Introduction
Non-small-cell lung cancer (NSCLC) is still the
leading cause for cancer related mortality worldwide (1,2).
Treatment of EGFR-mutant NSCLC patients with specific tyrosine
kinase inhibitors (TKIs), such as gefitinib, erlotinib or afatinib,
has led to remarkable tumor shrinkage and improvement in
progression-free survival and quality of life compared with the
standard chemotherapy (3–5). However, not all patients respond to
TKI treatment, and patients who initially benefited from the TKI
treatment inevitably developed drug resistance (6,7). The
resistance mechanisms to TKI treatment can be grouped into four
categories: mutation of EGFR to a drug-resistance state, for
example, T790M mutation; activation of a bypass pathway, such as
MET amplification; impairment of a pathway that is essential for
TKI-induced apoptosis; and histologic transformation to small cell
lung cancer or epithelial-mesenchymal transition (8). Apart from these clues, accumulating
evidence indicate that macroautophagy (hereafter autophagy) plays a
vital role in the resistance to TKI.
Autophagy is a lysosomal degradative pathway
characterized by the formation of double-membrane autophagic
vesicles, which engulf portions of the cytosol, damaged organelles,
protein aggregates and bacteria. In virtually all cells, autophagy
occurs at basal levels to maintain homeostatic balance by updating
proteins and organelles (9).
Autophagy is upregulated when cells are under unfavorable
environments, such as starvation, hypoxia, and drug exposure, to
help cells survive in adverse conditions.
Cancer cells have increased metabolic demands due to
high levels of cellular proliferation and cellular stress, which
activates autophagy to maintain energy balance and to prevent cell
death (10). Theoretically,
autophagy may be a strategy to confer not only unfavorable
environments but also chemicals exposure. Many cancer therapeutics,
including TKIs, were reported to activate pro-survival autophagy
and blockage of autophagy facilitates cell death (11–14).
However, so far, the mechanisms underlying the synergistic effect
of EGFR-TKI and autophagy inhibition remains unclear.
The endoplasmic reticulum (ER) is an organelle
involved in protein folding and assembly, lipid and sterol
biosynthesis, and free calcium storage. ER stress is caused when
accumulation of unfolded and misfolded proteins exceed the capacity
of the ER process. Upon ER stress, protein kinase RNA (PKR)-like ER
kinase (PERK) is activated following dissociation from the ER
chaperone GRP78. Activated PERK transiently inhibits protein
synthesis by phosphorylating eukaryotic initiation factor 2α
(eIF2α). Activation of PERK can also lead to the induction of
CCAAT/enhancer binding protein homologous protein (CHOP), which
switches the ER stress response from proadaptive to proapoptotic
signaling (15–17). Given inhibition of autophagy would
cause accumulation of damaged proteins and excessive accumulation
of misfolded proteins would induce ER stress, we reasoned that
there may be a link between autophagy inhibition and ER stress
induced apoptosis.
In this study, we tested our hypothesis by a
combination of western blotting, transmission electron microscopy,
and fluorescent microscopy to detect autophagy in several types of
cancer cells after erlotinib treatment. We also explored the
mechanisms underlying the synergistic effect of autophagy
inhibition and erlotinib. Our findings suggest that autophagy acts
as a cell protect factor and contributes to the EGFR-TKI
resistance. The combination of autophagy inhibition and erlotinib
may be a promising route to enhance EGFR-TKI sensitivity and
overcome EGFR-TKI resistance.
Materials and methods
Reagents and antibodies
The chemicals used were erlotinib (Selleck),
chloroquine (CQ, Sigma), and 3-methyladenine (3-MA, Sigma).
Erlotinib was dissolved in dimethylsulfoxide (DMSO), while CQ and
3-MA were dissolved in cells culture medium. The primary antibodies
were antibodies against LC-3 (1:1,000, Novus, #NB100-2220),
beclin-1 (1:1,000, Cell Signaling Technology, #3495), caspase 3
(1:500, ProteinTech, #19677-1-AP), cleaved-caspase 3 (1:500, Cell
Signaling Technology, #9664), chop (1:1,000, Cell Signaling
Technology, #2895), p-EIF2α (1:1,000, Cell Signaling Technology,
#3398), EIF2α (1:1,000, Cell Signaling Technology, #5324), EGFR
(1:1,000, Cell Signaling Technology, #4267) and GAPDH (1:2,000,
Cell Signaling Technology, #5174).
Cell culture
The human NSCLC cell lines HCC827 (adenocarcinoma),
A549 (adenocarcinoma), H460 (large cell carcinoma) and H1975
(adenocarcinoma) were obtained from Cell Bank (Chinese Academy of
Sciences) and maintained in suggested complete medium. HCC827 has a
mutation in the EGFR tyrosine kinase domain (E746-A750 deletion).
H1975 has two mutations in the EGFR tyrosine kinase domain (L858R
and T790M). While A549 and H460 are EGFR wild-type cells. To
establish an EGFR-TKI-resistant cell line, HCC827 cells were
cultured in medium containing escalating concentrations of
erlotinib for 6 months to select cells which could grow in
micromolar concentrations of erlotinib. The selected cells were
confirmed erlotinib-resistant, and named HCC827-R.
Cell proliferation
The cytotoxicity of chemicals against cancer cells
were determined with CCK8 tests. Cells were seeded in 96-well
plates at a concentration of 1,500–2,000 cells per well, allowed to
attach overnight, and treated with various concentrations of
chemicals. After 72-h incubation, 10 μl of CCK8 (Boster, China) was
added into each well for another 1-h incubation. Then, the cell
optical density (OD) was measured at an absorbance wavelength of
450 nm.
Clonogenic assay
Cells were seeded in 6-well plates at a
concentration of 1,000 cells per well, allowed to attach overnight,
and treated with 10 μM CQ, 0.1 μM erlotinib, 1 μM erlotinib, alone
or in combination. Culture medium and chemicals were replaced every
72 h. After 10–14 days, cells were fixed with methanol and stained
with 1% crystal violet. Colonies were counted with ImageJ
software.
Western blotting
After chemical treatment, cells were lysed in a
lysis buffer containing protease and phosphatase inhibitors. Equal
amounts of protein were separated by SDS-PAGE and transferred to
PVDF membrane (Millipore, Billerica, MA, USA). After blocking with
5% non-fat milk, the membranes were then incubated overnight at 4°C
with primary antibodies. Then, after washing with TBST, the
membranes were incubated with corresponding secondary antibodies
conjugated with horseradish peroxidase, developed with ECL
solution, and exposed onto Hyperfilm (OptiCam600; UVP, USA).
cDNA and siRNA transfection
The GFP-tagged LC3 cDNA expression construct was
kindly provided by Professor Jinghua Ren (Union Hospital, Wuhan,
China) and transfected with Lipofectamine 2000 (Invitrogen). The
CHOP-siRNA and non-specific control of siRNA were from RiboBio
Technology (Guangzhou, China) and the targeted sequence of
CHOP-siRNA was GGCTCAAGCAGGAAATCGA. Cells were transfected with
siRNA using Interferin (Plosplus) according to the manufacturer's
instructions and incubated 24 h prior to further treatments.
Apoptosis assay
Apoptosis events were measured using FITC Annexin V
Apoptosis Detection kit (BD) that quantitatively measures Annexin V
or PI (propidium iodide)-positive cells. Western blot analyses were
used to detect the protein level of cleaved caspase 3 after various
treatments.
Transmission electron microscopy
Transmission electron microscopy was performed as
previously described (18). In
brief, H1975 cells were treated with erlotinib (1 μM) for 48 h,
washed 3 times with PBS, and fixed with glutaraldehyde overnight.
After washing with PBS, cells were fixed in 1% OsO4 and
embedded in polybed resin. The ultrathin sections were doubly
stained with uranyl acetate and lead citrate and analyzed by
transmission electron microscopy (Hitachi, Japan).
Quantification and statistics
Differences between groups were compared using the
Student's t-test or two-way ANOVA followed by the Bonferroni test.
The criterion for statistical significance was P<0.05.
Results
Autophagy involves EGFR-TKI
sensitivity
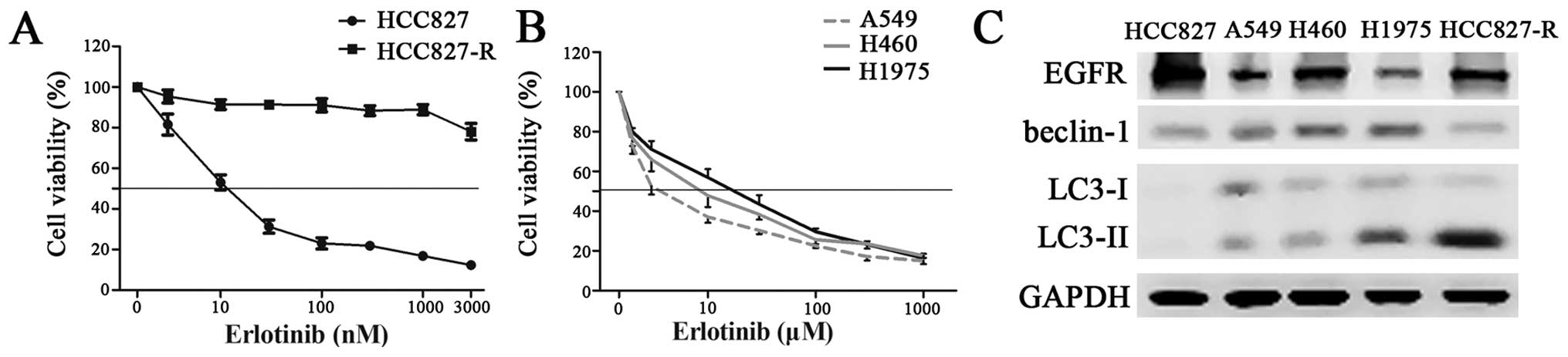
With CCK8 tests, we examined the sensitivity of
HCC827, A549, H460, H1975, and HCC827-R cells to erlotinib. HCC827
cells, with a mutation in the EGFR tyrosine kinase domain
(E746-A750 deletion), was the most sensitive to erlotinb. In
contrast, HCC827-R cells showed significant resistance to
erlotinib. A549 and H460 cells, both of EGFR wild-type, were
relatively resistant to erlotinib. H1975 cells, with two mutations
in the EGFR tyrosine kinase domain (L858R and T790M), showed
further resistance to erlotinb than A549 or H460 cells (Fig. 1A and B). In western blotting
results, HCC827 cells showed abundant basal EGFR expression, while
the basal EGFR expression of A549, H460 and HCC827-R cells was
moderate. H1975 cells showed only slight EGFR expression. We tested
the basal autophagy level by measuring the conversion of the
LC3/Atg8 from the cytoplasmic form LC3-I, to the autophagosomic
form LC3-II. The results showed that HCC827-R cells expressed
considerable level of LC3-II, indicating an active basal autophagy
activity. H1975 cells also showed easily detectable amounts of
conversion from LC3-I to LC3-II, while the other three cells showed
little conversion (Fig. 1C). In
line with the expression of LC3-II, HCC827, A549, H460, and H1975
cells showed increasing protein level of beclin-1, which was
negatively correlated with the cell sensitivity to erlotinib.
However, HCC827-R cells showed slight beclin-1 expression,
similarly to HCC827 cells. Given that autophagy is an inducible
process under survival stress and the active autophagy activity of
HCC827-R and H1975 was generated in a well-cultured environment,
just like the other three cell lines, we reasoned that the high
basal autophagy levels of HCC827-R and H1975 were not
stress-induced, but an intrinsic character and involved in their
resistance to erlotinib.
Autophagy is induced in cancer cells
after erlotinib treatment
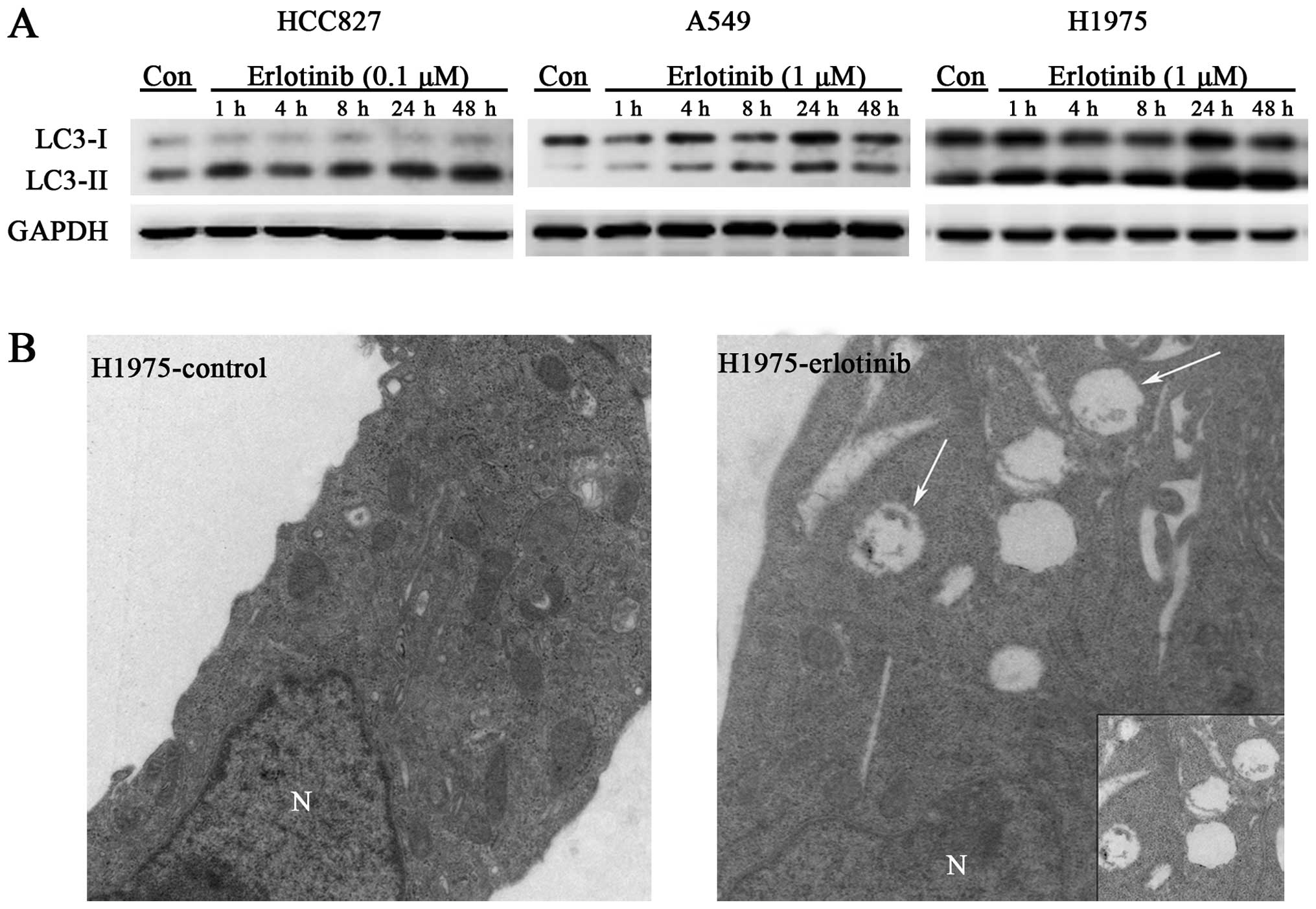
Autophagy was induced under treatment of erlotinib
in three cell lines, HCC827, A549, and H1975. With HCC827 cells,
0.1 μM of erlotinib induced marked autophagy just 1 h after
exposure and lasted for >48 h. Of note, after 4 h of erlotinib
exposure, HCC827 cells showed less conversion of LC3 compared with
that of 1-h exposure, but then the conversion of LC3 turned to
increase from 4 to 48 h persistently. With A549 cells, 1 μM of
erlotinib induced a time-dependent accumulation of autophagy, with
the highest level achieved at 24 h. Similar with A549, H1975 cells
also showed a time-dependent increase of autophagy under the
treatment of erlotinib (1 μM) (Fig.
2A). We also measured the punctate fluorescence after erlotinib
treatment in the cells transfected with a GFP-tagged LC3 construct.
In all three cell lines, erlotinib induced more punctate
fluorescence than did the control cells, which showed only slight
fluorescence (Fig. 3C). This
result was further confirmed with transmission electron microscopy,
which revealed autophagosomes in H1975 cells after 48-h erlotinib
treatment, whereas autophagosomes were scarce in untreated cells
(Fig. 2B). Together, these
findings suggest that erlotinb treatment induces autophagy in both
EGFR-TKI-sensitive and -resistant cancer cells.
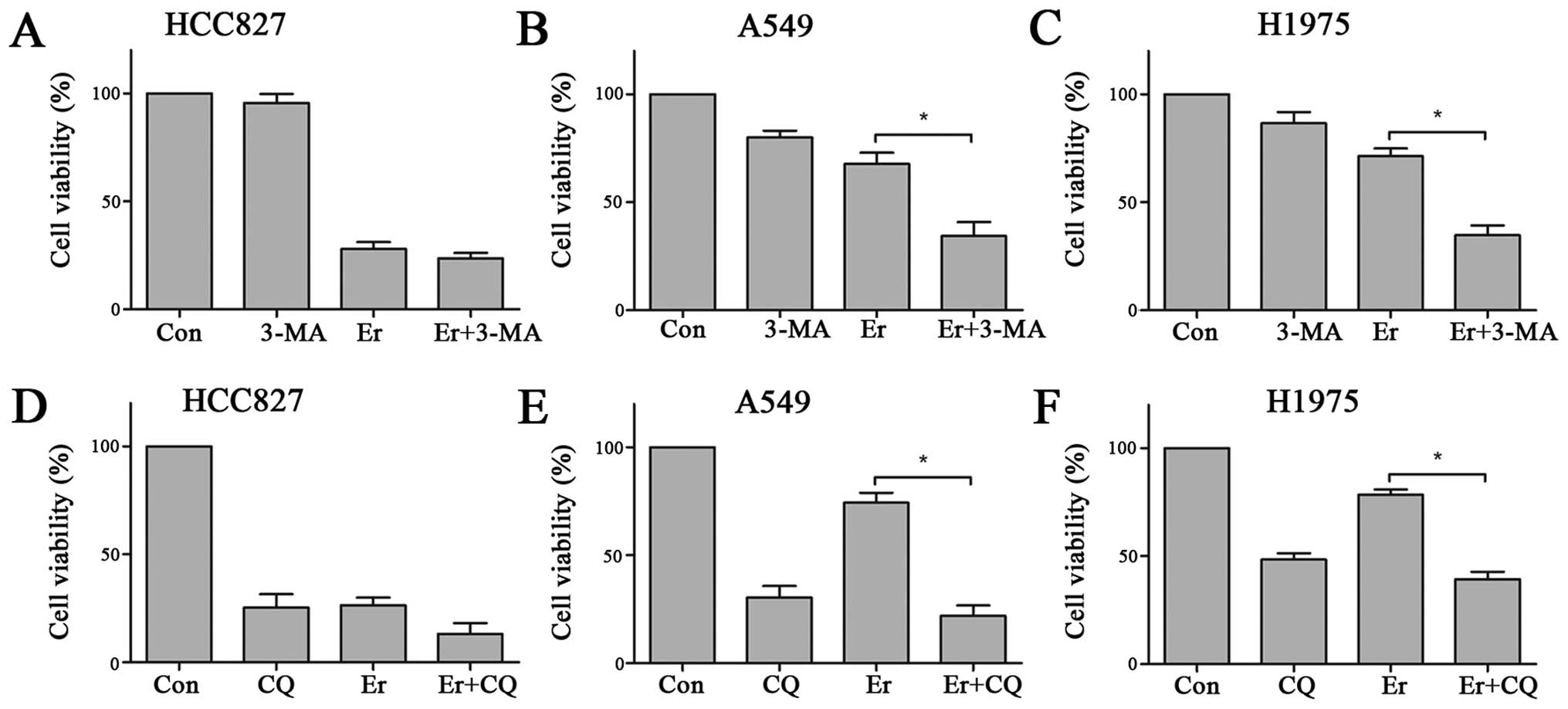
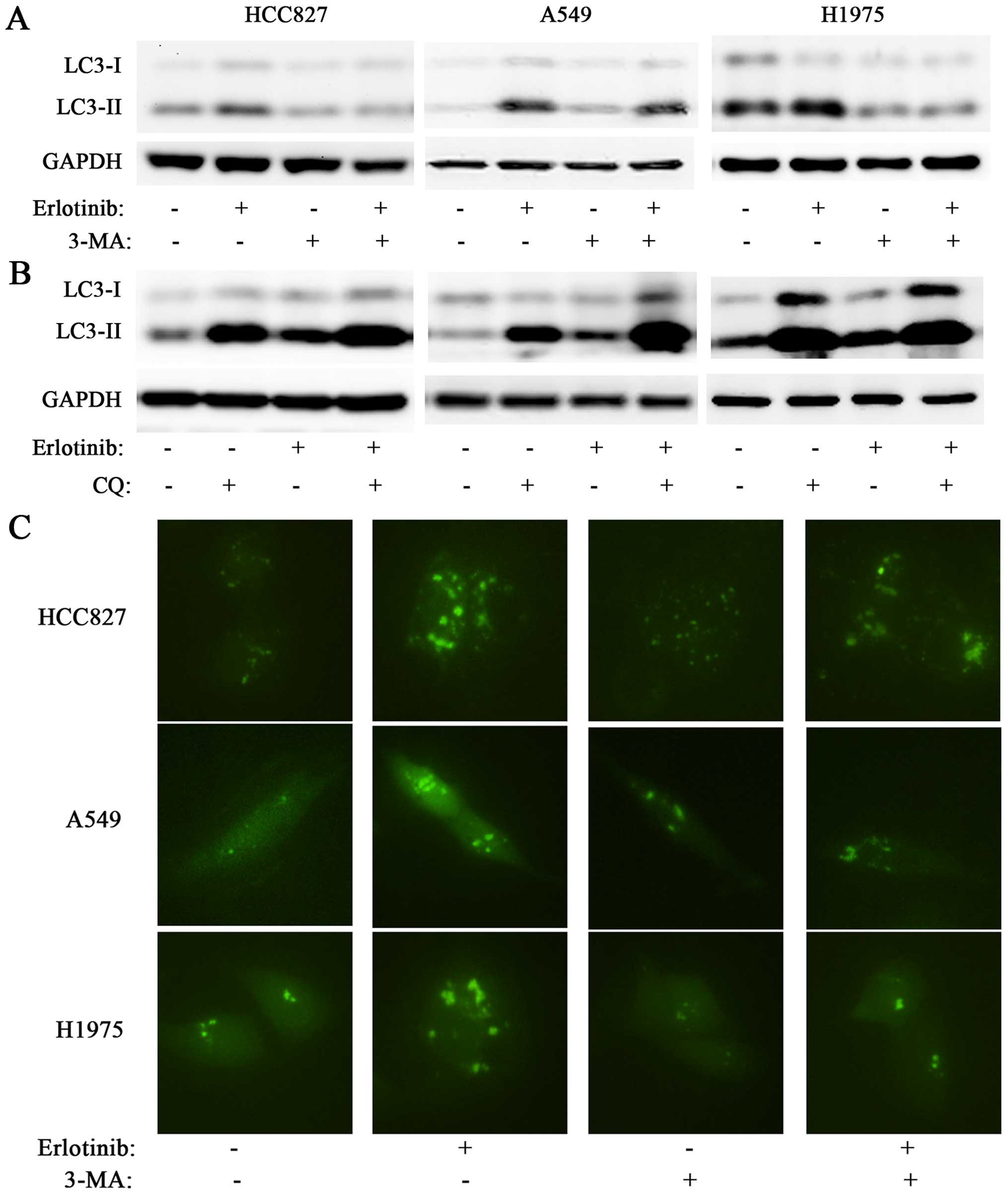 | Figure 3(A) HCC827, A549, and H1975 cells
were treated with erlotinib (0.1 μM for HCC827, 1 μM for A549 and
H1975), 3-MA (5 mM), or a combination of both for 48 h. Western
blotting showed that treatment with 3-MA reversed erlotinib-induced
increase of LC3-II in all three cell lines. (B) HCC827, A549, and
H1975 cells were treated erlotinib (0.1 μM for HCC827, 1 μM for
A549 and H1975), chloroquine (CQ, 10 μM), or a combination of both
for 48 h. In all three cell lines, western blotting showed that CQ
treatment significantly increased the LC3-II level, while the
combination of CQ and erlotinib induced a further accumulation of
LC3-II as compared with CQ single treatment. (C) HCC827, A549, and
H1975 cells were transfected with a GFP-LC3 construct. In all the
three cell lines, photographs from fluorescence microscopy showed
that 3-MA reversed the erlotinib-induced punctate fluorescence
increase. |
3-MA and CQ abates erlotinib-induced
autophagy
As a classic autophagy inhibitor, 3-MA was tested
for the ability to abate erlotinib-induced autophagy. With western
blotting, 3-MA was able to diminish the conversion from LC3-I to
LC3-II in HCC827, A549, and H1975 cells. The combination of 3-MA
and erlotinib markedly decreased the erlotinib-induced conversion
from LC3-I to LC3-II, nearly to the same level with 3-MA single
treatment (Fig. 3A). This result
was further confirmed by measuring the punctate fluorescence in the
cells transfected with a GFP-tagged LC3 construct. In all three
cell lines, 3-MA significantly downregulated the erlotinib-induced
LC3-GFP protein accumulation (Fig.
3C). We also used CQ, an autophagy-lysosomal inhibitor, to
block erlotinib-induced autophagy. While 3-MA is able to inhibit
autophagosome formation, CQ functions by blocking the degradation
of autophagosomes and thereby induces accumulation of LC3-II. In
this experiment, treatment of CQ induced abundant accumulation of
LC3-II in all three cell lines. The combination of CQ and erlotinib
showed still further LC3-II accumulation than CQ alone, indicating
that CQ blocked the degradation of erlotinib-induced autophagosomes
(Fig. 3B). Moreover, this result
illustrates that the erlotinib-induced LC3-II accumulation was a
reflection of increased autophay flux, rather than dysfunction of
lysosomes.
Combination with autophagy inhibition
enhances erlotinib-induced cytotoxicity
As autophagy might be involved in the resistance of
H1975 cells to erlotinib, we tested the synergistic effect of
autophagy inhibition and EGFR-TKI by combining 3-MA with erlotinib.
With CCK8 tests, 3-MA and erlotinib co-treatment showed significant
synergistic cytotoxicity to H1975 cells while 3-MA single treatment
showed only slight influence (Fig.
4C). Consistently, A549 cells showed much higher sensitivity to
the co-treatment of 3-MA and erlotinib than treatment of 3-MA or
erlotinib alone (Fig. 4B). HCC827
was naturally sensitive to erlotinb treatment, but resistant to
3-MA (Fig. 4A). The effect of CQ
was examined as well. To our surprise, HCC827, A549, and H1975
cells were all extraordinarily sensitive to CQ. Under CQ (10 μM)
exposure for 72 h, the cell viabilities decreased to 25.4±6.2% for
HCC827 cells, 30.4±5.3% for A549 cells, and 48.5±2.9% for H1975
cells. For H1975 and A549 cells, the combination of CQ and
erlotinib showed increased cytotoxicity compared with CQ single
exposure, but failed to reach statistical difference. However, when
compared with erlotinib single treatment, co-administration of CQ
and erlotinib significantly enhanced the inhibition of cell
proliferation. For HCC827 cells, erlotinib, with or without CQ, was
able to dramatically inhibit the cell proliferation, with the
combination treatment showed further significant effect (Fig. 4D–F).
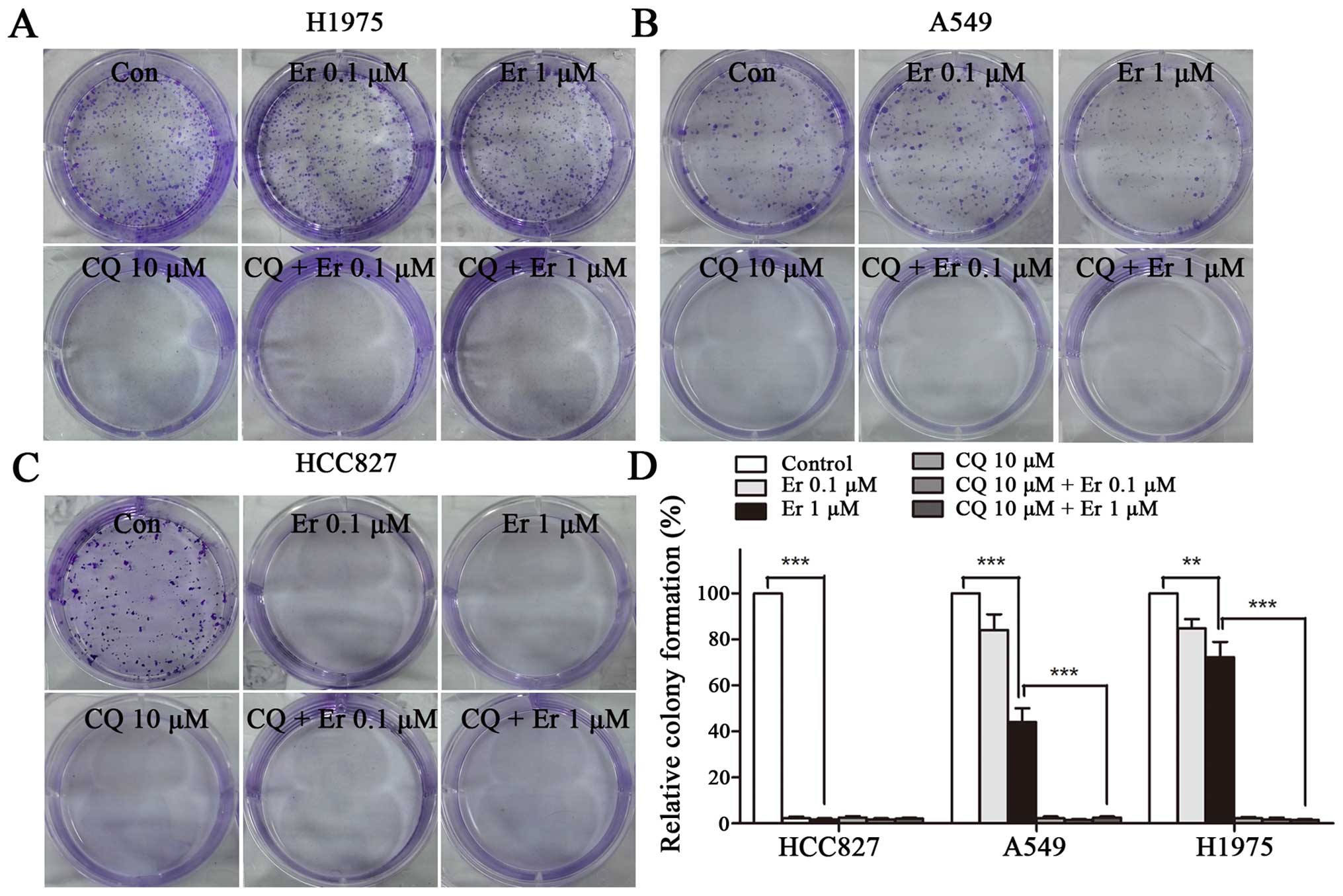
The impact of chemicals on colony formation was
further measured. HCC827, A549, and H1975 cells were treated with
10 μM CQ, 0.1 μM erlotinib, 1 μM erlotinib, or the combination of
CQ and erlotinib for 10–14 days. It was found that 0.1 μM erlotinib
caused 16 and 15% reduction of cell colonies in A549 and H1975
cells, respectively. While, 1 μM erlotinib, respectively caused 56
and 28% reduction of cell colonies in A549 and H1975 cells. In
HCC827 cells, 0.1 or 1 μM erlotinib showed nearly complete
elimination of colonies formation. Remarkably, CQ alone or in
combination with erlotinib caused nearly complete inhibition of
cell growth in all the three cell lines (Fig. 5). These findings indicate that
inhibition of autophagy can enhance erlotinib sensitivity in
sensitive NSCLC cells and overcome erlotinb resistance in resistant
NSCLC cells.
Combination of autophagy inhibition and
erlotinib induces ER stress and apoptosis
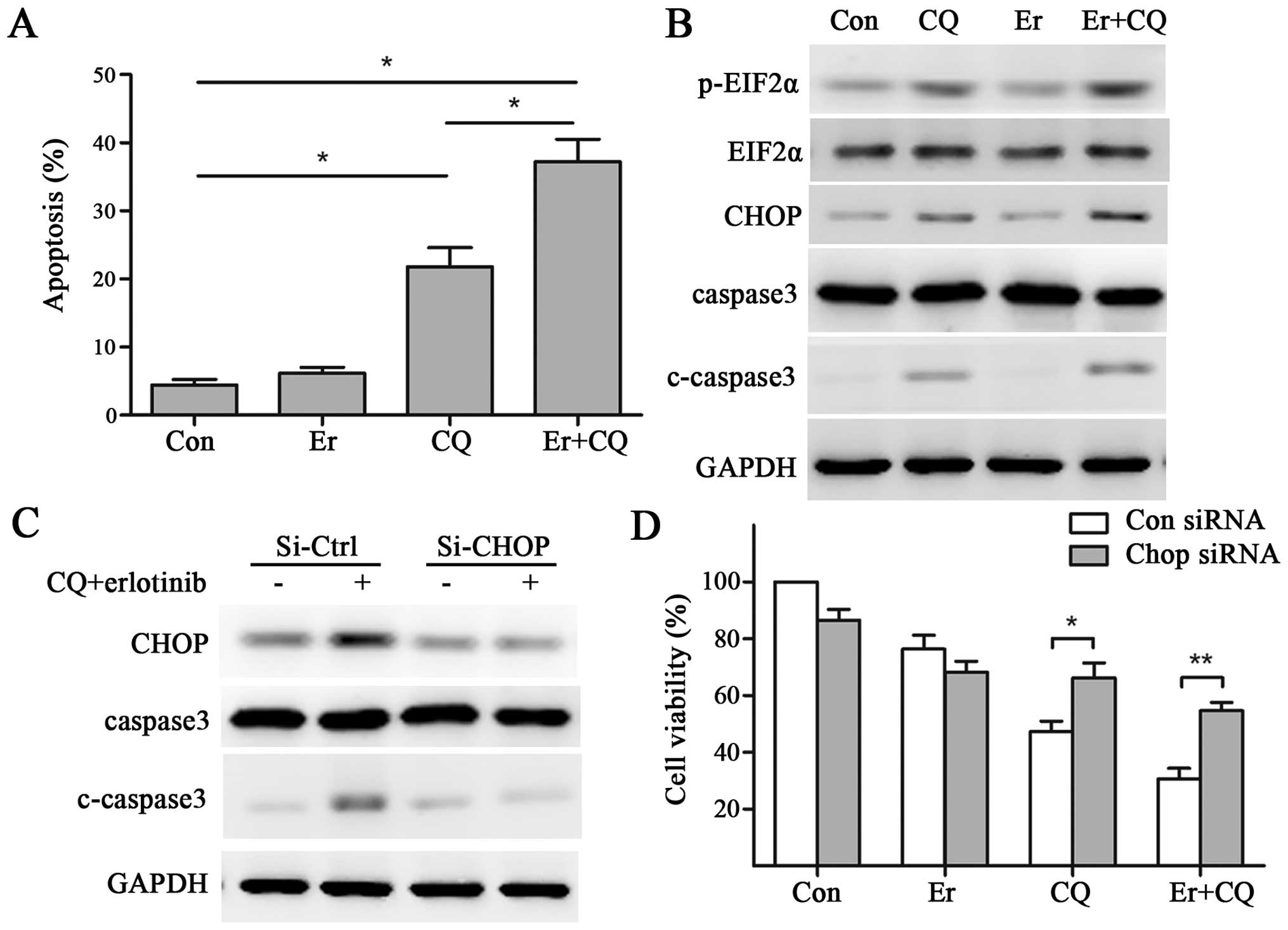
We tested whether combination of erlotinib and
inhibition of autophagy with CQ would induce apoptosis in
EGFR-TKI-resistant cells. To this aim, H1975 was treated with CQ,
erlotinib, alone or in combination for 48 h, and then measured for
apoptosis with flow cytometry by quantitatively measuring Annexin V
and PI. The results showed CQ induced statistically significant
induction of apoptosis, but erlotinib did not. The combination of
CQ and erlotinib also induced apoptosis, and this effect was
significantly greater than apoptosis seen with CQ single treatment
(Fig. 6A). Consistent with the
flow cytometry results, western blot analyses revealed that CQ
markedly induced the activation of caspase 3, a critical
executioner of apoptosis, which was further enhanced by combination
of CQ and erlotinib (Fig. 6B).
The impact of autophagy inhibition and erlotinib on
triggering ER stress was analysed by measuring its well-documented
markers, CHOP and the phosphorylated form of the initiation factor
2-α (p-eIF2α). CHOP is an ER-stress associated proapoptotic marker,
and eIF2α is a translation initiation factor phosphorylated by
protein kinase RNA-like endoplasmic reticulum kinase (PERK) in
response to ER stress. The protein levels of p-eIF2α and CHOP
increased in H1975 cells after treatment with CQ for 48 h, whereas
the 48-h treatment of erlotinib showed no change. By contrast,
co-administration of CQ and erlotinib induced a more significant
increase of p-eIF2α and CHOP compared with CQ single treatment
(Fig. 6B). These data suggest that
CQ with or without erlotinib treatment could induce apoptosis and
trigger ER stress.
CHOP knockdown counteracts the
cytotoxicity and apoptosis induced by co-treatment of CQ and
erlotinib
To gain further insight into the role of ER stress
in CQ and erlotinib co-treatment induced cytotoxicity, the effects
of knockdown of CHOP was tested. At 24 h after CHOP siRNA
transfection, H1975 cells was exposed to CQ, erlotinib, or the
combination of both chemicals for additional 48 h for western blot
analyses or 72 h for CCK8 tests. As a result, knockdown of CHOP
blocked the CQ-erlotinib co-treatment induced CHOP upregulation.
Also, the cleavage of caspase 3 induced by CQ-erlotinib
co-treatment was significantly diminished, indicating blockage of
apoptosis caused by CQ-erlotinib co-treatment (Fig. 6C). In CCK8 tests, knockdown of CHOP
significantly counteracted the cytotoxicity caused by CQ or
CQ-erlotinib co-treatment, while the effect of erlotinib single
treatment was not obviously affected (Fig. 6D).
Discussion
In this study, we found that erlotinib induced
autophagy in three lung cancer cell lines, HCC827, A549, and H1975
by detecting conversion from LC3-I to LC3-II with western blotting.
The GFP-LC3 construct and transmission electron microscopy were
used to further confirm the autolysosome formation after erlotinib
exposure. Two kinds of autophagy inhibitors, 3-MA and CQ, were used
to ablate the induction of autophagy under exposure of erlotinib.
Synergistic effect between erlotinib and 3-MA/CQ were found in all
three cell lines, with CQ single treatment presenting much more
cytotoxicity than 3-MA, indicating different mechanisms for
blocking autophagy may associate with their distinct cytotoxicity.
Co-treatment of CQ and erlotinib induced ER stress in H1975 cells,
which evoked apoptosis through modulation of ER stress. Inhibition
of CHOP upregulation alleviated apoptosis and counteracted
cytotoxicity caused by CQ or CQ-erlotinib co-administration.
EGFR-EKI induces autophagy
Accumulating studies have reported that anticancer
therapies, including radiotherapy, chemotherapy and targeted
therapies, could induce autophagy in different tumor cell lines
(19–22). Though the correlation between
anticancer therapies and autophagy has been proposed, the role of
autophagy in cancer treatment is complex. On the one hand, the role
of autophagy is pro-survival and contributes to therapy resistance
through removal of damaged cell components and proteins to help
cancer cells survive under stress. On the other hand, autophagy is
pro-death and contributes to therapy effect by enhancing induction
of apoptosis or mediating ‘autophagic cell death’. One theory to
explain this double-faced role of autophagy in cancer treatment is
that there is a checkpoint in therapy induction of autophagy. As a
balance keeper, modest autophagy induction could eliminate damage
and recycle energy to promote cancer cells survival. While
excessive autophagy may induce vital protein or organelle lysis,
triggering apoptosis or autophagic cell death, known as the second
programmed cell death.
EGFR-TKIs, was reported to induce marked autophagy
in sensitive cancer cells, like HCC827 and PC-9 (14,18,22).
In EGFR-TKI-resistant cancer cells, most authors presented that
EGFR-TKI was able to induce autophagy, but Li et al reported
that EGFR-TKI failed to induce autophagy in resistant cells
(23). Apart from targeting EGFR,
other TKIs, such as imatinib (targeting BCR ABL, KIT, and PDGFR),
dasatinib (targeting BCRABL, KIT, and SRC), and sorafenib (a
multi-tyrosine-kinase inhibitor), could induce autophagy in
distinct tumor cell lines as well (24–26).
Furthermore, the anti-EGFR antibody cetuximab was reported to
induce autophagy through inhibiting the class I PI3K/Akt/mTOR
pathway (13). In this study, we
found that erlotinib, a well-used EGFR-TKI, elicited a rapid and
robust induction of autophagy in HCC827, and caused a relatively
slow but persistent induction of autophagy in A549 and H1975 cells,
as demonstrated by the increased conversion of LC3. We also found
that EGFR-TKI resistance was positively correlated with baseline
autophagy level. As compared with the parental HCC827 cells,
HCC827-R cells showed significant resistance to erlotinib and much
more conversion of LC3. Consistently, A549, H460, and H1975 cells
showed increasing conversion of LC3 and exaggerating resistance to
erlotinib. In line with this point, Guo et al observed that
patients with low LC3 expression had a higher objective response
rates in advanced colorectal cancer patients treated with
cetuximab-containing chemotherapy, indicating that autophagy may
reduce cetuximab efficacy in patients with colorectal cancer
(27).
Autophagy and EGFR-TKI resistance
EGFR-TKI acquired resistance is the major failure in
treatment of NSCLC, in which half of resistance can be attributed
to EGFR T790M mutation (3).
However, inhibition of signaling molecules downstream of EGFR did
not enhance the sensitivity of NSCLC cells with EGFR T790M mutation
to EGFR-TKIs, indicating that there may be another pathway
mediating EGFR-TKI resistance (2,28).
Several studies demonstrated that autophagy inhibition acted
synergistically with EGFR-TKIs to induce lung cancer cells death
(18,22,29).
For example, Zou et al (18) reported that erlotinib, at a
clinically relevant concentration (2 μM) induced autophagy in NSCLC
cells with wild-type EGFR and inhibition of autophagy with CQ
enhanced erlotinib cytotoxicity in vitro and in vivo.
In this study, we confirmed the synergistic effect of autophagy
inhibition and erlotinib with 3-MA and CQ, two distinct inhibitors
of autophagy, in three NSCLC cell lines with different types of
EGFR mutation. 3-MA, a PtdIns3K inhibitor that effectively blocks
an early stage of autophagy, reversed erlotinib-induced autophagy,
demonstrated by decline of LC3 conversion. 3-MA was able to
statistically significantly increase the cytotoxicity of erlotinib
in A549 and H1975 cells.
CQ is an FDA approved drug used for malaria,
rheumatoid arthritis and other autoimmune diseases, with an
established history of generally well-tolerated clinical use. CQ is
a weak base with hydrophobic characteristics that diffuses into the
lysosomes of cells, where it becomes protonated and trapped, thus
leading to increase in lysosomal pH. These CQ-loaded lysosomes can
no longer fuse with autophagosomes, thus blocking autophagy at a
late stage (30,31). In this study, CQ not only enhanced
erlotinib cytotoxicity, but also induced significant cell death in
HCC827, A549, and H1975 cells by single treatment, which was quite
different from the effect of the another autophagy inhibitor 3-MA.
The distinct mechanisms for blocking autophagy may account for this
inconformity. However, studies on cytotoxicity of CQ on cancer
cells were not consistent, some claimed that CQ single treatment
was able to induce marked cytotoxicity to cancer cells (32–36),
but others reported that CQ single treatment showed only modest
cytotoxicity (13,29). To explain this contradiction, cell
line difference may be an element, while distinct CQ concentration
and exposure time may be the main cause.
ER stress, apoptosis and autophagy
ER responds to ER stress through translational
attenuation, enhancement of protein folding ability, and
degradation of unfolded proteins by a quality-control system.
However, when the stress exceeds the ability of ER to process,
apoptotic signals are elicited (37–39).
CHOP plays an important role in ER stress-induced apoptosis as the
fact that CHOP−/− mice exhibit reduced apoptosis in
response to ER stress (40). A
link between autophagy and ER stress has been substantiated by the
finding that the PERK-eIF2α-CHOP pathway is essential for autophagy
induction under ER stress, in which CHOP was able to
transcriptionally regulate more than a dozen ATG genes (41,42).
On the contrary, elicitation of ER stress demonstrated by increased
IRE1 activity was observed in ATG-knockout mice, indicating
dysregulated autophagy may also trigger ER stress as a feedback
mechanism (43,44). In this study, we found that
autophagy inhibition by CQ elicited a slight induction of ER
stress, demonstrated by increased eIF2α phosphorylation.
Combination of CQ and erlotinib further increased eIF2α
phosphorylation, which induced apoptosis through upregulation of
CHOP. This autophagy inhibition-induced ER stress offered a further
validation for the ER stress-autophagy feedback paradigm and a new
interpretation for the synergistic effect of autophagy inhibition
and erlotinib.
In conclusion, the role of autophagy in cancer
biology and cancer treatment is complex, and related studies have
shown consistent, but also contradictory, conclusions. In this
study, we confirmed the ability of erlotinib to induce autophagy in
not only EGFR-TKI-sensitive but also EGFR-TKI-resistant cancer
cells. Inhibition of autophagy acts synergistically with erlotinib
to overcome erlotinib resistance through modulation of ER stress
induced apoptosis, in which CHOP acts as a key effector molecule.
Together, autophagy is elicited as a cytoprotective process when
cancer cells are facing erlotinib exposure, and inhibition of
autophagy may represent a promising therapeutic strategy to
overcome erlotinib resistance in NSCLC.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (no. 81302020).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sui X, Kong N, Zhu M, Wang X, Lou F, Han W
and Pan H: Cotargeting EGFR and autophagy signaling: A novel
therapeutic strategy for non-small-cell lung cancer. Mol Clin
Oncol. 2:8–12. 2014.PubMed/NCBI
|
|
3
|
Camidge DR, Pao W and Sequist LV: Acquired
resistance to TKIs in solid tumours: Learning from lung cancer. Nat
Rev Clin Oncol. 11:473–481. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee JY, Lim SH, Kim M, Kim S, Jung HA,
Chang WJ, Choi MK, Hong JY, Lee SJ, Sun JM, et al: Is there any
predictor for clinical outcome in EGFR mutant NSCLC patients
treated with EGFR TKIs? Cancer Chemother Pharmacol. 73:1063–1070.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Park S, Langley E, Sun JM, Lockton S, Ahn
JS, Jain A, Park K, Singh S, Kim P and Ahn MJ: Low EGFR/MET ratio
is associated with resistance to EGFR inhibitors in non-small cell
lung cancer. Oncotarget. 6:30929–30938. 2015.PubMed/NCBI
|
|
6
|
Presutti D, Santini S, Cardinali B, Papoff
G, Lalli C, Samperna S, Fustaino V, Giannini G and Ruberti G: MET
gene amplification and MET receptor activation are not sufficient
to predict efficacy of combined MET and EGFR inhibitors in EGFR
TKI-resistant NSCLC cells. PLoS One. 10:e01433332015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ricciuti B, Mecca C, Cenci M, Leonardi GC,
Perrone L, Mencaroni C, Crinò L, Grignani F, Baglivo S, Chiari R,
et al: miRNAs and resistance to EGFR-TKIs in EGFR-mutant non-small
cell lung cancer: Beyond ‘traditional mechanisms’ of resistance. E
Cancer Med Sci. 9:5692015.
|
|
8
|
Chong CR and Jänne PA: The quest to
overcome resistance to EGFR-targeted therapies in cancer. Nat Med.
19:1389–1400. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Amaravadi RK, Lippincott-Schwartz J, Yin
XM, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT and White E:
Principles and current strategies for targeting autophagy for
cancer treatment. Clin Cancer Res. 17:654–666. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang ZJ, Chee CE, Huang S and Sinicrope F:
Autophagy modulation for cancer therapy. Cancer Biol Ther.
11:169–176. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Paglin S, Hollister T, Delohery T, Hackett
N, McMahill M, Sphicas E, Domingo D and Yahalom J: A novel response
of cancer cells to radiation involves autophagy and formation of
acidic vesicles. Cancer Res. 61:439–444. 2001.PubMed/NCBI
|
|
12
|
Ertmer A, Huber V, Gilch S, Yoshimori T,
Erfle V, Duyster J, Elsässer HP and Schätzl HM: The anticancer drug
imatinib induces cellular autophagy. Leukemia. 21:936–942.
2007.PubMed/NCBI
|
|
13
|
Li X and Fan Z: The epidermal growth
factor receptor antibody cetuximab induces autophagy in cancer
cells by downregulating HIF-1alpha and Bcl-2 and activating the
beclin 1/hVps34 complex. Cancer Res. 70:5942–5952. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Han W, Pan H, Chen Y, Sun J, Wang Y, Li J,
Ge W, Feng L, Lin X, Wang X, et al: EGFR tyrosine kinase inhibitors
activate autophagy as a cytoprotective response in human lung
cancer cells. PLoS One. 6:e186912011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Suh DH, Kim MK, Kim HS, Chung HH and Song
YS: Unfolded protein response to autophagy as a promising druggable
target for anticancer therapy. Ann NY Acad Sci. 1271:20–32. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Biol. 8:519–529. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Harding HP, Zhang Y, Bertolotti A, Zeng H
and Ron D: Perk is essential for translational regulation and cell
survival during the unfolded protein response. Mol Cell. 5:897–904.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zou Y, Ling YH, Sironi J, Schwartz EL,
Perez-Soler R and Piperdi B: The autophagy inhibitor chloroquine
overcomes the innate resistance of wild-type EGFR non-small-cell
lung cancer cells to erlotinib. J Thorac Oncol. 8:693–702. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hu YL, Jahangiri A, Delay M and Aghi MK:
Tumor cell autophagy as an adaptive response mediating resistance
to treatments such as antiangiogenic therapy. Cancer Res.
72:4294–4299. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jo GH, Bogler O, Chwae YJ, Yoo H, Lee SH,
Park JB, Kim YJ, Kim JH and Gwak HS: Radiation-induced autophagy
contributes to cell death and induces apoptosis partly in malignant
glioma cells. Cancer Res Treat. 47:221–241. 2015. View Article : Google Scholar :
|
|
21
|
Sui X, Chen R, Wang Z, Huang Z, Kong N,
Zhang M, Han W, Lou F, Yang J, Zhang Q, et al: Autophagy and
chemotherapy resistance: A promising therapeutic target for cancer
treatment. Cell Death Dis. 4:e8382013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sugita S, Ito K, Yamashiro Y, Moriya S,
Che XF, Yokoyama T, Hiramoto M and Miyazawa K: EGFR-independent
autophagy induction with gefitinib and enhancement of its cytotoxic
effect by targeting autophagy with clarithromycin in non-small cell
lung cancer cells. Biochem Biophys Res Commun. 461:28–34. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li YY, Lam SK, Mak JC, Zheng CY and Ho JC:
Erlotinib-induced autophagy in epidermal growth factor receptor
mutated non-small cell lung cancer. Lung Cancer. 81:354–361. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gupta A, Roy S, Lazar AJ, Wang WL,
McAuliffe JC, Reynoso D, McMahon J, Taguchi T, Floris G,
Debiec-Rychter M, et al: Autophagy inhibition and antimalarials
promote cell death in gastrointestinal stromal tumor (GIST). Proc
Natl Acad Sci USA. 107:14333–14338. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Milano V, Piao Y, LaFortune T and de Groot
J: Dasatinib-induced autophagy is enhanced in combination with
temozolomide in glioma. Mol Cancer Ther. 8:394–406. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Martin AP, Park MA, Mitchell C, Walker T,
Rahmani M, Thorburn A, Häussinger D, Reinehr R, Grant S and Dent P:
BCL-2 family inhibitors enhance histone deacetylase inhibitor and
sorafenib lethality via autophagy and overcome blockade of the
extrinsic pathway to facilitate killing. Mol Pharmacol. 76:327–341.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Guo GF, Jiang WQ, Zhang B, Cai YC, Xu RH,
Chen XX, Wang F and Xia LP: Autophagy-related proteins Beclin-1 and
LC3 predict cetuximab efficacy in advanced colorectal cancer. World
J Gastroenterol. 17:4779–4786. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Moreira-Leite FF, Harrison LR, Mironov A,
Roberts RA and Dive C: Inducible EGFR T790M-mediated gefitinib
resistance in non-small cell lung cancer cells does not modulate
sensitivity to PI103 provoked autophagy. J Thorac Oncol. 5:765–777.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tang MC, Wu MY, Hwang MH, Chang YT, Huang
HJ, Lin AM and Yang JC: Chloroquine enhances gefitinib cytotoxicity
in gefitinib-resistant nonsmall cell lung cancer cells. PLoS One.
10:e01191352015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Klionsky DJ, Abeliovich H, Agostinis P,
Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA,
Ballabio A, et al: Guidelines for the use and interpretation of
assays for monitoring autophagy in higher eukaryotes. Autophagy.
4:151–175. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Morgan MJ, Gamez G, Menke C, Hernandez A,
Thorburn J, Gidan F, Staskiewicz L, Morgan S, Cummings C, Maycotte
P, et al: Regulation of autophagy and chloroquine sensitivity by
oncogenic RAS in vitro is context-dependent. Autophagy.
10:1814–1826. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kimura T, Takabatake Y, Takahashi A and
Isaka Y: Chloroquine in cancer therapy: A double-edged sword of
autophagy. Cancer Res. 73:3–7. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hu T, Li P, Luo Z, Chen X, Zhang J, Wang
C, Chen P and Dong Z: Chloroquine inhibits hepatocellular carcinoma
cell growth in vitro and in vivo. Oncol Rep. 35:43–49. 2016.
|
|
34
|
Fan C, Wang W, Zhao B, Zhang S and Miao J:
Chloroquine inhibits cell growth and induces cell death in A549
lung cancer cells. Bioorg Med Chem. 14:3218–3222. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zheng Y, Zhao YL, Deng X, Yang S, Mao Y,
Li Z, Jiang P, Zhao X and Wei Y: Chloroquine inhibits colon cancer
cell growth in vitro and tumor growth in vivo via induction of
apoptosis. Cancer Invest. 27:286–292. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang A, Rajeshkumar NV, Wang X, Yabuuchi
S, Alexander BM, Chu GC, Von Hoff DD, Maitra A and Kimmelman AC:
Autophagy is critical for pancreatic tumor growth and progression
in tumors with p53 alterations. Cancer Discov. 4:905–913. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar
|
|
38
|
Wolff S, Weissman JS and Dillin A:
Differential scales of protein quality control. Cell. 157:52–64.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Buchberger A, Bukau B and Sommer T:
Protein quality control in the cytosol and the endoplasmic
reticulum: Brothers in arms. Mol Cell. 40:238–252. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Oyadomari S, Koizumi A, Takeda K, Gotoh T,
Akira S, Araki E and Mori M: Targeted disruption of the Chop gene
delays endoplasmic reticulum stress-mediated diabetes. J Clin
Invest. 109:525–532. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
B'chir W, Maurin AC, Carraro V, Averous J,
Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P and Bruhat
A: The eIF2α/ATF4 pathway is essential for stress-induced autophagy
gene expression. Nucleic Acids Res. 41:7683–7699. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Senft D and Ronai ZA: UPR, autophagy, and
mitochondria crosstalk underlies the ER stress response. Trends
Biochem Sci. 40:141–148. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Deegan S, Saveljeva S, Gorman AM and
Samali A: Stress-induced self-cannibalism: On the regulation of
autophagy by endoplasmic reticulum stress. Cell Mol Life Sci.
70:2425–2441. 2013. View Article : Google Scholar
|
|
44
|
Adolph TE, Tomczak MF, Niederreiter L, Ko
HJ, Böck J, Martinez-Naves E, Glickman JN, Tschurtschenthaler M,
Hartwig J, Hosomi S, et al: Paneth cells as a site of origin for
intestinal inflammation. Nature. 503:272–276. 2013.PubMed/NCBI
|