Introduction
Presently, acute promyelocytic leukemia is
characterized by the differentiation arrest during the
promyelocytic stage and elevation of the hematopoietic stem cells
(1,2). Chemotherapy is the initial treatment
of choice. Due to the abnormal collection of immature precursors,
as well as the suppression of normal hemopoiesis, acute
promyelocytic leukemia represents as a medical emergency, which
causes a high level of early fatalities from the massive hemorrhage
(3–5). Most patients with acute promyelocytic
leukemia will receive a combination of medications. There are no
surgical options owing to the body-wide distribution of malignant
cells (6,7). Therefore, finding effective and new
therapeutic strategies and revealing the underlying molecular
mechanisms regulating leukemia cells are necessary and could be
beneficial and useful for patients with acute promyelocytic
leukemia.
Actein is a tetracyclic triterpenoid compound,
isolated from the rhizome of Cimicifuga foetida (8). Cimicifuga species has a long
history for medicine to protect people suffering from rheumatism,
sore throat, and diarrhea in North America (9). In Asia, Cimicifuga species are
used owing to its various bioactivities, such as antidiabetic,
anti-osteoporosis and antiviral (10). In addition, the extracts from
rhizome of Cimicifuga species have been applied to prevent
female-related diseases clinically (11). Hence, Cimicifuga species
could be considered as a natural medicinal herb with promising
medicinal values. Moreover, actein was selective for human breast
tumor cells, which could synergize with other chemotherapy agents
to inhibit tumor growth (12).
Moreover, p53 signaling pathway was revealed to be modulated by
actein (11). P53, as a tumor
suppressor, plays an essential role in apoptosis induction through
regulating caspases (13–15). However, the effects of actein on
modulating tumor growth, including human leukemia, is poorly
understood. Herein, we attempted to explore if actein could
suppress human leukemia development though inducing apoptosis and
to reveal the underlying molecular mechanism.
Rho-associated kinase (ROCK) is reported as a
serine/threonine kinase and one of the major downstream effectors
of the small GTPase RhoA (16).
The RhoA/ROCK signaling pathway is closely associated with the
pathogenesis of various disorders and is also involved in a number
of aspects of tumors, including human leukemia (17,18).
Rho could modulate the cell actin cytoskeleton through its
down-stream effective factor of ROCK, which is highly included in
the biological processes of cell movement, cell migration, gene
transcription, nerve regeneration, and apoptosis (19,20).
Also, elevation of RhoA/ROCK has been reported in tumors and
associated with cancer development (21). Therefore, targeting RhoA/ROCK
signaling pathway might be a potential therapeutic strategy for
human leukemia treatment. Though it has been reported in leukemia
progression, further study is still required to further reveal the
underlying molecular mechanism. In this study, we attempted to
explore the role of actein in modulating human leukemia cell
progression through apoptosis induction, which relied on regulation
of RhoA/ROCK1. In vivo, the U937-bearing tumor growth was
inhibited by actein treatment. The suppressive role of actein in
human leukemia included its effects on AKT dephosphorylation,
phosphatase and tensin homolog (PTEN) activation, pro-apoptotic
signal promotion as well as anti-apoptotic molecule reduction. The
results suggested that actein could be an effective candidate for
human leukemia.
Materials and methods
Cells and culture
Human leukemia cell lines, U937, K562 and NB4, were
purchased from American Type Culture Collection (ATCC, USA). Human
hepatocyte cell line L02 and human tubular epithelial cells HK2
were purchased from KeyGen Biotech Co., Ltd. (Nanjing, China). All
cells were cultured in RPMI-1640 medium, which is supplemented with
10% fetal bovine serum (FBS) (Gibco, Invitrogen, USA), 100 U/ml
penicillin and 100 μg/ml streptomycin at 37°C in a 5%
CO2 humidified environment. The peripheral blood samples
in our studies were isolated from 10 patients with acute
promyelocytic leukemia after acquiring the informed consent: two
patients are M2, four are M4, and four are M5 following the
French-American-British (FAB) classification system. Approval for
the study was obtained from Huai'an First People's Hospital,
Nanjing Medical University (Jiangsu, China). Acute promyelocytic
leukemia blasts were extracted using Histopaque-1077 density
gradient centrifugation (Sigma-Aldrich, USA) for 15 min at 600 g.
The isolated mononuclear cells were then suspended in RPMI-1640
medium at 8×105/ml. Peripheral blood mononuclear cells
from blood samples collected from the healthy volunteers were also
isolated by Histopaque 1077 density gradient centrifugation.
As for the gene knockdown, the cells were
transfected with (100 nM) nonsense control, siRNA against caspase-3
(#1:5′-UGU AGG AGA GUU GAG GUC GAG GU) and siRNA against ROCK1
(#1:5′-GAU UAU AGA GUG GUG GUG ACG GGU A) for 24 h using
Lipofectamine 2000 (Invitrogen) according to the manufacturer's
protocol. All small interfering RNAs were synthesised by GenePharma
(Shanghai, China). After various treatments, further experiments
were conducted. Actein (CAS:18642-44-9, 98% HPLC) used in our study
was purchased from Shanghai Yuanye Bio-Technology Co., Ltd.
(Shanghai, China). AKT inhibitor, LY294002, and caspase-3
inhibitor, Z-VAD-FMK, were purchased from Sigma-Aldrich, and ROCK1
inhibitor, Y-27632 dihydrochloride was obtained from Tocris
Bioscience (Bristol, UK). RhoA inhibitor, C3 exoenzyme, was
purchased from Alexis Biochemicals (USA).
Flow cytometric analysis
After treatment under various conditions, flow
cytometric analysis was used to determine apoptosis levels in U937
cells using Annexin V/PI staining kit (Roche, Switzerland)
following the manufacturer's protocol. In order to analyze the
mitochondria injury, 2×105 U937 cells were cultured with
3,3-dihexyloxacarbocynine at the dose of 40 nM (DiOC6,
Sigma-Aldrich) in PBS for 20 min at 37°C. The results acquisition
and analysis were performed using a Becton-Dickinson FACSCalibur
flow cytometer with Cell Quest software.
Mitochondrial and cytosolic
fractions
Mitochondria/cytosol fractionation kits (ab65320,
Abcam, USA) were used to isolate the mitochondrial fraction from
cytosolic fraction. Then, the enriched mitochondrial and cytosolic
fractions were used for western blot analysis.
Hoechst 33258 staining
Hoechst 33258 staining of the cells was performed to
evaluate the apoptosis induced by actein. The cells were seeded at
a concentration of 1×106 cells/ml in 6-well plates and
treated with the indicated concentration of actein. The cells were
harvested, washed twice with PBS, fixed with 4% formaldehyde for 10
min and stained with Hoechst 33258 (Sigma-Aldrich) staining
solution following the manufacturer's instructions. The images were
immediately photographed under a fluorescence microscope (Olympus,
Japan).
Evaluation of apoptosis
After induction of actein for 24 h, leukemia cell
apoptosis was measured using a commercial single-stranded DNA
(ssDNA) enzyme-linked immunosorbent assay kit (Millipore Chemicon,
USA), which detects ssDNA, corresponding to the most specific
apoptosis end-product.
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide
(MTT) analysis
MTT (Beyotime, Nanjing, China) was used to calculate
cell viability. Cells (2×103/well) were seeded on
96-well plates and treated under different conditions as indicated
and incubated at 37°C. MTT solution (300 μl/well) was added
after incubation. Following incubation at 37°C for an additional 4
h, the supernatants were removed and 200 μl dimethyl
sulfoxide (DMSO, Sigma-Aldrich) was added into each well to
dissolve the formazan crystals. The 96-well plates were then placed
in a microplate reader to determine the absorbance at 490 nm. Each
test was carried out in triplicate.
Animal treatments
Twenty, 5-week-old male nu/nu mice (15–18 g) were
injected with 5×106 U937 cells subcutaneously. All mice
were purchased from the Animal Experiment Center of Nanjing Medical
University (Nanjing, China). Before the experiments, all mice were
allowed to adapt to the environment for a week. All protocols were
in line with the Regulations of Experimental Animal Administration
issued by the Ministry of Science and Technology of the People's
Republic of China. Mouse care and usage were performed according to
the ethical guidelines of Huai'an First People's Hospital, Nanjing
Medical University. The mice were raised in air-conditioned
pathogen-free rooms (25±2°C, 50±10% humidity) under controlled
lighting (12 h light/day) and fed with water and standard
laboratory food. When the tumors were visible, the mice were
randomly divided into two groups (10 mice per group). The control
group received the vehicle (PBS) injection i.p., and the treatment
group was administered with actein i.p. every day at a dose of 15
mg/kg (11). Sixty days later, all
mice were sacrificed, tumors were excised and measured, and tumor
tissues were fixed in 10% formalin. After embedding in paraffin,
immunohistochemical analysis was performed.
Immunofluorescent analysis
U937 cells were treated with or without 40 μM
actein for 24 h. Cells were then harvested through centrifugation,
and fixed with 4% paraformaldehyde for 10 min, permeabilized using
0.1% Triton X-100 for 10 min, and finally blocked with 1% bovine
serum albumin for 30 min at room temperature. Cells were further
incubated with a primary antibody (Cyto-c, 1:200, Abcam) at
4°C overnight, and followed by a secondary goat anti-mouse IgG
H&L (Alexa Fluor® 488) (Abcam) for 1 h at room
temperature. After washing with PBS, the images were captured with
a confocal microscope (Olympus, Japan).
Immunohistochemical assays
Paraffin-embedded tumor sections were used for the
blinded assessment of caspase-3, Ki-67, ROCK1 and apoptosis levels,
respectively. Mouse tumors were sectioned at 3 μM thickness,
and terminal deoxynucleotidyl transferase (TdT) dUTP nick-end
labeling (TUNEL) assay was carried out using light and electron
microscopy-based kits (R&D Systems, USA) for detecting DNA
fragments. For staining of ROCK1 (1:200, Abcam, UK), caspase-3
(1:200, Abcam) and Ki-67 (1:200, Abcam), the tumor sections were
analyzed using a microscope. Images were arranged using the
TissueFAXs (Tissue-Gnostics) software.
Western blot analysis
For western blotting, cells and tissue samples after
various treatments were lysed in RIPA lysis buffer (150 mM NaCl,
0.1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM
Tris-HCl, pH 8.0) to yield a homogenate. Also, the final
supernatants were obtained by centrifugation at 12,000 g for 15
min. The protein concentration was calculated using bicinchoninic
acid (BCA) protein assay kit (Thermo Scientific, USA) with bovine
serum albumin as a standard. The total protein extract was later
used for western blot analysis. Total protein (40 μg) was
loaded and proteins were separated using SDS-PAGE and
electrophoretically transferred to polyvinylidene difluoride
membranes (Millipore, USA). The membranes were then blocked with 5%
non-fat dry milk in Tris buffered saline (20 mM Tris, pH 7.6, 137
mM NaCl) with 0.1% Tween-20, washed, and then incubated with
primary antibody. The primary antibodies were as follows: rabbit
anti-p-AKT (1:1,000, Cell Signaling Technology), rabbit anti-AKT
(1:1,000, Cell Signaling Technology), rabbit anti-Mcl-1 (1:1,000,
Abcam), rabbit anti-Bcl-xl (Abcam, USA), rabbit anti-Bax (1:1,000,
Abcam), rabbit anti-PTEN (1:1,000, Abcam), rabbit anti-p-PTEN
(1:1,000, Abcam), rabbit anti-caspase-3 (1:1,000, Abcam), mouse
anti-Bcl-2 (1:1,000, Cell Signaling Technology), rabbit anti-PARP
(1:1,000, Cell Signaling Technology), rabbit anti-p-Bad (1:1,000,
Abcam), rabbit anti-Bad (1:1,000, Abcam), rabbit anti-RhoA
(1:1,000, Abcam), rabbit anti-ROCK1 (1:1,000, Abcam), rabbit
anti-Cox IV (1:1,000, Abcam), mouse anti-Cyto-c (1:1,000,
Abcam) and anti-GAPDH (1:500, Santa Cruz Biotechnology, Inc.).
Immunoreactive bands were visualized by ECL Immunoblot Detection
system (Pierce Biotechnology, Inc., Rockford, IL, USA) and exposed
to Kodak (Eastman Kodak Co., USA) X-ray film. Each protein
expression level was defined as grey value (Version 1.4.2b, Mac OS
X, ImageJ, National Institutes of Health, USA) and standardized to
housekeeping gene of GAPDH and expressed as a fold of control.
Statistical analysis
Results are represented as the mean ± SEM of
triplicate experiments. Statistically significant values were
compared by use of the ANOVA and the Dunnett's post hoc test, and
P-values of <0.05 were considered to indicate a statistically
significant result.
Results
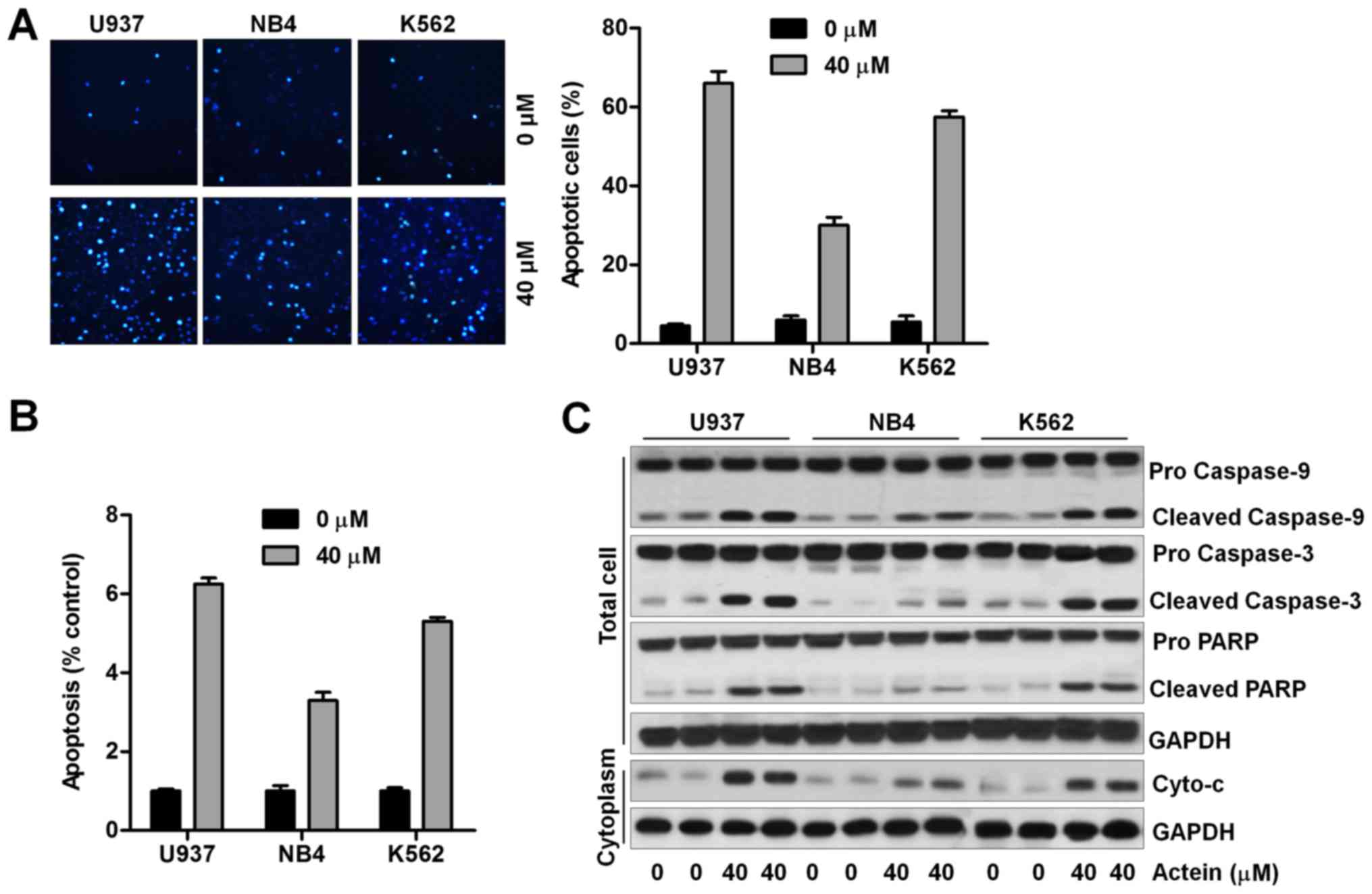
Actein triggers apoptosis and
mitochondrial damage in human leukemia cells
Actein has been investigated in human breast cancer
though apoptosis induction (10,11).
Thus, here we first examined the role of actein in apoptosis and
the mitochondrial injury in human leukemia cells, U937. Treating
U937 cells with different concentrations of actein for 24 h
significantly enhanced apoptosis (Fig.
1A, left) and mitochondrial injury in a dose-dependent manner
(Fig. 1B, left). Furthermore, U937
cells were treated with 40 μM actein for the indicated time,
ranging from 0 to 24 h. Flow cytometric analysis indicated that
apoptosis was highly induced (Fig.
1A, right). Also, the mitochondria injury was observed in
actein-treated cells, which was time-dependent (Fig. 1B, right). Following, the proteins
isolated from total cell and cytoplasm, respectively, were used for
western blot analysis. The results indicated that caspase-9,
caspase-3 and PARP cleavage was dramatically increased after actein
administration dose-dependently. Similarly, Cyto-c was found
to be upregulated with the increasing of actein treatment in
cytoplasm, indicating actein administration promoted Cyto-c
release into the cytoplasm (Fig.
1C, left). In line with the results of flow cytometry,
caspase-9, caspase-3 and PARP activation was significantly improved
after actein treatment, which was shown in a time-dependent manner
(Fig. 1C, right). Furthermore,
Hoechst 33258 staining indicated that the number of apoptosis was
significantly high in U937 and K562 cells after actein treatment
(Fig. 2A). Cell apoptosis was also
evaluated using ssDNA detection kit. Untreated leukemia cells
served as the control. As shown in Fig. 2B, we found that treatment of actein
significantly increased the apoptosis proportion in leukemia cell
lines, and consistently, U937 cells were more sensitive to actein
treatment. Also, western blot analysis indicated that caspase-9,
caspase-3, and PARP cleavage and Cyto-c in cytoplasm were
apparently induced by actein in 937 and K562 cells (Fig. 2C). The results here indicated that
U937 and K562 are likely to be more sensitive to actein. Taken
together, the data above indicated that actein could potentiate
apoptosis in human leukemia cells.
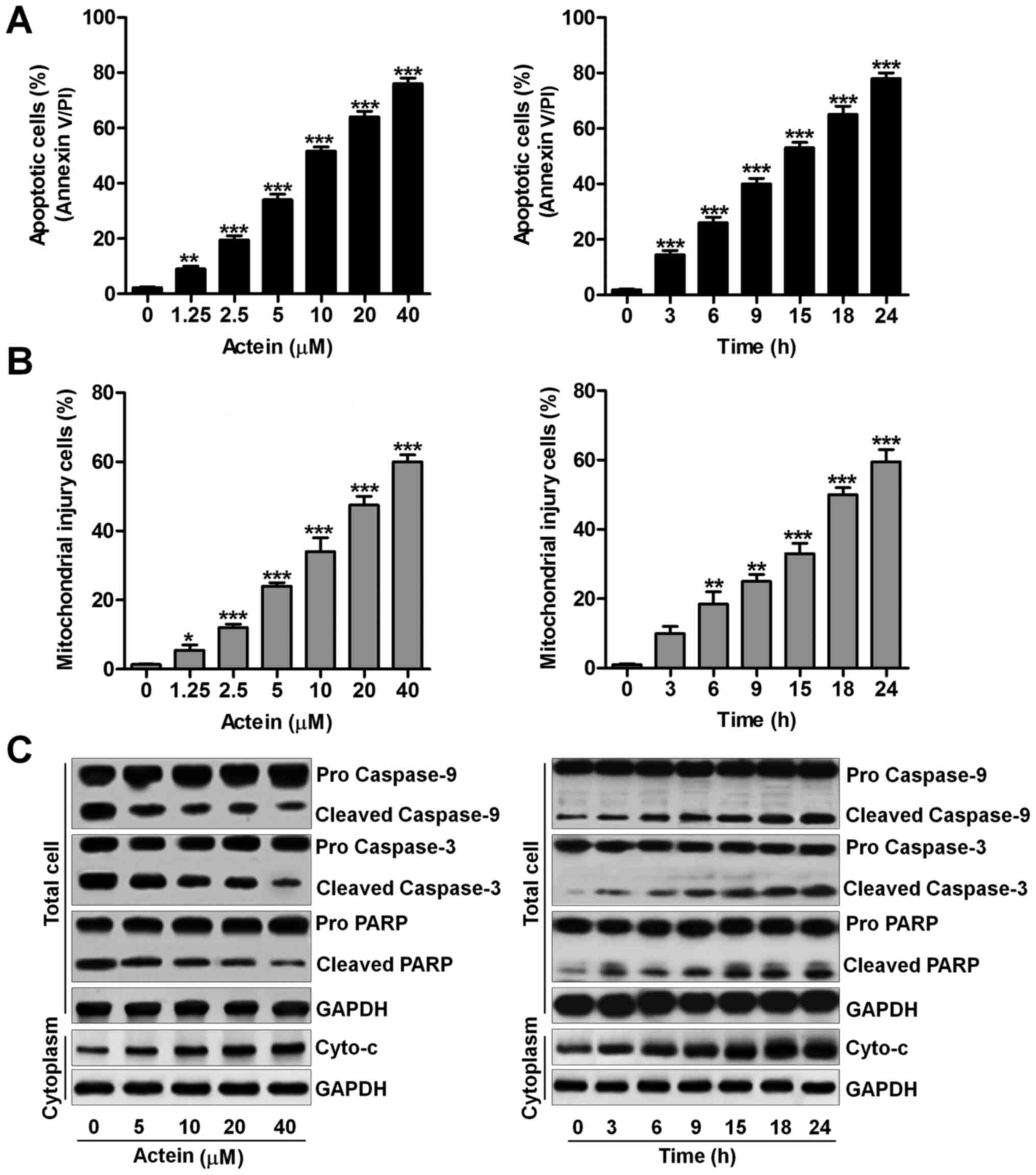 | Figure 1Actein triggers apoptosis and
mitochondrial damage in human leukemia cells. (A) Left, the human
leukemia cells of U937 were exposed to actein at the indicated
concentrations, ranging from 0 to 40 μM as indicated for 24
h. After treatments, all cells were harvested for apoptotic
analysis using flow cytometry. Right, U937 cells were cultured with
40 μM actein for, respectively, 0, 3, 6, 9, 15, 18 or 24 h.
Then, all cells were collected for calculating the number of
apoptotic cells using flow cytometry. (B) Left, U937 cells were
treated at different doses of actein as described for 24 h and
stained with DiOC6. Then, flow cytometric analysis was
used to evaluate the number of cells experiencing mitochondrial
injury. Right, U937 cells were treated with 40 μM actein for
the indicated time, followed by DiOC6 staining. Next,
all cells were harvested for flow cytometric assays. (C) All U937
cells were treated as described. The extracts from the whole cells
and the cytoplasm were used for western blot analysis using primary
antibodies of caspase-9, caspase-3, PARP and Cyto-c. Data
are analyzed as mean ± SEM, n=8. *P<0.05,
**P<0.01 and ***P<0.001 versus the Con
group in the absence of any treatments. |
Actein induces apoptosis response in
primary human leukemia cells
We attempted to evaluate if actein could induce
apoptosis in human primary leukemia cells. The human primary
leukemia cells were extracted from 10 patients with acute myeloid
leukemia. Then, actein was administered to cells. As shown in
Fig. 3A, we found that actein
significantly induced apoptosis in human primary leukemia cells.
Western blot analysis further indicated that the activation of
caspase-9, caspase-3 and PARP was markedly induced in
actein-treated primary leukemia cells obtained from two patients.
Also, Cyto-c releasing in the cytoplasm of human primary
leukemia cells was also induced by using actein (Fig. 3B). Next, the effects of actein on
peripheral blood mononuclear cells obtained from normal healthy
people were explored. As shown in Fig.
3C, cells exposed to 40 μM actein for various times
showed no significant difference on apoptosis induction. In
addition, western blot analysis in peripheral blood mononuclear
cells and cytoplasm indicated that caspase-9, caspase-3, PARP and
Cyto-c were not alternative either in the group with actein
exposure or not (Fig. 3D).
Peripheral blood mononuclear cells, human hepatocyte cell line L02
and human tubular epithelial cells HK2 were exposed to actein for
24 h at the indicated concentartions. Then, all cells were
harvested for MTT analysis. From the results in Fig. 3E, we found that there was no
significant difference between the groups of cells. The results
indicated that actein at the indicated concentrations used in our
study showed no cytotoxicity to normal cells. The findings above
indicated that actein could trigger apoptosis in human primary
leukemia cells without any changes in peripheral blood mononuclear
cells from normal people.
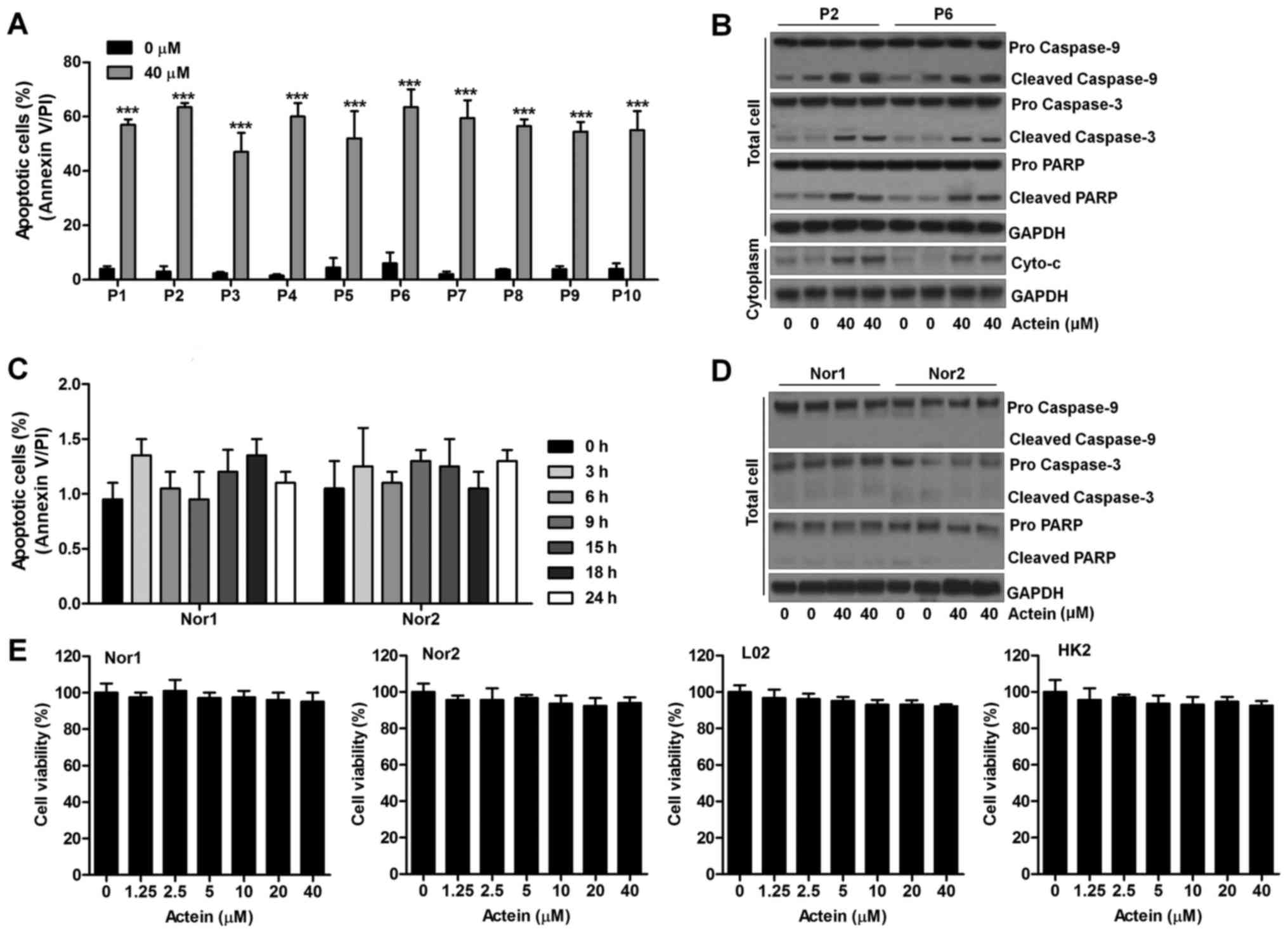 | Figure 3Actein induces apoptotic response in
primary human leukemia cells. (A) Primary human leukemia cells were
derived from peripheral blood of 10 patients. Immediate after
treatment with actein (40 μM) for 24 h, the percentage of
apoptotic cells was calculated through flow cytometry. (B) The
lysates from the whole cell and the cytosolic fractions in leukemia
cells extracted from two patients with acute myeloid leukemia were
collected for western blot analysis of caspase-9, caspase-3, PARP
and Cyto-c. (C) Peripheral blood mononuclear cells isolated
from normal human were cultured with 40 μM actein for 0, 3,
6, 9, 15, 18 and 24 h. Then, flow cytometry was carried out to
evaluate the number of apoptosis. (D) Peripheral blood mononuclear
cells obtained from normal healthy people were exposed to 40
μM actein for 24 h, followed by assessment of caspase-9,
caspase-3, PARP and Cyto-c using western blot analysis. (E)
Peripheral blood mononuclear cells, human hepatocyte cell line L02
and human tubular epithelial cells HK2 were exposed to actein for
24 h at the indicated concentrations. Then, all cells were
harvested for MTT analysis. Data are analyzed as mean ± SEM, n=8.
***P<0.001 versus the Con group in the absence of any
treatment. |
Actein causes apoptosis through
regulating pro- and anti-apoptotic signals
Anti-apoptotic and pro-apoptotic signals are well
characterized in regulating apoptosis (22,23).
Thus, in order to explore the underlying molecular mechanism
regarding to actein-induced apoptosis in human leukemia. Protein
levels in total cellular, cytoplasm and mitochondria were
calculated using western blot analysis. As shown in Fig. 4A, we found that Bcl-2, Bcl-xl and
Mcl-1 expressed highly in actein-treated U937 cells. Bad
phosphorylation, suppressing apoptosis, was reduced by actein. In
contrast, Bad was observed with increased levels after actein
administration in whole cells. Bax in cytoplasm was discovered with
reduced protein abundance in actein-treated cells dose-dependently.
Accordingly, in mitochondria, Bax was elevated, which indicated the
ability of actein in inducing apoptosis. The results above were
also triggered by actein in a time-dependent manner in U937 cells
(Fig. 4B). Furthermore,
immunofluorescent analysis confirmed that the expression of Bax,
proved by the fluorescent intensity, in cytoplasm was suppressed by
actein (Fig. 4C). Also, primary
leukemia cells isolated from patient indicated that anti-apoptotic
molecules of Bcl-2, Bcl-xl and Mcl-1 were suppressed by actein
administration, as well as phosphorylated Bad, while Bad was
significantly triggered. Additionally, actein reduced
Bcl-2-associated X protein (Bax) expression in cytoplasm, and
elevated its levels in mitochondria (Fig. 4D). Finally, western blot analysis
of peripheral blood mononuclear cells extracted from normal people
showed no difference of these signals in expression (Fig. 4E).
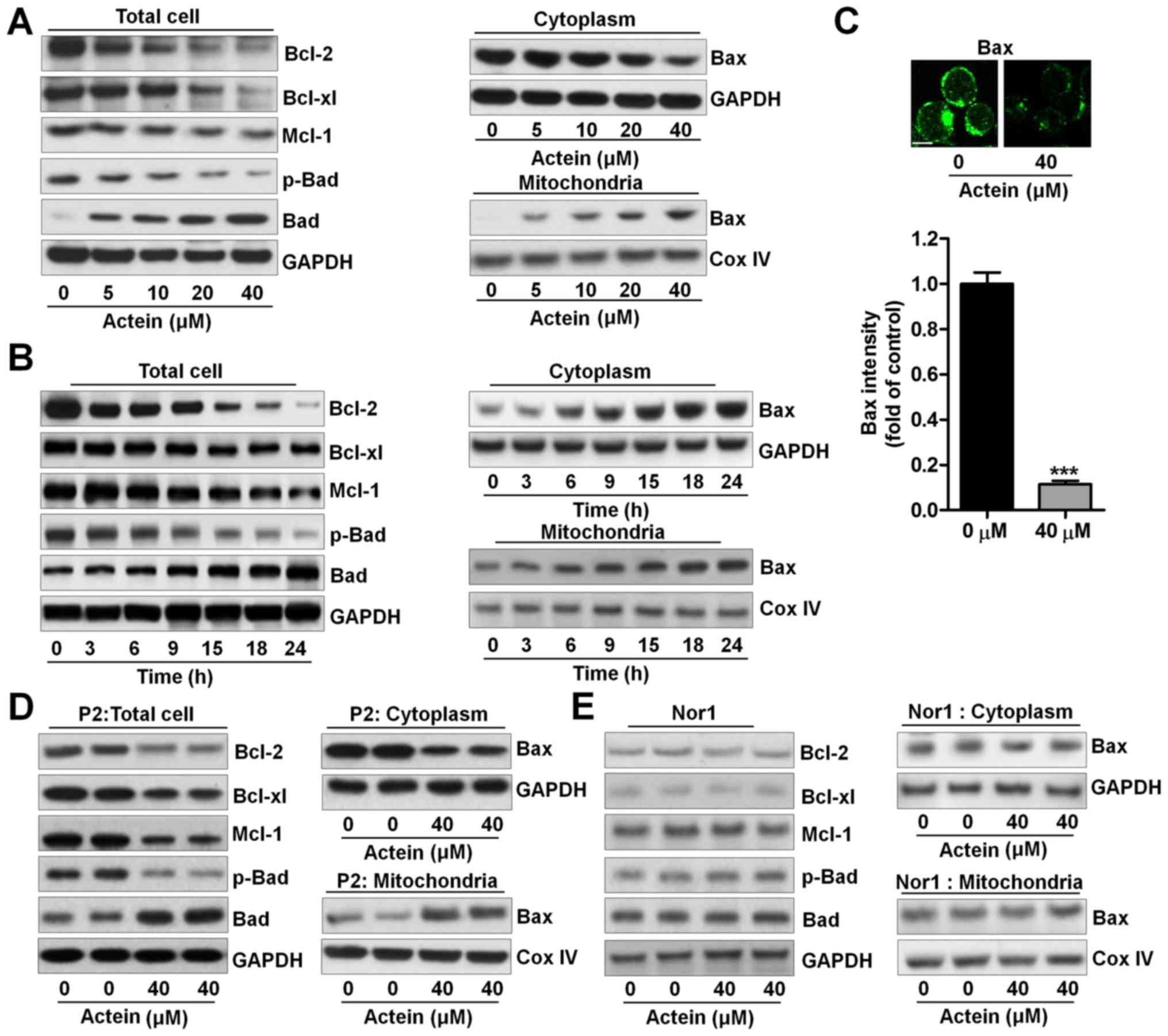 | Figure 4Actein causes apoptosis through
regulating pro- and anti-apoptotic signals. (A) U937 cells were
treated with actein (0, 5, 10, 20 and 40 μM) for 24 h,
followed by western blot analysis of Bcl-2, Bcl-xl, Mcl-1, p-Bad
and Bad in the total cells. Also, protein extracted from cytoplasm
and mitochondria was used for Bax using western blot assays. (B)
U937 cells were treated with 40 μM actein for the indicated
time. Next, western blot analysis was used to assess the whole
cellular Bcl-2, Bcl-xl, Mcl-1, p-Bad and Bad. Protein extracted
from cytoplasm and mitochondria was used for Bax abundance from
protein levels through western blot assays. (C) Actein (40
μM) was exposed to U937 cells for 24 h. After treatments,
all cells were performed for immunofluorescent analysis. (D) Acute
myeloid leukemia blasts and (E) peripheral blood mononuclear cells
from normal people were exposed to 40 μM actein for 24 h,
followed by western blot analysis of the whole cellular Bcl-2,
Bcl-xl, Mcl-1, p-Bad and Bad, as well as Bax in cytoplasm and
mitochondria. Data are analyzed as mean ± SEM, n=8.
***P<0.001 versus the Con group in the absence of any
treatment. |
Actein treatment suppresses AKT and
RhoA/ROCK1 signaling pathway
Referring to previous studies, PTEN and PI3K/AKT
signaling pathway is involved in regulating apoptosis (24). PTEN is reported to negatively
modulate the activation of PI3K/AKT (25,26).
In this regard, we found that phosphorylated PTEN was highly
induced by actein treatment in a dose- and time-dependent manner,
subsequently reducing AKT phosphorylated levels (Fig. 5A and B). Similar findings were
observed in primary human leukemia cells but not found in
peripheral blood mononuclear cells from normal human (Fig. 5C and D). Thus, we supposed that
PTEN phosphorylation and AKT dephosphorylation were related to
actein-induced apoptosis in leukemia cells. Following this,
RhoA/ROCK1 signaling pathway was investigated in our study. ROCK1
is suggested to be an important target for RhoA, which modulates
PTEN activation and regulates apoptotic response. As shown in
Fig. 5A and B, actein induced RhoA
and ROCK1 downregulation dose- and time-dependently. In agreement
with the results above, in primary human leukemia cells, marked
reduction of RhoA and ROCK1 were discovered in actein-treated
groups, while no significant difference was observed in peripheral
blood mononuclear cells extracted from normal people (Fig. 5C and D). In conclusion, the results
above indicated that RhoA/ROCK1 was also involved in
actein-modulated apoptosis.
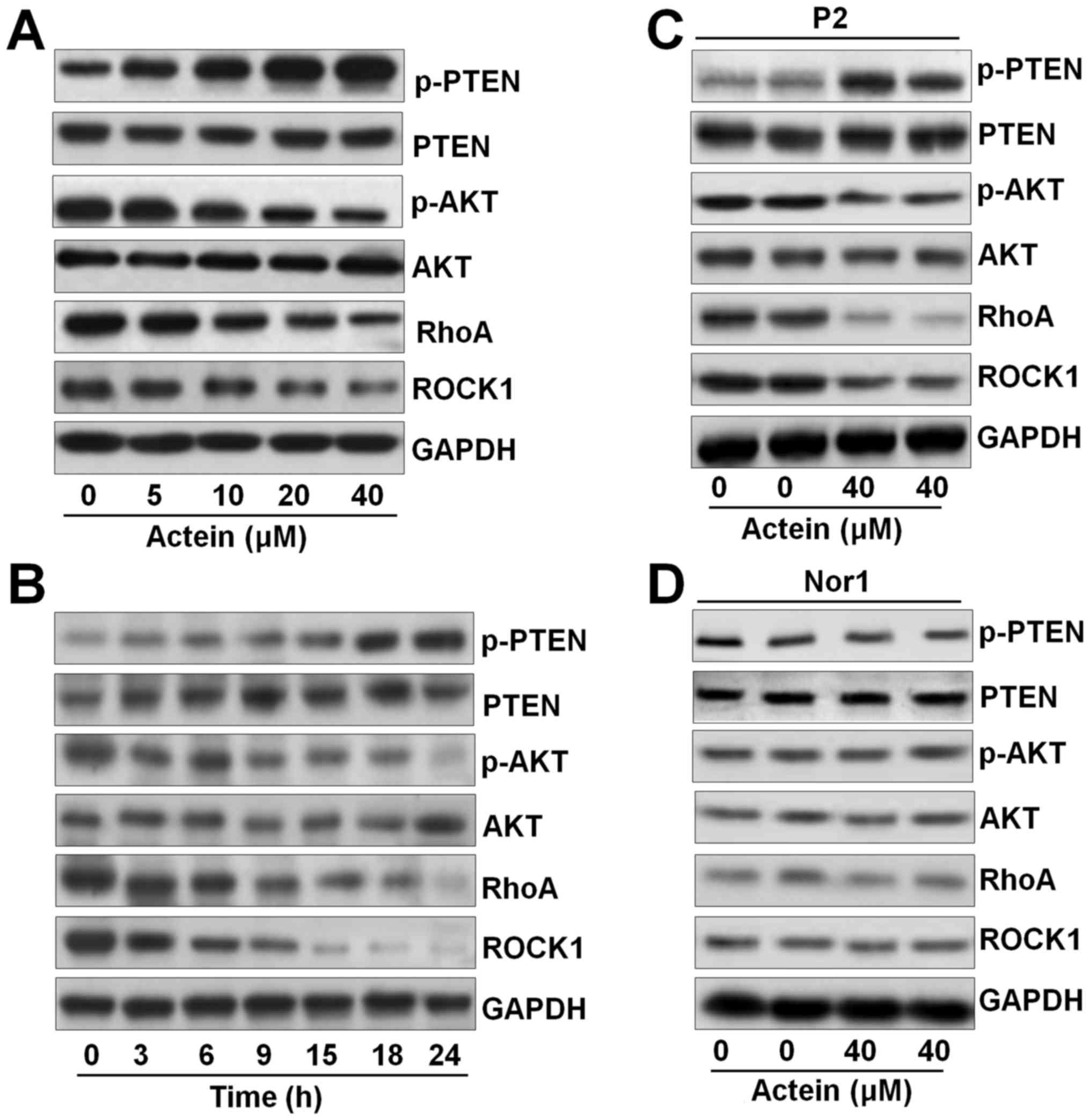 | Figure 5Actein treatment suppresses AKT and
RhoA/ROCK1 signaling pathway. (A) U937 cells were exposed to actein
at the described concentrations (0–40 μM) for 24 h or (B)
treated for the indicated time, ranging from 0 to 24 h, with 40
μM actein. Then, the cells were harvested for western blot
analysis using primary antibodies of p-PTEN, PTEN, p-AKT, AKT, RhoA
and ROCK1. (C) Acute myeloid leukemia blasts and (D) peripheral
blood mononuclear cells from normal people were treated with 40
μM actein for 24 h. Next, western blot analysis was carried
out to evaluate the protein levels of p-PTEN, PTEN, p-AKT, AKT,
RhoA and ROCK1. Data are analyzed as mean ± SEM, n=8. |
Actein-induced apoptosis has a close
relationship with RhoA/ROCK1 signaling pathway
Caspase-3 is a key during apoptosis induction,
having a close relationship with ROCK1 expression (27). Thus, in this regard, we attempted
to explore the role of caspase-3 in actein-induced apoptosis, and
its association with ROCK1. Fig. 6A
and C indicated that U937 cells treated with caspase-3
inhibitor, ZVAD, dramatically reduced apoptosis and diminished
caspase-3 activation. Also, caspase-3 knockdown with specific siRNA
significantly eliminated apoptosis and the cleavage of caspase-3
(Fig. 6B and D). Of note, we found
that both suppression of caspase-3 and knockdown of caspase-3
showed no effects on ROCK1 expression via western blot analysis
(Fig. 6C and D). Together, the
data above indicated that caspase-3 was not in our study included
in the regulation of ROCK1 expression during the actein-triggered
apoptosis. Thus, the expression of ROCK1 regulated by RhoA possibly
was explored using RhoA inhibitor. U937 cells with RhoA inhibitor
treatment enhanced apoptosis and cleaved caspase-3, caspase-9 and
PARP expression, especially in actein-cotreatment group. In
contrast, ROCK1 was markedly reduced, indicating that ROCK1
suppressed by actein was dependent on RhoA expression (Fig. 7A). Then, ROCK1 expression was
suppressed using its specific inhibitor and siRNA with the specific
sequence. As shown in Fig. 7B and
C, we found that suppression of ROCK1 apparently augmented
apoptotic response, as well as accelerated caspase-9, caspase-3 and
PARP cleavage, which was further upregulated by co-culture with
actein. AKT phosphorylated levels could be diminished by using
actein. Therefore, its inhibitor was further used here to calculate
its effects in apoptosis induced by actein. Fig. 7D suggested that AKT suppression
using its inhibitor significantly elevated apoptosis in U937 cells,
and its combination with actein further improved apoptotic
response, accompanied with upregulated caspase-9, caspase-3 and
PARP cleavage. The results above suggested that ROCK1 expression
was dependent on RhoA to modulate apoptosis induced by actein in
human leukemia cells.
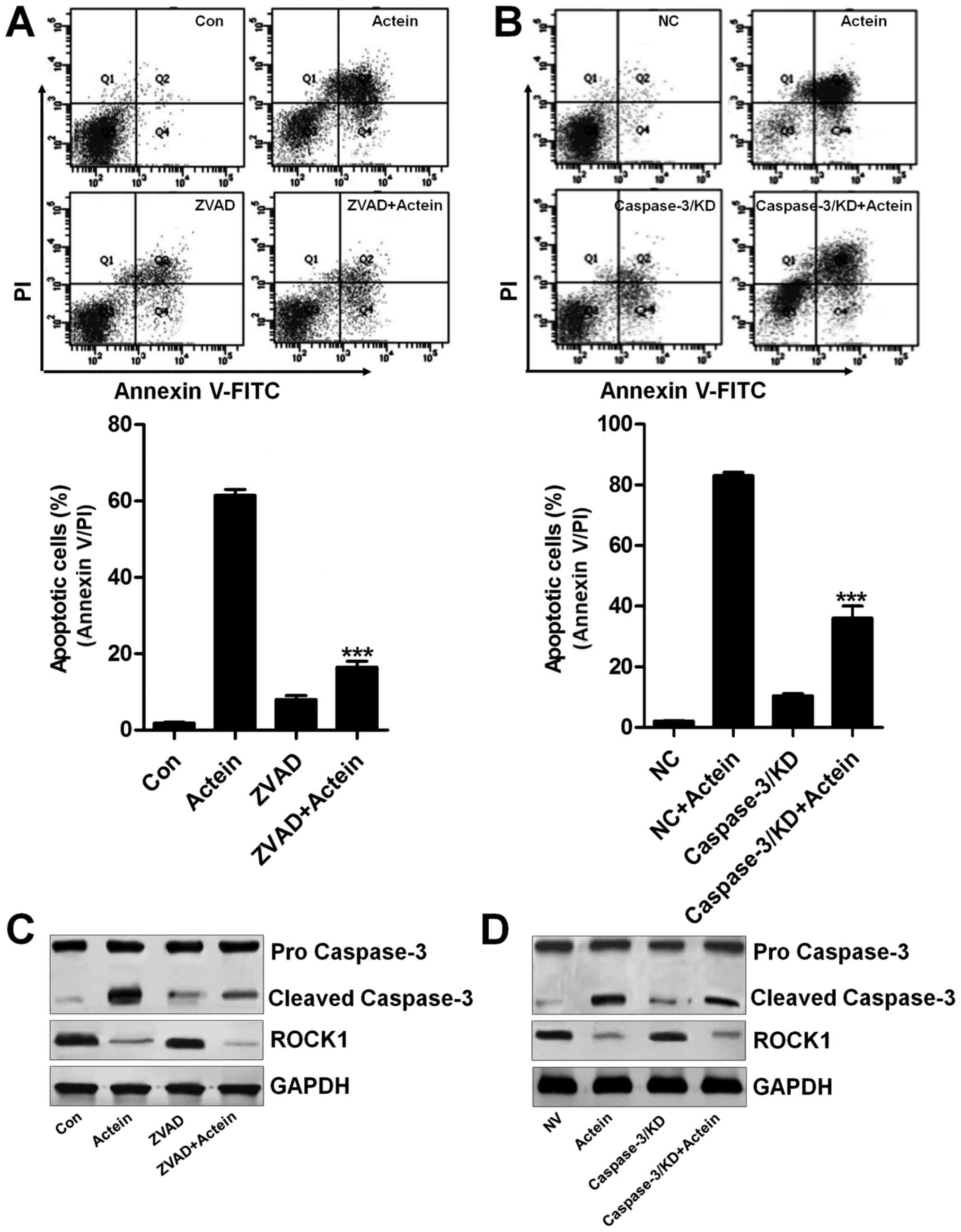 | Figure 6Caspase-3-independent ROCK1
activation is involved in actein-induced apoptosis. (A) U937 cells
were pre-treated with 20 μM caspase-3 inhibitor, ZVAD, for 2
h and then exposed to 40 μM actein for 24 h, followed by
flow cytometric analysis. (B) U937 cells were transfected with
caspase-3 siRNA for 24 h to knockdown caspase-3 expression,
followed by exposure to 40 μM actein for 24 h, and then flow
cytometric analysis was used to calculate apoptotic cells. (C) U937
cells were treated with caspase-3 inhibitor for 2 h to suppress
caspase-3 expression, followed by exposure to 40 μM actein
for another 24 h, and then western blot analysis was performed to
calculate cleaved caspase-3 and ROCK1 expression. (D) Caspase-3 was
knocked down using specific siRNA sequence for 24 h, followed by
exposure to 40 μM actein for another 24 h. Then, all cells
were harvested for western blot analysis. Data are analyzed as mean
± SEM, n=8. ***P<0.001 versus the actein group. |
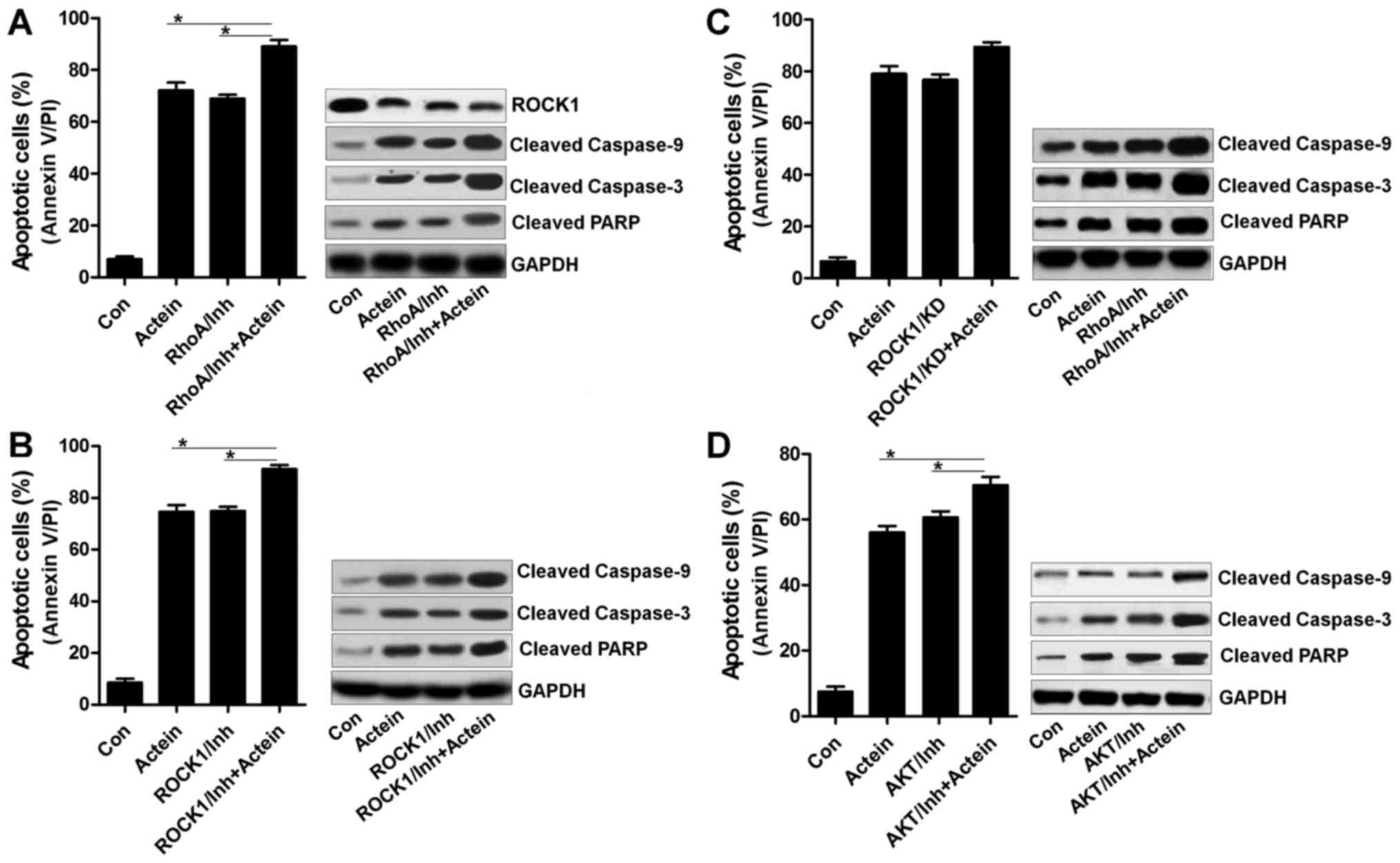 | Figure 7Actein-induced apoptosis has a close
relationship with RhoA/ROCK1 signaling pathway. (A) U937 cells were
pre-treated with RhoA inhibitor, C3 exoenzyme (10 μM), for 2
h, followed by 24-h treatment of actein (40 μM). Then, flow
cytometry and western blot assays were used to evaluate the
apoptosis levels. (B) The inhibitor of ROCK1, Y-27632
dihydrochloride (10 μM), was used in U937 cells for
suppressing ROCK1 expression, and actein was used to treat cells
for another 24 h. Apoptosis was calculated using flow cytometry and
western blot analysis. (C) ROCK1 was knocked down using specific
siRNA for 24 h, which was then exposed to actein for another 24 h.
After various treatments, all cells were harvested for apoptosis
analysis using flow cytometry and western blot assays. (D) The
inhibitor of AKT, LY294002 (5 μM), was used to treat U937
cells for 2 h, and then cells were exposed to actein for another 24
h, followed by flow cytometry and western blot analysis. Data are
analyzed as mean ± SEM, n=8. ***P<0.001 versus the
actein group. |
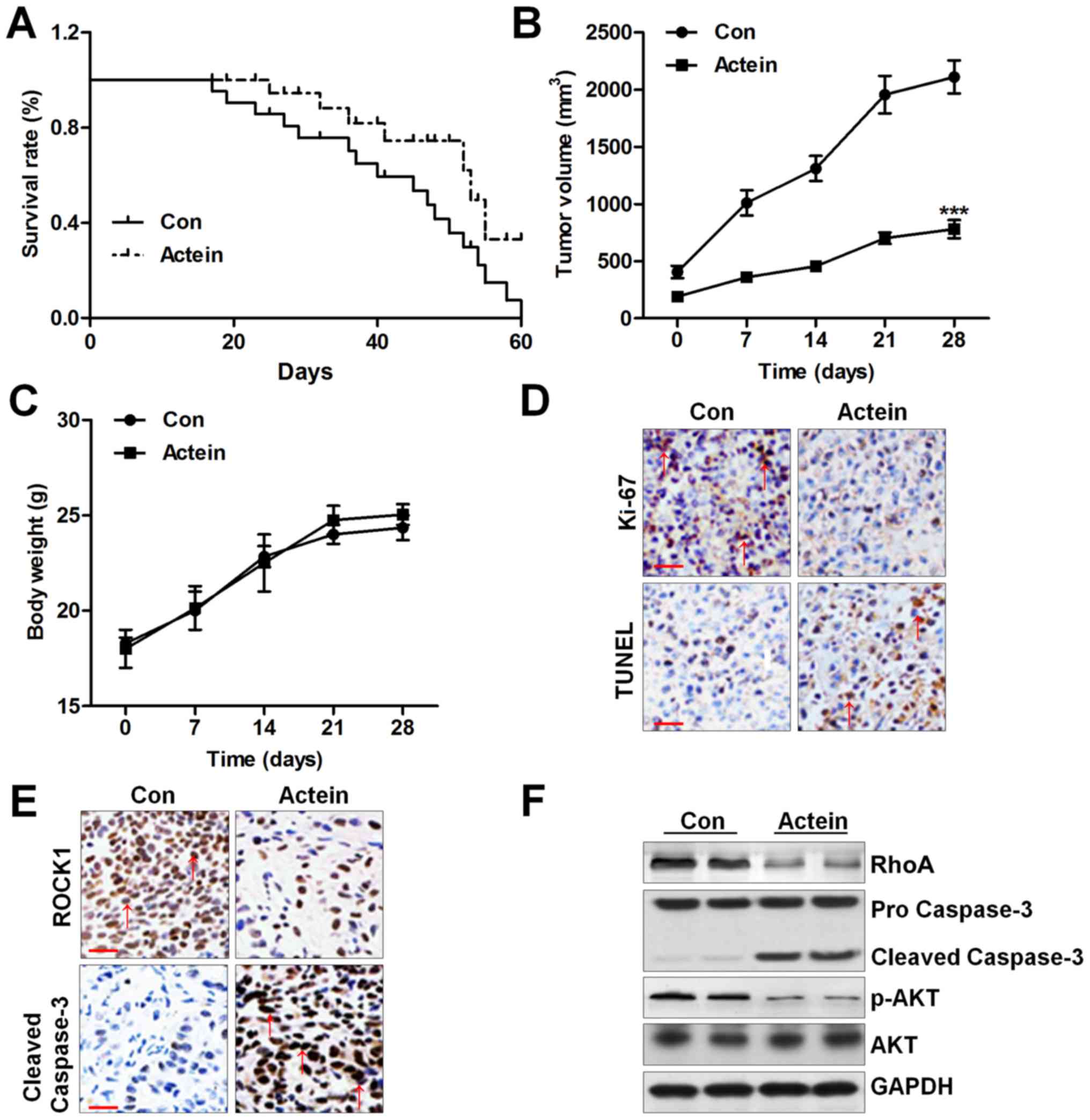
Actein suppresses U937-tumor growth and
triggers apoptosis in xenograft mouse models
In vitro, actein was evidenced to be
important in suppression of human leukemia cells. In order to
further confirm our data above, in vivo experiments were
conducted. The male nu/nu mice were subcutaneously injected with
U937 cells. After inoculation, half of the total number of mice was
administered with actein intraperitoneally for two months. As shown
in Fig. 8A, we found that actein
administration apparently prolonged the survival rate of mice
bearing U937 tumors, which was comparable to the control group. The
tumor volume of mice was also dramatically reduced by actein
treatment (Fig. 8B). However, no
significant difference of body weight was observed between the two
groups (Fig. 8C). Next, TUNEL
assay and Ki-67 levels were performed to further reveal the role of
actein in vivo. Ki-67-positive areas were reduced by actein
treatment. In contrast, the number of TUNEL-positive cells was
upregulated by using actein, indicating apoptosis induced by actein
in vivo (Fig. 8D).
Furthermore, actein reduced ROCK1 expression in the tumor sections,
while caspase-3 cleavage was found to be enhanced (Fig. 8E). Finally, western blot analysis
indicated that RhoA and phosphorylated AKT had low expression in
actein-treated group, and caspase-3 cleavage was elevated after
actein administration, which were indicative of apoptotic response
(Fig. 8F). Thus, the results
indicated that actein apparently suppressed U937 tumor growth
through triggering apoptosis in vivo.
Discussion
According to previous studies, RhoA/ROCK1 signaling
pathway plays an essential role in cell survival, which has been
investigated in various functional states (18,28,29).
For instance, several reports indicate evidence that inhibition of
the Rho/ROCK signaling pathway induces hepatocellular cancer (HCC)
cell apoptosis and endothelial cells via a mitochondrial apoptosis
pathway (30,31). Thus, suppression of RhoA/ROCK1
signaling pathway affects the transformation and proliferation of
progenitor cells (32). In
addition, as previously reported, RhoA/ROCK1 signaling pathway
suppression in human primary leukemia cells enhances cell death,
revealing that targeting RhoA/ROCK1 might be of potential value as
therapy to prevent human leukemia. In our study, we found that, in
line with previous studies, RhoA/ROCK1 pathway was involved in
apoptosis of acute promyelocytic leukemia. Apoptosis is an
evolutionarily conserved process, which irreversibly eliminates
injured or potentially harmful cells in order to protect the
organism. The caspases could be structurally divided according to
the absence or presence of an N-terminal pro-domain (33). The caspases containing long
pro-domains are the first to be activated responding to various
apoptotic stimuli (34). The
activated caspases damage the cellular architecture and eventually
lead to cell death. Caspase-3 is reported as the most essential
member of caspase family (35,36).
According to studies before, caspase-3 activation has an
association with ROCK1 expression (37). Caspase-3 suppression reduced ROCK1
expression (38). However, there
was also a study, which indicated that caspase-3-independent
regulation of ROCK1 was observed during apoptosis induction
(39). In the present study, U937
cells were exposed to caspase-3 inhibitor of ZVAD and actein
diminished caspase-3 cleavage, and apoptosis, while ROCK1 showed no
difference, which illustrated that ROCK1 expression was
caspase-3-independent, at least in our study, and that other
factors might be involved in regulating ROCK1 expression.
Attributing to the role of RhoA in modulating ROCK1, it was
investigated in our study. Actein exposure induced RhoA
downregulation. Also, pre-treatment with RhoA inhibitor in U937
cells, obviously eliminated apoptosis and caspases cleavage
triggered by actein. Thus, from the data of our study, RhoA, but
not caspase-3, stimulated the expression of ROCK1 in
actein-triggered apptosis.
In this study, we found that actein-induced
apoptosis in human leukemia cells was observed, which was
associated with PTEN and AKT activation. PTEN and AKT
phosphorylation play an important role in modulating apoptosis
responding to actein treatment. Accumulating evidence elucidated
that PTEN, a ROCK1 substrate, is suggested to be a negative
modulator of PI3K/AKT signaling pathway (40). Consistent with previous studies,
PTEN negatively regulated AKT activation during actein-induced
apoptosis in our study. AKT phosphorylation was decreased by actein
treatment, and its suppression using specific inhibitor abrogated
apoptosis and caspase activation.
Bcl-2 family proteins, including anti-apoptotic
members (Bcl-2, Mcl-1, and Bcl-xl) and pro-apoptotic members (Bax),
play a crucial role in apoptosis (41,42).
Bcl-2 has been known to form a heterodimeric complex with the
pro-apoptotic member Bax, neutralizing its pro-apoptotic effects.
Thus, the Bcl-2/Bax ratio is a decisive factor and plays a
significant role in determining if cells are likely to undergo
survival or death (43).
Similarly, in our study, Bcl-2, Bcl-xl and Mcl-1 were highly
reduced by using actein, indicating apoptosis induction and
contributing to cell death. Also, the high expression of Bax in
mitochondria contributes to apoptosis formation through releasing
Cyto-c into cytoplasm (44). Also, we found that Bax was
expressed highly in actein-treated U937 cells. In addition,
accordingly, the release of Cyto-c into the cytoplasm was
observed, which was in line with a previous study (45).
In conclusion, this study indicated that actein
selectively triggered apoptotic response and mitochondrial damage
in human leukemia cell lines, as well as in human primary leukemia
cells, and showed no effects on peripheral blood mononuclear cells
isolated from normal healthy humans. Furthermore in vivo,
actein suppressed the growth of U937 tumor through activating
PTEN/caspases and inactivating RhoA/ROCK1/AKT signaling pathways.
Thus, we supposed that actein could be applied as a safe and
effective candidate to treat human leukemia through inducing
apoptosis.
Glossary
Abbreviations
Abbreviations:
|
PARP
|
poly(ADP-ribose) polymerase
|
|
Cyto-c
|
cytochrome c
|
|
Mcl-1
|
myeloid cell leukemia-1
|
|
Bcl-2
|
B cell CLL/lymphoma 2
|
|
Bad
|
Bcl-2-associated death promoter
|
|
AKT
|
protein kinase B
|
|
ROCK1
|
Rho kinase 1
|
|
PTEN
|
phosphatase and tensin homolog
|
|
FBS
|
fetal bovine serum
|
|
TUNEL
|
terminal deoxynucleotidyl transferase
(TdT) dUTP nick-end labeling
|
|
MTT
|
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium
bromide
|
|
Bax
|
Bcl-2-associated X protein
|
|
BCA
|
bicinchoninic acid
|
References
|
1
|
Shivarov V and Bullinger L: Expression
profiling of leukemia patients: Key lessons and future directions.
Exp Hematol. 42:651–660. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ross K, Gillespie-Twardy AL, Agha M,
Raptis A, Hou JZ, Farah R, Redner RL, Im A, Duggal S, Ding F, et
al: Intensive chemotherapy in patients aged 70 years or older newly
diagnosed with acute myeloid leukemia. Oncol Res. 22:85–92. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang ES: Treating acute myeloid leukemia
in older adults. Hematology (Am Soc Hematol Educ Program).
2014:14–20. 2014.
|
|
4
|
Mayer J, Arthur C, Delaunay J, Mazur G,
Thomas XG, Wierzbowska A, Ravandi F, Berrak E, Jones M, Li Y, et
al: Multivariate and subgroup analyses of a randomized,
multinational, phase 3 trial of decitabine vs treatment choice of
supportive care or cytarabine in older patients with newly
diagnosed acute myeloid leukemia and poor- or intermediate-risk
cytogenetics. BMC Cancer. 14:692014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kantarjian HM, Thomas XG, Dmoszynska A,
Wierzbowska A, Mazur G, Mayer J, Gau JP, Chou WC, Buckstein R,
Cermak J, et al: Multicenter, randomized, open-label, phase III
trial of decitabine versus patient choice, with physician advice,
of either supportive care or low-dose cytarabine for the treatment
of older patients with newly diagnosed acute myeloid leukemia. J
Clin Oncol. 30:2670–2677. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dombret H, Seymour JF, Butrym A,
Wierzbowska A, Selleslag D, Jang JH, Kumar R, Cavenagh J, Schuh AC,
Candoni A, et al: International phase 3 study of azacitidine vs
conventional care regimens in older patients with newly diagnosed
AML with >30% blasts. Blood. 126:291–299. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhou T, Hasty P, Walter CA, Bishop AJ,
Scott LM and Rebel VI: Myelodysplastic syndrome: An inability to
appropriately respond to damaged DNA? Exp Hematol. 41:665–674.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Einbond LS, Shimizu M, Nuntanakorn P,
Seter C, Cheng R, Jiang B, Kronenberg F, Kennelly EJ and Weinstein
IB: Actein and a fraction of black cohosh potentiate
antiproliferative effects of chemotherapy agents on human breast
cancer cells. Planta Med. 72:1200–1206. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Einbond LS, Mighty J, Redenti S and Wu HA:
Actein induces calcium release in human breast cancer cells.
Fitoterapia. 91:28–38. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yue GGL, Xie S, Lee JKM, Kwok HF, Gao S,
Nian Y, Wu XX, Wong CK, Qiu MH and Lau CB: New potential beneficial
effects of actein, a triterpene glycoside isolated from Cimicifuga
species, in breast cancer treatment. Sci Rep. 6:352632016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang ZC and Ma J: Actein enhances TRAIL
effects on suppressing gastric cancer progression by activating
p53/caspase-3 signaling. Biochem Biophys Res Commun. Nov
30–2016.Epub ahead of print. View Article : Google Scholar
|
|
12
|
Einbond LS, Shimizu M, Ma H, Wu HA,
Goldsberry S, Sicular S, Panjikaran M, Genovese G and Cruz E:
Actein inhibits the Na+-K+-ATPase and
enhances the growth inhibitory effect of digitoxin on human breast
cancer cells. Biochem Biophys Res Commun. 375:608–613. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Harris SL and Levine AJ: The p53 pathway:
Positive and negative feedback loops. Oncogene. 24:2899–2908. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin MW, Wu CT, Shih JY, Chang YL and Yang
PC: Clinicopathologic characteristics and prognostic significance
of EGFR and p53 mutations in surgically resected lung
adenocarcinomas ≤2 cm in maximal dimension. J Surg Oncol.
110:99–106. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu Y, Wang L, Zheng X, Liu G, Wang Y, Lai
X and Li J: Positive expression of p53, c-erbB2 and MRP proteins is
correlated with survival rates of NSCLC patients. Mol Clin Oncol.
1:487–492. 2013. View Article : Google Scholar
|
|
16
|
Lee JH, Katakai T, Hara T, Gonda H, Sugai
M and Shimizu A: Roles of p-ERM and Rho-ROCK signaling in
lymphocyte polarity and uropod formation. J Cell Biol. 167:327–337.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Osiak AE, Zenner G and Linder S:
Subconfluent endothelial cells form podosomes downstream of
cytokine and RhoGTPase signaling. Exp Cell Res. 307:342–353. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Benitah SA, Valerón PF and Lacal JC: ROCK
and nuclear factor-kappaB-dependent activation of cyclooxygenase-2
by Rho GTPases: Effects on tumor growth and therapeutic
consequences. Mol Biol Cell. 14:3041–3054. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cai L, Threadgill MD, Wang Y and Li M:
Effect of poly (ADP-ribose) polymerase-1 inhibition on the
proliferation of murine colon carcinoma CT26 cells. Pathol Oncol
Res. 15:323–328. 2009. View Article : Google Scholar
|
|
20
|
Ratnam K and Low JA: Current development
of clinical inhibitors of poly(ADP-ribose) polymerase in oncology.
Clin Cancer Res. 13:1383–1388. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Takeba Y, Matsumoto N, Watanabe M,
Takenoshita-Nakaya S, Ohta Y, Kumai T, Takagi M, Koizumi S, Asakura
T and Otsubo T: The Rho kinase inhibitor fasudil is involved in
p53-mediated apoptosis in human hepatocellular carcinoma cells.
Cancer Chemother Pharmacol. 69:1545–1555. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tortora G, Caputo R, Damiano V, Caputo R,
Troiani T, Veneziani BM, De Placido S, Bianco AR,
Zangemeister-Wittke U and Ciardiello F: Combined targeted
inhibition of bcl-2, bcl-XL, epidermal growth factor receptor, and
protein kinase A type I causes potent antitumor, apoptotic, and
antiangiogenic activity. Clin Cancer Res. 9:866–871.
2003.PubMed/NCBI
|
|
23
|
Riedl SJ and Shi Y: Molecular mechanisms
of caspase regulation during apoptosis. Nat Rev Mol Cell Biol.
5:897–907. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang JY, Della-Fera MA, Rayalam S and
Baile CA: Enhanced effects of xanthohumol plus honokiol on
apoptosis in 3T3-L1 adipocytes. Obesity (Silver Spring).
16:1232–1238. 2008. View Article : Google Scholar
|
|
25
|
Roy S, Yu Y, Padhye SB, Sarkar FH and
Majumdar AP: Difluorinated-curcumin (CDF) restores PTEN expression
in colon cancer cells by down-regulating miR-21. PLoS One.
8:e685432013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mueller S, Phillips J, Onar-Thomas A,
Romero E, Zheng S, Wiencke JK, McBride SM, Cowdrey C, Prados MD,
Weiss WA, et al: PTEN promoter methylation and activation of the
PI3K/Akt/mTOR pathway in pediatric gliomas and influence on
clinical outcome. Neurooncol. 14:1146–1152. 2012.
|
|
27
|
Liu Y, Minze LJ, Mumma L, Li XC, Ghobrial
RM and Kloc M: Mouse macrophage polarity and ROCK1 activity depend
on RhoA and non-apoptotic caspase 3. Exp Cell Res. 341:225–236.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gilkes DM, Xiang L, Lee SJ, Chaturvedi P,
Hubbi ME, Wirtz D and Semenza GL: Hypoxia-inducible factors mediate
coordinated RhoA-ROCK1 expression and signaling in breast cancer
cells. Proc Natl Acad Sci USA. 111:E384–E393. 2014. View Article : Google Scholar :
|
|
29
|
Wang Y, Wang D and Guo D: miR-124 promote
neurogenic transdifferentiation of adipose derived mesenchymal
stromal cells partly through RhoA/ROCK1, but not ROCK2 signaling
pathway. PLoS One. 11:e01466462016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang JG, Li XY, Wang YZ, Zhang QD, Gu SY,
Wu X, Zhu GH, Li Q and Liu GL: ROCK is involved in vasculogenic
mimicry formation in hepatocellular carcinoma cell line. PLoS One.
9:e1076612014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Peng H, Luo P, Li Y, Wang C, Liu X, Ye Z,
Li C and Lou T: Simvastatin alleviates hyperpermeability of
glomerular endothelial cells in early-stage diabetic nephropathy by
inhibition of RhoA/ROCK1. PLoS One. 8:e800092013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lin SC, Gou GH, Hsia CW, Ho CW, Huang KL,
Wu YF, Lee SY and Chen YH: Simulated microgravity disrupts
cytoskeleton organization and increases apoptosis of rat neural
crest stem cells via upregulating CXCR4 expression and
RhoA-ROCK1-p38 MAPK-p53 signaling. Stem Cells Dev. 25:1172–1193.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Altieri DC: Survivin and IAP proteins in
cell-death mechanisms. Biochem J. 430:199–205. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mohan S, Abdul AB, Abdelwahab SI,
Al-Zubairi AS, Sukari MA, Abdullah R, Elhassan Taha MM, Ibrahim MY
and Syam S: Typhonium flagelliforme induces apoptosis in CEMss
cells via activation of caspase-9, PARP cleavage and cytochrome c
release: Its activation coupled with G0/G1 phase cell cycle arrest.
J Ethnopharmacol. 131:592–600. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu X, Zou H, Slaughter C and Wang X: DFF,
a heterodimeric protein that functions downstream of caspase-3 to
trigger DNA fragmentation during apoptosis. Cell. 89:175–184. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Grütter MG: caspases: Key players in
programmed cell death. Curr Opin Struct Biol. 10:649–655. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang Y, Xu J and Gu Y: 37 1, 25 (OH) 37
1,25(OH)2D3 suppresses oxidative stress-induced microparticle
release by placental trophoblasts: Placenta and decidua. Pregnancy
Hyperens. 6:1542016. View Article : Google Scholar
|
|
38
|
Shen K, Wang Y, Zhang Y, Zhou H, Song Y,
Cao Z, Kou J and Yu B: Cocktail of four active components derived
from Sheng Mai San inhibits hydrogen peroxide-induced PC12 cell
apoptosis linked with the caspase-3/ROCK1/MLC pathway. Rejuvenation
Res. 18:517–527. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li G, Zhou T, Liu L, Chen J, Zhao Z, Peng
Y, Li P and Gao N: Ezrin dephosphorylation/downregulation
contributes to ursolic acid-mediated cell death in human leukemia
cells. Blood Cancer J. 3:e1082013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Peng H, Cao J, Yu R, Danesh F, Wang Y,
Mitch WE, Xu J and Hu Z: CKD stimulates muscle protein loss via
rho-associated protein kinase 1 activation. J Am Soc Nephrol.
27:509–519. 2016. View Article : Google Scholar :
|
|
41
|
Heath-Engel HM, Chang NC and Shore GC: The
endoplasmic reticulum in apoptosis and autophagy: Role of the BCL-2
protein family. Oncogene. 27:6419–6433. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Martinou JC and Youle RJ: Mitochondria in
apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev
Cell. 21:92–101. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Levine B, Sinha S and Kroemer G: Bcl-2
family members: Dual regulators of apoptosis and autophagy.
Autophagy. 4:600–606. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Arnoult D, Parone P, Martinou JC,
Antonsson B, Estaquier J and Ameisen JC: Mitochondrial release of
apoptosis-inducing factor occurs downstream of cytochrome c release
in response to several proapoptotic stimuli. J Cell Biol.
159:923–929. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sheridan C, Delivani P, Cullen SP and
Martin SJ: Bax- or Bak-induced mitochondrial fission can be
uncoupled from cytochrome c release. Mol Cell. 31:570–585. 2008.
View Article : Google Scholar : PubMed/NCBI
|