Introduction
Diabetes mellitus has become one of the major
diseases that is hazardous to human health and is caused by the
change in lifestyle and western style diet in recent years
(1). Diabetes is a systemic
disease of metabolic disorders and leads to an increasing number of
deaths (2). Poor insulin activity
and inadequate insulin secretion can cause carbohydrate metabolism
problems, finally resulting in patients developing hyperglycemia
and hyperinsulinemia (3). In
addition, there is growing evidence that there is a connection
between type 2 diabetes and the increased appearance of new tumor
cases as shown in retrospective and prospective epidemiological
studies (4,5). Patients with type 2 diabetes have a
high risk of cancer, and colorectal cancer is the third leading
cause of cancer death according to the cancer statistics in Taiwan
(6). The principal source of
energy upon the glycolysis of glucose has been directly monitored
in migrating tumor cells (7).
However, in the insulin-free condition, the high blood glucose
fails to promote tumor growth due to insulin deficiency (8,9),
which might be a mediator in the induction of hyperinsulinemia to
increase the factors of cancer progression (10).
Colorectal cancer patients with diabetes have a
higher mortality rate than those with type 2 diabetes alone
(11,12). In addition, human evidence has also
demonstrated that high levels of insulin with an increased risk of
colon cancer and increased circulating concentrations of insulin
are associated with a higher risk of colonic neoplasia (13). Furthermore, chronic
hyperinsulinemia might stimulate insulin receptor (IR) signaling to
induce tumor growth in breast and pancreatic cancers (14,15).
Currently, there is no available information regarding a link
between colorectal cancer and type 2 diabetes in patients, and the
molecular mechanism of high insulin-induced signaling in colon
cancer remains unclear. Herein, we focused on the role of insulin
in the proliferation and metastatic effects on human colorectal
cancer cells.
We further investigated how the addition of insulin
promoted cell proliferation and migration by modifying the
expression of IR signaling, the phosphoinositide 3-kinase
(PI3K)/Akt/glycogen synthase kinase-3β (GSK3β) pathway and matrix
metalloproteinase-2 (MMP-2) regulation in treated colorectal cancer
cells. Therefore, our results confirm a link between insulin and
colorectal cancer cell progression and clarify how insulin acts as
a molecular modulator using a cultured cell model.
Materials and methods
Chemicals and reagents
Anti-β-actin antibody, fish gelatin and
poly(2-hydroxyethyl methacrylate) (poly-HEMA) were obtained from
Sigma-Aldrich (St. Louis, MO, USA). Penicillin-streptomycin
solution, insulin, sodium pyruvate, RPMI-1640 medium, trypsin-EDTA
and TRI Reagent Solution were purchased from Thermo Fisher
Scientific (Waltham, MA, USA). Fetal bovine serum (FBS) was
obtained from Biological Industries (Cromwell, CT, USA).
Nuclear/Cytosol Fractionation kit and anti-lamin B1 antibody were
from BioVision (Mountain View, CA, USA). Anti-IR antibody was
obtained from Abcam (Cambridge, UK). Anti-phospho-mTOR (Ser2448),
anti-mTOR, anti-cyclin D1, SB203580 (a p38 inhibitor) and
wortmannin (a PI3K inhibitor) were from EMD Millipore (Billerica,
MA, USA). Antibodies against JNK and p-JNK (Thr183/Tyr185) were
obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The
other antibodies used in this study were purchased from Cell
Signaling Technology (Beverly, MA, USA). Anti-rabbit and anti-mouse
secondary horseradish peroxidase (HRP) antibodies were obtained
from Bethyl Laboratories (Montgomery, TX, USA). PD98059 (an ERK
inhibitor) and SP600125 (a JNK inhibitor) were obtained from
Biosource International, Inc. (Camarillo, CA, USA).
Cell culture
Human colorectal carcinoma HCT-116 cell line (BCRC
no. 60349) was purchased from the Bioresource Collection and
Research Center (BCRC) (Hsinchu, Taiwan). Based on the information
of BCRC, the initial origin of this cell line is from male
colorectal carcinoma tissue. HCT-116 cells have been shown to
express Akt1 and Akt2 in the absence of Akt3 in insulin-stimulated
conditions (16,17). The cells were maintained in
RPMI-1640 medium supplemented with 10% FBS, 100 µg/ml
penicillin and 100 U/ml streptomycin at 37°C in a humidified
atmosphere of 95% air and 5% CO2. The culture medium was
renewed each day. Cells were subcultured every 3 days with 0.1%
trypsin-EDTA.
Cell viability assay
Cell viability was determined with a trypan blue
assay as previously described (18). HCT-116 cells (5×104
cells/well) were seeded onto a 24-well plate in RPMI-1640 medium
with 1% FBS. After a 24 h incubation, pretreatment with or without
20 µM of SP600125, PD98059, SB203580 and wortmanin,
respectively, for 1 h, and the cells were treated with or without
various concentrations (100, 150, 200, 250 and 300 nM) of insulin
in culture medium with 1% FBS for 48 h. Cells were harvested and
then stained with 0.4% trypan blue to count the live and dead cells
using an Olympus IX71 Inverted System Microscope (Olympus, Tokyo,
Japan).
Cell proliferation assay
The 96-well plate was coated with 50 µl of
12% poly-HEMA solution dissolved in 95% ethanol as described by
previous studies (19,20). HCT-116 cells (5×103
cells/well) were seeded onto 96-well plates coated with or without
poly-HEMA. After 24 h of incubation, the cells were treated with
various concentrations (100, 150 and 200 nM) of insulin in
RPMI-1640 medium with 1% FBS and further incubated for 48 h.
CellTiter 96 AQueous One Solution Cell Proliferation Assay kit
(Promega, Madison, WI, USA) was used, and the absorbance at 490 nm
was recorded with a FLUOstar galaxy spectrophotometer (BMG Labtech,
Ortenberg, Germany).
Western blotting
The treated HCT-116 cells were harvested and lysed
in RIPA Lysis Buffer (EMD Millipore). Nuclear and cytosolic
fractions were prepared utilizing the Nuclear/Cytosol Fractionation
kit (BioVision) as specified in the manufacturer's protocol. The
protein concentration was detected using the Bio-Rad protein assay
(Bio-Rad Laboratories, Hercules, CA, USA), and then the lysate was
mixed with protein loading dye and boiled, as previously described
(21). The sample (50 µg)
was subjected to sodium dodecyl sulfate (SDS)-polyacrylamide gel
electrophoresis, and the separated protein was transferred onto
cellulose nitrate membrane (Sartorius Stedim Biotech GmbH,
Goettingen, Germany). Each membrane was immunoblotted directly
against an appropriate antibody [anti-IR, anti-IRS-1,
anti-phospho-PI3K, anti-PI3K, anti-phospho-Akt (Ser473), anti-Akt,
anti-phospho-mTOR (Ser2448), anti-mTOR, anti-phospho-GSK3β (Ser9),
anti-GSK3β, anti-p53, anti-cyclin D1, anti-c-Myc, anti-heat shock
protein 27 (HSP27), anti-phospho-ERK1/2 (Thr202/Tyr204),
anti-ERK1/2, anti-phospho-JNK (Thr183/Tyr185), anti-JNK,
anti-phospho-p38 (Thr180/Tyr182) and anti-p38] overnight and then
further incubated with HRP-conjugated secondary antibody (1:10000
dilutions). The protein levels were normalized to lamin B1 or
β-actin. The signal was analyzed using Immobilon Western
Chemiluminescent HRP Substrate (EMD Millipore), and the signal
intensity was quantified using VisionWorks LS Image Acquisition and
Analysis Software (version 6.3.3, UVP, Upland, CA, USA).
Wound healing assay
The Culture-Insert 2 Well (Ibidi, Martinsried,
Germany) was placed onto a 12-well plate, and HCT-116 cells
(7×104 cells/well) were seeded into the culture-insert.
After 12 h of incubation, the culture-insert was removed, and cells
were treated with various concentrations (100, 150 and 200 nM) of
insulin in RPMI-1640 medium with 1% FBS and further incubated for
24, 48 and 72 h. Photographs of the wound adjacent to reference
lines scraped on the bottom of the plate were taken using an
Olympus IX71 Inverted System Microscope. The cells were examined
and quantified to measure relative to a control well.
Transwell migration assay
Cell migration was determined with a Transwell
migration assay. Transwell polycarbonate membrane cell culture
inserts (Corning, Lowell, MA, USA) were placed onto a 24-well
plate, and HCT-116 cells (1×105 cells/well) were treated
at various concentrations (100, 150 and 200 nM) of insulin in
serum-free RPMI-1640 medium. A serum-containing medium (650
µl) was added to the lower chambers. After incubating for 48
h, filter inserts were removed from the wells, and the cells that
migrated through the membrane were fixed with methanol and stained
with crystal violet, as previously described (22). Photographs were taken using an
Olympus Power IX71 microscope, and the migrated cells were
quantified by ImageJ 1.47 program for Windows from the National
Institute of Health (NIH) (Bethesda, MD, USA).
Reverse transcription PCR
HCT-116 cells were treated with 100, 150 and 200 nM
of insulin for 48 h before being collected to extract total RNA
using TRIzol Reagent (Thermo Fisher Scientific), as previously
described (21) and reverse
transcribed to produce cDNA using the SuperScript III First-Strand
Synthesis SuperMix for qRT-PCR kit (Thermo Fisher Scientific)
according to the manufacturer's protocols. The primers used for PCR
to amplify the target genes were as follows: MMP-2, forward:
TACACCGGGCCTGGAGAACTAG, reverse: GCTCTGAGGGTTGGTGGGATTG; MMP-9,
forward: CTGGAGGTTCGACGTGAAGG, reverse: AGGTCACGTAGCCCACTTGG; 18S
rRNA, forward: GTAACCCGTTGAACCCCATT, reverse: CCATCCAATCGGTAGTAGCG.
The PCR products were separated by electrophoresis on a 1.5%
agarose gel, and the DNA bands were detected using the SYBR Safe
DNA gel stain (Thermo Fisher Scientific). The gel was then
photographed using a BioDoc-It system (UVP). The results were
expressed as the ratio of each DNA signal relative to the
corresponding 18S rRNA signal.
Gelatin zymography
HCT-116 cells (1×105 cells/well) were
seeded onto 24-well plates in serum-free RPMI-1640 medium and
thereafter treated with various concentrations (100, 150 and 200
nM) of insulin for 48 h. Thereafter, the conditioned media were
collected, and the unboiled sample was separated by electrophoresis
on 8% SDS-polyacrylamide gels containing 0.1% gelatin
(Sigma-Aldrich). After electrophoresis, the gels were washed twice
in washing buffer and then incubated in reaction buffer at 37°C for
16 h as previously described (22,23).
Bands corresponding to activity were visualized by negative
staining using Coomassie Brilliant Blue R-250 (Bio-Rad
Laboratories), and quantitative data were analyzed utilizing
VisionWorks LS Image Acquisition and Analysis Software (version
6.3.3, UVP).
Statistical analysis
The data are expressed as the mean ± SD of three
independent experiments. Kruscal-Wallis H test was subsequently
followed up with Bonferroni method to evaluate the statistical
significance. P-value <0.05 was considered to indicate a
statistically significant difference.
Results
Insulin induces human colorectal
carcinoma HCT-116 cell proliferation in vitro
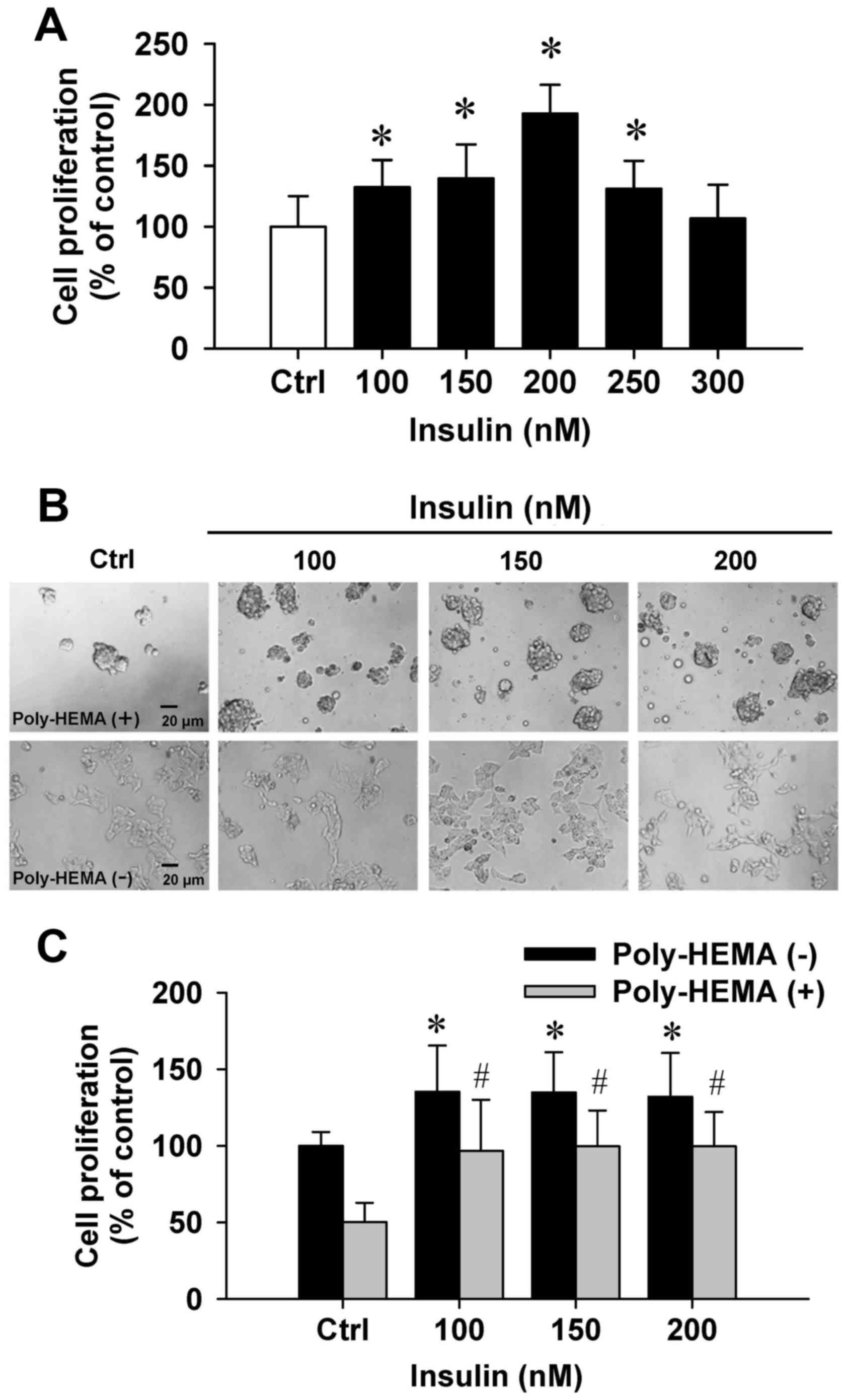
To examine the effects of insulin on cytotoxicity,
the trypan blue method was performed. Insulin at 100–300 nM showed
no cytotoxic influence on the number of dye-excluding HCT-116 cells
(Fig. 1A). Importantly, insulin at
100–250 nM significantly increased cell proliferation (Fig. 1A). Next, the effect of cell
proliferation on insulin-induced cells was examined by the
poly-HEMA-coated plate assay. Our data demonstrated that cells grew
in aggregates and showed sphere formation in a poly-HEMA-precoated
plates (Fig. 1B, top panel). A
comparison of adherent and aggregated cells revealed that cells
floated in the media. In addition, HCT-116 cell growth was notably
increased by insulin in coated and uncoated poly-HEMA samples
compared with their untreated counterparts (Fig. 1C). Thus, these data indicated that
insulin effectively promoted HCT-116 cell growth and proliferation
in vitro.
Insulin modulates the IR pathway via
PI3K/Akt signaling in HCT-116 cells
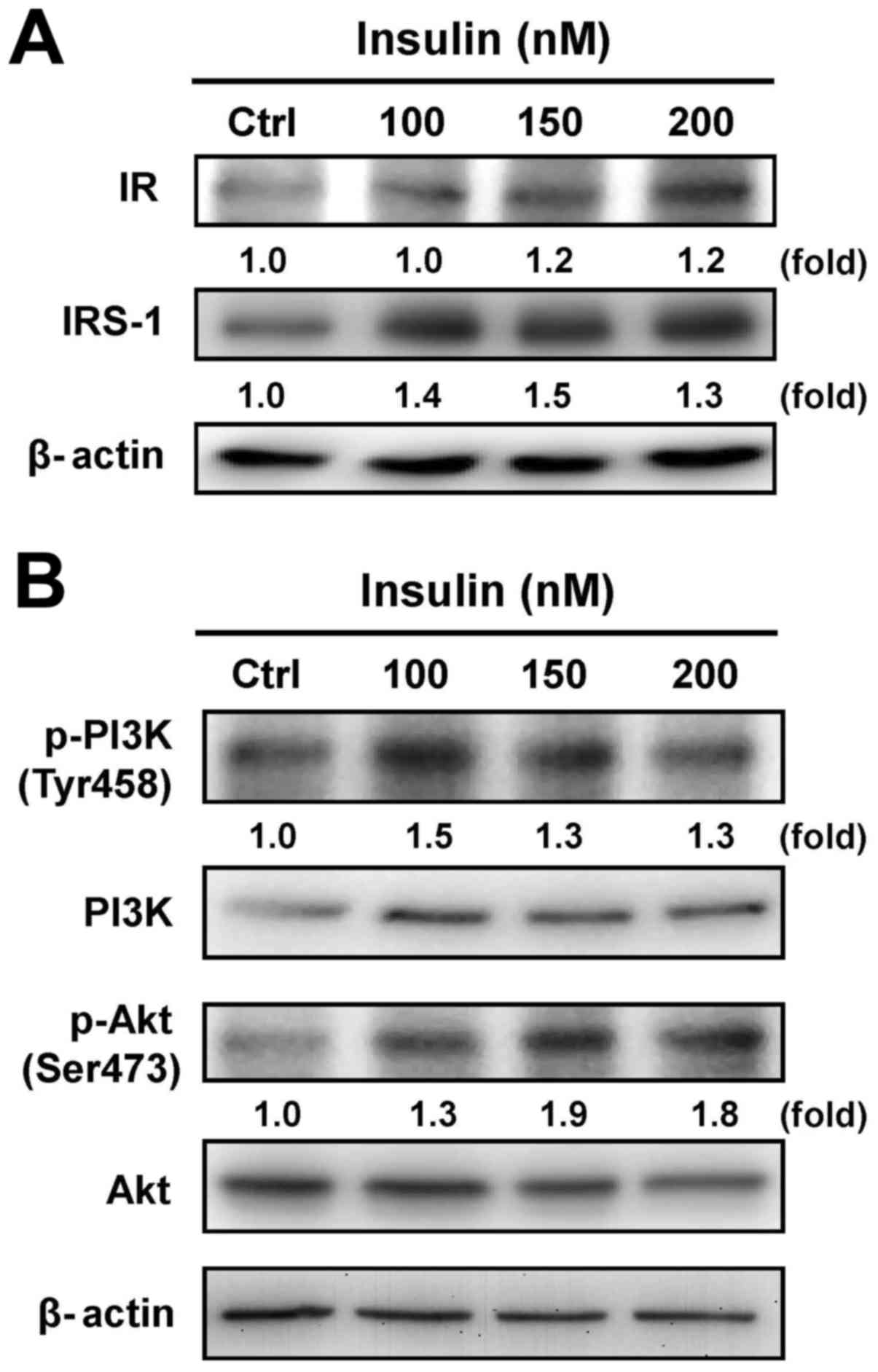
Cells were exposed to various concentrations of
insulin, and the protein levels of IR and IRS-1 were then assessed
by immunoblotting analysis. Both the IR and IRS-1 levels were
increased after treatment with insulin (Fig. 2A). Moreover, treatment with insulin
induced the phosphorylation of PI3K and Akt (Ser473) in HCT-116
cells, but no effect appeared in the protein levels of PI3K and Akt
in insulin-treated cells (Fig.
2B). These results suggest that insulin induced the IR signal
by regulating the PI3K/Akt pathway, showing that IR functionally
enhances pro-tumorigenic signals in HCT-116 cells.
Insulin regulates downstream PI3K/Akt
signaling in HCT-116 cells
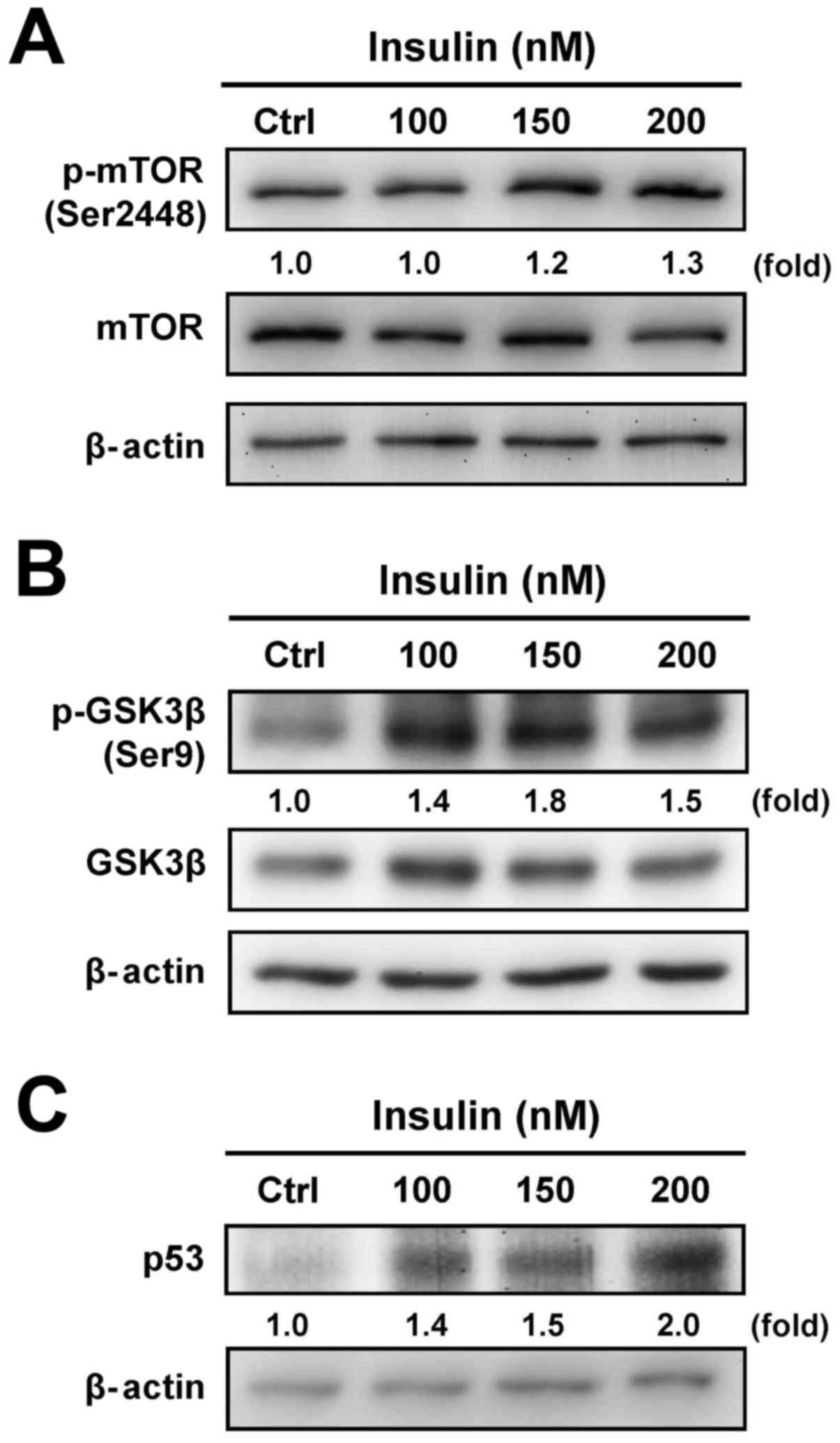
To determine whether the downstream signals are
involved in the regulation of insulin-treated HCT-116 cells through
the PI3K/Akt signaling pathway, we further tested the protein
levels of mTOR, GSK3β and p53 to specifically clarify the
functional regulation. Insulin treatment induced the
phosphorylation of mTOR (Ser2448) (Fig. 3A) and GSK3β (Ser9) (Fig. 3B). In addition, treatment with
insulin induced the p53 level in a concentration-dependent manner
in treated cells (Fig. 3C). These
results demonstrate that the enhancement of insulin-induced IR
signaling was observed via the PI3K/Akt pathway by activation of
mTOR/GSK3β, an activity that was not due to the suppression of the
p53 signal in HCT-116 cells.
Insulin affects cell cycle-associated
protein expression of c-Myc and cyclin D1, as well as HSP27 in
HCT-116 cells
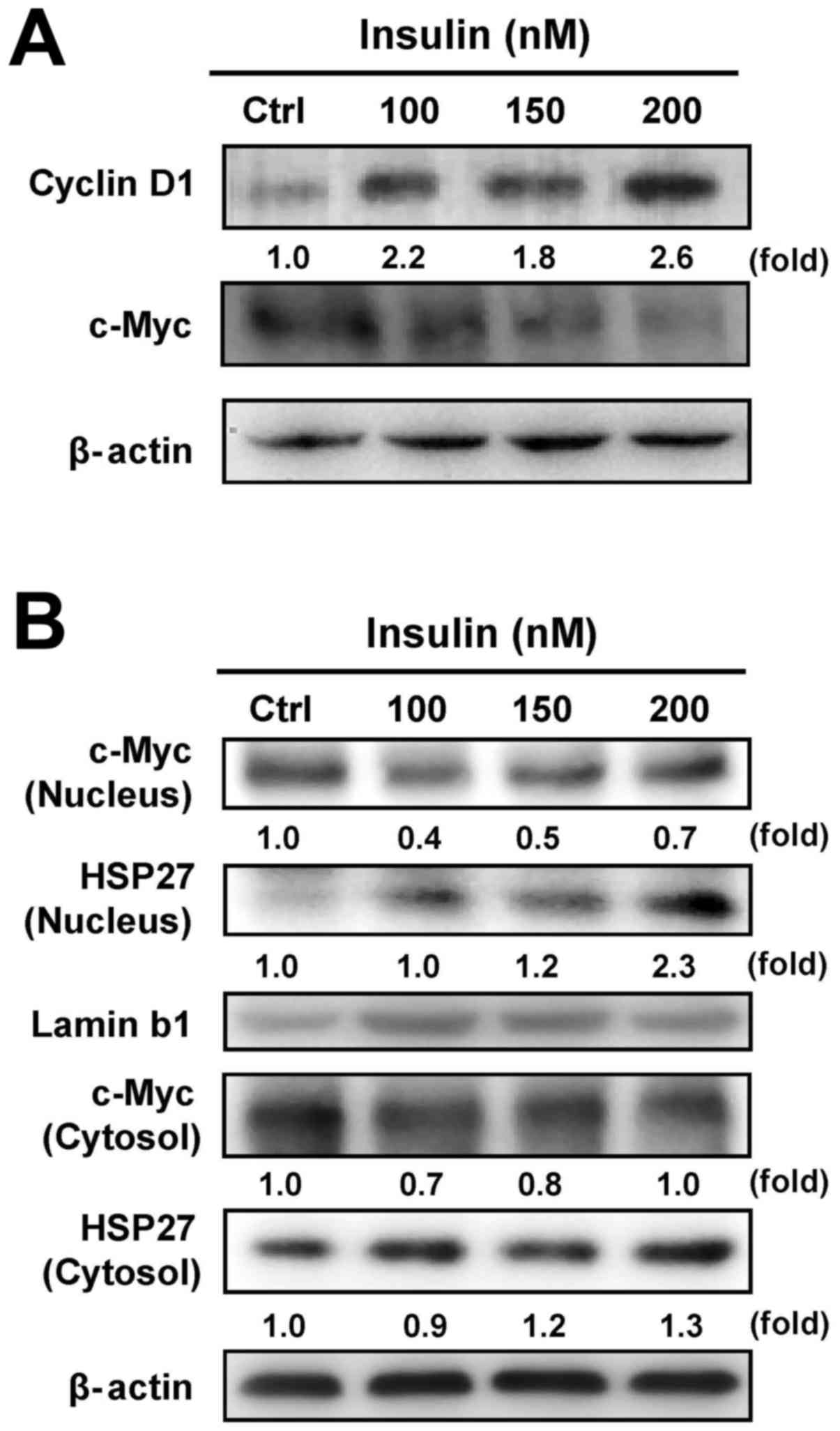
Our results have shown that insulin increased
HCT-116 cell proliferation (Fig.
1). To verify whether c-Myc and cyclin D1 are involved in
insulin-induced HCT-116 cell proliferation, we examined the levels
of c-Myc and cyclin D1 by western blot analysis. Insulin increased
the cyclin D1 level, but it decreased c-Myc protein expression in
HCT-116 cells (Fig. 4A). It seems
that insulin stimulated cell proliferation through regulating c-Myc
and cyclin D1 levels in HCT-116 cells. To further detect whether
insulin causes the translocation of HSP27 and c-Myc, we further
analyzed the protein expression of c-Myc and HSP27 in nuclear and
cytosolic fractions, respectively. We observed a decrease in c-Myc
nuclear translocation and an increase in HSP27 trafficking into the
nucleus of HCT-116 cells after insulin exposure, while insulin
attenuated cytosolic c-Myc and enhanced HSP27 in the cytosol
(Fig. 4B). These data implied that
the activity of c-Myc and HSP27 might be required in
insulin-induced HCT-116 cell proliferation.
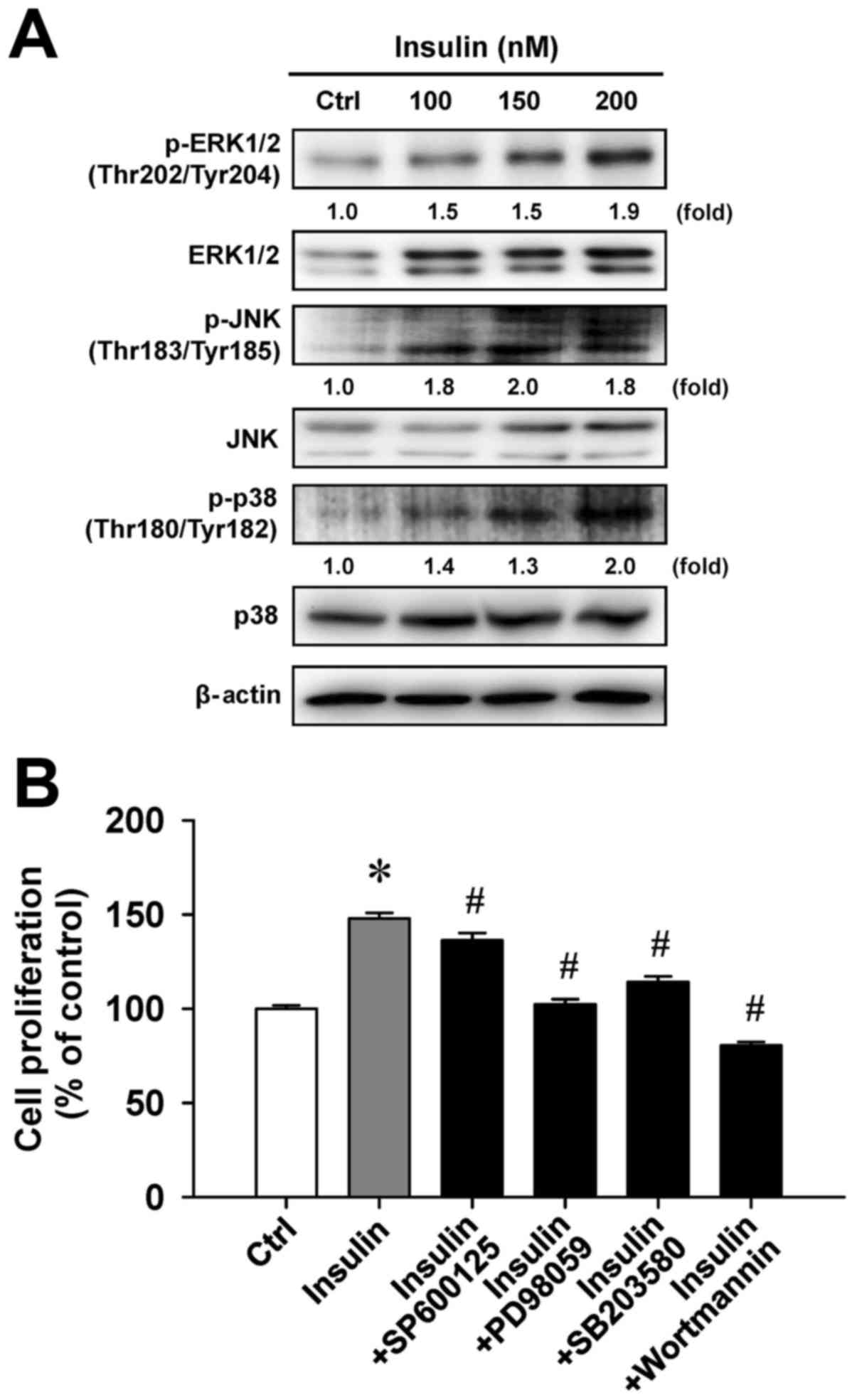
Insulin enhances MAPK signaling in
HCT-116 cells
To examine whether cell proliferation in response to
insulin challenge is associated with the MAPK pathway, we
determined the level of MAPK (ERK, JNK and p38) molecules in
insulin-treated HCT-116 cells. Insulin challenge resulted in an
upregulation of phosphorylated MAPK-associated protein levels,
including the phosphorylation of ERK1/2 on Thr202/Tyr204, JNK on
Thr183/Tyr185 and p38 on Thr180/Tyr182 (Fig. 5A). To confirm the roles of
MAPK-triggered cell proliferation by insulin, we individually
pretreated HCT-116 cells with or without SP600125 (a JNK-specific
inhibitor), PD98059 (an ERK-specific inhibitor), SB203580 (a
p38-specific inhibitor) or wortmannin (a PI3K-specific inhibitor)
before exposure to insulin to investigate cell viability. These
four inhibitors were effective on the inhibition of
insulin-enhanced cell proliferation (Fig. 5B). Specifically, PD98059 and
wort-mannin significantly attenuated insulin-induced viability by
up to 45.5% and 67.4%, respectively, compared with insulin
treatment alone. Therefore, we provide bimolecular evidence
regarding the influence of MAPK and PI3K signaling that contribute
to insulin-provoked proliferation in HCT-116 cells.
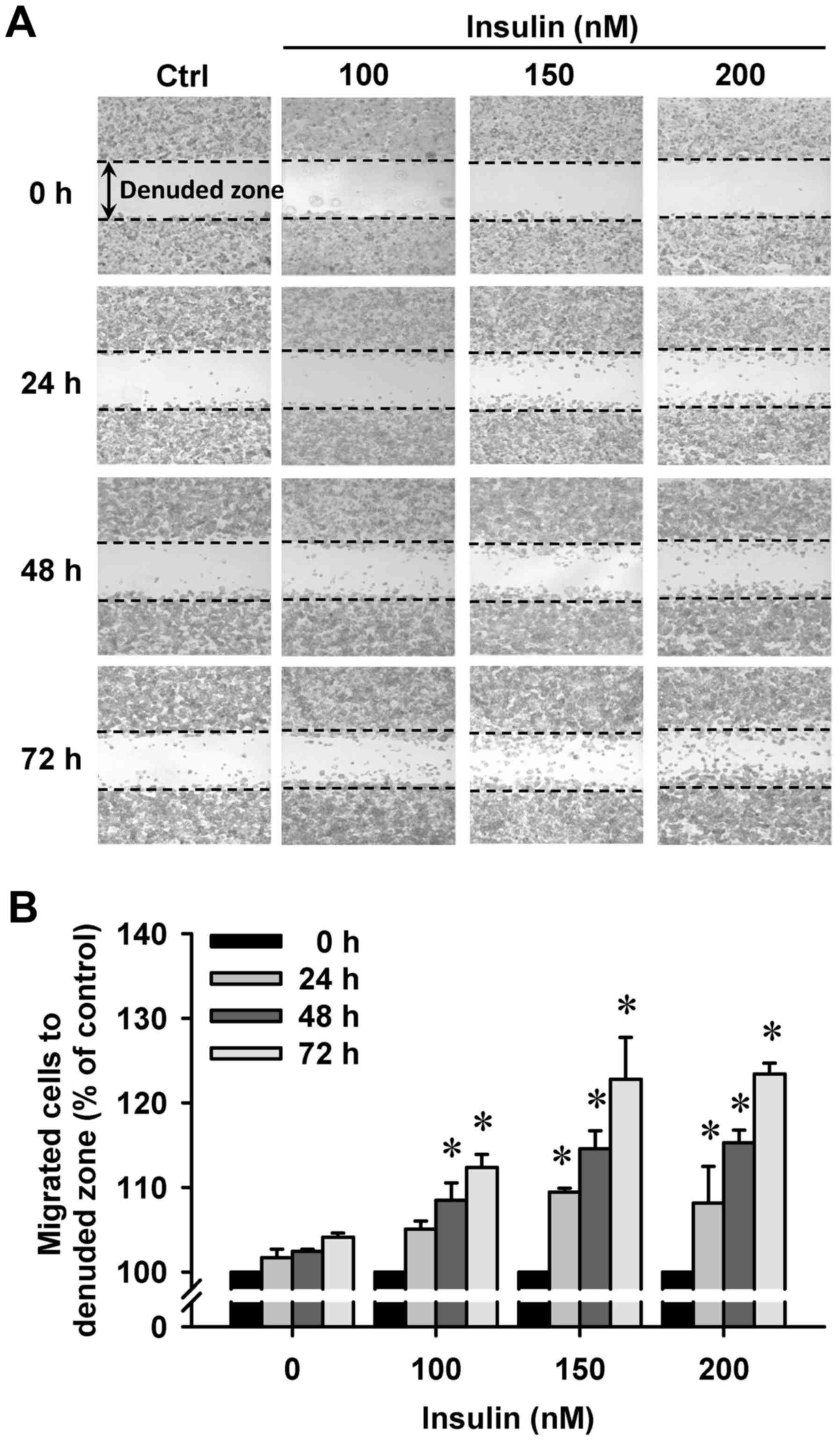
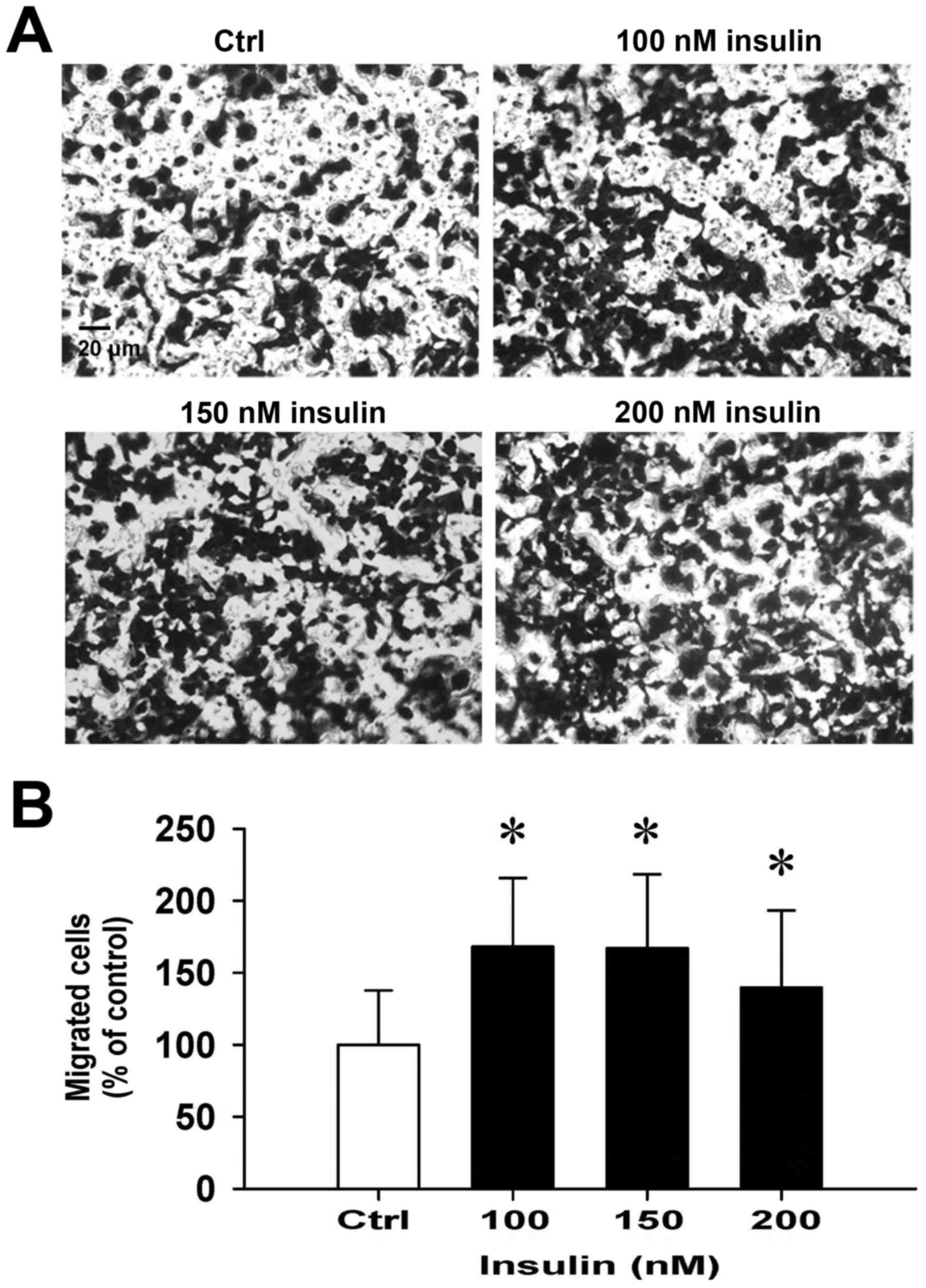
Insulin promotes HCT-116 cell motility
and migration in vitro
We then further investigated whether insulin has an
impact on cell migration in HCT-116 cells by the wound-healing
assay. Insulin treatment significantly enhanced cell migration to
the denuded zone (Fig. 6A) and
stimulated cell motility in a time-and concentration-dependent
manner (Fig. 6B). After 48 h of
insulin exposure, the cells on the lower surface of the filter were
examined under a microscope in HCT-116 cells. Insulin markedly
elicited the number of migrated cells as measured by the Transwell
migration assay at 48 h after insulin treatment (Fig. 7A). The migration ability was
increased by insulin treatment of (Fig. 7B). These findings demonstrate that
insulin promoted the migration ability of HCT-116 cells.
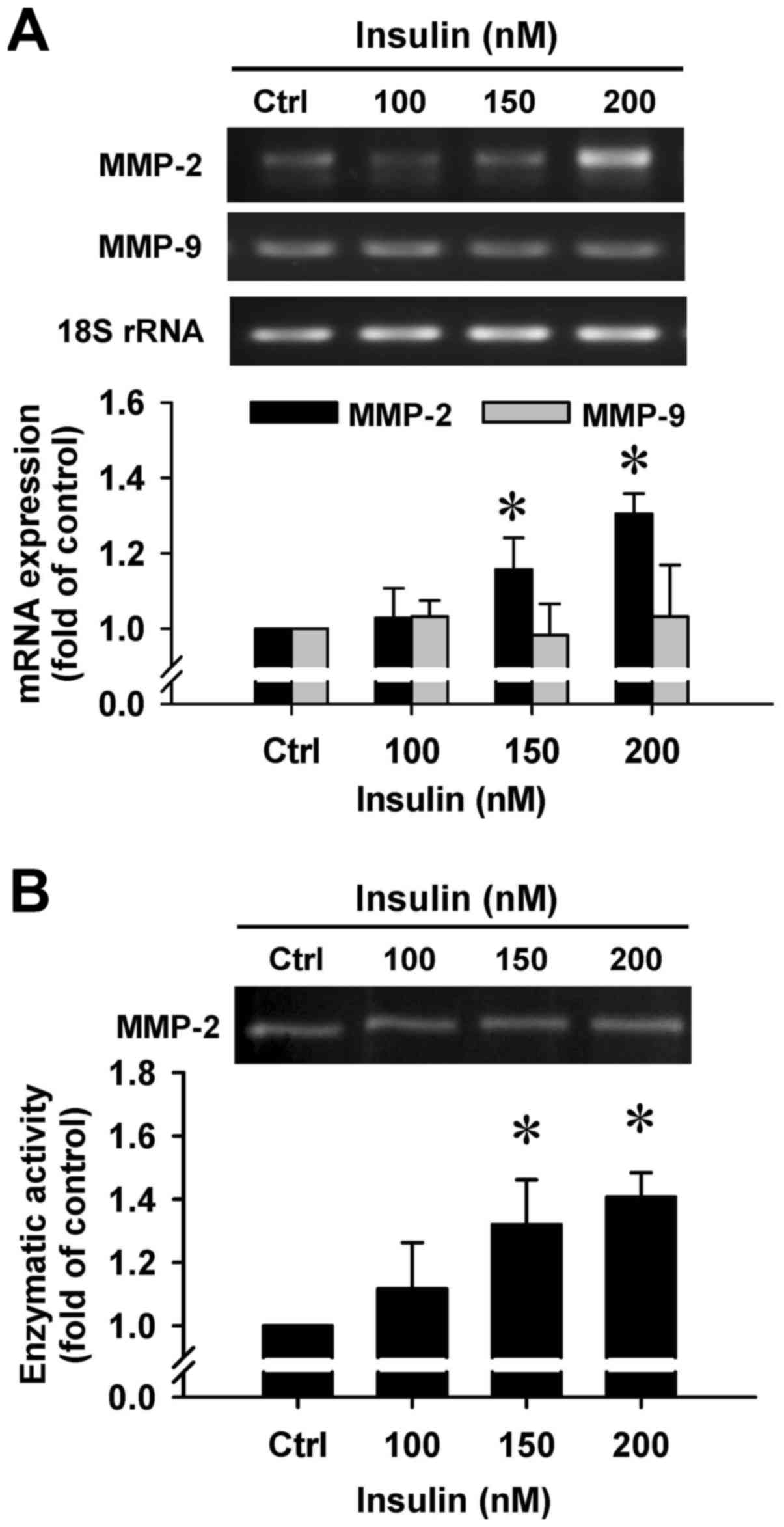
Insulin upregulates gene expression and
gelatinolytic activity of MMP-2 in HCT-116 cells
To clarify whether insulin regulates the secretion
of MMP-2 by HCT-116 cells, we employed RT-PCR and gelatin
zymography assays. There was a noticeable induction in MMP-2 gene
expression after insulin treatment at 150 and 200 nM for 48 h
(Fig. 8A). However, insulin had no
effect on the MMP-9 mRNA level in treated cells (Fig. 8A). In addition, a gelatinolytic
band corresponding to MMP-2 was significantly increased by insulin
in the culture medium that was stimulated, and quantitative
analysis of these results was performed to show that enzymatic
activity of MMP-2 was increased in HCT-116 cells (Fig. 8B). Based on the data, insulin leads
to cell migration through increasing the MMP-2 levels in HCT-116
cells.
Discussion
Diabetes is a systemic metabolic disorder caused by
defects in insulin secretion and insulin activity, resulting in
patients with abnormal carbohydrate metabolism inducing
hyperglycemia and hyperinsulinemia, which are the major causes of
the systemic inflammatory response (2,24).
It has been noted that the recurrence rate and mortality of
patients were substantially increased with the coexistence of
diabetes and some cancers (10,25).
Regarding the lack of insulin, tumor cell growth cannot directly be
promoted in diabetes patients with hyperglycemia (9). However, the high concentration of
insulin has been reported to activate IR signaling to enhance tumor
growth, which acts an important mediator (15,26).
Currently, the mortality of patients with diabetes and colorectal
cancer is higher than that in patients with colorectal cancer only;
among them, the patients with type 2 diabetes possess a higher risk
of colorectal cancer (12,27,28).
Therefore, this study focused on exploring the effect of insulin on
colorectal cancer cell proliferation and migration, as well as
clarifying the underlying molecular mechanism in human colorectal
cancer cells in vitro.
In this study, the human colon cancer cell line
HCT-116 was used to assess the effects of different concentrations
of insulin on the cytotoxicity of the tumor cells. A previous study
has shown that insulin treatment at 100 nM interacted with optimal
defensing expression (29) and was
involved in β-cell function during fasting plasma insulin
concentrations in man (30). We
found that insulin at 100–300 nM had no significant cytotoxic
effect and exhibited an increase in cell proliferation in HCT-116
cells (Fig. 1), agreeing with that
in a previously published study (31). In addition, the process of attached
cell adhesion to the extracellular matrix (ECM) is critical during
cancer cell proliferation and progression. The transformation of
cancer cells may be dependent on the behavior of continued growth,
which can serve as an important clue for the malignant progression
of cancer (32).
We further investigated the impact of insulin on
anchorage-independent cancer cell growth. Our findings demonstrate
that insulin significantly increased cell proliferation and
markedly promoted the growth of cancer cells by the poly-HEMA
coated assay to prevent cell attachment (Fig. 1B). Moreover, for long-term analysis
(7 days), there was a similar effect on HCT-116 cell proliferation
(data not shown). Previous studies have shown that mitogenic and
oncogenic stimulation enhance tumor cell growth and alter protein
kinase activity and nuclear localization (19,33).
Thus, it can be speculated that insulin promotes the
characteristics of cancer cell proliferation to stimulate cancer
progression in HCT-116 cells.
Insulin might play a vital role in the patients with
diabetes and cancer progression (34). Increasing evidence has suggested
that the activation of IR signaling is mediated through the binding
of insulin and IR to cause cancer cell survival and mitotic actions
(10). It has been recognized that
the activation of insulin signaling stimulates IRS-1 to further
affect the PI3K/Akt pathway and to induce ERK phosphorylation,
finally leading to cancer cell survival and mitotic effects
(35–37). Our data showed that insulin
increased the levels of IR and IRS-1 expression in HCT-116 cells
(Fig. 2), a finding that agrees
with a previously published study regarding insulin enhancing IR
expression and then dramatically altering the downstream signaling
pathway in breast cancer LCC6 cells (36). Therefore, we suggest that it is
unnecessary that a dramatic increase in the IR signal caused by
insulin occurs, but their downstream proteins are important after
IR activation in colon cancer cells.
Many studies have provided consistent evidence that
the phosphorylation of PI3K/Akt and ERK signaling can be
upregulated upon insulin pathway activation (36,38).
Our experimental results showed that the induction of the protein
levels of phosphorylated PI3K/Akt and ERK signals was observed in
insulin-treated HCT-116 cells (Figs.
2B and 5A). In addition,
insulin increased the phosphorylated proteins of JNK and p38 MAPKs
in the examined cells (Fig. 5A).
Importantly, preincubation with MAPK inhibitors (SP600125, PD98059
and SB203580) after insulin exposure diminished HCT cell
proliferation (Fig. 5B).
Therefore, the event of the MAPK signaling response for cell
proliferation requires the accumulation of insulin in human
colorectal cancer cells. Furthermore, the roles of the PI3K/Akt
pathway and its downstream signaling are vital during cancer
progression (39). For example,
the mTOR signal is involved in cell proliferation; GSK3β can
regulate the cell cycle-associated proteins (c-Myc and cyclin D1);
p53 can alter the effect of apoptosis and cell cycle-related
signals (39). It has been
reported that mRNA transcription, cell growth and cell
proliferation can modulate the phosphorylation of mTOR by
activating the PI3K/Akt/mTOR pathway (40,41).
Our current study provides information that the
insulin-promoted cell proliferation occurred partly through mTOR
signaling (Fig. 3A), indicating
that mTOR signaling is a minor mediator, and the other crucial
regulators might exist in insulin-treated HCT-116 cells. It was
reported that the activation of PI3K/Akt/GSK3β signaling promotes
the phosphorylated GSK3β on Ser9 to increase cyclin D1 expression,
which accelerates the entry from G1 phase into S phase (42,43).
Our results clearly showed that insulin significantly increased
phosphorylated GSK3β (Ser9) (Fig.
3B) and cyclin D1 (Fig. 4A) in
HCT-116 cells, suggesting that insulin activated the Akt signal to
phosphorylate GSK3β following the induction of cell proliferation
through increasing cyclin D1 expression, an activity that is
required for cell cycle progression. Furthermore, the growth
factors can suppress p53 expression through stimulating PI3K/Akt
signaling to promote cell cycle progression. However, the
regulation of PTEN expression by p53 can inhibit Akt activation
(44). In the present study, our
results showed that insulin treatment markedly promoted p53
expression in HCT-116 cells (Fig.
3C). A previous study demonstrated that the p53-dependent
effect contributing to the proliferation in uterine serous
carcinoma USPC-1 cells carrying wild-type p53 was induced by
metformin through the downregulation of insulin/IGF-1 signaling
(45). These results suggest that
the insulin-stimulated Akt signal could not suppress p53 expression
due to HCT-116 cells carrying wild-type p53 (46), creating a feedback effect to reach
the balance of dramatic cell proliferation caused by insulin.
MAPK family members can regulate cell proliferation,
cell differentiation and cancer progression and play vital roles in
cell apoptosis (47,48). Insulin signaling induces cell
proliferation through the activation of the ERK/MAPK pathway, and
the JNK and p38 MAPK had similar effects (36,38).
Our findings showed that insulin significantly increased the
phosphorylation of ERK, JNK and p38 MAPKs in HCT-116 cells
(Fig. 5A). Additionally, it is
well known that HSP27 and the transcription factor c-Myc are
regulated by MAPK signaling (47,49).
We found that the transcription factor c-Myc was decreased in the
nuclear fraction, but HSP27 was translocated to the nucleus in
cells after insulin challenge (Fig.
4B). This finding is also in agreement with other reports
showing that MAPK signaling can modulate c-Myc expression, and the
cancer cell invasive ability can be regulated by p38 MAPK (47,49).
Based on our functional study, it was demonstrated
that insulin triggered cell proliferation and metastatic effects
through MAPK signaling, as well as decreasing c-Myc expression and
increasing HSP27 signaling in HCT-116 cells. Evidence has shown
that the function of the oncogene c-Myc exhibits stimulated cell
proliferation and apoptosis, and cyclin D1 can drive cell cycle
progression and acts on cell growth to integrate the cell cycle
machinery (42,50). Our results revealed that insulin
increased the amount of cyclin D1 expression and reduced the
protein levels of c-Myc expression (Fig. 4A), suggesting that cancer cell
apoptosis was evaded by insulin and showed a high ability of drug
resistance (50). Cancer cell
metastasis has been shown to be involved in the vital events of the
intercellular degradation of the extracellular matrix, invasion and
migration ability during tumor progression (22,51).
Furthermore, chronic hyperinsulinemia promotes primary tumor growth
and the progression to lung metastasis in a mouse model of type 2
diabetes. We further investigated the effect of insulin on the
metastatic effects of HCT-116 cells. Our data demonstrated that
insulin enhanced HCT-116 cell migration, and MMP-2 might be
involved in the metastatic process caused by insulin (Figs. 6Figure 7–8), eventually leading to cancer cell
progression. This finding is also in agreement with the report by
Qi et al (52), proving
that insulin/protein kinase B signaling upregulates
metastasis-related phenotypes and molecules in human
hepatocarcinoma cells.
In conclusion, our study showed, for the first time,
that insulin triggers cell proliferation and the induction of
metastatic effects on human colorectal cancer HCT-116 cells that is
modulated by IR signaling and the PI3K/Akt/GSK3β pathway as well as
MMP-2 regulation. We report a concept and insight addressing how
insulin interacts with cell proliferation and migration in colon
cancer cells. Therefore, these data support our hypothesis and
offer an approach for insulin induction. The existing evidence
indicates that colorectal cancer progression might be involved in
insulin induction, and diabetes patients might need to pay close
attention to this issue.
Abbreviations:
|
ECM
|
extracellular matrix
|
|
IR
|
insulin receptor
|
|
IRS-1
|
insulin receptor substrate 1
|
|
MAPKs
|
mitogen-activated protein kinases
|
|
MMP-2
|
matrix metalloproteinase-2
|
|
PI3K
|
phosphoinositide 3-kinase
|
|
poly-HEMA
|
poly(2-hydroxyethyl methacrylate)
|
Acknowledgments
This work was supported in part by the Ministry of
Education, Taiwan, under the ATU plan and in part by the grant
MOST103-2313-B-038-003-MY3 from the Ministry of Science and
Technology, Taiwan.
References
|
1
|
Zimmet P, Alberti KG and Shaw J: Global
and societal implications of the diabetes epidemic. Nature.
414:782–787. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tsai TY, Cheng JF and Lai YM: Prevalence
of metabolic syndrome and related factors in Taiwanese high-tech
industry workers. Clinics (Sao Paulo). 66:1531–1535. 2011.
View Article : Google Scholar
|
|
3
|
Maria Rotella C, Pala L and Mannucci E:
Role of insulin in the type 2 diabetes therapy: Past, present and
future. Int J Endocrinol Metab. 11:137–144. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Krone CA and Ely JT: Controlling
hyperglycemia as an adjunct to cancer therapy. Integr Cancer Ther.
4:25–31. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stattin P, Björ O, Ferrari P, Lukanova A,
Lenner P, Lindahl B, Hallmans G and Kaaks R: Prospective study of
hyperglycemia and cancer risk. Diabetes Care. 30:561–567. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hsieh MC, Lee TC, Cheng SM, Tu ST, Yen MH
and Tseng CH: The influence of type 2 diabetes and glucose-lowering
therapies on cancer risk in the Taiwanese. Exp Diabetes Res.
2012:4137822012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Beckner ME, Stracke ML, Liotta LA and
Schiffmann E: Glycolysis as primary energy source in tumor cell
chemotaxis. J Natl Cancer Inst. 82:1836–1840. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Heuson JC and Legros N: Influence of
insulin deprivation on growth of the
7,12-dimethylbenz(a)anthracene-induced mammary carcinoma in rats
subjected to alloxan diabetes and food restriction. Cancer Res.
32:226–232. 1972.PubMed/NCBI
|
|
9
|
Masur K, Vetter C, Hinz A, Tomas N,
Henrich H, Niggemann B and Zänker KS: Diabetogenic glucose and
insulin concentrations modulate transcriptome and protein levels
involved in tumour cell migration, adhesion and proliferation. Br J
Cancer. 104:345–352. 2011. View Article : Google Scholar
|
|
10
|
Giovannucci E, Harlan DM, Archer MC,
Bergenstal RM, Gapstur SM, Habel LA, Pollak M, Regensteiner JG and
Yee D: Diabetes and cancer: A consensus report. CA Cancer J Clin.
60:207–221. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Will JC, Galuska DA, Vinicor F and Calle
EE: Colorectal cancer: Another complication of diabetes mellitus?
Am J Epidemiol. 147:816–825. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Saydah SH, Platz EA, Rifai N, Pollak MN,
Brancati FL and Helzlsouer KJ: Association of markers of insulin
and glucose control with subsequent colorectal cancer risk. Cancer
Epidemiol Biomarkers Prev. 12:412–418. 2003.PubMed/NCBI
|
|
13
|
Giovannucci E: Insulin, insulin-like
growth factors and colon cancer: A review of the evidence. J Nutr.
131(Suppl): 3109S–3120S. 2001.PubMed/NCBI
|
|
14
|
Zhang H, Pelzer AM, Kiang DT and Yee D:
Down-regulation of type I insulin-like growth factor receptor
increases sensitivity of breast cancer cells to insulin. Cancer
Res. 67:391–397. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dinchuk JE, Cao C, Huang F, Reeves KA,
Wang J, Myers F, Cantor GH, Zhou X, Attar RM, Gottardis M, et al:
Insulin receptor (IR) pathway hyperactivity in IGF-IR null cells
and suppression of downstream growth signaling using the dual
IGF-IR/IR inhibitor, BMS-754807. Endocrinology. 151:4123–4132.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guo H, Gao M, Lu Y, Liang J, Lorenzi PL,
Bai S, Hawke DH, Li J, Dogruluk T, Scott KL, et al: Coordinate
phosphorylation of multiple residues on single AKT1 and AKT2
molecules. Oncogene. 33:3463–3472. 2014. View Article : Google Scholar :
|
|
17
|
Ericson K, Gan C, Cheong I, Rago C,
Samuels Y, Velculescu VE, Kinzler KW, Huso DL, Vogelstein B and
Papadopoulos N: Genetic inactivation of AKT1, AKT2, and PDPK1 in
human colorectal cancer cells clarifies their roles in tumor growth
regulation. Proc Natl Acad Sci USA. 107:2598–2603. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Strober W: Trypan blue exclusion test of
cell viability. Curr Protoc Immunol. 111:A3.B.1–3. 2015. View Article : Google Scholar
|
|
19
|
Fukazawa H, Mizuno S and Uehara Y: A
microplate assay for quantitation of anchorage-independent growth
of transformed cells. Anal Biochem. 228:83–90. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Folkman J and Moscona A: Role of cell
shape in growth control. Nature. 273:345–349. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lu CC, Yang SH, Hsia SM, Wu CH and Yen GC:
Inhibitory effects of Phyllanthus emblica L. on hepatic steatosis
and liver fibrosis in vitro. J Funct Foods. 20:20–30. 2016.
View Article : Google Scholar
|
|
22
|
Lu CC, Yang JS, Chiang JH, Hour MJ,
Amagaya S, Lu KW, Lin JP, Tang NY, Lee TH and Chung JG: Inhibition
of invasion and migration by newly synthesized quinazolinone MJ-29
in human oral cancer CAL 27 cells through suppression of MMP-2/9
expression and combined down-regulation of MAPK and AKT signaling.
Anticancer Res. 32:2895–2903. 2012.PubMed/NCBI
|
|
23
|
Lu CC, Yang JS, Huang AC, Hsia TC, Chou
ST, Kuo CL, Lu HF, Lee TH, Wood WG and Chung JG: Chrysophanol
induces necrosis through the production of ROS and alteration of
ATP levels in J5 human liver cancer cells. Mol Nutr Food Res.
54:967–976. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kitabchi AE, Umpierrez GE, Miles JM and
Fisher JN: Hyper-glycemic crises in adult patients with diabetes.
Diabetes Care. 32:1335–1343. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Barone BB, Yeh HC, Snyder CF, Peairs KS,
Stein KB, Derr RL, Wolff AC and Brancati FL: Long-term all-cause
mortality in cancer patients with preexisting diabetes mellitus: A
systematic review and meta-analysis. JAMA. 300:2754–2764. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lawlor MA and Alessi DR: PKB/Akt: A key
mediator of cell proliferation, survival and insulin responses? J
Cell Sci. 114:2903–2910. 2001.PubMed/NCBI
|
|
27
|
Le Marchand L, Wilkens LR, Kolonel LN,
Hankin JH and Lyu LC: Associations of sedentary lifestyle, obesity,
smoking, alcohol use, and diabetes with the risk of colorectal
cancer. Cancer Res. 57:4787–4794. 1997.PubMed/NCBI
|
|
28
|
Kono S, Honjo S, Todoroki I, Nishiwaki M,
Hamada H, Nishikawa H, Koga H, Ogawa S and Nakagawa K: Glucose
intolerance and adenomas of the sigmoid colon in Japanese men
(Japan). Cancer Causes Control. 9:441–446. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Barnea M, Madar Z and Froy O: Glucose and
insulin are needed for optimal defensin expression in human cell
lines. Biochem Biophys Res Commun. 367:452–456. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Matthews DR, Hosker JP, Rudenski AS,
Naylor BA, Treacher DF and Turner RC: Homeostasis model assessment:
Insulin resistance and beta-cell function from fasting plasma
glucose and insulin concentrations in man. Diabetologia.
28:412–419. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ayiomamitis GD, Notas G, Zaravinos A,
Drygiannakis I, Georgiadou M, Sfakianaki O, Mastrodimou N, Thermos
K and Kouroumalis E: Effects of octreotide and insulin on colon
cancer cellular proliferation and correlation with hTERT activity.
Oncoscience. 1:457–467. 2014. View Article : Google Scholar
|
|
32
|
DeBerardinis RJ, Lum JJ, Hatzivassiliou G
and Thompson CB: The biology of cancer: Metabolic reprogramming
fuels cell growth and proliferation. Cell Metab. 7:11–20. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lv L, Xu YP, Zhao D, Li FL, Wang W, Sasaki
N, Jiang Y, Zhou X, Li TT, Guan KL, et al: Mitogenic and oncogenic
stimulation of K433 acetylation promotes PKM2 protein kinase
activity and nuclear localization. Mol Cell. 52:340–352. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Duan W, Shen X, Lei J, Xu Q, Yu Y, Li R,
Wu E and Ma Q: Hyperglycemia, a neglected factor during cancer
progression. BioMed Res Int. 2014:4619172014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Saltiel AR and Pessin JE: Insulin
signaling pathways in time and space. Trends Cell Biol. 12:65–71.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang H, Fagan DH, Zeng X, Freeman KT,
Sachdev D and Yee D: Inhibition of cancer cell proliferation and
metastasis by insulin receptor downregulation. Oncogene.
29:2517–2527. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lau MT and Leung PC: The PI3K/Akt/mTOR
signaling pathway mediates insulin-like growth factor 1-induced
E-cadherin down-regulation and cell proliferation in ovarian cancer
cells. Cancer Lett. 326:191–198. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ulanet DB, Ludwig DL, Kahn CR and Hanahan
D: Insulin receptor functionally enhances multistage tumor
progression and conveys intrinsic resistance to IGF-1R targeted
therapy. Proc Natl Acad Sci USA. 107:10791–10798. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jiang BH and Liu LZ: PI3K/PTEN signaling
in tumorigenesis and angiogenesis. Biochim Biophys Acta.
1784:150–158. 2008. View Article : Google Scholar
|
|
40
|
Dancey J: mTOR signaling and drug
development in cancer. Nat Rev Clin Oncol. 7:209–219. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Laplante M and Sabatini DM: mTOR signaling
in growth control and disease. Cell. 149:274–293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yang K, Guo Y, Stacey WC, Harwalkar J,
Fretthold J, Hitomi M and Stacey DW: Glycogen synthase kinase 3 has
a limited role in cell cycle regulation of cyclin D1 levels. BMC
Cell Biol. 7:332006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Takahashi-Yanaga F and Sasaguri T:
GSK-3beta regulates cyclin D1 expression: A new target for
chemotherapy. Cell Signal. 20:581–589. 2008. View Article : Google Scholar
|
|
44
|
Maddocks OD and Vousden KH: Metabolic
regulation by p53. J Mol Med (Berl). 89:237–245. 2011. View Article : Google Scholar
|
|
45
|
Sarfstein R, Friedman Y, Attias-Geva Z,
Fishman A, Bruchim I and Werner H: Metformin downregulates the
insulin/IGF-I signaling pathway and inhibits different uterine
serous carcinoma (USC) cells proliferation and migration in
p53-dependent or -independent manners. PLoS One. 8:e615372013.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zawacka-Pankau J, Issaeva N, Hossain S,
Pramanik A, Selivanova G and Podhajska AJ: Protoporphyrin IX
interacts with wild-type p53 protein in vitro and induces cell
death of human colon cancer cells in a p53-dependent and
-independent manner. J Biol Chem. 282:2466–2472. 2007. View Article : Google Scholar
|
|
47
|
Zhang W and Liu HT: MAPK signal pathways
in the regulation of cell proliferation in mammalian cells. Cell
Res. 12:9–18. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wada T and Penninger JM: Mitogen-activated
protein kinases in apoptosis regulation. Oncogene. 23:2838–2849.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Huang C, Jacobson K and Schaller MD: MAP
kinases and cell migration. J Cell Sci. 117:4619–4628. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Liao DJ, Thakur A, Wu J, Biliran H and
Sarkar FH: Perspectives on c-Myc, Cyclin D1, and their interaction
in cancer formation, progression, and response to chemotherapy.
Crit Rev Oncog. 13:93–158. 2007. View Article : Google Scholar
|
|
51
|
Lu Z, Lu N, Li C, Li F, Zhao K, Lin B and
Guo Q: Oroxylin A inhibits matrix metalloproteinase-2/9 expression
and activation by up-regulating tissue inhibitor of
metalloproteinase-2 and suppressing the ERK1/2 signaling pathway.
Toxicol Lett. 209:211–220. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Qi HL, Zhang Y, Ma J, Guo P, Zhang XY and
Chen HL: Insulin/protein kinase B signalling pathway upregulates
metastasis-related phenotypes and molecules in H7721 human
hepatocarcinoma cell line. Eur J Biochem. 270:3795–3805. 2003.
View Article : Google Scholar : PubMed/NCBI
|