Introduction
Pyrogallol (PG; benzene-1,2,3-triol) exists widely
as a disintegration product of hydrolysable tannins and it
possesses anti-psoriatic and anti-fungal properties (1). In addition, PG triggers mutagenesis,
carcinogenesis and impairment of the immune system (1). Since PG generates free radicals,
especially superoxide anion (O2•−), it has
been commonly utilized as a photographic developing agent and in
hair dying industry (1). PG also
has been employed to investigate the role of
O2•− in the biological system. PG induces the
O2•−-mediated death of various cell types
such as cervical cancer cells (2),
lymphoma cells (3), gastric cancer
cells (4), reninoma cells
(5) and lung cancer cells
(6). Despite the beneficial
effects of PG, its toxicity has been a main concern for the
individuals exposed to it. The molecular mechanism to understand
the toxicity of PG is still only partially understood.
The O2•− belongs to the
reactive oxygen species (ROS) with hydrogen peroxide
(H2O2) and hydroxyl radical (•OH).
ROS are involved in a variety of cellular events such as gene
expression, differentiation, cell proliferation and cell death
(7,8). They are primarily generated during
the mitochondrial respiration and are specifically made by various
oxidases such as nicotin-amide adenine dinucleotide phosphate
(NADPH) oxidase and xanthine oxidase (XO) (9). In relation to the principal metabolic
pathways, superoxide dismutases (SODs) converts
O2•− to H2O2 (10). Further metabolism yields
O2 and H2O through catalase (CAT) or
glutathione (GSH) peroxidase (GPX) (11). Especially, thioredoxin (TXN) system
consists of TXN, TXN reductase (TXNR) and NADPH, which is
critically implicated in maintaining cellular redox homeostasis
(12). TXN as a thiol reductase is
a potent anti-oxidant and acts as a scavenger of ROS (12). Oxidative stress due to the
overproduction of ROS and/or the accumulation of them can initiate
events that lead to cell death via the oxidation of DNA, lipid and
protein.
PG reduces the growth of Calu-6 and A549 lung cancer
cells via apoptosis and cell cycle arrest (13–15).
PG also induces GSH depletion in lung cancer cells (6,13).
However, little is known about the cellular effects of PG on normal
primary lung cells. Therefore, this study investigated the effects
of PG on cell growth and death in human pulmonary fibroblast (HPF)
cells in relation to ROS and GSH levels, and examined the effects
of N-acetylcysteine (NAC) and vitamin C (well known antioxidants)
or L-buthionine sulfoximine (BSO; an inhibitor of GSH synthesis) on
PG-induced HPF cell death.
Materials and methods
Cell culture
HPF cells purchased from PromoCell GmbH (Heidelberg,
Germany) were cultured in RPMI-1640 supplemented with 10% fetal
bovine serum (Sigma-Aldrich Chemical Co., St. Louis, MO, USA) and
1% penicillin-streptomycin (Gibco BRL, Grand Island, NY, USA). The
cells between passages four and eight were utilized for the
experiments.
Reagents
PG was purchased from Sigma-Aldrich Chemical Co. and
it was dissolved in water. Pan-caspase inhibitor (Z-VAD-FMK;
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone) was obtained from
R&D Systems, Inc. (Minneapolis, MN, USA). NAC and BSO were
obtained from Sigma-Aldrich Chemical Co. NAC was dissolved in the
buffer [20 mM HEPES (pH 7.0)]. BSO was dissolved in water. Vitamin
C purchased from Riedel-de Haen (Hannover, Germany) was also
dissolved in water. Based on a previous study (16), cells were pretreated with or
without 15 μM Z-VAD, 2 mM NAC, 10 μM BSO or 0.4 mM vitamin C for
one hour prior to the treatment of PG.
Cell growth inhibition assays
Cell growth changes were determined by measuring the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT,
Sigma-Aldrich Chemical Co.) dye absorbance as previously described
(17). Cells were exposed to the
indicated amounts of PG (5–200 μM) with or without Z-VAD, NAC,
vitamin C or BSO for 24 h.
Cell cycle and sub-G1 analysis
Cell cycle and sub-G1 cells were determined by
propidium iodide (PI, Ex/Em=488/617 nm; Sigma-Aldrich) staining as
previously described (13). Cells
were incubated with the indicated amounts of PG (5–100 μM) for 24
h. Cellular DNA content was measured using a FACStar flow cytometer
(Becton-Dickinson, Franklin Lakes, NJ, USA).
Annexin V/PI staining for cell death
detection
Apoptosis was determined by staining cells with
Annexin V-fluorescein isothiocyanate (FITC, Ex/Em=488/519 nm;
Invitrogen Molecular Probes, Eugene, OR, USA) and propidium iodide
(PI, Ex/Em=488/617 nm; Sigma-Aldrich) as previously described
(18). Cells were incubated with
the indicated amounts of PG (5–100 μM) in the presence or absence
of Z-VAD, NAC, vitamin C or BSO for 24 h. Annexin V/PI staining was
analyzed with a FACStar flow cytometer (Becton-Dickinson).
Western blot analysis
The changes of proteins related to apoptosis and
antioxidant system were determined by western blotting as
previously described (18). Cells
were incubated with 100 μM PG for 24 h. Samples containing 10 μg
total protein were resolved by 12.5% SDS-PAGE gels. The antibodies
against Bcl-2, Bax, p53, procaspase-3, CAT, SOD1, TXN, TXNR1 and
β-actin were obtained from Santa Cruz Biotechnology (Santa Cruz,
CA, USA).
Measurement of MMP (ΔΨm)
MMP (ΔΨm) levels were measured using a
rhodamine 123 fluorescent dye (Sigma-Aldrich; Ex/Em=485/535 nm) as
previously described (17,18). Cells were incubated with the
indicated amounts of PG (5–100 μM) in the presence or absence of
Z-VAD, NAC, vitamin C or BSO for 24 h. The absence of rhodamine 123
from cells indicated the loss of MMP (ΔΨm) in HPF cells.
The MMP (ΔΨm) levels in the cells excluding MMP
(ΔΨm) loss cells were expressed as mean fluorescence
intensity (MFI), which was calculated by CellQuest software
(Becton-Dickinson).
Quantification of caspase-3 and caspase-8
activities
The activities of caspase-3 and -8 were assessed
using the caspase-3 and -8 colorimetric Assay kits (R&D
Systems, Inc.). Cells were incubated with 100 μM PG for 24 h. The
activities of caspase-3 and -8 were expressed in arbitrary
absorbance units, as previously described (18).
Detection of intracellular ROS
levels
Intracellular ROS were detected by a fluorescent
probe dye, 2′,7′-dichlorodihydrofluorescein diacetate
(H2DCFDA, Ex/Em=495/529 nm; Invitrogen Molecular Probes)
as previously described (18).
Dihydroethidium (DHE, Ex/Em=518/605 nm; Invitrogen Molecular
Probes) is a fluorogenic probe that is highly selective for
O2•− among ROS. Mitochondrial
O2•− level was detected using MitoSOX™ Red
mitochondrial O2•− indicator (Ex/Em=510/580
nm; Invitrogen Molecular Probes) as previously described (19). Cells were incubated with the
indicated doses of PG (5–100 μM) in the presence or absence of NAC,
BSO or vitamin C for the indicated times. DCF, DHE and MitoSOX Red
fluorescences were detected using a FACStar flow cytometer
(Becton-Dickinson). ROS levels were expressed as mean fluorescence
intensity.
Detection of the intracellular
glutathione (GSH)
Cellular GSH levels were analyzed using a
5-chloromethylfluorescein diacetate dye (CMFDA, Ex/Em=522/595 nm;
Invitrogen Molecular Probes) as previously described (17,18).
Cells were incubated with the indicated doses of PG (5–100 μM) in
the presence or absence of NAC, BSO or vitamin C for the indicated
times. CMF fluorescence intensity was determined using a FACStar
flow cytometer (Becton-Dickinson). Negative CMF staining (GSH
depleted) cells were expressed as the percent of (-) CMF cells.
Statistical analysis
The data were analyzed using Instat software
(GraphPad Prism4, San Diego, CA, USA). The Student's t-test or
one-way analysis of variance (ANOVA) with post hoc analysis using
Tukey's multiple comparison test was used for parametric data.
Statistical significance was defined as p<0.05.
Results
Effects of PG on cell growth and cell
cycle distributions in HPF cells
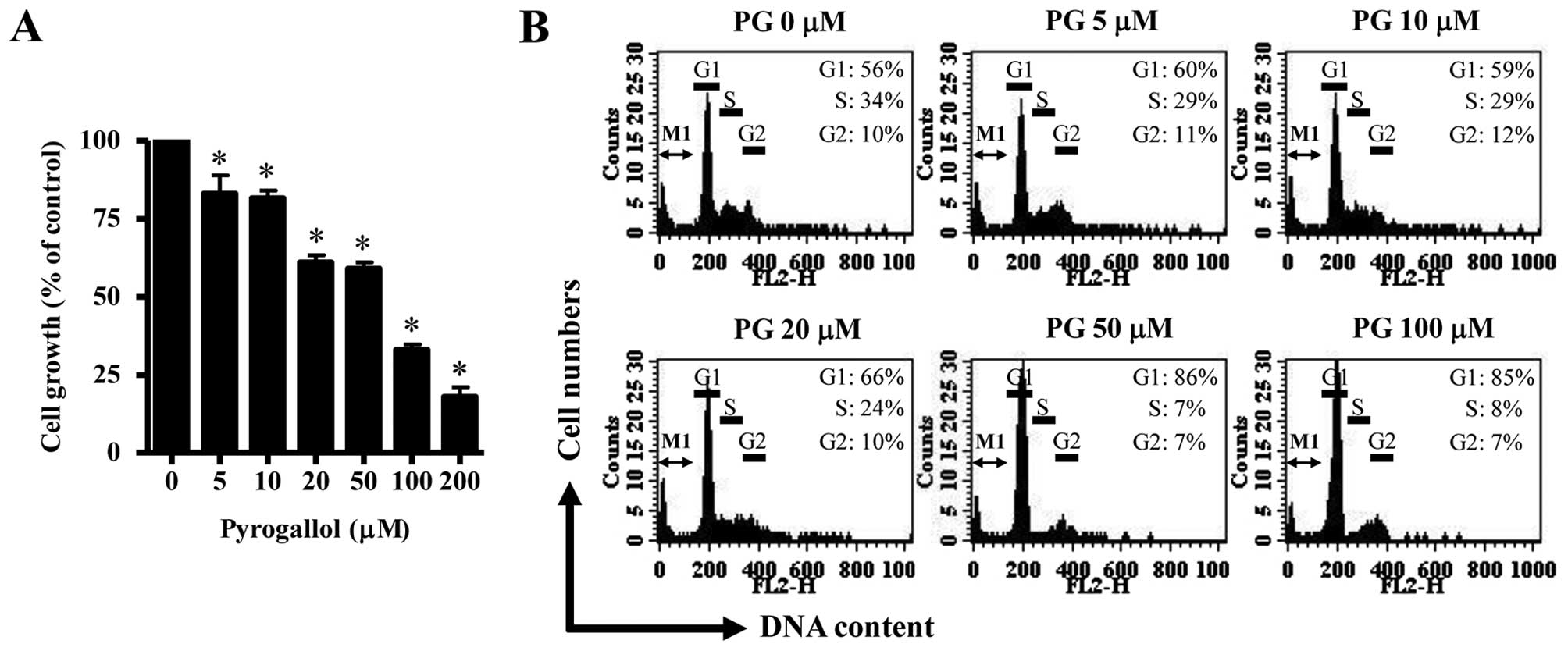
Based on MTT assays, PG dose-dependently decreased
HPF cell growth with an IC50 of ~50–100 μM at 24 h
(Fig. 1A). When cell cycle
distributions were investigated in PG-treated HPF cells, 5 μM PG
seemed to induce a G1 phase arrest of the cell cycle as compared
with control cells and the higher doses of 50 or 100 μM PG strongly
increased the proportion of G1 phase (Fig. 1B). However, an increase in sub-G1
DNA content cells was not observed in PG-treated HPF cells
(Fig. 1B).
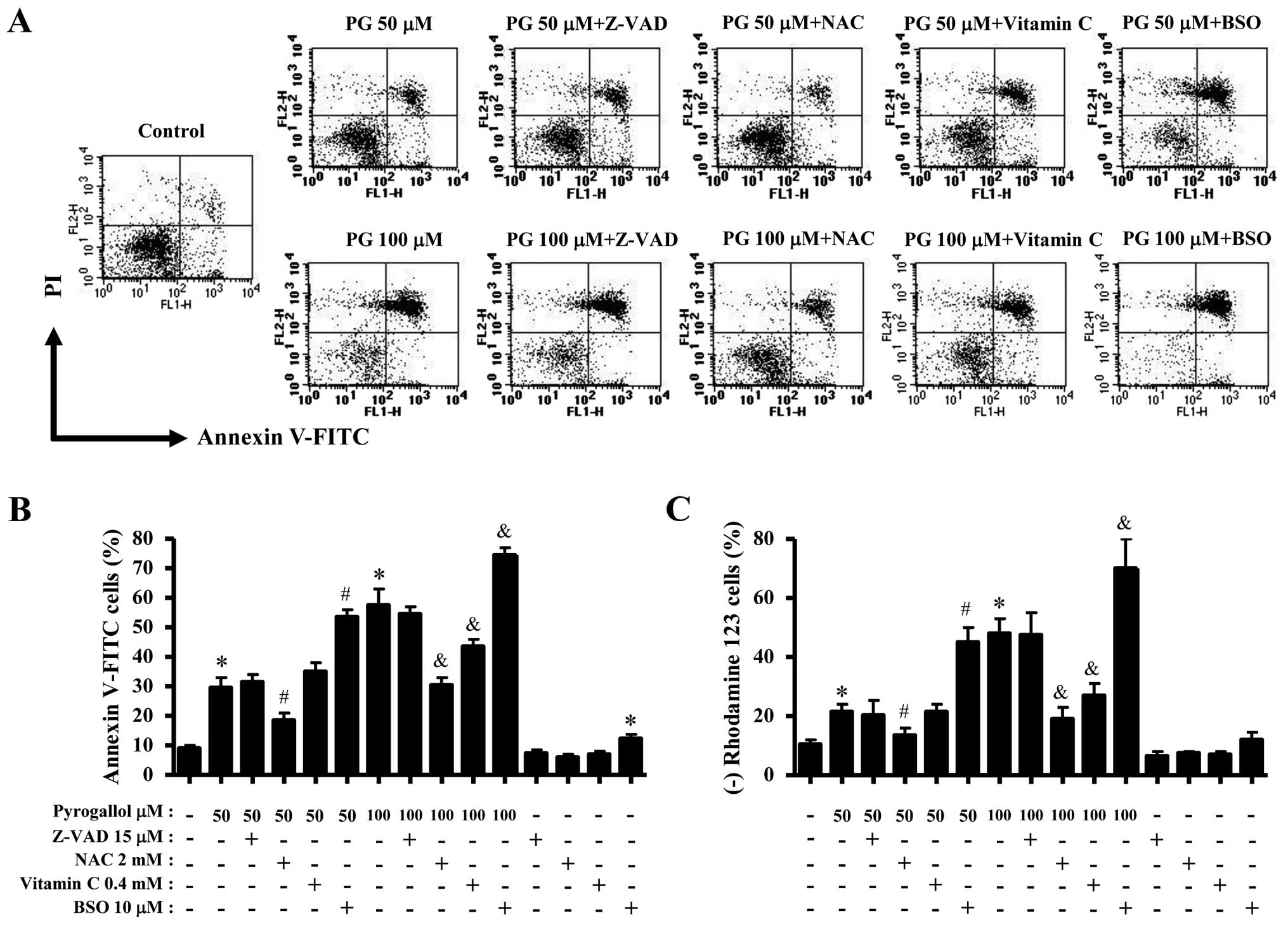
Effects of PG on cell death,
apoptosis-related proteins and MMP (ΔΨm) in HPF
cells
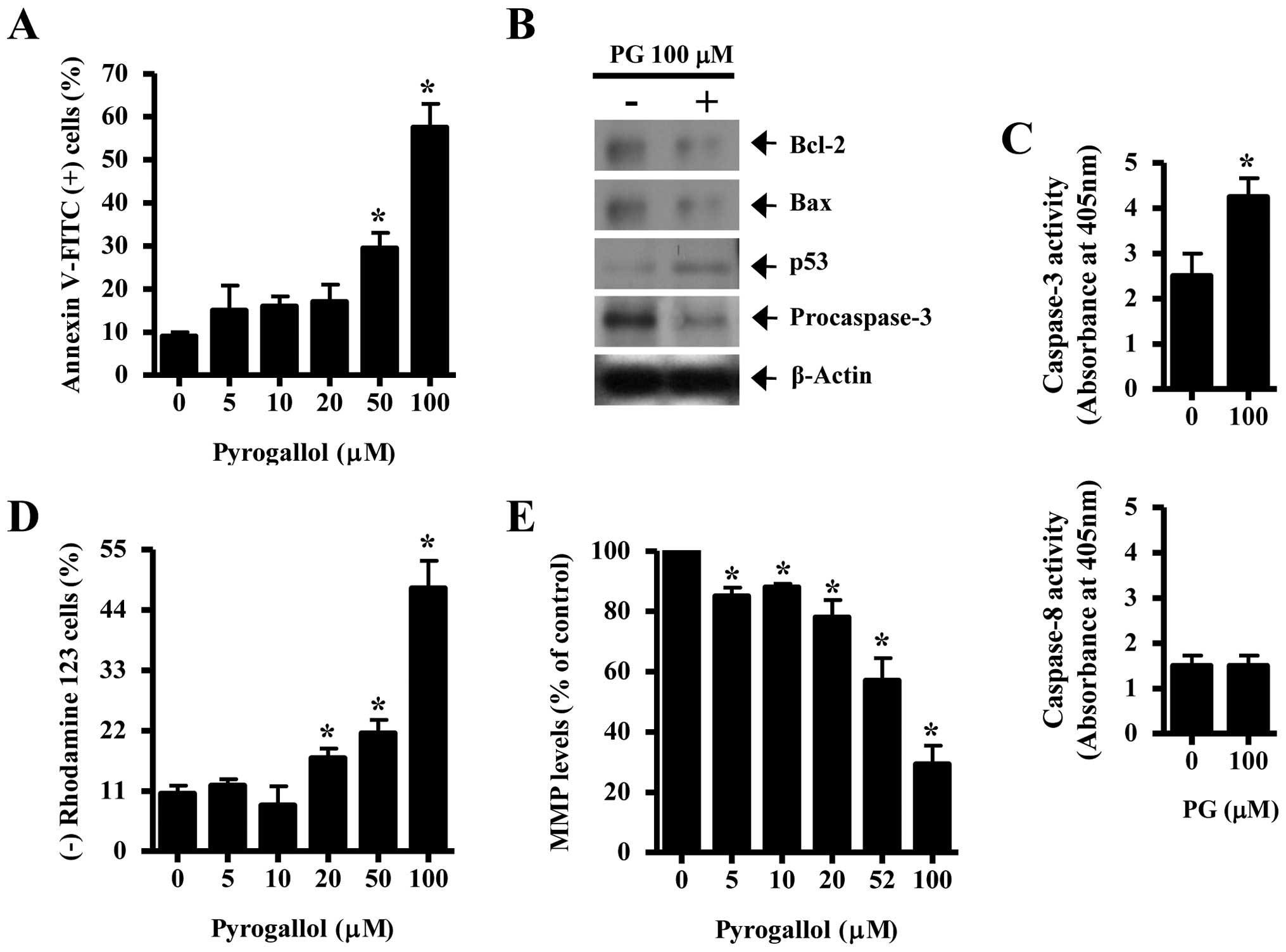
As shown in Fig.
2A, PG increased the numbers of Annexin V-FITC positive cells
in a dose-dependent manner. Concerning the relationship between
Bcl-2 and Bax regulation in PG-treated HPF cells, both proteins
were clearly decreased by PG (Fig.
2B). p53, which regulates the expression of Bcl-2, Bax or
cyclin-dependent kinase inhibitor (CDKI) in response to DNA damage
(20), was increased by PG
(Fig. 2B). Caspase-3 plays an
essential role as an executor in apoptosis (21). A 32-kDa precursor (procaspase-3)
obviously disappeared in PG-treated HPF cells (Fig. 2B) and the activity of caspase-3 was
increased by PG (Fig. 2C).
However, the activity of caspase-8, which is involved in receptor-
or extrinsic-mediated apoptosis (22), was not affected by PG (Fig. 2C). In addition, 20–100 μM PG
significantly induced the loss of MMP (ΔΨm) in HPF cells
(Fig. 2D). The levels of MMP
(ΔΨm) in HPF cells except for MMP (ΔΨm) loss
cells were decreased by PG in a dose-dependent manner (Fig. 2E).
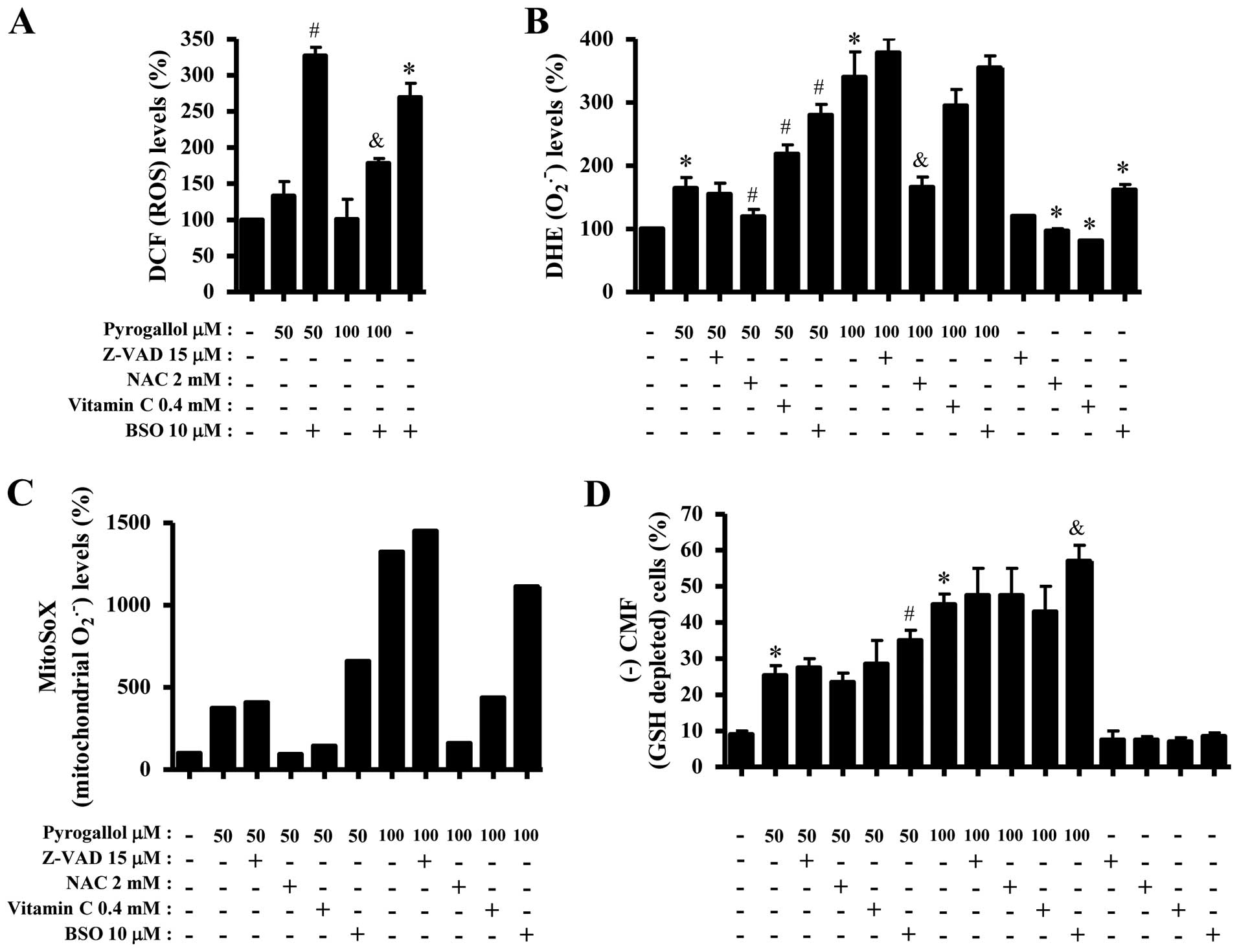
Effects of PG on ROS, GSH and
antioxidant-protein levels in HPF cells
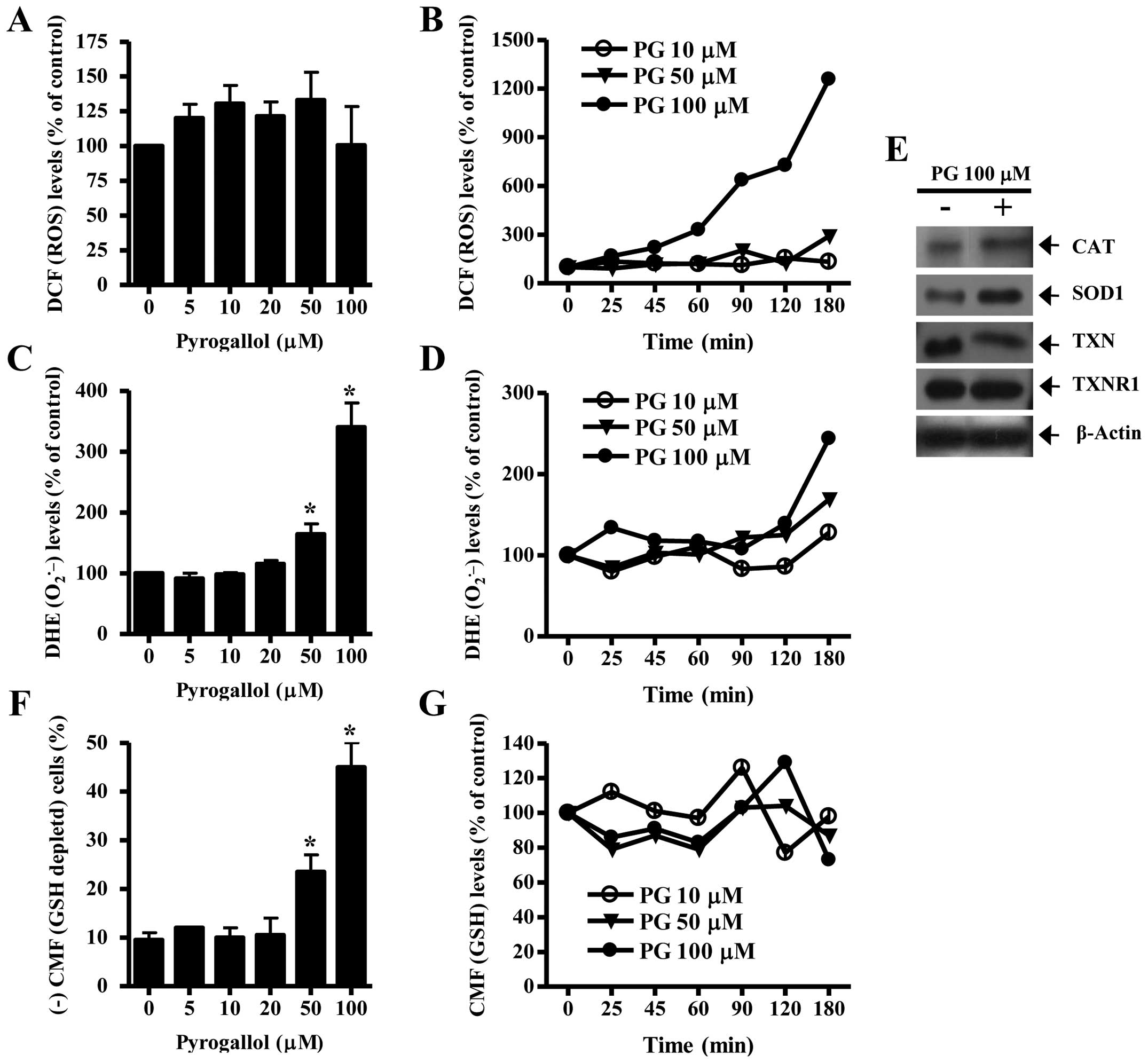
As shown in Fig.
3A, 5–50 μM PG increased ROS (DCF) levels in HPF cells at 24 h
but 100 μM PG did not significantly affect the level. While 10 and
50 μM PG did not alter ROS (DCF) levels from 25 to 120 min, 50 μM
PG increased the level at 180 min (Fig. 3B). Treatment with 100 μM PG
augmented ROS (DCF) levels from the early time of 25 min and the
gradual increases continued for the tested times (25–180 min)
(Fig. 3B). Intracellular
O2•− (DHE) level significantly increased in
50 or 100 μM PG-treated HPF cells whereas the level was not clearly
changed by 5–20 μM PG treatment (Fig.
3C). While 10 or 50 μM PG transiently decreased
O2•− level in HPF cells at 25 min, 100 μM PG
increased the level at this time (Fig.
3D). At 180 min, all the tested doses of PG increased
O2•− levels in HPF cells and 100 μM PG showed
a strong effect on the level (Fig.
3D). When antioxidant-protein levels were assessed in 100 μM
PG-treated HPF cells, the expression of CAT and SOD1 was increased
by PG treatment (Fig. 3E). In
addition, PG clearly downregulated the expression of TXN in HPF
cells and did not change that of TXNR1 (Fig. 3E).
In relation to GSH levels, 50 or 100 μM PG
significantly increased GSH depleted cell number in HPF cells
whereas lower doses of 5, 10 or 20 μM PG did not induce GSH
depletion in HPF cells, as compared with control HPF cells
(Fig. 3F). At the early time of 25
min in PG-treated HPF cells, 10 μM PG transiently increased GSH
level whereas 50 or 100 μM PG decreased the level (Fig. 3G). Although there were transient
increases in GSH levels at 90 or 120 min in PG treated-HPF cells,
the GSH levels generally decreased in these cells at 180 min
(Fig. 3G).
Effects of Z-VAD, NAC, vitamin C or BSO
on cell growth, cell death and MMP (ΔΨm) in PG-treated
HPF cells
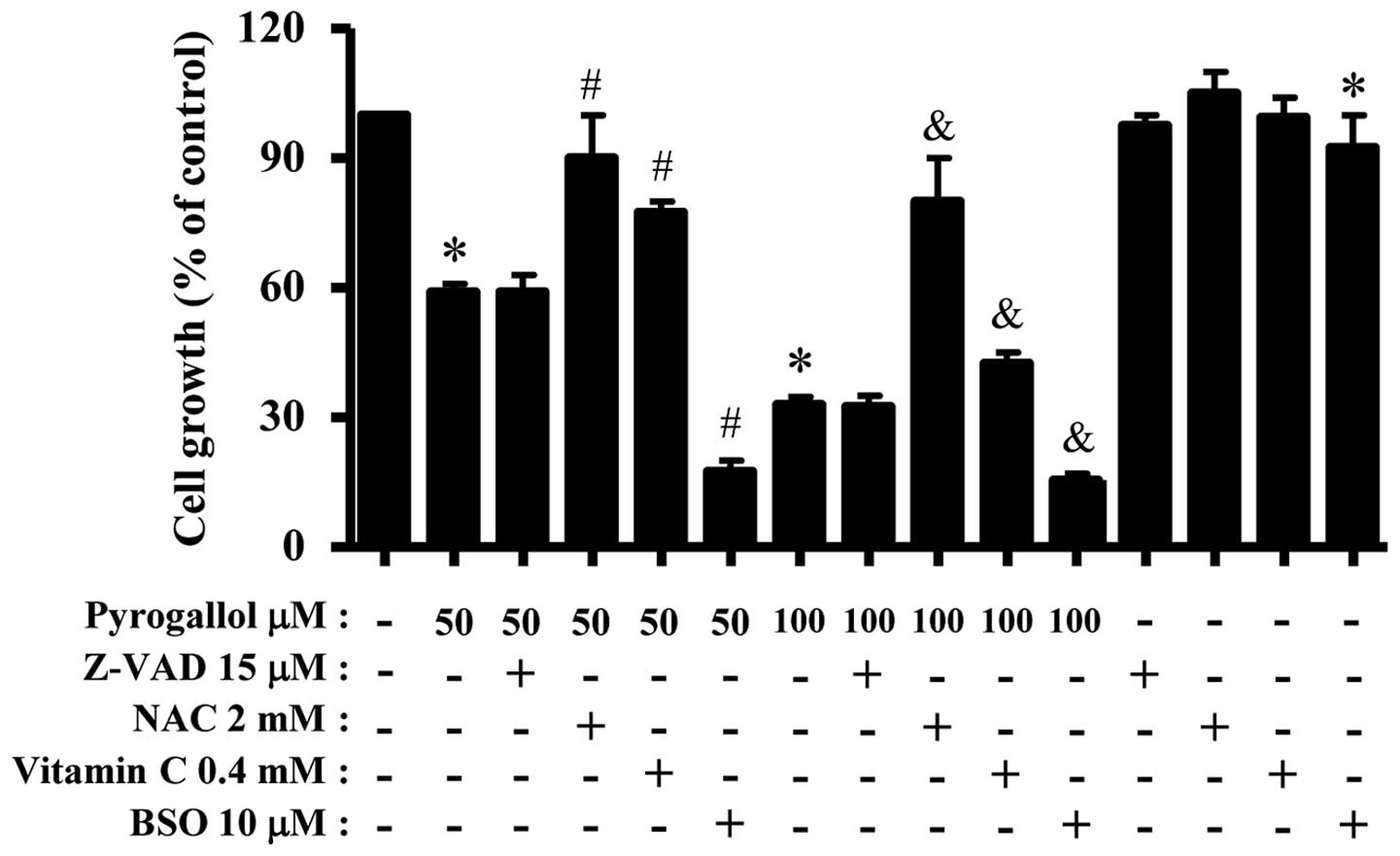
For this experiment, 50 or 100 μM PG was used as a
suitable dose to differentiate the levels of cell growth inhibition
and death. Treatment with 15 μM Z-VAD did not significantly affect
growth inhibition in PG-treated or -untreated HPF cells (Fig. 4). Both antioxidants of NAC and
vitamin C significantly prevented the growth inhibition by 50 or
100 μM PG whereas BSO enhanced the growth inhibition (Fig. 4). BSO alone slightly inhibited HPF
cell growth (Fig. 4). In relation
to cell death, Z-VAD did not influence HPF cell death by PG.
However, NAC significantly rescued HPF cells from the insult of PG
(Fig. 5A and B). While vitamin C
did not attenuate cell death in 50 μM PG-treated HPF cells, this
agent significantly prevented the death induced by 100 μM PG
(Fig. 5A and B). BSO enhanced HPF
cell death by PG (Fig. 5A and B).
Similar to the results of Annexin V staining cells, Z-VAD did not
change the loss of MMP (ΔΨm) in PG-treated HPF cells and
NAC decreased the loss in these cells (Fig. 5C). In addition, vitamin C
significantly prevented the loss of MMP (ΔΨm) induced by
100 μM PG (Fig. 5C). BSO enhanced
MMP (ΔΨm) loss in PG-treated HPF cells (Fig. 5C).
Effects of Z-VAD, NAC, vitamin C or BSO
on ROS and GSH levels in PG-treated HPF cells
As shown in Fig.
6A, ROS (DCF) levels in PG-treated or -untreated HPF cells were
significantly increased by BSO. However, Z-VAD, NAC or vitamin C
did not strongly change ROS (DCF) levels in PG-treated HPF cells
(data not shown). Z-VAD also did not significantly affect
O2•− level in PG-treated HPF cells whereas
NAC strongly decreased O2•− level in these
cells (Fig. 6B). While vitamin C
increased O2•− level in 50 μM PG-treated HPF
cells, it decreased the level in 100 μM PG-treated cells (Fig. 6B). Both NAC and vitamin C decreased
basal ROS levels including O2•− in HPF
control cells (Fig. 6B). In
contrast, BSO significantly increased O2•−
levels in 50 μM PG-treated or -untreated HPF cells but it slightly
increased the level in 100 μM PG-treated cells (Fig. 6B). Furthermore, MitoSOX Red
fluorescence levels strongly increased in 50 or 100 μM PG-treated
HPF cells at 24 h (Fig. 6C). Z-VAD
did not alter mitochondrial O2•− level in
PG-treated HPF cells whereas NAC and vitamin C decreased the level
in these cells (Fig. 6C). BSO
increased mitochondrial O2•− level in 50
PG-treated HPF cells but it decreased the level in 100 μM
PG-treated HPF cells (Fig. 6C).
When GSH depletion levels in PG-treated HPF cells were assessed in
the presence or absence of Z-VAD, NAC, vitamin C or BSO, only BSO
significantly increased GSH depleted cell number in PG-treated HPF
cells and the others did not change GSH depletion levels (Fig. 6D).
Discussion
This study elucidated the cellular effect of PG on
cell growth and death in HPF cells in relation to ROS and GSH
levels. PG decreased HPF cell growth with an IC50 of
~50–100 μM. According to previous reports, the IC50 of
PG in Calu-6 and A549 lung cancer cells was ~20 and 50 μM at 24 h,
respectively (13,15). Therefore, the susceptibility to PG
in HPF cells was lower than other lung cancer cells. In addition,
the growth of calf pulmonary artery endothelial cells is inhibited
by PG with an IC50 of ~50 μM (23). The difference of susceptibility to
PG between these lung cells is probably due to the different basal
activities of mitochondria and antioxidant enzymes depending on
cell type, tissue origin and species (24). In addition, PG induced a G1 phase
arrest of the cell cycle in HPF cells, which might be mediated by
the increase of p53 protein, a CDKI inducer. It is reported that PG
induces G1 and G2 phase arrests in A549 (14) and Calu-6 cells (14), respectively. PG also inhibits the
growth of As4.1 juxtaglomerular cells via a G2 phase arrest
(25). Alternatively, PG
non-specifically induces cell cycle arrests in SNU-484 gastric
cancer cells (4) and HeLa cervical
cancer cells (2). Taken together,
these results suggest that the molecular mechanism of cell cycle
arrest by PG has great variety depending on the cell types.
PG induces apoptosis through the downregulation of
Bcl-2 protein in cancer cells (2,25,26).
Similarly, HPF cell death by PG was accompanied by a decrease in
Bcl-2 protein. In addition, p53 protein, which regulates the
expressions of Bcl-2, increased in PG-treated HPF cells. In
contrast, Bax level is reported to be upegulated in PG-treated lung
cancer cells (26). However, Bax
protein in HPF cells was decreased by PG. In addition, Bax level is
not changed in PG-treated As4.1 and HeLa cells (2,25).
The changes of Bax levels by PG seem to be different depending on
cell types. PG-mediated HPF apoptosis probably resulted from the
downregulation of Bcl-2 and the upegulation of p53. In addition, PG
induces the collapse of MMP (ΔΨm) during apoptosis in
cancer cells (4,13–15).
Correspondingly, PG induced the loss of MMP (ΔΨm) in HPF
cells and decreased its level in viable HPF cells. The activation
of caspase-3 is important for the process of PG-induced cell death
(2,15,25).
Likewise, PG apparently increased the activity of caspase-3 and
decreased the precursor form of this caspase. Caspase-8 is
activated in PG-treated Calu-6 lung cells (15). However, the activity of caspase-8
was not altered in PG-treated HPF cells, implying that PG-mediated
HPF apoptosis was not related to receptor- or extrinsic-mediated
apoptosis. Its activation by PG can be differently regulated
depending on cell types. Interestingly, an increase in sub-G1 cells
was not observed in PG-treated HPF cells. Therefore, PG seemed to
induce HPF cell death via necrosis as well as apoptosis. In fact,
100 μM PG increased the lactate dehydrogenase activity released
from HPF cells (unpublished data). In addition, Z-VAD did not
affect cell growth inhibition, death and MMP (ΔΨm) loss
in PG-treated HPF cells. However, Z-VAD including other caspase
inhibitors strongly prevented apoptosis in PG-treated cells
(19,23,25).
It is possible that the mechanism of cell death induced by PG can
be different between cancer and normal cells.
ROS level (as determined by DCF) increased in HPF
cells treated with 5–50 μM PG, but not 100 μM PG at 24 h. However,
100 μM PG strongly augmented ROS (DCF) levels at the early time
phases of 25–180 min whereas 10 or 50 μM PG did not increase at
these times. Because 100 μM PG increased CAT protein at 24 h, it is
feasible that ROS such as H2O2 in 100 μM
PG-treated HPF cells was rapidly converted into O2 and
H2O by the increased CAT. In fact, PG increased the
activity of CAT in Calu-6 cells and it decreases ROS (DCF) levels
(6). O2•−
level (as determined by DHE) significantly increased in HPF cells
treated with 50–100 μM, but not 5–20 μM PG. Treatment with 100 μM
PG transiently increased O2•− level at 25 min
whereas 10 or 50 μM PG decreased the level at this time. At 180
min, all the tested doses of PG increased the
O2•− levels. Increased
O2•− levels in PG-treated HPF cells at 24 h
seemed to result from the enhanced production of
O2•− itself rather than the reduction of SOD
activity since MMP (ΔΨm) loss and mitochondrial
O2•− level increased in these cells and the
expression of SOD1 was not downregulated by PG. Especially, PG
relatively showed a strong increased effect on mitochondrial
O2•− level in HPF cells as compared with
O2•− level. It is possible that 100 μM PG
damaged mitochondria and generated O2•− level
at 25 min, consequently increasing ROS (DCF) levels at the early
time phases of 25–180 min. PG did not affect the level of TXNR1
protein in HPF cells but it downregulated TXN level. Because TXN
and TXNR1 induce resistance to anticancer drugs (27), the downregulation of TXN by PG may
render HPF cells sensitive to this agent.
NAC significantly attenuated
O2•− level including mitochondrial
O2•− in PG-treated or -untreated HPF cells.
It also attenuated cell growth inhibition, cell death and MMP
(ΔΨm) loss in PG-treated HPF cells. In contrast, BSO
strongly enhanced cell growth inhibition, cell death and MMP
(ΔΨm) loss in PG-treated HPF cells and augmented ROS
levels in these cells. In addition, Z-VAD did not significantly
alter O2•− levels in PG-treated HPF cells.
Therefore, PG is likely to induce HPF cell death through oxidative
stresses. BSO alone induced cell growth inhibition and cell death
in HPF control cells and it strongly increased ROS levels.
Therefore, ROS increased by BSO might be related to HPF cell death.
Vitamin C did not prevent cell death and MMP (ΔΨm) loss
in 50 μM PG-treated HPF cells but this agent significantly
decreased cell death and MMP (ΔΨm) loss by 100 μM PG. In
addition, vitamin C increased O2•− level in
50 μM PG-treated HPF cells whereas it decreased the level in 100 μM
PG-treated cells. Vitamin C strongly decreased mitochondrial
O2•− in 50 or 100 μM PG-treated HPF cells.
Therefore, vitamin C showed different effects on cell death in HPF
cells depending on the exposed doses of PG. In addition, vitamin C
played a role as a strong antioxidant in reducing mitochondrial
O2•− level in PG-treated HPF cells.
The intracellular GSH content has a decisive effect
on PG-induced apoptosis (4–6,13,23,28).
Likewise, PG increased the number of GSH-depleted cells in HPF
cells. As expected, BSO as a GSH synthesis inhibitor increased the
numbers of GSH depleted cells in PG-treated HPF cells. In addition,
Z-VAD did not change the numbers in these cells. NAC as a GSH
precursor attenuates GSH depletion in PG-treated cells (28,29).
However, NAC did not affect GSH depletion in PG-treated HPF cells,
implying that this agent was not used as a GSH precursor in HPF
cells. In addition, vitamin C did not attenuate GSH depletion in
100 μM PG-treated HPF cells and BSO alone did not induce GSH
depletion in control HPF cells. These results suggest that an
intracellular GSH content is a decisive role in PG-induced HPF cell
death but its content change is not enough to estimate cell death
precisely.
In conclusion, PG inhibited the growth of HPF cells
via the cell death (apoptosis and/or necrosis) as well as a G1
phase arrest of the cell cycle. PG-induced HPF cell death was
related to increases in ROS level and GSH depletion. These results
provide useful information to understand the cellular effect of PG
on normal lung cells in relation to ROS and GSH.
Acknowledgements
The author would like to thank Dr Bo Ra You for
helping with western blot analysis. This study was supported by a
grant from the National Research Foundation of Korea (NRF) funded
by the Korean government (MSIP; No. 2008-0062279 and
2016R1A2B4007773).
Abbreviations:
|
HPF
|
human pulmonary fibroblast
|
|
PG
|
pyrogallol
|
|
ROS
|
reactive oxygen species
|
|
MMP (ΔΨm)
|
mitochondrial membrane potential
|
|
SOD
|
superoxide dismutase
|
|
CAT
|
catalase
|
|
GSH
|
glutathione
|
|
GPX
|
GSH peroxidase
|
|
TXN
|
thioredoxin
|
|
TXNR
|
TXN reductase
|
|
PI
|
propidium iodide
|
|
H2DCFDA
|
2′,7′-dichlorodihydrofluorescein
diacetate
|
|
DHE
|
dihydroethidium
|
|
CMFDA
|
5-chloromethylfluorescein
diacetate
|
|
NAC
|
N-acetylcysteine
|
|
BSO
|
L-buthionine sulfoximine
|
References
|
1
|
Upadhyay G, Gupta SP, Prakash O and Singh
MP: Pyrogallol-mediated toxicity and natural antioxidants: Triumphs
and pitfalls of preclinical findings and their translational
limitations. Chem Biol Interact. 183:333–340. 2010. View Article : Google Scholar
|
|
2
|
Kim SW, Han YW, Lee ST, Jeong HJ, Kim SH,
Kim IH, Lee SO, Kim DG, Kim SH, Kim SZ, et al: A superoxide anion
generator, pyrogallol, inhibits the growth of HeLa cells via cell
cycle arrest and apoptosis. Mol Carcinog. 47:114–125. 2008.
View Article : Google Scholar
|
|
3
|
Saeki K, Hayakawa S, Isemura M and Miyase
T: Importance of a pyrogallol-type structure in catechin compounds
for apoptosis-inducing activity. Phytochemistry. 53:391–394. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Park WH, Park MN, Han YH and Kim SW:
Pyrogallol inhibits the growth of gastric cancer SNU-484 cells via
induction of apoptosis. Int J Mol Med. 22:263–268. 2008.PubMed/NCBI
|
|
5
|
Park WH, Han YW, Kim SH and Kim SZ: A
superoxide anion generator, pyrogallol induces apoptosis in As4.1
cells through the depletion of intracellular GSH content. Mutat
Res. 619:81–92. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Han YH, Kim SZ, Kim SH and Park WH:
Apoptosis in pyrogallol-treated Calu-6 cells is correlated with the
changes of intracellular GSH levels rather than ROS levels. Lung
Cancer. 59:301–314. 2008. View Article : Google Scholar
|
|
7
|
Gonzalez C, Sanz-Alfayate G, Agapito MT,
Gomez-Niño A, Rocher A and Obeso A: Significance of ROS in oxygen
sensing in cell systems with sensitivity to physiological hypoxia.
Respir Physiol Neurobiol. 132:17–41. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Baran CP, Zeigler MM, Tridandapani S and
Marsh CB: The role of ROS and RNS in regulating life and death of
blood monocytes. Curr Pharm Des. 10:855–866. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial ROS-induced ROS release: An update and review.
Biochim Biophys Acta. 1757:509–517. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zelko IN, Mariani TJ and Folz RJ:
Superoxide dismutase multigene family: A comparison of the CuZn-SOD
(SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures,
evolution, and expression. Free Radic Biol Med. 33:337–349. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wilcox CS: Reactive oxygen species: Roles
in blood pressure and kidney function. Curr Hypertens Rep.
4:160–166. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Marks PA: Thioredoxin in cancer - role of
histone deacetylase inhibitors. Semin Cancer Biol. 16:436–443.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Han YH, Kim SH, Kim SZ and Park WH:
Pyrogallol inhibits the growth of human pulmonary adenocarcinoma
A549 cells by arresting cell cycle and triggering apoptosis. J
Biochem Mol Toxicol. 23:36–42. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Han YH, Kim SZ, Kim SH and Park WH:
Pyrogallol inhibits the growth of human lung cancer Calu-6 cells
via arresting the cell cycle arrest. Toxicol In Vitro.
22:1605–1609. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Han YH, Kim SZ, Kim SH and Park WH:
Pyrogallol inhibits the growth of lung cancer Calu-6 cells via
caspase-dependent apoptosis. Chem Biol Interact. 177:107–114. 2009.
View Article : Google Scholar
|
|
16
|
Park WH and You BR: Antimycin A induces
death of the human pulmonary fibroblast cells via ROS increase and
GSH depletion. Int J Oncol. 48:813–820. 2016.
|
|
17
|
You BR, Kim SH and Park WH: Reactive
oxygen species, glutathione, and thioredoxin influence suberoyl
bishydroxamic acid-induced apoptosis in A549 lung cancer cells.
Tumour Biol. 36:3429–3439. 2015. View Article : Google Scholar
|
|
18
|
You BR, Shin HR, Han BR and Park WH: PX-12
induces apoptosis in Calu-6 cells in an oxidative stress-dependent
manner. Tumour Biol. 36:2087–2095. 2015. View Article : Google Scholar
|
|
19
|
Han YH, Kim SH, Kim SZ and Park WH:
Caspase inhibitor decreases apoptosis in pyrogallol-treated lung
cancer Calu-6 cells via the prevention of GSH depletion. Int J
Oncol. 33:1099–1105. 2008.PubMed/NCBI
|
|
20
|
Coutts AS and La Thangue N: The p53
response during DNA damage: Impact of transcriptional cofactors.
Biochem Soc Symp. 73:181–189. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Porter AG and Jänicke RU: Emerging roles
of caspase-3 in apoptosis. Cell Death Differ. 6:99–104. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ashkenazi A and Dixit VM: Death receptors:
Signaling and modulation. Science. 281:1305–1308. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Han YH and Park WH: Pyrogallol-induced
calf pulmonary arterial endothelial cell death via
caspase-dependent apoptosis and GSH depletion. Food Chem Toxicol.
48:558–563. 2010. View Article : Google Scholar
|
|
24
|
Oberley LW and Oberley TD: Role of
antioxidant enzymes in cell immortalization and transformation. Mol
Cell Biochem. 84:147–153. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Park WH, Han YH, Kim SH and Kim SZ:
Pyrogallol, ROS generator inhibits As4.1 juxtaglomerular cells via
cell cycle arrest of G2 phase and apoptosis. Toxicology.
235:130–139. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang CJ, Wang CS, Hung JY, Huang HW, Chia
YC, Wang PH, Weng CF and Huang MS: Pyrogallol induces G2-M arrest
in human lung cancer cells and inhibits tumor growth in an animal
model. Lung Cancer. 66:162–168. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim SJ, Miyoshi Y, Taguchi T, Tamaki Y,
Nakamura H, Yodoi J, Kato K and Noguchi S: High thioredoxin
expression is associated with resistance to docetaxel in primary
breast cancer. Clin Cancer Res. 11:8425–8430. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Han YH, Kim SZ, Kim SH and Park WH:
Pyrogallol as a glutathione depletor induces apoptosis in HeLa
cells. Int J Mol Med. 21:721–730. 2008.PubMed/NCBI
|
|
29
|
Han YH, Moon HJ, You BR, Kim SZ, Kim SH
and Park WH: Pyrogallol-induced endothelial cell death is related
to GSH depletion rather than ROS level changes. Oncol Rep.
23:287–292. 2010.
|