Introduction
Hepatocellular carcinoma (HCC), a common solid
tumor, is the third most frequent cause of cancer-related death in
the world. Hepatitis B virus (HBV) infection is the main cause of
HCC in China (1). Individuals with
chronic HBV infection, especially those who have progressed to
chronic liver disease and cirrhosis, are at high risk of developing
HCC (2,3). Most HCC patients are diagnosed at
their advancing stage and refractory to chemotherapy and
radiotherapy (4,5). Even if the patients receive liver
transplantation, the recurrence rate is still high (6).
Epigenetic modifications are found to play important
roles in various biological processes especially in cancer
development (7). Methylation of
DNA at 5-position of cytosines (5-mC) is a key epigenetic mark that
has been extensively studied in many types of malignancies
(8). Aberrant DNA methylation of
promoter CpG islands has been associated with global
hypomethylation and specific loci hypermethylation, which has the
potential to become diagnostic markers for the progression of
malignant tumors (9). 5-mC can be
converted to 5-hydroxymethylcytosine (5-hmC) by the ten-eleven
translocation (TET) family proteins. In mammals, 5-hmC is detected
in almost all tissues and cell types (10,11).
Emerging evidence has shown that 5-hmC and TET family might serve
unique biological roles in many biological processes such as gene
expression regulation, gene transcription and DNA methylation
regulation (12,13). Several studies have found 5-hmC
alternations in the epigenetic regulation of various diseases,
including cancer (14).
Studies of DNA methylation changes in HCC have led
to the identification of several candidate methylated genes as
potential tumor biomarkers (15,16),
yet, little is known about hydroxymethylation distribution in HCC.
In previous studies, DNA methylation was determined using
methylation sensitive polymerase chain reaction combined bisulfite
restriction analysis (COBRA) or bisulfite sequencing techniques.
With the development of high-throughput sequencing technologies,
the whole-genome DNA (hydroxy)methylation profiling in cancer has
generated data with significantly higher quantity and quality
(17,18). However, most existing studies of
DNA methylation in HCC employed Infinium HumanMethylation BeadChip
Arrays or Methylation Microarray (19,20),
which may have some limitations on resolution and scope. A novel
method termed (hydroxy)methylated DNA immunoprecipitation
sequencing [(h)MeDIP-Seq], combining DNA immunoprecipitation with
high-throughput sequencing, has emerged as an advantageous tool for
identifying (hydroxy) methylated CpG-rich sequences in a much
faster and more sensitive manner than ever before.
In an attempt to explore the 5-mC/5-hmC changes in
HCC, we performed a genome-wide mapping of 5-mC/5-hmC in four
paired HCC tissues and adjacent peritumor tissues (APTs) using
MeDIP-Seq/hMeDIP-Seq.
Materials and methods
Clinical samples
Total of 4 fresh-frozen primary HCC tissues and
paired APTs were included in MeDIP-Seq/hMeDIP-Seq. The collected
cancer tissues were excised within the margins of the cancer
lesion, and the APTs were collected from a location at least 3 cm
distant from the tumor boundaries. All the collections followed the
same protocol. All of the cancerous tissues were diagnosed as
primary hepatocellular carcinoma, provided by two independent and
experienced pathologists. Fresh-frozen HCC tissues and APTs were
collected during the surgical resection.
The four HCC patients were HBV surface
antigen-positive without hepatitis C virus (HCV) infection and
exhibited the same cirrhosis etiology. Retrospectively data were
collected including demographic, preoperative laboratory and
pathological parameters from electronic medical records, and are
summarized in Table I.
 | Table IClinicopathological features for the
four HCC patients included in the study. |
Table I
Clinicopathological features for the
four HCC patients included in the study.
| Variables | SAM 1 | SAM 2 | SAM 3 | SAM 4 |
|---|
| Case number | 560 | 629852 | 716677 | 717323 |
| Age (years) | 41 | 53 | 40 | 57 |
| Gender | Male | Male | Male | Male |
| ALT (U/l) | 35 | 30 | 54 | 34 |
| AFP(ng/ml) | 2 | >50000 | 12628.5 | 8241 |
| HBV-DNA | 104 | 103 | 103 | 103 |
| Tumor size
(cm) | 2×2 | 7.5×8 | 3.5×3 | 8.5×8.5 |
| Tumor number | Single | Multiple | Single | Single |
| PVTT | No | Yes | Yes | Yes |
| Grade | Moderate | Poor | Moderate | Moderate |
DNA extraction
Genomic DNA was extracted from frozen HCC tissues
and paired APTs using the DNeasy Blood and Tissue kit (Qiagen;
69504) according to the manufacturer's protocol. Briefly, tissues
were homogenized using a hand-held homogenizer, digested with
Proteinase K (Qiagen; 69504) and RNase A (Qiagen; 19101) overnight
at 56°C, precipitated and washed. Concentration and purity of DNA
were measured using a NanoDrop 1000 Spectrophotometer (Thermo
Fisher Scientific, Waltham, MA, USA).
MeDIP-seq and hMeDIP-seq
As previously described (21,22),
the genomic DNA was fragmented using a Covaris sonication system
(Covaris, Woburn, MA, USA) according to the parameters. After
sonication, the fragments were denatured to produce single stranded
DNA (ssDNA). Following denaturation, the ssDNA was incubated with
anti-5-mC antibody or anti-5-hmC antibody. The antibody-DNA
complexes were captured by protein A/G beads, and the MeDIP and
hMeDIP products were collected for sequencing with HiSeq™ 2000
sequencing system (Illumina, Inc., San Diego, CA, USA).
Identification of differential
methylation/hydroxymethylation regions (DMR/DHMR)
DMR and DHMR identification are based on reads per
kilo base of transcript per million mapped reads (RPKM)-normalized
to 5-mC and 5-hmC density, used model-based analysis of ChIP-Seq
(MACS).
Functional enrichment analysis
Functional enrichment analysis is for the genes
associated with DMRs and DHMRs. Gene Ontology (GO) (https://david.ncifcrf.gov/) is a standard
classification system inferring gene function and gene products.
PANTHER website (http://go.pantherdb.org/) and Kyoto Encyclopedia of
Genes and Genomes (KEGG) pathway analysis (Web-based Gene Set
Analysis Tool Kit and http://www.kegg.jp/kegg/pathway.html) were also used
suggesting physiological functions of these genes.
Ethics statement
All experimental protocols and study methods were
approved by the Ethics Committee of the First Affiliated Hospital,
School of Medicine, Zhejiang University. The written consent was
received from each participant in the present study at the time of
surgery.
Results
Global DNA (hydroxy)methylation changes
in HCC tissues and APTs
We isolated total genomic DNA from the 4 HCC tumor
tissues and paired APTs, and employed (h)MeDIP-seq to explore
genome-wide 5-mC and 5-hmC profiles for the 8 samples. In total,
MACS identified 4.52 million reads and 6.0 million reads of
sequencing data for 5-mC and 5-hmC, respectively (Table II). Differential 5-mC and 5-hmC
peaks between HCC tumor tissues and paired APTs are shown in
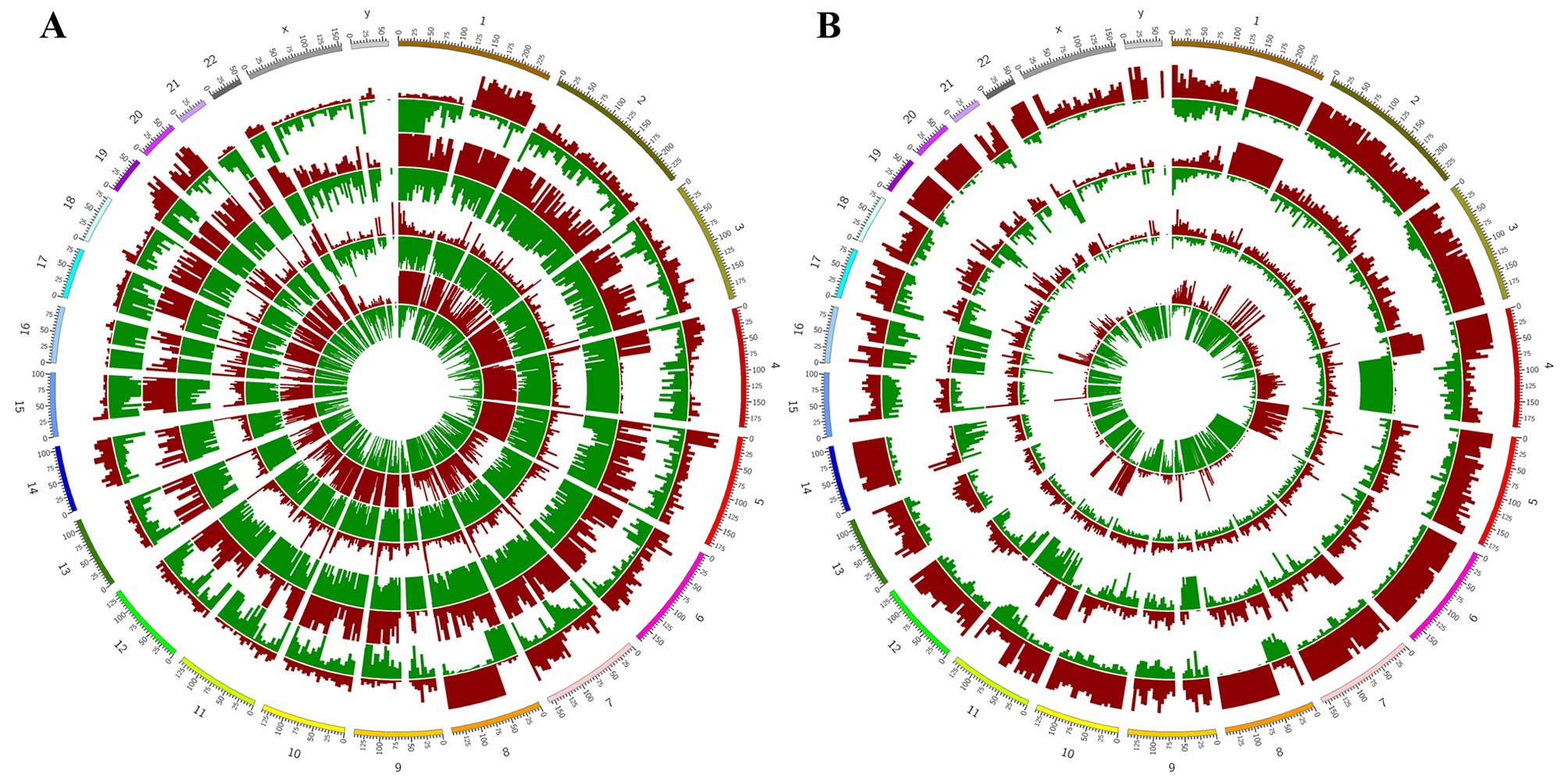
Table III. Density distribution
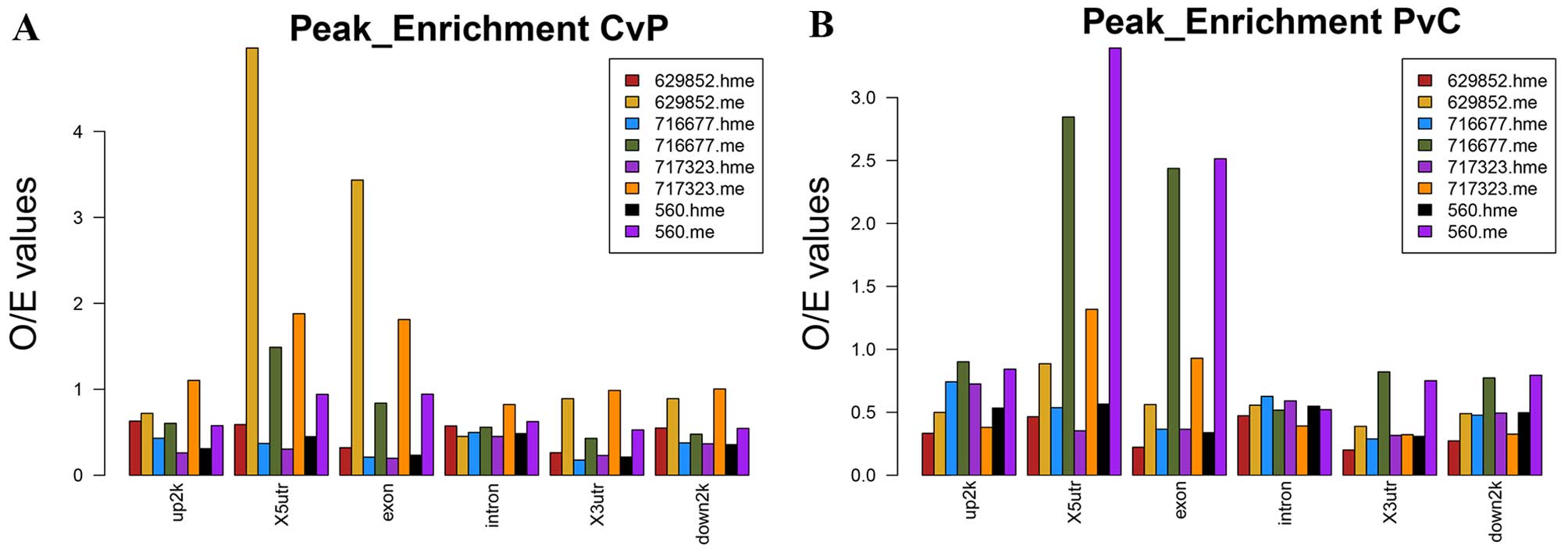
of these peaks on the whole genome is shown in Fig. 1. 5-mC and 5-hmC peak enrichment
profiles of HCC tumor tissues compared with APTs in genomic areas
were shown in Fig. 2. Both CvP and
PvC differential peaks of 5-mC were enriched in X5UTR and Exon.
5-hmC peak enrichment seemed average in each genomic features.
| Table IINumber of reads generated by
(h)MeDIP-Seq for each sample. |
Table II
Number of reads generated by
(h)MeDIP-Seq for each sample.
| | 560 (P/T) | 629852 (P/T) | 716677 (P/T) | 717323 (P/T) |
|---|
| MeDIP-Seq |
| Total number of
reads | P | 178,832,47 | 288,179,93 | 22939802 | 20338428 |
| T | 176,540,07 | 204,678,83 | 21452236 | 31407738 |
| Total number of
mapped read | P | 129,391,26 | 222,400,94 | 21408527 | 16080416 |
| T | 152,462,39 | 160,719,42 | 19448683 | 28980186 |
| Mapping rate
(%) | P | 72.35% | 77.17% | 93.32% | 79.06% |
| T | 86.36% | 78.52% | 90.66% | 92.27% |
| hMeDIP-Seq |
| Total number of
reads | P | 251,088,30 | 299,964,13 | 340,096,74 | 316,801,82 |
| T | 340,606,22 | 314,499,77 | 265,629,54 | 269,660,39 |
| Total number of
mapped reads | P | 191,931,19 | 248,284,70 | 265,222,32 | 239,597,42 |
| T | 266,231,98 | 247,343,23 | 213,428,28 | 214,664,23 |
| Mapping rate
(%) | P | 76.44% | 82.77% | 77.98% | 75.63% |
| T | 78.16% | 78.65% | 80.35% | 79.60% |
| Table IIIDifferentially expressed peaks of
5-mC and 5-hmC MACS of each paired samples. |
Table III
Differentially expressed peaks of
5-mC and 5-hmC MACS of each paired samples.
| Samples | MeDIP-CvP | MeDIP-PvC | hMeDIP-CvP | hMeDIP-PvC |
|---|
| 560 | 51475 | 44772 | 14853 | 53739 |
| 629852 | 9137 | 35823 | 9061 | 5748 |
| 717323 | 70363 | 87400 | 16464 | 17036 |
| 716677 | 17408 | 23586 | 42497 | 9332 |
Analysis of DMR and hDMR-associated genes
in promoter regions
Promoter region is an important gene regulation
region, and the methylation or demethylation at this region plays a
key regulatory role in gene expression. In the present study, we
carried out further bioinformatics analysis to identify
locus-specific DMRs and hDMRs between HCC tumor tissues and APTs in
the promoter region (−3.5K to +1.5K of TSS). The total differential
5-mC peaks (DMRs) that exhibited significant difference between the
two groups (>2-fold, P<0.05) were associated with nearly 4097
genes in terms of RefSeq ID. Of the four samples, 1924, 788, 4521
and 734 genes had hypermethylation (5-mC-CvP) at their promoters
while 2956, 1667, 2490 and 1310 genes had hypomethylation
(5-mC-PvC), respectively (Table
IV).
| Table IVNumbers of DMRs and hDMRs associated
genes. |
Table IV
Numbers of DMRs and hDMRs associated
genes.
| Samples | 5-mC-CvP | 5-mC-PvC | 5-hmC-CvP | 5-hmC-PvC |
|---|
| 560 | 1924 | 2957 | 385 | 2061 |
| 629852 | 734 | 1310 | 457 | 171 |
| 717323 | 4521 | 2490 | 391 | 897 |
| 716677 | 788 | 1667 | 1506 | 507 |
The same analysis was carried out to search for
differential 5-hmC peaks (DHMRs) between HCC tumor tissues and APTs
in the promoter. An average of 1593 genes were associated. Genes
(385, 1506, 391 and 457) showed higher 5-hmC levels (5-hmC-CvP) in
the promoter in HCC tissues compared with APTs of the four samples,
while 2061, 507, 897 and 171 genes showed lower 5-hmC levels
(5-hmC-PvC), respectively. (Table
IV).
Functional enrichment analysis of 5-mC
and 5-hmC associated genes
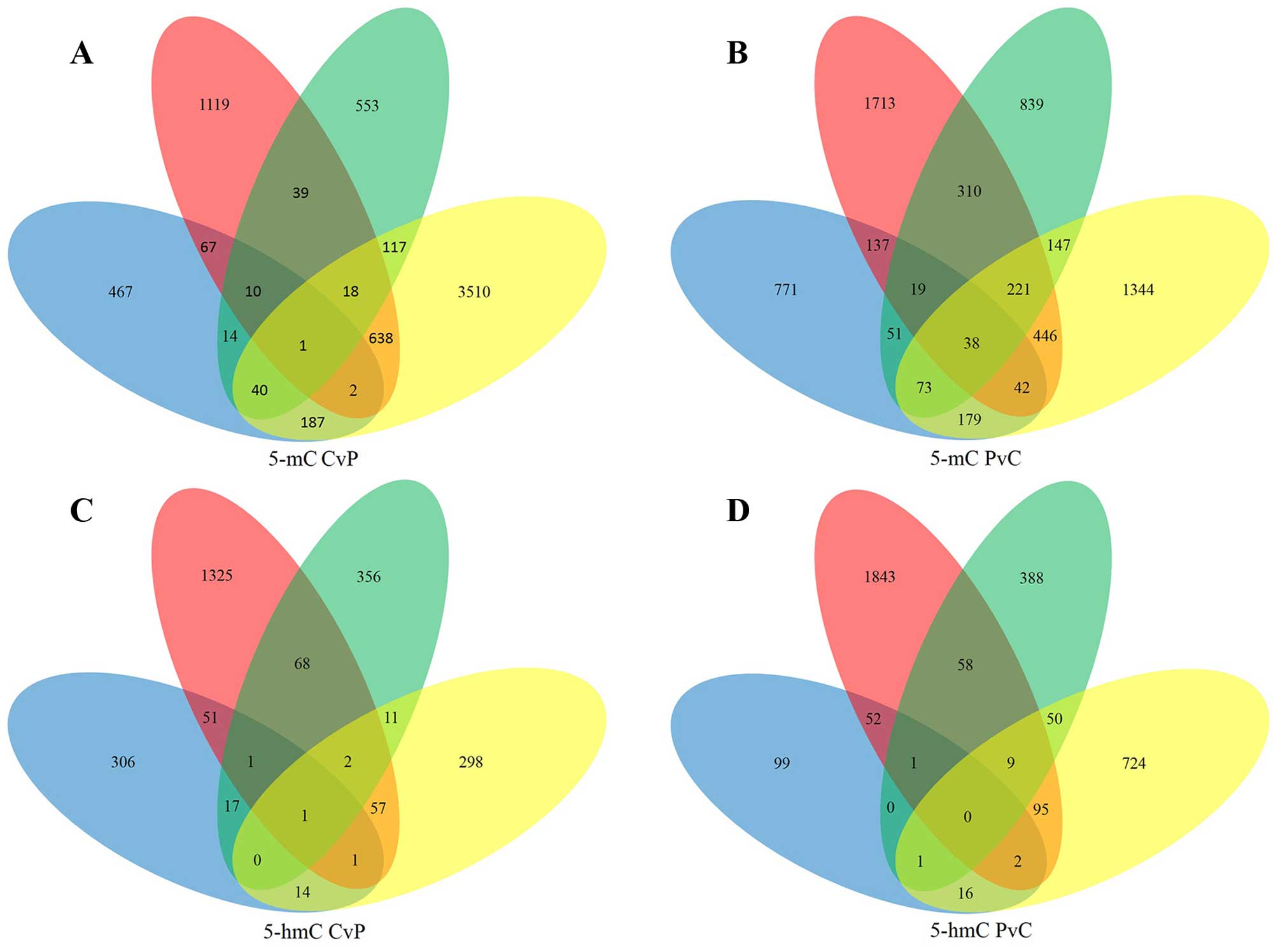
A total of 1133 5-mC-CvP genes and 1663 5-mC-PvC
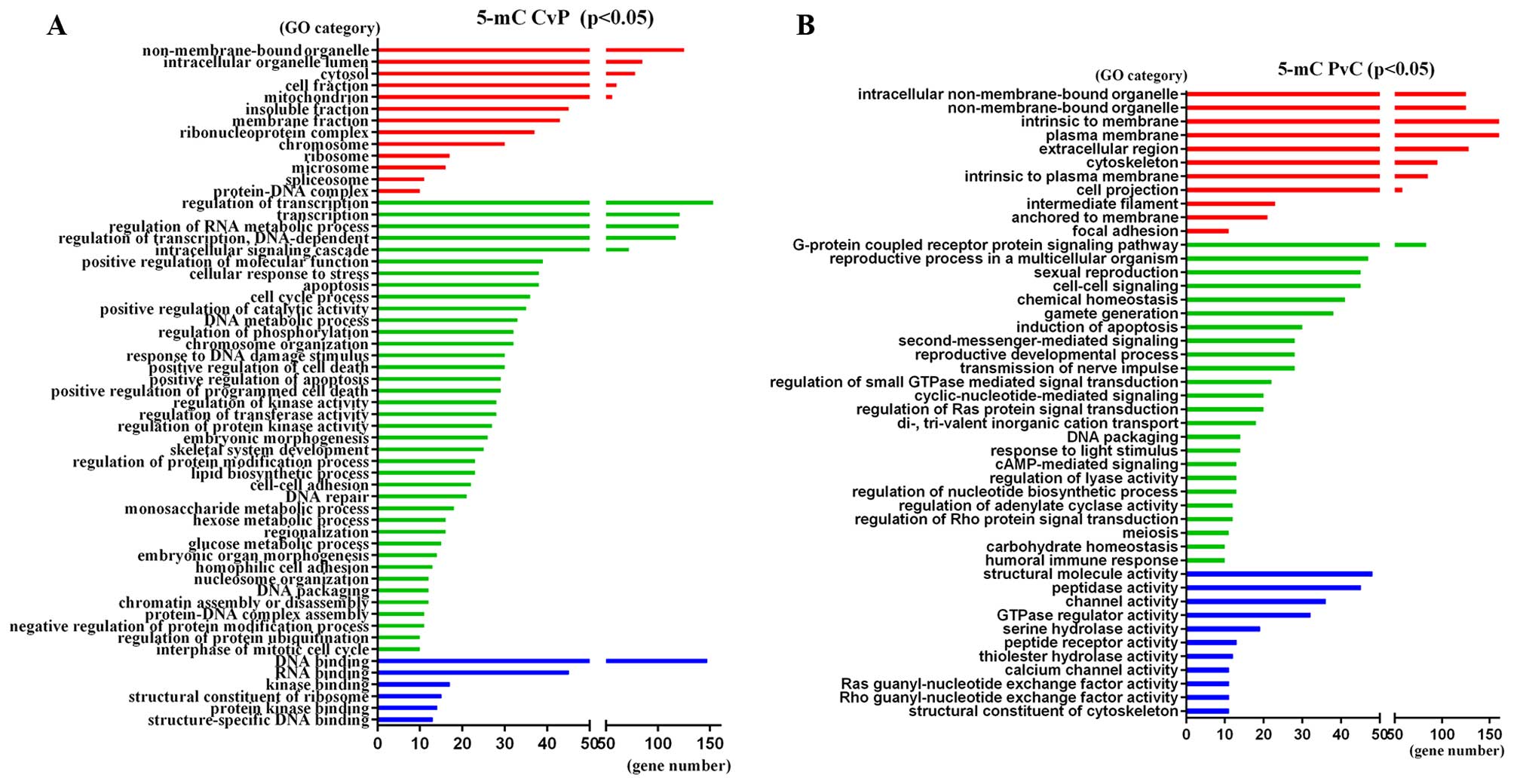
genes were found in at least two samples (Fig. 3A and B). The significant GO
categories of these genes are shown in Fig. 4 (P<0.05). The most enriched term
of 5-mC-CvP genes is ‘Transition metal ion binding’ (GO:0046914,
P=7.20E-06), ‘Regulation of RNA metabolic process’ (GO:0051252,
P=4.10E-05) and ‘DNA binding’ (GO:0003677, P=4.60E-05), while the
5-mC-PvC genes were enriched in ‘Plasma membrane’ (GO:0005886,
P=2.80E-05), ‘Keratin filament’ (GO:0045095, P=6.50E-05) and
‘Cyclic-nucleotide-mediated signaling’ (GO:0019935,
P=8.70E-05).
The KEGG pathway analysis showed that the
significantly differential hypermethylated genes were enriched in
several pathways such as ‘Metabolic pathways’ (adjP=0.0021) (such
as GBE1, GALNT6, NDUFS6, HEXB, RRM1 and CKMT2) and ‘Pathways in
cancer’ (adjP=0.0027) (such as CDKN2A, CDKN2B, APC, GSTP1, DAPK3,
FADD, FGF4 and FGF19), while differential hypomethylated genes were
enriched in ‘Neuroactive ligand-receptor interaction’ (adjP=0.0011)
(such as P2RY4, SSTR5, AVPR2, MAS1, NTSR1 PRSS3, GHSR and CALCRL)
and ‘Calcium signaling pathway’ (adjP=0.0140) (such as ATP2B3,
RYR1, NTSR1, NOS1, HTR5A, SLC25A31, GNAS and DRD5). In Table V all significant KEGG pathways
(adjp<0.05) are listed. For protein class by PANTHER website,
both hypermethylated and hypomethylated genes were mainly
associated with ‘Nucleic acid binding’, ‘Hydrolase’ and ‘Receptor’
(Fig. 5).
| Table VKEGG pathway analysis of
hypermethylated and hypomethylated genes. |
Table V
KEGG pathway analysis of
hypermethylated and hypomethylated genes.
| A, KEGG pathway
analysis of hypermethylated genes |
|---|
|
|---|
| Pathway name | ID | Gene | EntrezGene | Statistic |
|---|
| Regulation of actin
cytoskeleton | 04810 | 20 | WASF2, GNG12, NRAS,
FGF4, APC, CYFIP2, PDGFB, FGF19, MAP2K2, TIAM2, ARHGAP35, GNA12,
PIK3R5, TMSB4Y, ARHGEF1, PPP1CA, ITGB4, RRAS2, ACTN3, MYL12B |
C=213;O=20;E=5.37;R=3.72;
rawP=5.64e-07;adjP=6.26e-05 |
| Systemic lupus
erythematosus | 05322 | 15 | HIST1H4F, HIST1H4K,
HIST1H2BM, H2AFY, HIST1H2BI, ELANE, HIST1H2AL, HIST3H2BB, HIST1H3G,
ACTN3, TROVE2, HIST3H2A, HIST1H3J, HIST1H2AJ, HIST1H4H |
C=136;O=15;E=3.43;R=4.37;
rawP=1.96e-06;adjP=0.0001 |
| Metabolic
pathways | 01100 | 51 | GBE1, GALNT6,
NDUFS6, HEXB, RRM1, CKMT2, GDA, BCAT1, RPE, GLUL, SDHA, HYAL2,
CYP51A1, NDUFV1, HMGCS1, NDUFA2, CYP4F11, CTPS1, SUCLG2, POLR3C,
DGAT1, LDHB, HMGCR, B3GALT6, ALOX12, MGAT4B, SMPD1, TBXAS1, AK4,
BST1, POLG2, HYAL4, DGKE, POLR3G, GALNT3, AK1, ATP6V1D, SGSH,
TCIRG1, B3GAT2, PC, DCXR, DHRS9, CEPT1, PLCB4, SQLE, ACADM, GPI,
PTDSS1, UGT2B7, ALOX15B |
C=1130;O=51;E=28.51;R=1.79;
rawP=5.73e05;adjP=0.0021 |
| Pathways in
cancer | 05200 | 21 | CDKN2B, NRAS,
LAMC1, FGF4, DVL3, CTBP2, APC, PDGFB, GSTP1, FGF19, MAP2K2, DAPK3,
FADD, FZD7, CSF3R, BMP2, LEF1, PIK3R5, MSH3, BMP4, CDKN2A |
C=326;O=21;E=8.22;R=2.55;
rawP=9.84e05;adjP=0.0027 |
| Gastric acid
secretion | 04971 | 8 | ADCY6, PLCB4,
SLC4A2, CFTR, ADCY5, KCNJ1, KCNJ15, CALML6 |
C=74;O=8;E=1.87;R=4.29;
rawP=0.0006;adjP=0.0133 |
| Purine
metabolism | 00230 | 12 | AK1, PDE4A, ADCY5,
PDE7A, RRM1, AK4, ADCY6, GDA, PDE4D, POLR3C, PDE3B, POLR3G |
C=162;O=12;E=4.09;R=2.94;
rawP=0.0009;adjP=0.0148 |
| Pancreatic
secretion | 04972 | 9 | ADCY6, PLCB4,
CELA2A, SLC4A2, CFTR, CELA3B, BST1, ADCY5, ATP2B1 |
C=101;O=9;E=2.55;R=3.53;
rawP=0.0011;adjP=0.0148 |
| Melanoma | 05218 | 7 | NRAS, FGF4, PDGFB,
PIK3R5, FGF19, MAP2K2, CDKN2A |
C=71;O=7;E=1.79;R=3.91;
rawP=0.0021;adjP=0.0194 |
| Tight junction | 04530 | 10 | NRAS, PRKCZ, MPP5,
CTTN, RRAS2, ACTN3, CLDN14, MYL12B, TJAP1, CLDN11 |
C=132;O=10;E=3.33;R=3.00;
rawP=0.0020;adjP=0.0194 |
| Basal cell
carcinoma | 05217 | 6 | BMP2, DVL3, APC,
LEF1, BMP4, FZD7 |
C=55;O=6;E=1.39;R=4.32;
rawP=0.0026;adjP=0.0222 |
| Cell cycle | 04110 | 9 | CDKN2B, PCNA,
STAG1, YWHAZ, ORC1, TFDP2, CDC23, CDKN2A, ORC6 |
C=124;O=9;E=3.13;R=2.88;
rawP=0.0043;adjP=0.0341 |
| Glioma | 05214 | 6 | NRAS, PDGFB,
PIK3R5, MAP2K2, CDKN2A, CALML6 |
C=65;O=6;E=1.64;R=3.66;
rawP=0.0059;adjP=0.0409 |
| Vascular smooth
muscle contraction | 04270 | 8 | GNA12, ADCY5,
ARHGEF1, CALML6, PLCB4, ADCY6, PPP1CA, MAP2K2 |
C=116;O=8;E=2.93;R=2.73;
rawP=0.0093;adjP=0.0492 |
| Glutathione
metabolism | 00480 | 5 | OPLAH, GPX7, GSTP1,
GSTM4, RRM1 |
C=50;O=5;E=1.26;R=3.96;
rawP=0.0084;adjP=0.0492 |
| Insulin signaling
pathway | 04910 | 9 | PRKAG2, NRAS,
PIK3R5, PRKCZ, CALML6, PPP1CA, PDE3B, MAP2K2, PRKAR1A |
C=138;O=9;E=3.48;R=2.59;
rawP=0.0085;adjP=0.0492 |
| Oocyte meiosis | 04114 | 8 | SPDYA, ADCY5, SLK,
CALML6, ADCY6, YWHAZ, PPP1CA, CDC23 |
C=112;O=8;E=2.83;R=2.83;
rawP=0.0076;adjP=0.0492 |
|
| B, KEGG pathway
analysis of hypomethylated genes |
|
| Pathway name | ID | Gene | EntrezGene | Statistic |
|
| Neuroactive
ligand-receptor interaction | 04080 | 26 | P2RY4, SSTR5,
AVPR2, MAS1, NTSR1, PRSS3, GHSR, CALCRL, CHRM4, F2RL3, HTR5A, GRM8,
HTR1D, SSTR3, DRD5, GABRB3, P2RX6, CNR1, GRM4, LEP, UTS2R, SSTR4,
MC5R, ADRA2B, PARD3, HRH1 |
C=272;O=26;E=10.05;R=2.59;
rawP=1.10e05;adjP=0.0011 |
| Dilated
cardiomyopathy | 05414 | 11 | GNAS, SGCA, TPM2,
CACNA1C, ADCY5, CACNB2, ADCY9, TPM4, ADCY6, CACNA2D4, ADCY7 |
C=90;O=11;E=3.33;R=3.31;
rawP=0.0005;adjP=0.0098 |
| Bile secretion | 04976 | 10 | GNAS, SLC4A5,
KCNN2, ADCY5, ATP1A4, ADCY9, ADCY6, HMGCR, ADCY7, AQP8 |
C=71;O=10;E=2.62;R=3.81;
rawP=0.0003;adjP=0.0098 |
| Calcium signaling
pathway | 04020 | 16 | ATP2B3, RYR1,
NTSR1, NOS1, HTR5A, SLC25A31, GNAS, DRD5, CACNA1C, CALML5, P2RX6,
ADCY9, CACNA1B, CALML3, ADCY7, HRH1 |
C=177;O=16;E=6.54;R=2.45;
rawP=0.0009;adjP=0.0140 |
| Pathogenic
Escherichia coli infection | 05130 | 8 | FYN, TUBA3C,
ARPC1B, NCK2, ARPC2, TUBB8, TUBA3E, TUBB3 |
C=56;O=8;E=2.07;R=3.87;
rawP=0.0010;adjP=0.0140 |
| Chemokine signaling
pathway | 04062 | 16 | CXCR5, CX3CR1,
BCAR1, TIAM2, SHC1, GNGT2, ADCY5, ADCY9, IL8, ADCY6, GRK1, ARRB2,
TIAM1, PARD3, ADCY7, GRK7 |
C=189;O=16;E=6.99;R=2.29;
rawP=0.0019;adjP=0.0169 |
| Gap junction | 04540 | 10 | GNAS, TUBA3C,
ADCY5, ADCY9, GJD2, ADCY6, TUBB8, TUBA3E, TUBB3, ADCY7 |
C=90;O=10;E=3.33;R=3.01;
rawP=0.0018;adjP=0.0169 |
| Gastric acid
secretion | 04971 | 9 | GNAS, CALML5,
ADCY5, ATP1A4, ADCY9, ADCY6, ATP4B, CALML3, ADCY7 |
C=74;O=9;E=2.74;R=3.29;
rawP=0.0016;adjP=0.0169 |
| Melanogenesis | 04916 | 10 | GNAS, CALML5,
ADCY5, FZD9, ADCY9, POMC, ADCY6, CALML3, TCF7L2, ADCY7 |
C=101;O=10;E=3.73;R=2.68;
rawP=0.0042;adjP=0.0317 |
| Pancreatic
secretion | 04972 | 10 | GNAS, CTRB1,
CELA3A, ATP2B3, ADCY5, PRSS3, ATP1A, ADCY9, ADCY6, ADCY7 |
C=101;O=10;E=3.73;R=2.68;
rawP=0.0042;adjP=0.0317 |
Next, DHMRs between HCC tumor tissues and APTs in
the promoter were subjected to the same analysis. A total of 223
5-hmC-CvP genes and 284 5-hmC-PvC genes were found in at least two
samples (Fig. 3C and D). The most
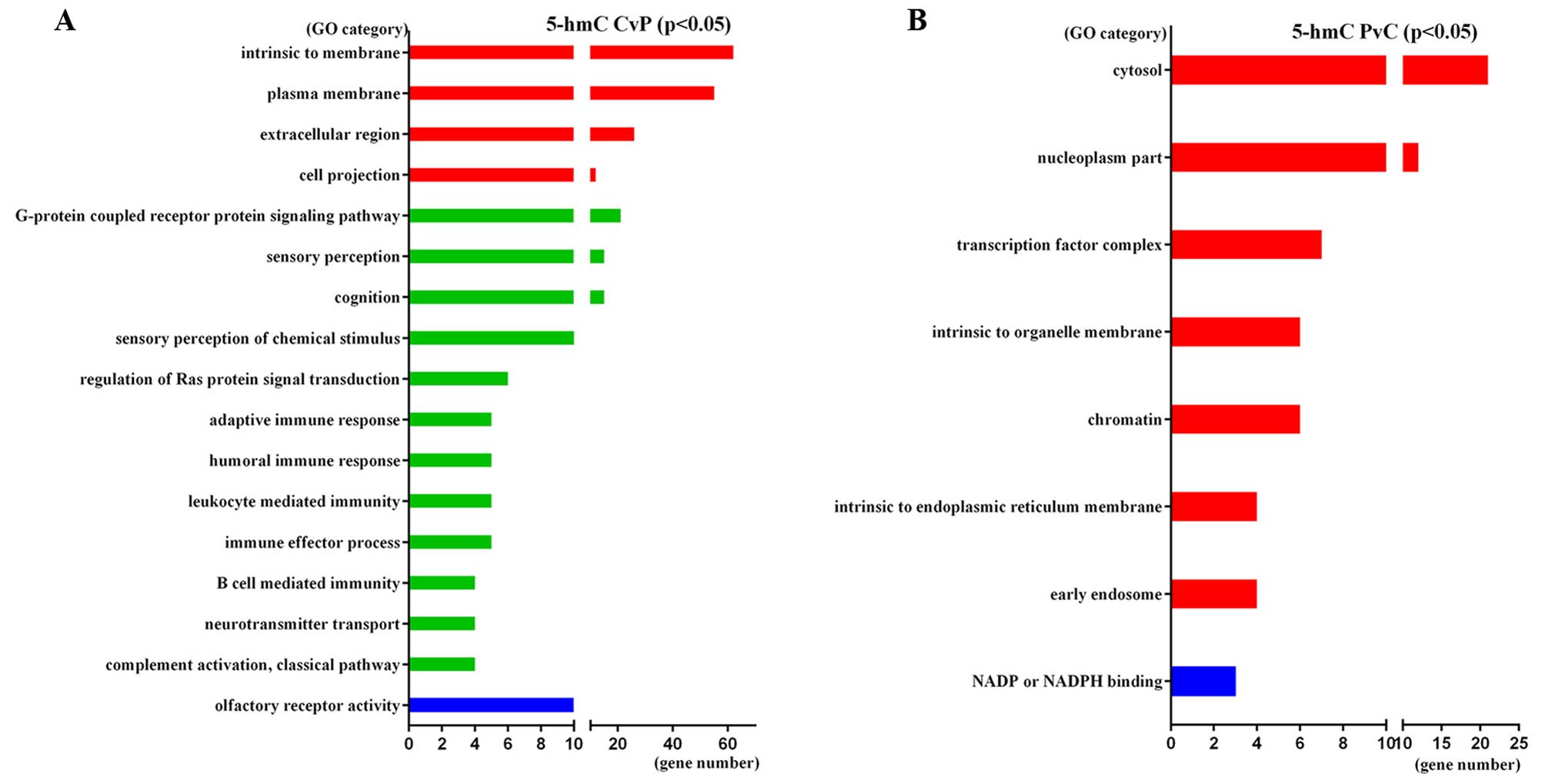
significant GO categories of 5-hmC-CvP genes were ‘Plasma membrane’
(GO:0005886, p=3.10E-05), ‘G-protein coupled receptor protein
signaling pathway’ (GO:0007186, P=3.60E-03) and ‘Intrinsic
component of membrane’ (GO:0031224, P=1.10E-02). 5-hmC-PvC genes
were enriched in ‘Cytosol’ (GO:0005829, P=3.10E-02), ‘Nucleoplasm
part’ (GO:0044451, P=1.90E-02) and ‘Transcription factor complex’
(GO:0005667, P=1.60E-02). The significant GO categories of these
genes are shown in Fig. 6.
KEGG pathway analysis revealed that 5-hmC-CvP genes
were enriched in ‘MAPK signaling’ (such as FGF4, FGF19, MEF2C and
FGF3) and ‘Pathway in cancer’ (such as MMP9, SMAD4, FGF19 and
FGF3), while 5-hmC-PvC genes were enriched in ‘Cell cycle’ (such as
MDM2, STAG1 and E2F4) and ‘Metabolic pathways’ (such as ALG9,
FLAD1, ST3GAL4, NDUFC2, POLR2J3, DHRS9 and G6PD). All significant
pathways are listed in Table VI.
The PANTHER classification system identified that DHMRs associated
genes were mainly enriched in ‘Nucleic acid binding’ and
‘Hydrolase’, the same as the DMRs (Fig. 5).
| Table VIKEGG pathway analysis of upregulated
and downregulated 5-hmC related genes. |
Table VI
KEGG pathway analysis of upregulated
and downregulated 5-hmC related genes.
| A, KEGG pathway
analysis of upregulated 5-hmC related genes |
|---|
|
|---|
| Pathway name | ID | Gene | EntrezGene | Statistic |
|---|
| Olfactory
transduction | 04740 | 9 | OR2T3, OR51V1,
OR2L3, OR51F2, OR56A1 OR2M3, OR52N5, OR4M2, OR4N4 |
C=388;O=9;E=1.65;R=5.47;
rawP=4.63e-05;adjP=0.0005 |
| Antigen processing
and presentation | 04612 | 3 | KIR3DL1, KIR3DL2,
KIR3DL3 |
C=76;O=3;E=0.32;R=9.30;
rawP=0.0042;adjP=0.0092 |
| Melanoma | 05218 | 3 | FGF4, FGF19,
FGF3 |
C=71;O=3;E=0.30;R=9.96;
rawP=0.0035;adjP=0.0092 |
| Natural killer cell
mediated cytotoxicity | 04650 | 4 | KIR3DL1, ICAM2,
KIR3DL2, KIR3DL3 |
C=136;O=4;E=0.58;R=6.93;
rawP=0.0028;adjP=0.0092 |
| Pathways in
cancer | 05200 | 5 | MMP9, FGF4, SMAD4,
FGF19, FGF3 |
C=326;O=5;E=1.38;R=3.61;
rawP=0.0130;adjP=0.0238 |
| MAPK signaling
pathway | 04010 | 4 | FGF4, FGF19, MEF2C,
FGF3 |
C=268;O=4;E=1.14;R=3.52;
rawP=0.0278;adjP=0.0382 |
| RNA transport | 03013 | 3 | NUP62, GEMIN4,
NXT2 |
C=151;O=3;E=0.64;R=4.68;
rawP=0.0268;adjP=0.0382 |
|
| B, KEGG pathway
analysis of downregulated 5-hmC related genes |
|
| Pathway name | ID | Gene | EntrezGene | Statistics |
|
| Cell cycle | 04110 | 5 | MDM2, STAG1, E2F4,
CDK4, TFDP1 |
C=124;O=5;E=0.78;R=6.37;
rawP=0.0012;adjP=0.0140 |
| TGF-β signaling
pathway | 04350 | 4 | DCN, GDF6, E2F4,
TFDP1 |
C=84;O=4;E=0.53;R=7.52;
rawP=0.0020;adjP=0.0140 |
| Staphylococcus
aureus infection | 05150 | 3 | C1QB, FCGR3B,
C3AR1 |
C=55;O=3;E=0.35;R=8.62;
rawP=0.0052;adjP=0.0243 |
| Epithelial cell
signaling in Helicobacter pylori infection | 05120 | 3 | F11R, ATP6V1G2,
ATP6V0A4 |
C=68;O=3;E=0.43;R=6.97;
rawP=0.0093;adjP=0.0254 |
| Ubiquitin mediated
proteolysis | 04120 | 4 | RHOBTB2, UBE2Q1,
UBE3B, MDM2 |
C=135;O=4;E=0.85;R=4.68; raw
P=0.0109;adjP=0.0254 |
| Protein processing
in endoplasmic reticulum | 04141 | 4 | HSP90AA1, ERP29,
DNAJA2, TXNDC5 |
C=165;O=4;E=1.04;R=3.83;
rawP=0.0212;adjP=0.0371 |
| Metabolic
pathways | 01100 | 13 | ALG9, FLAD1,
ST3GAL4, NDUFC2, POLR2J3, DHRS9, G6PD, ATP6V1G2, SMPD1, ASMT,
ATP6V0A4, NOS1, AKR1A1 |
C=1130;O=13;E=7.15;R=1.82;
rawP=0.0290;adjP=0.0451 |
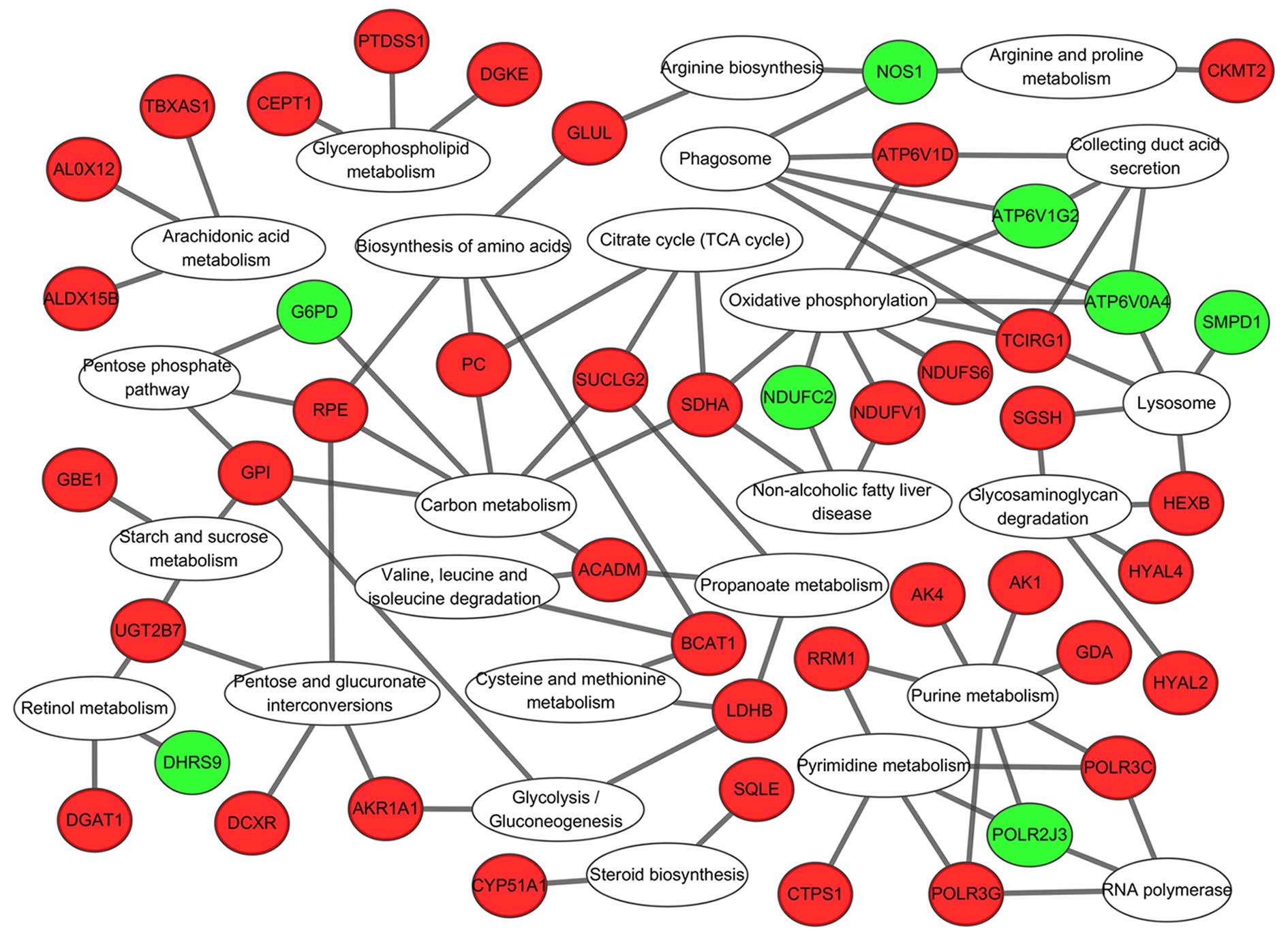
KEGG pathway analysis of ‘metabolic
pathway’-associated genes
There were several 5-mC and 5-hmC changed genes
enriched in ‘Metabolic pathways’ further KEGG pathway analysis for
these genes revealed that they were gathered in glucose metabolism
[including ‘Glycolysis/gluconeogenesis’ (00010), ‘Pentose and
glucuronate interconversions’ (00030), ‘Starch and sucrose
metabolism’ (00500), ‘Glycosaminoglycan degradation’ (00531)],
energy metabolism [including ‘Oxidative phosphorylation’ (00190),
‘Citrate cycle (TCA cycle)’ (00020), ‘Carbon metabolism’ (01200)],
and amino acid metabolism, [including ‘Biosynthesis of amino acids’
(01230), ‘Cysteine and methionine metabolism’ (00270), ‘Arginine
and proline metabolism’ (00330), ‘Arachidonic acid metabolism’
(00590)] ‘Purine metabolism’ and ‘Pyrimidine metabolism’ (Fig. 7).
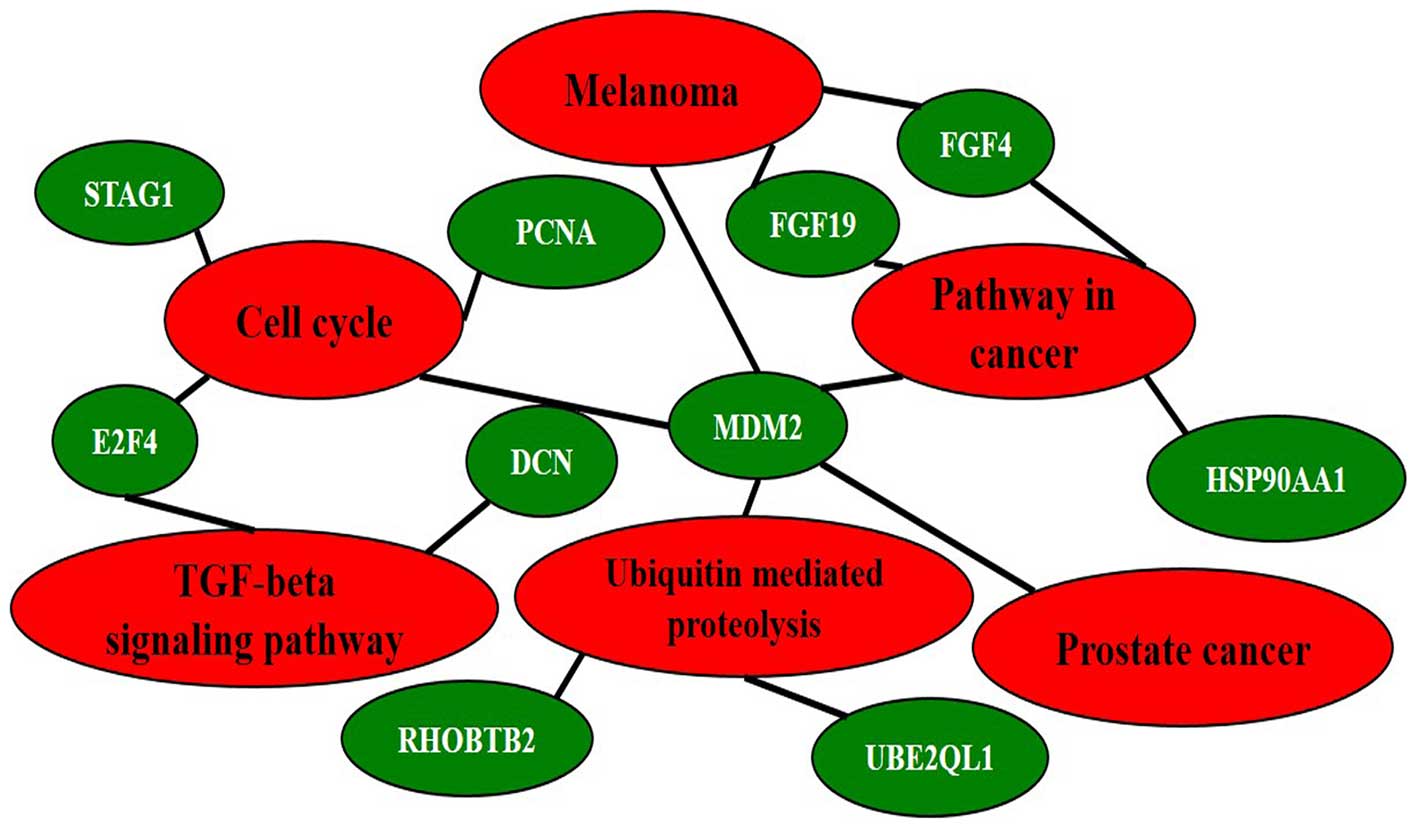
Both DMR and DHMR-associated genes
A total of 141 genes were found with both 5-mC and
5-hmC changes in at least two patients. KEGG pathway analysis for
these 141 genes identified five major pathways involved (‘Cell
cycle’, ‘Pathway in cancer’, ‘Ubiquitin mediated proteolysis’,
‘Melanoma’ and ‘Prostate cancer’) were enriched (adjp<0.05). Ten
interconnected and enriched genes (PCNA, MDM2, STAG1, E2F4, FGF4,
FGF19, RHOBTB2, UBE2QL1, DCN and HSP90AA1) were revealed (Fig. 8).
Discussion
DNA methylation is one of the major epigenetic
mechanisms that regulate gene expression in humans, and the
alterations of methylation profiles are regarded as one of the
major molecular aberrations in malignancies (23,24).
Several studies of genome-wide DNA methylation have shown that the
characteristic features of CpGs and certain microRNAs had
differences in methylation levels between HCC and non-cancerous
livers (25,26). Thus far, only a few studies have
reported that DNA hydroxymethylation is associated with several
human cancers (14,27), yet, the biological significance of
5-hmC in tumorigenesis remains unclear. In the present study we
showed widespread alterations in DNA methylation and
hydroxymethylation in HCC tumor tissues and paired APTs. 5-mC and
5-hmC levels exhibited no significant differences between the
groups, which might be due to the huge variation between HCC
individuals. The strong features of this study include the highly
sensitive method and high-throughput sequencing used, which enabled
the non-biased mapping of aberrant (hydroxy) methylation sites
between the tumor tissues and APTs, and distinguished the
alternation of 5-mC from that of 5-hmC. To the best of our
knowledge, this is the first report on the genome-wide profiling of
5-mC and 5-hmC in HCC using this technique.
As is known, in the mammalian genome, methylation
takes place only at cytosine bases that are located 5′ to a
guanosine in a CpG dinucleotide, known as CpG islands. Most CpG
islands are found in the proximal promoter regions (28). Methylation and demethylation in
promoter region may regulate gene expression, playing important
roles in biology process, especially in the development of tumors
(25,29,30).
The identification of genes that are specifically hypermethylated
(which results in gene silencing) or hypomethylated (which results
in increased transcription) might lead to the discovery of new
factors that are important for tumor initiation and progression. In
this study, we identified 1133 hypermethylated genes and 1663
hypomethylated genes in the promoter regions, many more than
reported in a previous study (19), proving that MeDIP-seq has a better
sensitivity than microarray, and more methylated sites can be found
by using this novel technology. GO analysis showed they were
enriched in various biological processes. Hypermethylation mostly
gathered in ‘Regulation’-related biology processes, including
‘Regulation of RNA metabolic process’ (P=4.10E-05), ‘Regulation of
kinase activity’ (P=8.80E-03) and ‘Regulation of transcription’
(P=9.40E-04). Although it is widely accepted that ‘DNA methylation
suppresses gene expression’, this statement is an
over-simplification. Methylation at the promoter regions can change
the interactions between proteins and DNA, which can lead to the
alterations in chromatin structure and either a decrease or an
increase in the rate of transcription (31,32).
We indeed found hypermethylation genes enriched in ‘protein-DNA
complex assembly’ (P=9.40E-03). Furthermore, the position of the
methylation change relative to the transcription start site is
critical to the outcome (23).
‘Binding’ is another category where the hypermethylated genes are
enriched, and it includes ‘DNA binding’ (P=4.60E-05), ‘Ion binding’
(P=1.20E-03) and ‘RNA binding’ (P=3.60E-02), which are accordant
with the results of Zhai et al (33) In contrast, hypomethylation genes
are enriched in totally different categories, such as ‘Plasma
membrane’ (p=2.80E-05), ‘Cytoskeleton’ (P=9.70E-03) and ‘G-protein
coupled receptor protein signaling pathway’ (P=4.60E-03).
Our KEGG pathway analysis identified some
interesting pathways for hypermethylation. ‘Pathway in cancer’
contained genes such as CDKN2A and CDKN2B (cyclin-dependent kinase
inhibitor 2A/2B) that are recognized as a tumor suppressor genes.
The inactivation of CDKN2A/2B have been reported in several primary
tumors (34–36). There might be three different
molecular mechanisms resulting in the loss of the CDKN2A/2B gene
functions, namely homozygous deletions, point mutations, and
transcriptional silencing by methylation at CpG islands.
Methylation of CDKN2 has been observed in cell lines and cancer
specimens derived from glioma, breast, colonic, head and neck
cancers, hepatoblastoma, and in transitional cell carcinomas of the
bladder (37–39). Shen et al (40) used Illumina Methylated Arrays and
pyrosequencing technique and indicated that CDKN2A may be a
potential biomarker for early HCC diagnosis. Another important gene
in this pathway is APC (adenomatous polyposis coli) which also is
an important tumor associated gene. The profiling of gene promoter
hypermethylation across human tumor types showed that APC promoter
hypermethylation occurred in tumors including colon, breast,
kidney, bladder, esophagus, stomach, pancreas and liver tumors
(41). Furthermore, studies showed
that high-level APC promoter methylation is a useful biomarker and
predictor in esophageal adenocarcinoma, breast and prostate cancer
(42–44). Methylation of APC in HCC is
frequent and occurs in a gene-specific and disease-specific manner.
It was detected more frequently in hepatitis C virus-positive HCC
(45,46). Other genes such as GSTP1
(Glutathione S-transferase P1) have also been found to be
epigenetically silenced by promoter methylation, and associated
with increased risk and shortened survival in patients with various
tumors, including HCC, breast and prostate cancer (47–49).
Promoter methylation and epigenetic silencing of DAPK3
(death-associated protein kinase 3) and FADD [Fas
(TNFRSF6)-associated via death domain] have not been studied in
depth to the extent of those genes, and only a few studies
implicated their participation in cancers such as oral squamous
cell carcinoma and non-muscle invasive bladder carcinoma (50–52).
Their promoter methylation could also be a potential marker for
HCC, and further studies are needed to confirm this. Other
identified pathways such as ‘Cell cycle’ (PCNA, STAG1, YWHAZ, ORC1,
TFDP2 and CDC23) and ‘Chemokine signaling pathway’ (GNG12, NRAS,
ELMO1, PIK3R5, ADCY5, PRKCZ and PLCB4) are also considered to be
important for the development and progression of malignant
carcinoma, and the involved genes with their methylation status may
provide potential novel biomarkers for HCC.
5-hmC is usually found in human embryonic stem (ES)
cells and particularly abundant in certain genomic regions such as
enhancers associated with histone modifications and other
protein-DNA interaction sites based both on the information of gene
expression and sequence composition (53). However, in human HCC tumor tissues
and paired APTs, the locations of DHMRs seemed not to be
significantly different among the whole genomic region. Although
there is substantial evidence indicating that hydroxymethylation
may be associated with actively transcribed genes, the exact
biochemical mechanisms still remain enigmatic (54,55).
Our GO analysis showed that compared with APTs, both high-level
hydroxymethylated genes and hypomethylated genes in HCC tissues
were enriched in the same pathways, namely the ‘Plasma membrane’
(P=3.10E-05) and ‘G-protein coupled receptor protein signaling
pathway’ (P=3.60E-03), indicating that hydroxymethylation as a kind
of demethylation may play similar or related roles with
hypomethylation. In contrast, the downregulated hydroxymethylated
genes are found mainly gathered in the ‘Cytosol’ (P=3.10E-02).
Although the KEGG pathway analysis for 5-hmC did not
come up with as many enriched genes as that for 5-mC, they can
still be categorized into a number of meaningful pathways. For
instance, high level hydroxymethylated genes such as MMP9, SMAD4,
FGF19, FGF3 and MEF2C were enriched in ‘Pathway in cancer’ and
‘MAPK signaling pathway’, while low level hydroxymethylated genes
were mostly enriched in ‘Metabolic pathways’ and ‘Cell cycle’.
‘TGF-β signaling pathway’ related genes DCN, E2F4, TFDP1 also have
strong correlations with tumors (56–58).
Since there have been few studies on the hydroxymethylation of
these genes, more work is needed to fully elucidate the potential
roles of hydroxy-methylation. Protein class analysis by PANTHER
website showed that both DMR and DHMR-associated genes were in
similar category, indicating that genes with hypermethylation or
demethylation epigenetic changes tend to be coordinately regulated
to participate in similar or related biology processes. Further
work is warranted to test this hypothesis.
Over half a century ago, Warburg linked metabolism
and cancer through enhanced aerobic glycolysis (59). This metabolic switch places the
emphasis on producing intermediates for cell growth and division.
The most rapidly growing tumor cell lines obtain up to 50% of their
total ATP production from glycolytic metabolism, with a
corresponding decrease in oxidative phosphorylation and in cell
mitochondrial content (60,61).
With numerous in-depth studies, the multi-faceted links between
metabolism and cancer have now been revealed. Cellular metabolism
is regulated by both oncogenes and tumor suppressor genes in a
number of key signaling pathways. Metabolism generates oxygen
radicals, which contribute to oncogenic mutations. Activated
oncogenes and loss of tumor suppressors in turn alter metabolism
and induce aerobics (62). In the
present study, we found several 5-mC and 5-hmC changed genes
enriched in ‘Metabolic pathways’, and further analysis showed they
were specifically clustered in ‘Glycolysis/gluconeogenesis’,
‘Oxidative phosphorylation’ and ‘Citrate cycle (TCA cycle)’
(Fig. 7), metabolic pathways were
proven to be critical in controlling cancer cell survival and
proliferation. Although the regulatory mechanisms underlying
aerobic and glycolytic metabolic pathways are complex, our findings
indicate that (hydroxy)methylation-based epigenetic modifications
may affect the development of HCC through the regulation of
cellular metabolism.
DNA methylation as a characterized epigenetic
mechanism, its relationship with other biochemical pathways
represents a critical stage in the elucidation of biological
information processing. Some amino acid metabolism has been related
to DNA methylation in tumors, such as homo-cysteine metabolism and
the dynamics of methionine cycle (63,64).
Accordingly, this study also found several 5-mC and 5-hmC changed
genes that are associated with amino acid metabolism, including
‘Arginine biosynthesis’, ‘Cysteine and methionine metabolism’,
‘Valine, leucine and isoleucine degradation’ and ‘Arginine and
proline metabolism’ which may provide new clues for studying the
relationship between (hydroxy)methylation and metabolism in
HCC.
The present study found that a total of 141 genes
have both 5-mC and 5-hmC changes in at least two of the HCC
patients. KEGG pathway analysis showed five pathways (‘Cell cycle’,
‘Pathway in cancer’, ‘Ubiquitin mediated proteolysis’, ‘Melanoma’
and ‘Prostate cancer’) including ten genes (PCNA, MDM2, SAG1, E2F4,
FGF4, FGF19, RHOBTB2, UBE2QL1, DCN and HSP90AA1) are enriched
(Fig. 8). It is known for decades
that, PCNA (proliferating cell nuclear antigen) acts as a central
coordinator of DNA transactions by providing a multivalent
interaction surface for factors involved in DNA replication,
repair, chromatin dynamics, and cell cycle regulation (65), and is involved in the progression
of tumors and highly altered in some tumors (66). Furthermore, studies have shown that
the p21 protein negatively regulates targeting of DNA-MTase to the
replication associated PCNA. They proposed that the presence of p21
prevents DNA-MTase access to replicating DNA, thereby impeding
hypermethylation in normal cells (67). The present study indicated that
(hydroxy) methylation of PCNA might be associated with HCC, which
warrants further study. The 90-kDa heat shock protein HSP90AA1,
another p21 regulator, has been found highly expressed in many
cancers. Its mechanism in the tumorigenesis is varied (68,69).
Here we provided evidence that methylation or hydroxymethylation of
HSP90AA1 may play a crucial role in HCC. The epigenetic alterations
of other identified genes such as MDM2, SAG, FGF4, FGF19, RHOBTB2
and DCN in HCC and other cancers also deserve further research.
One of the potential limitations of the present
study is the sample size, which may not be sufficiently large. This
is mainly due to the high cost of (h)MeDIP-seq, which precludes its
application in a large scale. Nevertheless, we performed a rather
comprehensive methylation and hydroxymethylation profiling of human
HCC tumor tissues and paired APTs, and correlated multiple
(hydroxy)methylation-altered genes with a number of important
biological pathways. ‘Metabolic pathways’ are found to contain the
largest number of (hydroxy) methylation-altered genes, indicating
the crucial roles of metabolic processes (such as
glycolysis/gluconeogenesis, oxidative phosphorylation and citrate
cycle) in the occurrence and progression of HCC. Some of the
identified (hydroxy) methylation-altered genes may serve as
biomarkers for the diagnosis and prognosis of HCC. Future studies
with a larger sample size combined with a series of biochemical
approaches hold the promise of elucidating the specific roles of
epigenetic modifications in the pathogenesis of HCC.
Acknowledgements
We thank Professor Yingjie Wang for the helpful
comments and language supports on this manuscript. The present
study is supported by the Chinese High Tech Research and
Development (863) Program (grant nos. 2012AA020204 and
2013AA020102) and the National S&T Major Project (grant no.
2012ZZX10002004-001).
References
|
1
|
Bertino G, Demma S, Ardiri A, Proiti M,
Gruttadauria S, Toro A, Malaguarnera G, Bertino N, Malaguarnera M,
Malaguarnera M, et al: Hepatocellular carcinoma: Novel molecular
targets in carcinogenesis for future therapies. BioMed Res Int.
2014:2036932014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ishikawa T: Clinical features of hepatitis
B virus-related hepatocellular carcinoma. World J Gastroenterol.
16:2463–2467. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bréchot C, Gozuacik D, Murakami Y and
Paterlini-Bréchot P: Molecular bases for the development of
hepatitis B virus (HBV)-related hepatocellular carcinoma (HCC).
Semin Cancer Biol. 10:211–231. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ma S, Jiao B and Liu X, Yi H, Kong D, Gao
L, Zhao G, Yang Y and Liu X: Approach to radiation therapy in
hepatocellular carcinoma. Cancer Treat Rev. 36:157–163. 2010.
View Article : Google Scholar
|
|
5
|
Lo CM, Ngan H, Tso WK, Liu CL, Lam CM,
Poon RT, Fan ST and Wong J: Randomized controlled trial of
transarterial lipiodol chemoembolization for unresectable
hepatocellular carcinoma. Hepatology. 35:1164–1171. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Poon RT, Fan ST, Lo CM, Ng IO, Liu CL, Lam
CM and Wong J: Improving survival results after resection of
hepatocellular carcinoma: A prospective study of 377 patients over
10 years. Ann Surg. 234:63–70. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jones PA and Baylin SB: The epigenomics of
cancer. Cell. 128:683–692. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Paska AV and Hudler P: Aberrant
methylation patterns in cancer: A clinical view. Biochem Med
Zagreb. 25:161–176. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Robertson KD: DNA methylation and human
disease. Nat Rev Genet. 6:597–610. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Globisch D, Münzel M, Müller M, Michalakis
S, Wagner M, Koch S, Brückl T, Biel M and Carell T: Tissue
distribution of 5-hydroxymethylcytosine and search for active
demethylation intermediates. PLoS One. 5:e153672010. View Article : Google Scholar
|
|
11
|
Ruzov A, Tsenkina Y, Serio A, Dudnakova T,
Fletcher J, Bai Y, Chebotareva T, Pells S, Hannoun Z, Sullivan G,
et al: Lineage-specific distribution of high levels of genomic
5-hydroxymethylcytosine in mammalian development. Cell Res.
21:1332–1342. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tan L and Shi YG: Tet family proteins and
5-hydroxymethylcytosine in development and disease. Development.
139:1895–1902. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Branco MR, Ficz G and Reik W: Uncovering
the role of 5-hydroxymethylcytosine in the epigenome. Nat Rev
Genet. 13:7–13. 2011.PubMed/NCBI
|
|
14
|
Haffner MC, Chaux A, Meeker AK, Esopi DM,
Gerber J, Pellakuru LG, Toubaji A, Argani P, Iacobuzio-Donahue C,
Nelson WG, et al: Global 5-hydroxymethylcytosine content is
significantly reduced in tissue stem/progenitor cell compartments
and in human cancers. Oncotarget. 2:627–637. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Moribe T, Iizuka N, Miura T, Kimura N,
Tamatsukuri S, Ishitsuka H, Hamamoto Y, Sakamoto K, Tamesa T and
Oka M: Methylation of multiple genes as molecular markers for
diagnosis of a small, well-differentiated hepatocellular carcinoma.
Int J Cancer. 125:388–397. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lou C, Du Z, Yang B, Gao Y, Wang Y and
Fang S: Aberrant DNA methylation profile of hepatocellular
carcinoma and surgically resected margin. Cancer Sci. 100:996–1004.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mardis ER: Next-generation DNA sequencing
methods. Annu Rev Genomics Hum Genet. 9:387–402. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shendure J and Ji H: Next-generation DNA
sequencing. Nat Biotechnol. 26:1135–1145. 2008. View Article : Google Scholar
|
|
19
|
Shitani M, Sasaki S, Akutsu N, Takagi H,
Suzuki H, Nojima M, Yamamoto H, Tokino T, Hirata K, Imai K, et al:
Genome-wide analysis of DNA methylation identifies novel
cancer-related genes in hepatocellular carcinoma. Tumour Biol.
33:1307–1317. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shen J, Wang S, Zhang YJ, Wu HC, Kibriya
MG, Jasmine F, Ahsan H, Wu DP, Siegel AB, Remotti H, et al:
Exploring genome-wide DNA methylation profiles altered in
hepatocellular carcinoma using Infinium HumanMethylation 450
BeadChips. Epigenetics. 8:34–43. 2013. View Article : Google Scholar :
|
|
21
|
Iyer P, Zekri AR, Hung CW, Schiefelbein E,
Ismail K, Hablas A, Seifeldin IA and Soliman AS: Concordance of DNA
methylation pattern in plasma and tumor DNA of Egyptian
hepatocellular carcinoma patients. Exp Mol Pathol. 88:107–111.
2010. View Article : Google Scholar
|
|
22
|
Tan L, Xiong L, Xu W, Wu F, Huang N, Xu Y,
Kong L, Zheng L, Schwartz L, Shi Y, et al: Genome-wide comparison
of DNA hydroxymethylation in mouse embryonic stem cells and neural
progenitor cells by a new comparative hMeDIP-seq method. Nucleic
Acids Res. 41:e842013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jones PA and Takai D: The role of DNA
methylation in mammalian epigenetics. Science. 293:1068–1070. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Esteller M: Epigenetics in cancer. N Engl
J Med. 358:1148–1159. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shen J, Wang S, Zhang YJ, Kappil MA, Chen
Wu H, Kibriya MG, Wang Q, Jasmine F, Ahsan H, Lee PH, et al:
Genome-wide aberrant DNA methylation of microRNA host genes in
hepatocellular carcinoma. Epigenetics. 7:1230–1237. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nishida N, Nishimura T, Nakai T, Chishina
H, Arizumi T, Takita M, Kitai S, Yada N, Hagiwara S, Inoue T, et
al: Genome-wide profiling of DNA methylation and tumor progression
in human hepatocellular carcinoma. Dig Dis. 32:658–663. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kudo Y, Tateishi K, Yamamoto K, Yamamoto
S, Asaoka Y, Ijichi H, Nagae G, Yoshida H, Aburatani H and Koike K:
Loss of 5-hydroxymethylcytosine is accompanied with malignant
cellular transformation. Cancer Sci. 103:670–676. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bird A: DNA methylation patterns and
epigenetic memory. Genes Dev. 16:6–21. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jones PA and Laird PW: Cancer epigenetics
comes of age. Nat Genet. 21:163–167. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jones PA and Baylin SB: The fundamental
role of epigenetic events in cancer. Nat Rev Genet. 3:415–428.
2002.PubMed/NCBI
|
|
31
|
Jones PL, Veenstra GJ, Wade PA, Vermaak D,
Kass SU, Landsberger N, Strouboulis J and Wolffe AP: Methylated DNA
and MeCP2 recruit histone deacetylase to repress transcription. Nat
Genet. 19:187–191. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gonzalgo ML, Hayashida T, Bender CM, Pao
MM, Tsai YC, Gonzales FA, Nguyen HD, Nguyen TT and Jones PA: The
role of DNA methylation in expression of the p19/p16 locus in human
bladder cancer cell lines. Cancer Res. 58:1245–1252.
1998.PubMed/NCBI
|
|
33
|
Zhai JM, Yin XY, Hou X, Hao XY, Cai JP,
Liang LJ and Zhang LJ: Analysis of the genome-wide DNA methylation
profile of side population cells in hepatocellular carcinoma. Dig
Dis Sci. 58:1934–1947. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cairns P, Mao L, Merlo A, Lee DJ, Schwab
D, Eby Y, Tokino K, van der Riet P, Blaugrund JE and Sidransky D:
Rates of p16 (MTS1) mutations in primary tumors with 9p loss.
Science. 265:415–417. 1994. View Article : Google Scholar
|
|
35
|
Okamoto A, Demetrick DJ, Spillare EA,
Hagiwara K, Hussain SP, Bennett WP, Forrester K, Gerwin B, Serrano
M and Beach DH: Mutations and altered expression of p16INK4 in
human cancer. Proc Natl Acad Sci USA. 91:11045–11049. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rousseau E, Ruchoux MM, Scaravilli F,
Chapon F, Vinchon M, De Smet C, Godfraind C and Vikkula M: CDKN2A,
CDKN2B and p14ARF are frequently and differentially methylated in
ependymal tumours. Neuropathol Appl Neurobiol. 29:574–583. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gonzalez-Zulueta M, Bender CM, Yang AS,
Nguyen T, Beart RW, Van Tornout JM and Jones PA: Methylation of the
5′ CpG island of the p16/CDKN2 tumor suppressor gene in normal and
transformed human tissues correlates with gene silencing. Cancer
Res. 55:4531–4535. 1995.PubMed/NCBI
|
|
38
|
Colot V and Rossignol JL: Isolation of the
Ascobolus immersus spore color gene b2 and study in single cells of
gene silencing by methylation induced premeiotically. Genetics.
141:1299–1314. 1995.PubMed/NCBI
|
|
39
|
Iolascon A, Giordani L, Moretti A, Basso
G, Borriello A and Della Ragione F: Analysis of CDKN2A, CDKN2B,
CDKN2C, and cyclin Ds gene status in hepatoblastoma. Hepatology.
27:989–995. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shen J, Wang S, Zhang YJ, Kappil M, Wu HC,
Kibriya MG, Wang Q, Jasmine F, Ahsan H, Lee PH, et al: Genome-wide
DNA methylation profiles in hepatocellular carcinoma. Hepatology.
55:1799–1808. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Esteller M, Corn PG, Baylin SB and Herman
JG: A gene hypermethylation profile of human cancer. Cancer Res.
61:3225–3229. 2001.PubMed/NCBI
|
|
42
|
Kawakami K, Brabender J, Lord RV, Groshen
S, Greenwald BD, Krasna MJ, Yin J, Fleisher AS, Abraham JM, Beer
DG, et al: Hypermethylated APC DNA in plasma and prognosis of
patients with esophageal adenocarcinoma. J Natl Cancer Inst.
92:1805–1811. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Van De Voorde L, Speeckaert R, Van Gestel
D, Bracke M, De Neve W, Delanghe J and Speeckaert M: DNA
methylation-based biomarkers in serum of patients with breast
cancer. Mutat Res. 751:304–325. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Henrique R, Ribeiro FR, Fonseca D, Hoque
MO, Carvalho AL, Costa VL, Pinto M, Oliveira J, Teixeira MR,
Sidransky D, et al: High promoter methylation levels of APC predict
poor prognosis in sextant biopsies from prostate cancer patients.
Clin Cancer Res. 13:6122–6129. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Nishida N, Nagasaka T, Nishimura T, Ikai
I, Boland CR and Goel A: Aberrant methylation of multiple tumor
suppressor genes in aging liver, chronic hepatitis, and
hepatocellular carcinoma. Hepatology. 47:908–918. 2008. View Article : Google Scholar
|
|
46
|
Yang B, Guo M, Herman JG and Clark DP:
Aberrant promoter methylation profiles of tumor suppressor genes in
hepatocellular carcinoma. Am J Pathol. 163:1101–1107. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Fang C, Wei XM, Zeng XT, Wang FB, Weng H
and Long X: Aberrant GSTP1 promoter methylation is associated with
increased risk and advanced stage of breast cancer: A meta-analysis
of 19 case-control studies. BMC Cancer. 15:9202015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zelic R, Fiano V, Zugna D, Grasso C,
Delsedime L, Daniele L, Galliano D, Pettersson A, Gillio-Tos A,
Merletti F, et al: Global hypomethylation (LINE-1) and
gene-specific hypermethylation (GSTP1) on initial negative prostate
biopsy as markers of prostate cancer on a rebiopsy. Clin Cancer
Res. 22:984–992. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Liu D, Wu J, Liu M, Yin H, He J and Zhang
B: Downregulation of miRNA-30c and miR-203a is associated with
hepatitis C virus core protein-induced epithelial-mesenchymal
transition in normal hepatocytes and hepatocellular carcinoma
cells. Biochem Biophys Res Commun. 464:1215–1221. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Saberi E, Kordi-Tamandani DM, Jamali S and
Rigi-Ladiz MA: Analysis of methylation and mRNA expression status
of FADD and FAS genes in patients with oral squamous cell
carcinoma. Med Oral Patol Oral Cir Bucal. 19:e562–e568.
2014.PubMed/NCBI
|
|
51
|
Friedrich MG, Chandrasoma S, Siegmund KD,
Weisenberger DJ, Cheng JC, Toma MI, Huland H, Jones PA and Liang G:
Prognostic relevance of methylation markers in patients with
non-muscle invasive bladder carcinoma. Eur J Cancer. 41:2769–2778.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Brognard J, Zhang YW, Puto LA and Hunter
T: Cancer-associated loss-of-function mutations implicate DAPK3 as
a tumor-suppressing kinase. Cancer Res. 71:3152–3161. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Stroud H, Feng S, Morey Kinney S, Pradhan
S and Jacobsen SE: 5-Hydroxymethylcytosine is associated with
enhancers and gene bodies in human embryonic stem cells. Genome
Biol. 12:R542011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ficz G, Branco MR, Seisenberger S, Santos
F, Krueger F, Hore TA, Marques CJ, Andrews S and Reik W: Dynamic
regulation of 5-hydroxymethylcytosine in mouse ES cells and during
differentiation. Nature. 473:398–402. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ito S, D'Alessio AC, Taranova OV, Hong K,
Sowers LC and Zhang Y: Role of Tet proteins in 5mC to 5hmC
conversion, ES-cell self-renewal and inner cell mass specification.
Nature. 466:1129–1133. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Mlakar V, Berginc G, Volavsek M, Stor Z,
Rems M and Glavac D: Presence of activating KRAS mutations
correlates significantly with expression of tumour suppressor genes
DCN and TPM1 in colorectal cancer. BMC Cancer. 9:2822009.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Nevins JR: The Rb/E2F pathway and cancer.
Hum Mol Genet. 10:699–703. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yasui K, Okamoto H, Arii S and Inazawa J:
Association of over-expressed TFDP1 with progression of
hepatocellular carcinomas. J Hum Genet. 48:609–613. 2003.
View Article : Google Scholar
|
|
59
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Pedersen PL: Tumor mitochondria and the
bioenergetics of cancer cells. Prog Exp Tumor Res. 22:190–274.
1978. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Nakashima RA, Paggi MG and Pedersen PL:
Contributions of glycolysis and oxidative phosphorylation to
adenosine 5′-triphos-phate production in AS-30D hepatoma cells.
Cancer Res. 44:5702–5706. 1984.PubMed/NCBI
|
|
62
|
Levine AJ and Puzio-Kuter AM: The control
of the metabolic switch in cancers by oncogenes and tumor
suppressor genes. Science. 330:1340–1344. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ulrey CL, Liu L, Andrews LG and Tollefsbol
TO: The impact of metabolism on DNA methylation. Hum Mol Genet.
14:R139–R147. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Hoffman RM: Altered methionine metabolism,
DNA methylation and oncogene expression in carcinogenesis. A review
and synthesis. Biochim Biophys Acta. 738:49–87. 1984.PubMed/NCBI
|
|
65
|
Chiang CP, Lang MJ, Liu BY, Wang JT, Leu
JS, Hahn LJ and Kuo MY: Expression of proliferating cell nuclear
antigen (PCNA) in oral submucous fibrosis, oral epithelial
hyperkeratosis and oral epithelial dysplasia in Taiwan. Oral Oncol.
36:353–359. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Lv Q, Zhang J, Yi Y, Huang Y, Wang Y, Wang
Y and Zhang W: Proliferating cell nuclear antigen has an
association with prognosis and risks factors of cancer patients: A
systematic review. Mol Neurobiol. Nov 12–2015.(Epub ahead of
print). View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Chuang LS, Ian HI, Koh TW, Ng HH, Xu G and
Li BF: Human DNA-(cytosine-5) methyltransferase-PCNA complex as a
target for p21WAF1. Science. 277:1996–2000. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Whitesell L and Lindquist SL: HSP90 and
the chaperoning of cancer. Nat Rev Cancer. 5:761–772. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Workman P and Powers MV: Chaperoning cell
death: A critical dual role for Hsp90 in small-cell lung cancer.
Nat Chem Biol. 3:455–457. 2007. View Article : Google Scholar : PubMed/NCBI
|