation. Mol
Cancer. 13:1372014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Floor S, van Staveren WC, Larsimont D,
Dumont JE and Maenhaut C: Cancer cells in epithelial-to-mesenchymal
transition and tumor-propagating-cancer stem cells: Distinct,
overlapping or same populations. Oncogene. 30:4609–4621. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hills CE and Squires PE: The role of TGF-β
and epithelial-to mesenchymal transition in diabetic nephropathy.
Cytokine Growth Factor Rev. 22:131–139. 2011.PubMed/NCBI
|
|
16
|
Yilmaz M and Christofori G: EMT, the
cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev.
28:15–33. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huo X, Li S, Shi T, Suo A, Ruan Z and Yao
Y: Tripartite motif 16 inhibits epithelial-mesenchymal transition
and metastasis by down-regulating sonic hedgehog pathway in
non-small cell lung cancer cells. Biochem Biophys Res Commun.
460:1021–1028. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
van Zijl F, Zulehner G, Petz M, Schneller
D, Kornauth C, Hau M, Machat G, Grubinger M, Huber H and Mikulits
W: Epithelial-mesenchymal transition in hepatocellular carcinoma.
Future Oncol. 5:1169–1179. 2009. View Article : Google Scholar : PubMed/NCBI
|