Introduction
A number of reports have identified a putative
anticancer role for naltrexone when used at doses lower than those
conventionally administered (1).
In particular, it has been shown that these low doses of naltrexone
(LDN) are able to suppress tumour growth. A definitive mechanism of
action has yet to be established, but what is known is that the
effect can be via modifications to the host immune system rendering
it more anticancer in nature, or through direct antagonism of
tumour growth. Naltrexone, being a specific antagonist of the
opioid receptors, can influence the activity of intracellular
signalling pathways found downstream of the G protein-coupled
receptor. As the net actions of these pathways determine the fate
of cells, naltrexone, as well as other opiates, is able to alter
cellular function, and induce cell death (2–5).
Indeed, in addition to their universally accepted analgesic
qualities, the opioids have also been widely reported to elicit a
number of other cellular responses that lead to reductions in
tumours.
Studies investigating the intracellular effects of
naltrexone noted that the mechanism underlying the action of
opioids generally involved, in part, modulation of the PI3-kinase
cascade (6,7). This suggested that these agents may
be able to influence the growth and survival of cancer cells.
However, the literature surrounding this is still very unclear, and
can be contradictory. Reports have simultaneously shown the
archetypal opiate morphine is able to both inhibit the growth of
cancer cells as well as to stimulate them in vitro. As some
of these cancer cell lines express relatively low levels of the
opioid receptors (8), the effects
on growth may be independent of them. Furthermore, as both the pro-
and anticancer effects have been seen in animal model systems with
intact immunity, it has been postulated that morphine may affect
the immune response (9), although,
this remains inconclusive (10).
Naltrexone has been demonstrated to have a very
diverse range of effects on cells. Some are pro-survival in nature
such as the induction of proliferation and protection against cell
death, whereas others are associated with growth inhibition and the
induction of apoptosis. This diversity has made it difficult to
establish the principal mechanism of action. The ultimate
consequence of treatment with naltrexone however, appears to be
determined by dose and schedule (6). Essentially, naltrexone exhibits
non-cytotoxic anticancer activities and as such, may be best
combined with other modalities and combination schedules that
maximise the individual action of each partner (11). Furthermore, the use of drugs
concomitantly may allow the individual elements to prime for the
effects of another and result in a therapeutic synergy. These
schedules may also involve adaptations in the sequence in which the
agents are employed, and is especially true if the strategy
involves a combination of a chemotherapy and an immune adjuvant
(11). Determining the best
combinatorial partner for naltrexone requires careful
consideration, as drugs with characteristics that are unsuitable
together can result in antagonism, disruption to the effects of the
individual drugs and consequential loss of overall activity.
Here, we describe part of our ongoing studies that
explore the anticancer properties of agents that are not cytotoxic
nor typically anticancer in application. We have performed a gene
expression screen in a cancer cell line previously used to study
other known anticancer drugs to determine the profile of naltrexone
action. Specifically, we have studied the genetic impact when used
at a dose that is typically employed clinically as well as at a
lower concentration. Using this information, we then examined the
effect that different treatment strategies and schedules have on
the efficacy of this drug in vitro.
Materials and methods
Cell culture and drugs
All cell lines were purchased from the European
Collection of Cell Cultures (Salisbury, UK), and maintained and
grown in the culture medium specified by the depositor.
Specifically, the principal cell lines used in this study were
HCT116 (human colorectal cancer) and A549 (human lung cancer),
which were cultured in DMEM (Sigma) supplemented with 10% fetal
bovine serum (FBS: Life Technologies, Paisley, UK) and 2 mM
L-glutamine (Life Technologies). All cells were grown in a
humidified atmosphere with 5% CO2 in air at 37°C, and
discarded after ~12 passages. Authentication of the cell lines was
performed by the service providers using the AmpFISTR Identifier
Plus PCR amplification kit looking for the presence of <10 known
loci for each cell line.
Naltrexone hydrochloride (naltrexone),
cyclophosphamide (CPM), gemcitabine (GEM) and oxaliplatin (OXP)
(all Sigma Ltd., Dorset, UK) were dissolved in DMSO, with the final
DMSO concentration in individual tests being <0.05%. Preliminary
studies suggested the action of naltrexone was dependent upon
concentration; therefore, two concentration-ranges were assessed.
Conventional doses were >1 μM and designated as NTX, whilst low
doses of naltrexone (LDN) were <100 nM. Typical NTX
concentrations were 1 and 10 μM, whereas LDN concentrations were
3-log lower being 1 and 10 nM.
RNA extraction and microarray
analysis
The genomic arm of this study followed a similar
path to one we have published a number of times (12–15).
Briefly, exponentially growing cells were seeded into 6-well plates
(BD Biosciences, Oxford, UK) at a concentration of
2×105/well and were left to adhere overnight. Cells were
then treated for 4 h with NTX (10 μM) or LDN (10 nM), before RNA
was extracted and processed as described previously (13).
Samples were then processed for microarray analysis
according to the methodologies detailed previously (13). Briefly, equal amounts of
biotinylated cRNA was hybridised to the Illumina human HT12-v3
arrays (Applied Biosystems, Warrington, UK) for 18 h and
subsequently processed according to manufacturer's instructions
before scanning on an Illumina BeadArray Reader (Applied
Biosystems). The image data were processed using default values in
GenomeStudio v2009.1 with imputation of missing data, before
loading onto GeneSpring v9.0 for data normalisation and filtering.
A >0.25-fold change was used as our cut-off magnitude for gene
list compositions by using Excel software.
Proliferation assays
To study the effect of naltrexone on cell growth,
cells growing exponentially were added to 96-well plates at a
density of 3×104/well. LDN and/or NTX was then added to
the wells, ensuring an equal volume of 200 μl across the plate.
Cell number was measured at 72 h using a standard
methylthiazoltetrazolium (MTT)-based assay as described previously
(16).
Immunoblotting analysis
Following individual treatments, cells were then
harvested by scraping into lysis buffer (New England Biolabs,
Hitchin, UK), and standard western blot protocols were followed as
described previously (16).
Primary probing was performed with specific antibodies generated
against p21, cyclin D1, cyclin B1, BAD, BAX, phosphorylated (p)
AKT, AKT, pERK, ERK and tollip (all New England Biolabs).
Anti-GAPDH (New England Biolabs) was used as a loading control. All
antibodies were used at a dilution of 1:1,000, followed by the
appropriate HRP conjugated secondary antibodies (New England
Biolabs) also at a dilution of 1:1,000. Bands were visualised by
the SuperSignal chemiluminescent detection system (Thermo
Scientific, Northumberland, UK). Densitometry of band intensity was
determined using Adobe Photoshop CS3, v10.0 (Maidenhead, UK), and
normalised to the loading control.
Recovery studies
A549 and HCT116 cells were seeded into 6-well plates
at a density of 2×105/well and left to adhere overnight.
Cells were then cultured with NTX (1 and 10 μM) and LDN (1 and 10
nM). Drug-containing media was removed after 48 h, and cells were
rinsed gently with drug-free medium. Fresh culture medium was then
added to the cells with or without naltrexone at their matching
concentrations, and incubated for a further 24 h. Cell number and
viability were assessed at 48 h (pre-recovery) and 72 h
(post-recovery), with percentages of live and dead cells being
discriminated by trypan blue dye exclusion. Cells were also
processed for the determination of cell cycle distribution by flow
cytometry utilising the nucleic acid stain propidium iodide
(16).
Combination studies
The impact of combining LDN with other chemotherapy
agents was tested by culturing cells according to a treatment
schedule that involved two phases of treatment. The first phase
involved priming with LDN or NTX for 48 h, before treatment with
another drug for a further 48 h. A549 and HCT116 cells were seeded
into 6-well plates at a density of 2×105/well and left
to adhere overnight. Cells were then cultured with 10 nM LDN or 10
μM NTX. Drug-containing media was removed after 48 h, and cells
were rinsed gently with drug-free medium. Fresh culture medium that
contained CPM, GEM or OXP was then added to the cells. The
concentrations of the chemotherapy agents used were ~1/4
IC50 as established previously (17). Cells were then left for a further
48 h before cell counting and processing for flow cytometry and
western blotting.
Results
Different patterns of genes are affected
by NTX and LDN
Standard unsupervised microarray analysis of the
transcriptome of cells following treatment with NTX or LDN was
performed to understand the patterns of genes that were altered at
the different doses. It also served to possibly identify novel
targets of naltrexone, which could explain further the anticancer
effects associated with its use.
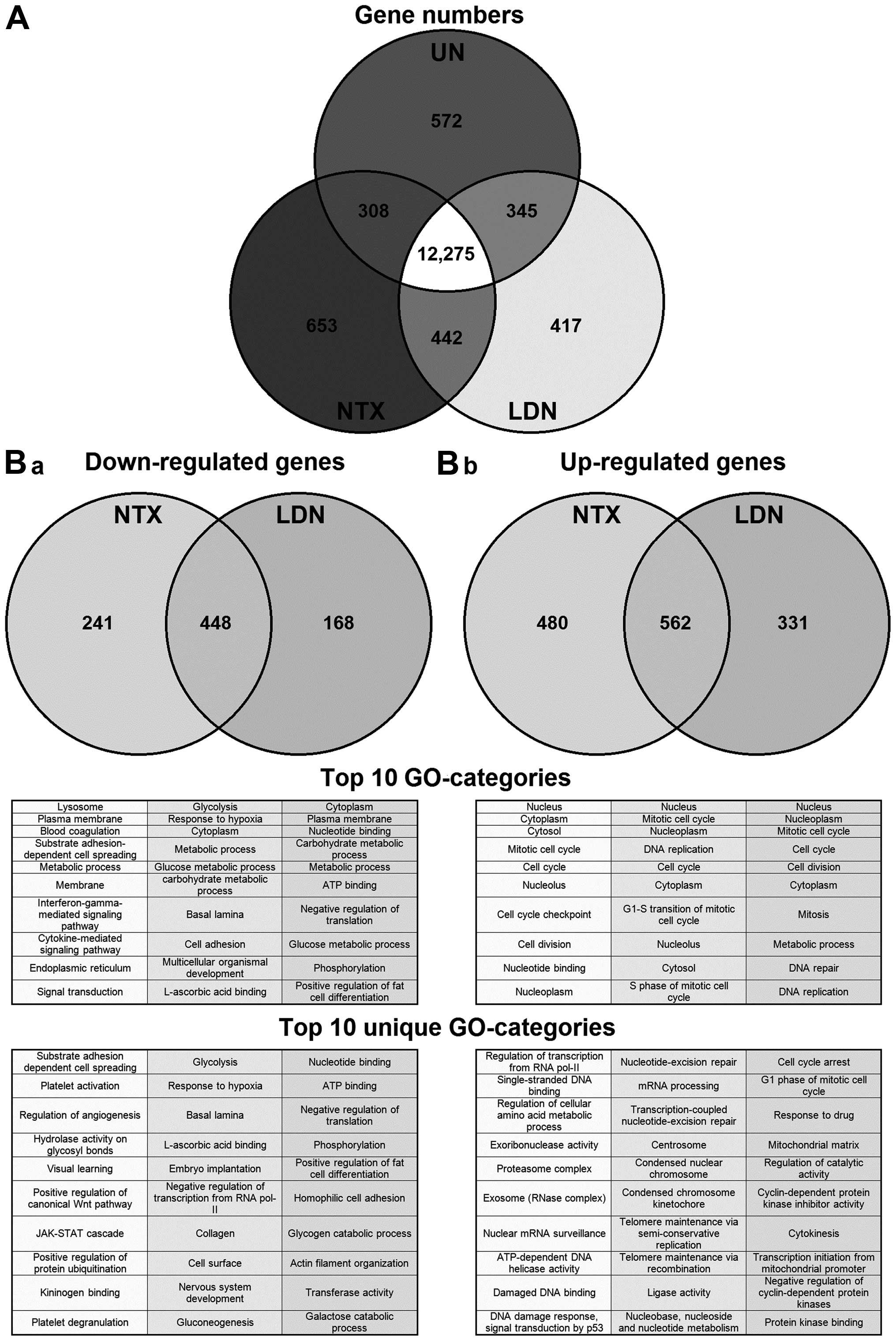
Following the initial filtering and removal of
nonsense genes from the gene microarray chip, 15,012/27,132 (55%)
genes were judged present in at least one of the treatment
conditions. Although a large proportion of these were present in
all conditions, 12,275/15,012 (82%) (Fig. 1A), some were unique to a particular
condition. We therefore focused next on those genes that, when
compared to untreated cells, were specifically altered following
treatment with NTX or with LDN (Fig.
1B). The majority of genes that were either down- or
upregulated were the same irrespective of the treatment being NTX
or LDN. However, there was a proportion of genes that changed
specifically in response to either NTX or LDN. For example,
concentrating on those genes (n=930) that were downregulated by
treatment, 448 were common to both NTX and LDN; however, 214 were
unique to NTX and 168 to LDN. Full gene lists are available at
ArrayExpress (www.ebi.ac.uk - accession no.
E-MTAB-4454).
A number of gene lists were made that recorded those
down- and upregulated by treatment with different doses. Specific
examination of the top 20 genes in these lists revealed a number of
genes that were of interest to us such as the paired
immunoglobin-like type 2 receptor α (PILRA), which is
involved in the regulation of the immune system, and the
pro-apoptotic gene BAK1. Both were increased significantly
by LDN but not NTX (Table I). This
highlighted the possibility that NTX and LDN affect different
groups of genes. Therefore, we next used gene ontology (GO) and
grouped the genes with changes in their expression according to
their molecular function and biological process. Results showed
some cross-over in the genes that were altered by the different
treatments. We next focussed specifically on the groups of genes
that were uniquely altered by NTX or LDN, and we saw that a number
of the GO-categories that were upregulated exclusively by LDN were
associated with cell cycling (Fig.
1B-b). This directed us to specifically examine genes that
directly regulated the cell cycle to see if there was any
divergence of changes in them caused by treatment with LDN or NTX.
Results showed they were generally altered by a similar magnitude
through treatment with both NTX and LDN; however, there were four
exceptions, in that CDK1, 4, 6 and cyclin B1 were affected
differently (Table II).
 | Table ITop 20 genes induced by treatment with
naltrexone.a |
Table I
Top 20 genes induced by treatment with
naltrexone.a
| Reduced in LDN
only | Increased in LDN
only |
|---|
|
|
|---|
| ILMN ID | Gene | UN | NTX | LDN | NTX:UN | LDN:UN | ILMN ID | Gene | UN | NTX | LDN | NTX:UN | LDN:UN |
|---|
| 1778691 | TIA1 | 429 | 428 | 312 | 1.00 | 0.73 | 2362858 | PILRA | 90 | 95 | 130 | 1.05 | 1.44 |
| 2084073 | UCN | 192 | 192 | 141 | 1.00 | 0.73 | 1746241 | SDHC | 622 | 585 | 789 | 0.94 | 1.27 |
| 2121816 | GPR137B | 303 | 260 | 188 | 0.86 | 0.62 | 1654217 | MPP2 | 131 | 123 | 164 | 0.93 | 1.25 |
| 2233366 | ASAP1 | 469 | 445 | 344 | 0.95 | 0.73 | 1733859 | DCAF15 | 117 | 117 | 157 | 1.00 | 1.34 |
| 1655614 | DSP | 779 | 701 | 544 | 0.90 | 0.70 | 2344373 | MVP | 201 | 193 | 257 | 0.96 | 1.28 |
| 1702835 | SH3BGRL | 1,337 | 1,214 | 953 | 0.91 | 0.70 | 1701621 | SCO2 | 633 | 602 | 799 | 0.95 | 1.26 |
| 1708611 | RDX | 969 | 900 | 722 | 0.93 | 0.75 | 2133799 | ACAT2 | x | 95 | 126 | 1.05 | 1.40 |
| 2111237 | MN1 | 169 | 142 | 111 | 0.84 | 0.66 | 1674337 | FKBP2 | 501 | 566 | 748 | 1.13 | 1.49 |
| 1748093 | PAFAH1B3 | 2,665 | 2,150 | 1,672 | 0.81 | 0.63 | 1768181 | TOR3A | 248 | 309 | 407 | 1.25 | 1.64 |
| 1756685 | DEPDC6 | 167 | 147 | 119 | 0.88 | 0.71 | 1700086 | DOK1 | x | x | 117 | x | 1.30 |
| 2292646 | GAD1 | 371 | 287 | 227 | 0.77 | 0.61 | 2410772 | KEAP1 | 678 | 669 | 866 | 0.99 | 1.28 |
| 1770293 | KLF5 | 174 | 157 | 130 | 0.90 | 0.75 | 1723087 | MDK | 121 | 117 | 151 | 0.97 | 1.25 |
| 3178302 | FNDC3B | 454 | 384 | 314 | 0.84 | 0.69 | 1714181 | MEGF8 | x | 102 | 1,301 | 1.14 | 1.45 |
| 2257833 | BBS7 | 371 | 331 | 274 | 0.89 | 0.74 | 2410262 | MTMR14 | 234 | 237 | 302 | 1.02 | 1.29 |
| 2347805 | EXOC1 | 418 | 375 | 312 | 0.90 | 0.75 | 1665884 | REP15 | 105 | 110 | 139 | 1.04 | 1.33 |
| 2287157 | DST | 546 | 471 | 392 | 0.86 | 0.72 | 1805990 | BAK1 | 101 | 122 | 155 | 1.20 | 1.53 |
| 1718063 | LIPA | 1,032 | 875 | 726 | 0.85 | 0.70 | 1765523 | TOLLIP | 115 | 118 | 149 | 1.02 | 1.29 |
| 2173004 | RAB8B | 655 | 545 | 451 | 0.83 | 0.69 | 1814200 | BMP2K | 91 | x | 114 | x | 1.25 |
| 1806667 | FRAS1 | 918 | 789 | 658 | 0.86 | 0.72 | 1788988 | THAP1 | 177 | 200 | 253 | 1.13 | 1.43 |
| 1758895 | CTSK | 168 | 136 | 113 | 0.81 | 0.67 | 1777584 | KARS | 792 | 824 | 1,038 | 1.04 | 1.31 |
| Table IIEffect of NTX and LDN on cell
cycle-related genes in HCT116 cells.a |
Table II
Effect of NTX and LDN on cell
cycle-related genes in HCT116 cells.a
| | Raw data | Relative to UN |
|---|
| |
|
|
|---|
| Process | Gene | UN | NTX | LDN | NTX/UN | LDN/UN | LDN/NTX |
|---|
| DNA damage | p53 | 271 | 257 | 251 | 0.95 | 0.93 | 0.98 |
| CDKi | p21 | 2,022 | 1,780 | 1,776 | 0.88 | 0.88 | 1.00 |
| p27 | 441 | 400 | 398 | 0.91 | 0.90 | 0.99 |
| p57 | 211 | 161 | 171 | 0.77 | 0.81 | 1.06 |
| p19 | 202 | 246 | 262 | 1.22 | 1.30 | 1.07 |
| p18 | 91 | 92 | - | 1.02 | - | - |
| p16 | 198 | 234 | 246 | 1.18 | 1.24 | 1.05 |
| p15 | 153 | 137 | 133 | 0.90 | 0.87 | 0.97 |
| G0 | cy C | 669 | 729 | 696 | 1.09 | 1.04 | 0.95 |
| CDK3 | 93 | 98 | 91 | 1.06 | 0.98 | 0.93 |
| G1 (early) | cy D1 | 6,580 | 6,773 | 6,574 | 1.03 | 1.00 | 0.97 |
| cy D2 | 95 | 96 | 91 | 1.01 | 0.96 | 0.95 |
| cy D3 | 1,247 | 1,542 | 1,511 | 1.24 | 1.21 | 0.98 |
| CDK4 | 3,528 | 3,541 | 4,345 | 1.00 | 1.23 | 1.23 |
| CDK6 | 550 | 682 | 768 | 1.24 | 1.40 | 1.13 |
| G1 (mid) | cy E1 | 357 | 453 | 455 | 1.27 | 1.27 | 1.00 |
| cy E2 | 491 | 804 | 766 | 1.64 | 1.56 | 0.95 |
| CDK2 | 765 | 974 | 989 | 1.27 | 1.29 | 1.02 |
| S | cy A2 | 1,473 | 1,933 | 2,040 | 1.31 | 1.38 | 1.06 |
| CDK2 | 765 | 974 | 989 | 1.27 | 1.29 | 1.02 |
| M | cy B1 | 1,528 | 1,715 | 1,919 | 1.12 | 1.26 | 1.12 |
| cy B2 | 2,530 | 2,686 | 2,806 | 1.06 | 1.11 | 1.04 |
| cy B3 | 105 | 97 | 96 | 0.93 | 0.92 | 0.99 |
| CDK1 | 1,016 | 1,378 | 1,207 | 1.36 | 1.19 | 0.88 |
NTX and LDN alter the expressions of key
proteins
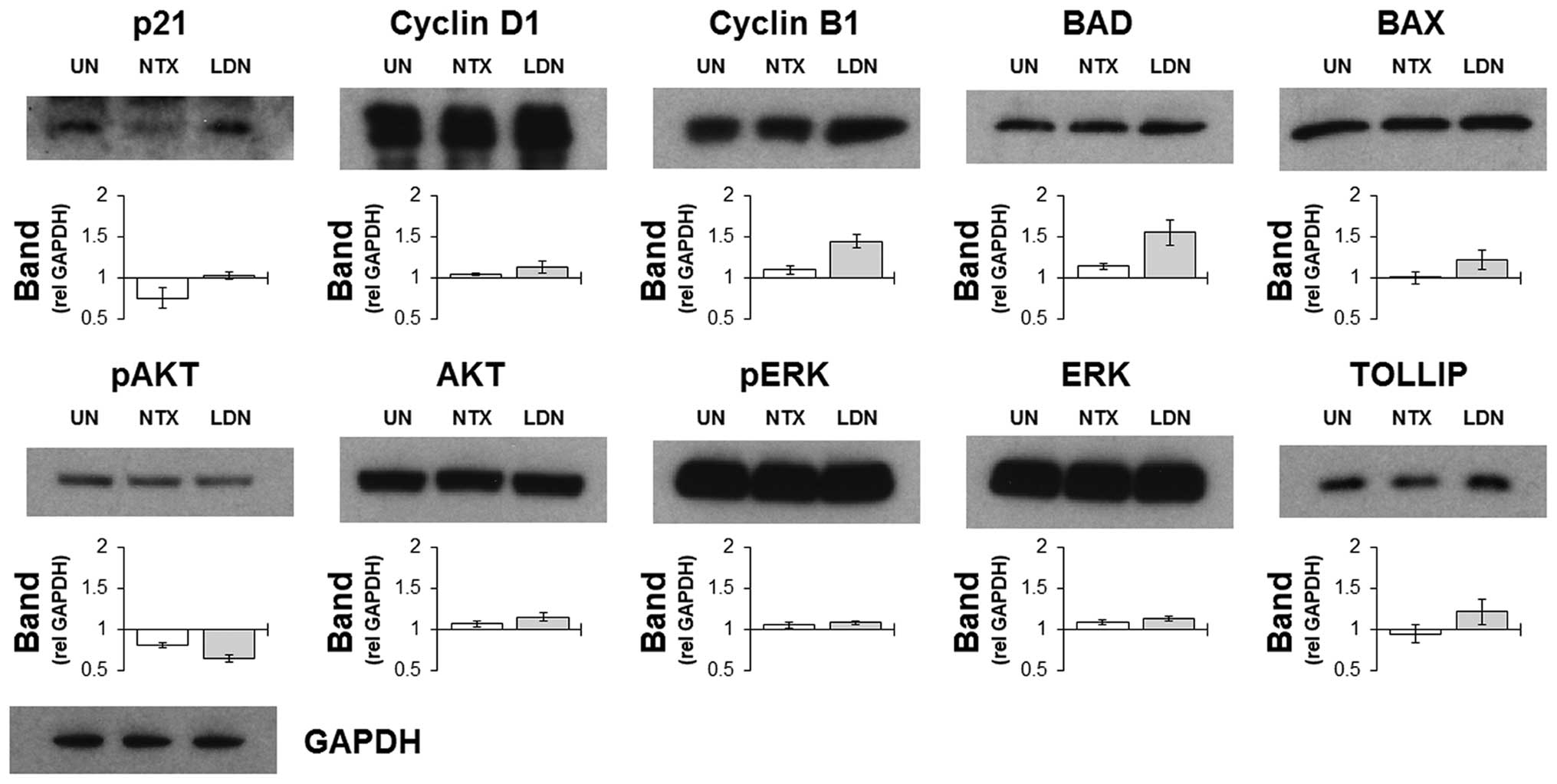
To confirm and assess the effects that treatments
had on targets identified by the microarray screen, western
blotting was performed on whole cell lysates harvested from HCT116
cells treated with NTX or LDN. The choice of proteins that were
assessed was guided by the lists from the microarray expression
data, and selected upon the basis of the magnitude of their
differential expressions. Results indicated that the selective
increases in some cell cycle related genes following treatment with
LDN were recapitulated by increased protein expression (Fig. 2). Other notable proteins that
corresponded with the gene data included BAD and TOLLIP, which were
increased following culture with LDN but not with NTX. These were
notable as both were involved in determining the level of cell
death by regulating apoptosis and autophagy, respectively. AKT and
ERK were also prospectively included as a way of assessing the
general signalling status of the cell, and results indicated that
they were impacted upon to a similar magnitude by NTX and LDN.
A break in treatment (recovery phase)
enhances the cytotoxic effect of LDN
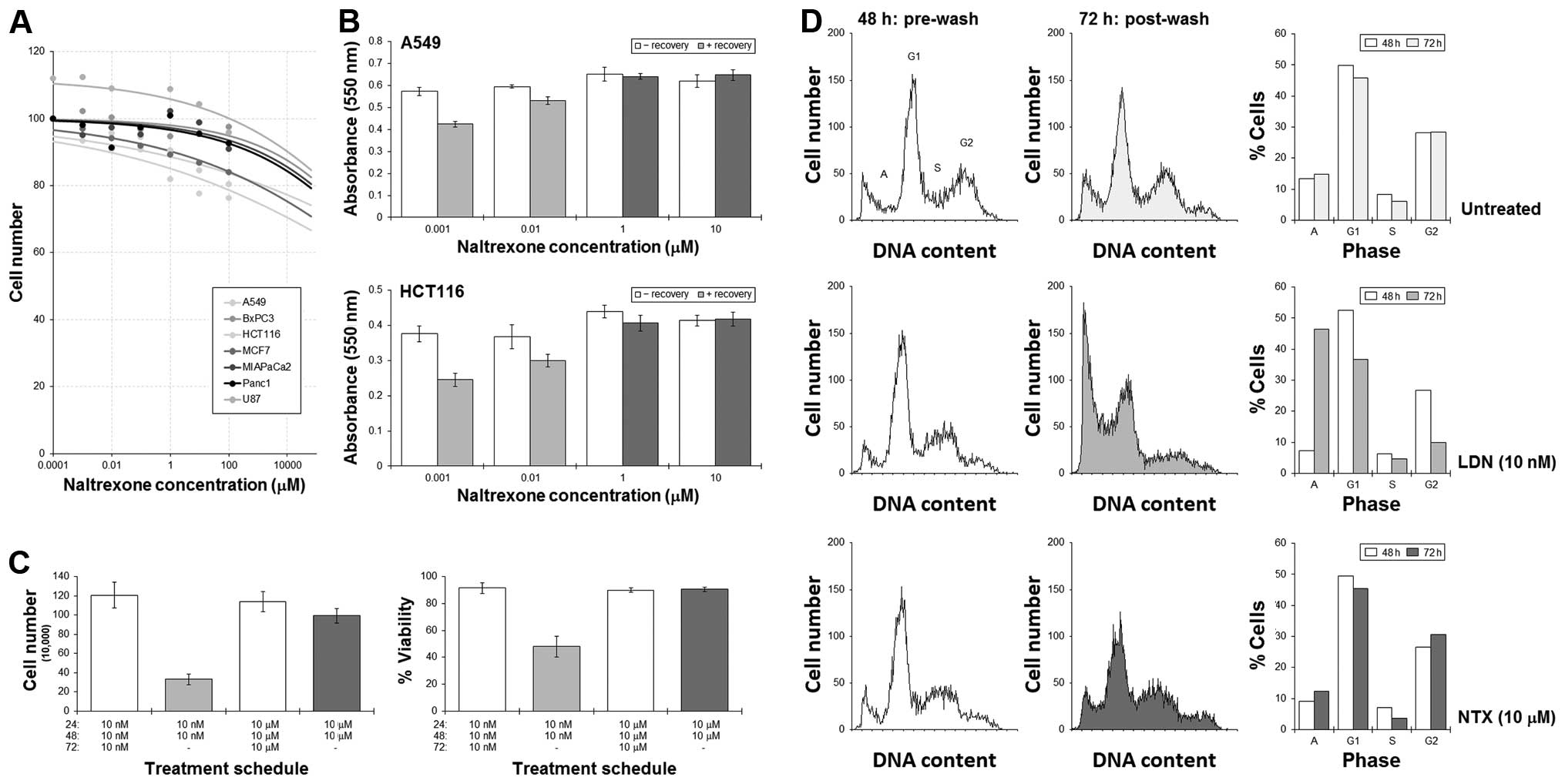
MTT assays were used to assess the effect of
naltrexone on a variety of cell lines from different tissues, and
results showed the effect on cell numbers was minimal. Extrapolated
IC50 concentrations for all the cell lines were >100
M (Fig. 3A). Our previous studies
have shown that the cytotoxicity of some agents can be increased by
employing a break in treatment, the effect of which is associated
with disturbances in cell cycling dynamics (16,18,19).
As cell cycling has been implicated as being important in this
study, we next tested the possibility that a recovery phase may
enhance cell killing. A549 and HCT116 cells were cultured with LDN
or NTX for 48 h, at which time, the drug-containing medium was
removed and the cells then allowed to recover in drug-free medium
for a further 24 h.
MTT assays showed significant reductions in the
number of A549 and HCT116 cells remaining when a ‘recovery’ phase
was adopted into the LDN treatment schedules (Fig. 3B). An initial culture with NTX did
not result in such a dramatic reduction in cell number. Cell
counting experiments in HCT116 cells revealed that the reduction in
cell number was associated with a fall in cell viability, which
suggested an active cytotoxic response was achieved (Fig. 3C). Additional flow cytometric
analysis of the cell cycle showed significant increases in the
sub-G1 (apoptosis) peak following a LDN-then-recovery schedule with
concomitant emptying of cells from G1 and G2. This was not as
pronounced with NTX (Fig. 3D).
Priming cells with LDN enhances the
activity of chemotherapy
Having shown that the pro-apoptotic proteins BAX and
BAD were upregulated following treatment with LDN, we next tested
the possibility that priming cells with LDN could sensitise cells
to common chemotherapy drugs. We assessed this by developing
treatment schedules that were made up of two separate treatment
phases. The first would last 48 h and comprise no treatment or
treatment with LDN. After this time, cells were removed from the
drug, and the second phase of treatment would be added. This would
also last 48 h, and be CPM, GEM or OXP. As a comparison, we also
performed parallel experiments in which we used NTX in the first
treatment phase.
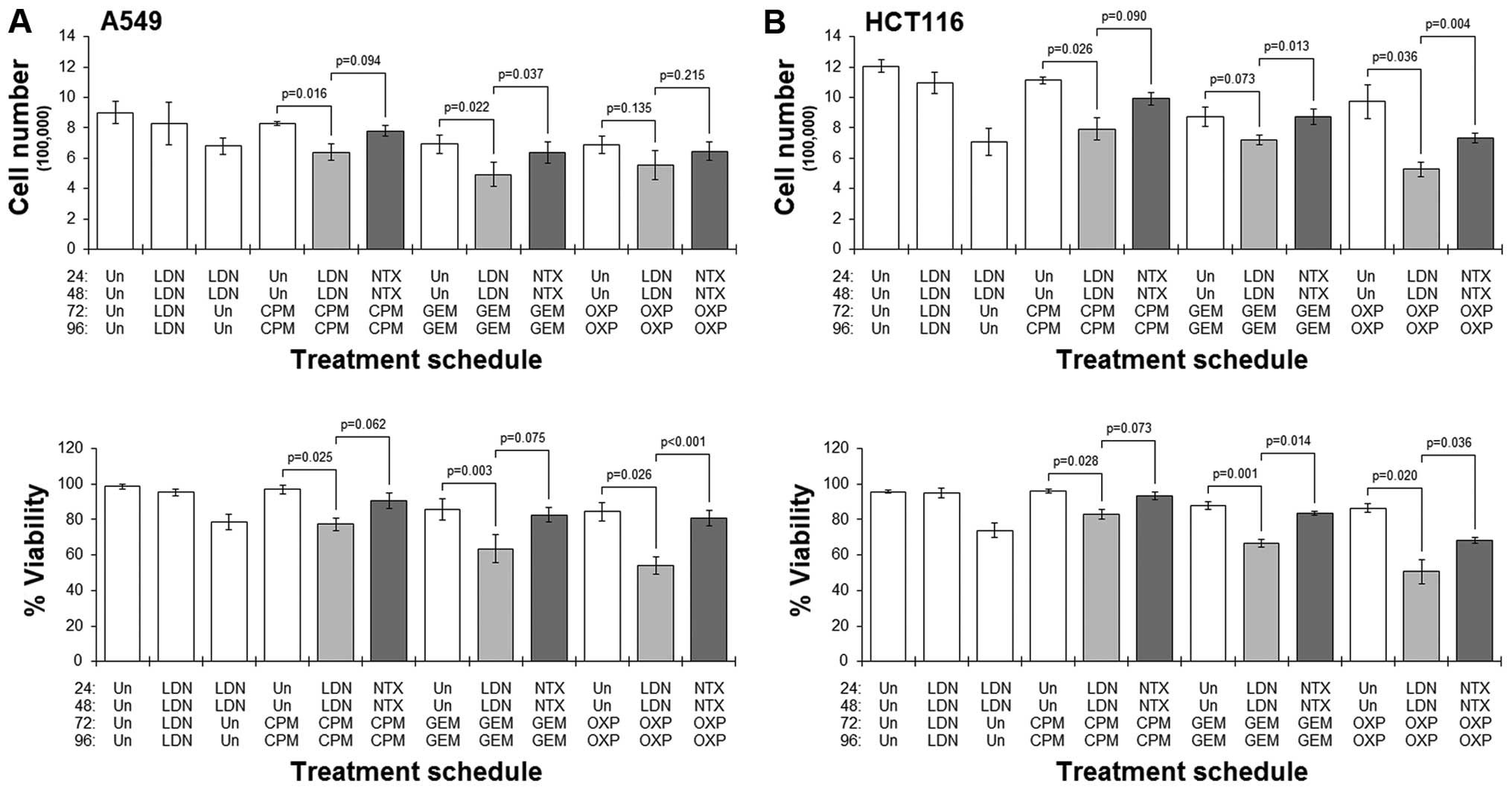
Results showed that in both cell lines studied,
pre-culture with LDN followed by culture with any of the drugs
always resulted in a greater reduction in cell number and viability
when compared to the corresponding schedule that did not have the
LDN pre-culture (Fig. 4).
Conversely, in those schedules where cells were pre-cultured with
standard NTX, treatment with any of the cytotoxic drugs did not
generally result in dramatic reductions in cell number or viability
(Fig. 4). Furthermore, comparing
LDN-priming schedules with their respective NTX-primed ones showed
significantly better cytotoxicity in the former schedules.
Discussion
Evidence of a role for naltrexone as an anticancer
agent has been described in the literature for some time. Studies
have highlighted an ability of this opioid antagonist to impede the
rate and way that cancer cells grow, and that these anticancer
action occur only at lower doses. However, the mechanism of this
action has yet to be fully defined, and as such, the true value of
this drug in the oncological setting has yet to be established in
the clinical community. In an attempt to shed more light on this
area, we have performed gene microarray studies to ascertain the
means by which naltrexone can disrupt tumour cell growth.
Additionally, we have compared the genetic fingerprint of tumour
cells treated either with LDN or NTX to evaluate whether there is a
divergence in the actions of the treatments. The significant
finding of this study has been the categories of genes that are
unique to LDN, which suggests that the mode of action differs
between LDN and NTX.
In the early 1960s, it was reported that morphine
possessed the capacity to disrupt the normal physiology of
tumour-bearing rats (20). The
study was undertaken primarily to explore the appetite-disruptive
nature of morphine on animals bearing tumours; however, in addition
to showing the morphine could reduce the body-weights of these
animals, it was also noticed that their tumour-weights were
concomitantly increased. The reason for this increase in tumour
size was unclear, but it was perceivable that morphine could have
had secondary effects on the immune system that supported tumour
progression (21). Alternatively,
the increases in tumour size could simply have been due to a direct
effect of morphine that resulted in enhanced cellular proliferation
in the tumours or reduced cell death. Indeed, morphine has been
shown to prevent the normal cell death in the ciliary ganglion of
the chick embryo, suggesting that in addition to modulating
neurotransmission, it and other endogenous opiates may also
regulate neurophysiology (22).
The concentration of morphine appeared to be important in which
effect it caused, as apoptosis in the chick embryo was only
disrupted when used at the higher doses, whilst no effect was seen
at the lower doses (6). Taken
together, this suggested the possibility that drugs of this class
could be used therapeutically to reduce tumour growth.
In fact, a similar growth-inhibitory action has been
reported for the antagonist naltrexone, which also exhibits a
similar dose-dependent quality (1). The mechanism by which it exerts its
anticancer effect has yet to be fully elucidated, but a number of
reports have indicated both direct-effects on tumour as well as
indirect modifications to immune function, which enhances host
immunity against tumours. Naltrexone has also been reported to
elicit a number of other cellular responses that lead to reductions
in tumours. Therefore, an agent that can alter survival and growth
characteristics of cancer cells directly, whilst simultaneously
promote an anticancer response by the immune system is attractive
as a putative therapy (23).
In the first part of the study, we employed gene
expression microarrays to identify the transcripts that were
altered following culture with naltrexone. As previous studies,
which included our own, acknowledged a possible divergence in
effects dependent upon dose, we assessed and compared the effects
of a low dose of naltrexone (LDN) with a more standard
concentration (NTX). Although LDN and NTX are essentially the same
drug, albeit at different concentrations, results showed
approximately half of the genes that were altered following
treatment with either were not the same. We thus focussed only on
these, and surveyed the profile more carefully by using gene
ontology. Results showed that the types of genes being
downregulated by treatment were variable, and no categories of
genes emerged as being prevalent. In contrast, the categories of
genes that were upregulated by treatment were associated with
regulation of the cell cycle. To get a perspective of whether
specific classes of genes were impacted upon by LDN and not NTX, we
next focussed on the genes that were altered uniquely by LDN.
Results showed that the common categories affected by LDN and not
NTX were those involved with the cell cycle.
A typical response to cellular and/or DNA damage as
a consequence of drug activity is to induce a cell cycle arrest
that allows for the cell to decide a response (24). Typically, this arrest is transient
and cells rapidly undergo repair or enter the apoptosis program.
However, in some cases, this arrest persists, and the cells enter a
protracted state of cytostasis (25), which can be inadvertently
maintained by the presence of the drug. This paradoxically reduces
the net sensitivity of the cell to the treatment. Cell death
systems are intricately linked with those that regulate cell
cycling, which means that the ability to perform one requires the
ability to perform the other (26). Therefore, a protracted blockade in
the cell cycle can minimise the ability of cells to undergo cell
death.
Naltrexone has been described to exhibit a similar
quality. One animal study from the 1980s reported the importance of
the naltrexone dose in determining the overall antitumour effect.
It was reported that, specifically for naltrexone, treatment in
mice with a clinically conventional dose (10 mg/kg) induces a
continuous occupancy of the opioid receptors, to which it is known
to bind, which resulted in increased tumour growth (27). However, if doses were reduced to 1
or 0.1 mg/kg, the receptor blockade was thought to be incomplete,
and binding sites remained open to ligands and thus activation of
antitumour processes.
Subsequent studies have also hinted at the
importance of treatment schedule in determining efficacy, with
intermittent administration of lower concentrations of naltrexone
achieving the greatest antitumour response (28). Antagonistic blockade of the opioid
receptors has been shown to result in a compensatory increase in
their amounts. The immediate consequence of this would be a boost
in receptors to which endogenous ligands such as the opioid growth
factor (OGF) could bind. OGF binding would ultimately result in an
inhibition of growth (1). This
feedback-like benefit is lost if naltrexone was left in culture, as
the continued presence of naltrexone occupies the de novo
binding sites and out-compete OGF. Thus no growth-inhibitory effect
would be achieved.
Taken together, we hypothesised that adopting a
recovery phase, during which time the cells would be devoid of
drug, could result in improved efficacy. Results of the current
experiments supported this, which showed that removing the cells
from the culture medium containing LDN and allowing them to grow in
drug-free medium significantly increased cell death. There are
precedents for this; in fact, we have recently shown with other
drugs that exhibit this protracted cell cycle blockade character
that cell death can be enhanced by introducing a drug-free phase in
the treatment schedule (16,19).
These studies showed that drugs such as artemisinin and cannabidiol
can elicit cell death in a number of cancer cells; however, in a
number of these cases, their use has been associated with an
absence of active cell killing. Instead, cells have arrested for
much longer. Although the accompanying reduction in cell number is
welcome, the lack of an active ‘cell killing’ reaction is not. One
way around this is to include a drug-free phase, which can
significantly enhance the cytotoxic nature of the treatments.
Our gene expression analysis also indicated a number
of pro-apoptotic genes were upregulated by LDN. For example, the
genes for bcl2-antagonist/killer 1 (BAK1) and the bcl2-associated X
protein (BAX) were both increased after treatment with LDN but not
after NTX. This disparity in gene expression was recapitulated at
the protein level, and offered the notion that treating tumour
cells with LDN may prime cells to apoptosis (29). We therefore tested this by
culturing cells with LDN before introducing them to a common
cytotoxic drug. Results indicated the cytotoxic effects of the
chemotherapies tested were significantly improved when cells were
pre-treated with LDN, whilst pre-treatment with NTX did not result
in such a drastic response. Parenthetically, as the act of removing
LDN could account for the increase in activity, paired t-test
analysis were performed, which showed significant differences when
comparing some of the schedules with the LDN:Un schedules.
The idea that LDN can prime a cancer cell to the
effect of an ‘old-school’ cytotoxic drug is attractive (30), and presents a way that treatment
regimens could be developed to exploit this potential mutualistic
effect. Indeed, we have illustrated how the essence of combination
therapy is to bring together drugs that have connected mechanisms
of action, which when used together, generates an effect that
surpasses what would have been achieved if the individual drugs
were used separately (11).
Improvements to outcome could equally be a consequence of a priming
effect as seen in this study. It is also worthwhile noting that the
drugs in the partnership do not need to be related, and one only
has to induce an effect that sensitises the cell to the other drug.
These combinations also usually involve a particular sequence of
administration, with the priming drug given first. Our results thus
suggest that LDN is a potential partner in drug-treatment regimens,
and should be given upfront before common cytotoxic agents. The
detailed molecular basis of this LDN-drug interaction needs to be
fully assessed to ensure that the most appropriate combinations are
identified. Unlike many other drugs used in oncology, LDN is
non-toxic and relatively cheap.
In conclusion, these data highlight the existence of
a fundamental difference in the mechanism by which naltrexone
elicits an effect. By using gene expression analysis, we showed
there was a difference in the gene-fingerprint of the drug when
used at two different concentration ranges. Specifically, LDN
resulted in explicit changes to genes involved in cell cycle
control, which were absent when doses were much higher. Further
experimentation that was steered by the gene data revealed the
efficacy of LDN to be enhanced by adaptations to treatment
schedules. These improvements were linked to our attempts to negate
a cell cycle and/or cell death blockade caused by the presence of
the drug. Additionally, by utilising the priming effect of LDN, the
cytotoxic effect of common chemotherapy drugs could be increased
through the sequential administration of the drugs. Overall, these
studies provide further evidence to support to role of LDN as an
anticancer agent.
Acknowledgements
The authors would like to acknowledge the use of
equipment located in the Medical Biomics Centre at St. George's,
University of London. Full data records for the gene data are
available at ArrayExpress (www.ebi.ac.uk -
accession no. E-MTAB-4454). W.M.L. was supported in part by the
Cancer Vaccine institute. W.M.L. and A.G.D. are listed as inventors
on a patent that describes the use of LDN as an anticancer
agent.
References
|
1
|
McLaughlin PJ and Zagon IS: Duration of
opioid receptor blockade determines biotherapeutic response.
Biochem Pharmacol. 97:236–246. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tegeder I and Geisslinger G: Opioids as
modulators of cell death and survival - unraveling mechanisms and
revealing new indications. Pharmacol Rev. 56:351–369. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Afsharimani B, Cabot P and Parat MO:
Morphine and tumor growth and metastasis. Cancer Metastasis Rev.
30:225–238. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gach K, Szemraj J, Stasikowska-Kanicka O,
Danilewicz M and Janecka A: Opioid-receptor gene expression and
localization in cancer cells. Cent Eur J Biol. 6:10–15. 2011.
|
|
5
|
Bimonte S, Barbieri A, Palma G and Arra C:
The role of morphine in animal models of human cancer: Does
morphine promote or inhibit the tumor growth? BioMed Res Int.
2013:2581412013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim MS, Cheong YP, So HS, Lee KM, Kim TY,
Oh J, Chung YT, Son Y, Kim BR and Park R: Protective effects of
morphine in peroxynitrite-induced apoptosis of primary rat neonatal
astrocytes: Potential involvement of G protein and
phosphatidylinositol 3-kinase (PI3 kinase). Biochem Pharmacol.
61:779–786. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cui Y, Zhang XQ, Cui Y, Xin WJ, Jing J and
Liu XG: Activation of phosphatidylinositol 3-kinase/Akt-mammalian
target of Rapamycin signaling pathway in the hippocampus is
essential for the acquisition of morphine-induced place preference
in rats. Neuroscience. 171:134–143. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gach K, Wyrębska A, Fichna J and Janecka
A: The role of morphine in regulation of cancer cell growth. Naunyn
Schmiedebergs Arch Pharmacol. 384:221–230. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ishikawa M, Tanno K, Kamo A, Takayanagi Y
and Sasaki K: Enhancement of tumor growth by morphine and its
possible mechanism in mice. Biol Pharm Bull. 16:762–766. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Boland JW, McWilliams K, Ahmedzai SH and
Pockley AG: Effects of opioids on immunologic parameters that are
relevant to anti-tumour immune potential in patients with cancer: A
systematic literature review. Br J Cancer. 111:866–873. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu WM: Enhancing the cytotoxic activity
of novel targeted therapies - is there a role for a combinatorial
approach? Curr Clin Pharmacol. 3:108–117. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu WM, Laux H, Henry JY, Bolton TB,
Dalgleish AG and Galustian C: A microarray study of altered gene
expression in colorectal cancer cells after treatment with
immunomodulatory drugs: Differences in action in vivo and in vitro.
Mol Biol Rep. 37:1801–1814. 2010. View Article : Google Scholar
|
|
13
|
Liu WM, Dennis JL, Fowler DW and Dalgleish
AG: The gene expression profile of unstimulated dendritic cells can
be used as a predictor of function. Int J Cancer. 130:979–990.
2012. View Article : Google Scholar
|
|
14
|
Liu WM, Dennis JL, Gravett AM,
Chanthirakumar C, Kaminska E, Coulton G, Fowler DW, Bodman-Smith M
and Dalgleish AG: Supernatants derived from chemotherapy-treated
cancer cell lines can modify angiogenesis. Br J Cancer.
106:896–903. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Scott KA, Dennis JL, Dalgleish AG and Liu
WM: Inhibiting heat shock proteins can potentiate the cytotoxic
effect of cannabidiol in human glioma cells. Anticancer Res.
35:5827–5837. 2015.PubMed/NCBI
|
|
16
|
Liu WM, Gravett AM and Dalgleish AG: The
antimalarial agent artesunate possesses anticancer properties that
can be enhanced by combination strategies. Int J Cancer.
128:1471–1480. 2011. View Article : Google Scholar
|
|
17
|
Liu WM, Fowler DW, Smith P and Dalgleish
AG: Pre-treatment with chemotherapy can enhance the antigenicity
and immunogenicity of tumours by promoting adaptive immune
responses. Br J Cancer. 102:115–123. 2010. View Article : Google Scholar :
|
|
18
|
Liu WM, Lawrence AJ and Joel SP: The
importance of drug scheduling and recovery phases in determining
drug activity. Improving etoposide efficacy in BCR-ABL-positive CML
cells. Eur J Cancer. 38:842–850. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Scott KA, Shah S, Dalgleish AG and Liu WM:
Enhancing the activity of cannabidiol and other cannabinoids in
vitro through modifications to drug combinations and treatment
schedules. Anticancer Res. 33:4373–4380. 2013.PubMed/NCBI
|
|
20
|
Sobel H and Bonorris G: Effect of morphine
on rats bearing Walker carcinosarcoma 256. Nature. 196:896–897.
1962. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sacerdote P: Opioids and the immune
system. Palliat Med. 20(Suppl 1): S9–S15. 2006.PubMed/NCBI
|
|
22
|
Meriney SD, Gray DB and Pilar G:
Morphine-induced delay of normal cell death in the avian ciliary
ganglion. Science. 228:1451–1453. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu WM and Dalgleish AG: The potential
beneficial effects of drugs on the immune response to vaccination.
Semin Oncol. 39:340–347. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pietenpol JA and Stewart ZA: Cell cycle
checkpoint signaling: Cell cycle arrest versus apoptosis.
Toxicology. 181–182:475–481. 2002. View Article : Google Scholar
|
|
25
|
Shapiro GI and Harper JW: Anticancer drug
targets: Cell cycle and checkpoint control. J Clin Invest.
104:1645–1653. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Waldman T, Zhang Y, Dillehay L, Yu J,
Kinzler K, Vogelstein B and Williams J: Cell-cycle arrest versus
cell death in cancer therapy. Nat Med. 3:1034–1036. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zagon IS and McLaughlin PJ: Naltrexone
modulates tumor response in mice with neuroblastoma. Science.
221:671–673. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Donahue RN, McLaughlin PJ and Zagon IS:
Low-dose naltrexone targets the opioid growth factor-opioid growth
factor receptor pathway to inhibit cell proliferation: Mechanistic
evidence from a tissue culture model. Exp Biol Med (Maywood).
236:1036–1050. 2011. View Article : Google Scholar
|
|
29
|
Tudor G, Aguilera A, Halverson DO, Laing
ND and Sausville EA: Susceptibility to drug-induced apoptosis
correlates with differential modulation of Bad, Bcl-2 and Bcl-xL
protein levels. Cell Death Differ. 7:574–586. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Makin G and Dive C: Recent advances in
understanding apoptosis: New therapeutic opportunities in cancer
chemotherapy. Trends Mol Med. 9:251–255. 2003. View Article : Google Scholar : PubMed/NCBI
|