Introduction
Statins, also known as 3-hydroxy-3-methylglutaryl
coenzyme A (HMG-CoA) reductase inhibitors, are a class of drugs
that inhibit the rate-limiting steps in the cholesterol
biosynthesis pathway (1,2). Any compound leading to the depletion
of cholesterol affects various cellular events, and also impairs
homeostasis and cholesterol-independent pleiotropic mechanisms,
which are likely a consequence of blocking intracellular signaling
(3).
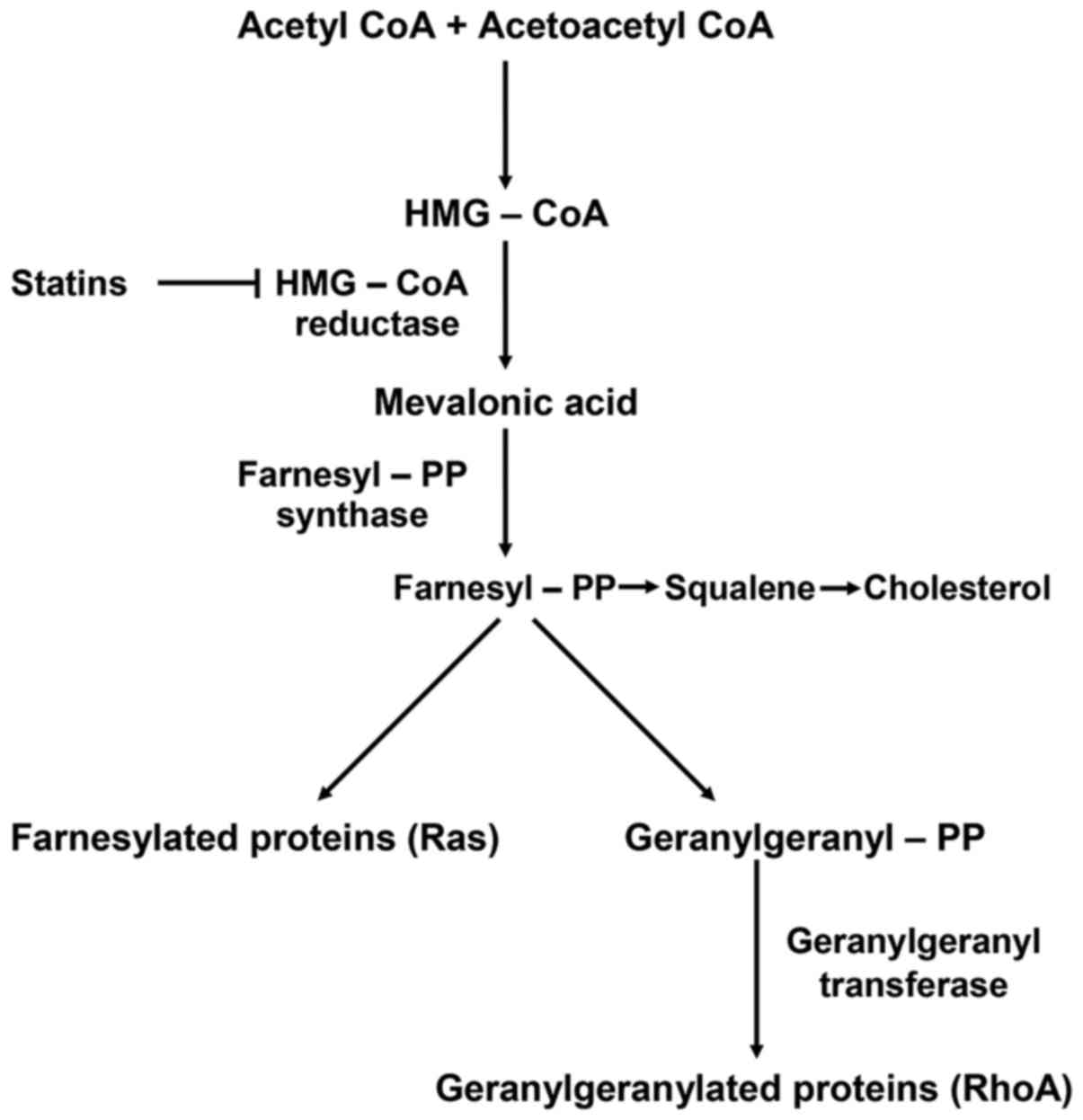
Statins, as described in the schematic diagram of
the biochemical pathway in Fig. 1,
act by inhibiting HMG-CoA reductase and therefore the conversion of
HMG-CoA to mevalonate (4).
Blocking mevalonate synthesis leads to the inhibition of farnesyl
and geranylgeranyl modifications of several functional proteins,
such as RhoA, a small guanosine triphosphate (GTP)-binding protein.
In particular, simvastatin selectively blocks RhoA
geranylgeranylation and its translocation to the cell membrane,
where it interacts with downstream effectors to control cell cycle
progression (5). Rho activation by
geranylgeranylation leads to a reduction in the inhibitory
phosphorylation of YAP/TAZ, and thus sustains YAP/TAZ nuclear
accumulation (6), thereby serving
a fundamental role in the control of tissue proliferation and organ
growth, as well as tumor onset and progression.
Considering the role of Rho, any inhibition of its
synthesis or modification may induce a reduction in proliferation
and thereby suppress tumor onset. Furthermore, tumors contain a
subset of cancer stem cells that drive metastatic spread and Rho
function may be associated with cancer stem cells. In these cells,
genes that confer the characteristics of multipotency to the cells,
associated with hyperproliferation and blocking of differentiation,
are overexpressed or re-expressed. Although several genes are
considered to contribute to maintaining hyperproliferative status,
we have focused on three representative stemness-related genes,
Oct4, Sox2 and Nanog (7–9), with the aim of investigating whether
simvastatin is able to modify, directly or indirectly, the
expression equilibrium of these genes or influence the translation
process of the respective proteins.
Oct4, Sox2 and Nanog are transcription factors that
regulate the pluripotency of human pluripotent stem cells (hPSCs)
and regulate mesendoderm and ectoderm differentiation (10–15).
It has been clearly demonstrated that, in embryonic stem cells
(ESCs), Sox2 interacts with Oct4 to form a regulatory dimer complex
(16–21), indicating that Sox2 is positively
auto-regulated by the Sox2-Oct4 complex. In human and mouse ESCs,
Oct4, Sox2 and Nanog form transcriptional regulatory circuitry to
activate the expression of pluripotency-related genes and repress
the expression of differentiation-related genes, and are implicated
in tumorigenesis in various organs (22–39).
Recently, it has been demonstrated that the inhibition of Sox2 can
induce the repression of YAP, TAZ and TEAD functions, exerting an
oncosuppressive effect (40–45).
In tumorigenesis, the epithelial-to-mesenchymal
transition (EMT) is an important developmental process in which
immotile epithelial cells acquire mesenchymal features (46–49),
along with the ability to spread through the extracellular matrix
and surrounding tissues. EMT can be induced by several signal
pathways, and principally by Wnt/β-catenin (50–53).
Sox2 and β-catenin have direct functions in tumor metastasis, and
the overexpression of Sox2 and β-catenin can stimulate EMT.
Therefore, inhibition of β-catenin expression may have a role in
inhibiting the EMT process and consequently repressing metastatic
tumor spread.
Migratory cancer cells remodel their cell-cell and
cell-matrix adhesions, which requires remodeling of the actin
cytoskeleton (54,55). Reorganization of the actin
cytoskeleton is one of the essential mechanisms involved in the
regulation of cell migration. Many factors promote actin
reorganization and cell motility, including small GTPases, such as
RhoA, Rho-associated protein kinase (ROCK) and Cdc42. These
proteins cycle between active and inactive states by binding to GTP
or GDP, respectively. With regard to the possible action mechanism,
simvastatin blocks the geranylgeranylation of RhoA, thereby
blocking the translocation of RhoA from the cytoplasm to the
membrane, which is where it binds a set of effector molecules that
are important for maintaining the undifferentiated status of the
cell; thus, we can also speculate that the inhibition of RhoA
function by simvastatin may induce differentiation-related
processes. From another point of view, the inhibition of RhoA
geranylation may account for the inhibition of ROCK2 activity,
which has an important role in mediating structural changes in the
actin cytoskeleton.
Another interesting aspect is the strain transfer
from the cytoskeleton to the nucleus. Evidence indicates that force
can be transmitted from the cytoskeleton to the nucleus, and that
these mechanical stimuli, which act on specific mechanosensory
complexes, are able to influence certain gene expression pathways.
It has been shown that the YAP/TAZ and ERK1/2 pathways can be
influenced by dynamic tensile loading, with consequent
transcriptional regulation of specific genes involved in the
proliferation/differentiation equilibrium. The inhibition of ROCK,
which directly regulates actin polymerization and
de-polymerization, decreases the nuclear strain transfer and leads
to loss of the cytoskeletal-to-nucleoskeletal connectivity required
for the transmission of regulatory signals. On the basis of these
studies, inhibition of ROCK may induce the downregulation of
stemness-related genes, which may be considered cytoskeleton- and
nucleoskeleton-associated genes.
It has also been shown that RhoA controls the
formation of stress fibers, while Cdc42 and Rac proteins influence
the production of filopodia and lamellipodia (56). Various mechanisms or factors that
influence the function of Rho appear to have fundamental roles in
the regulation of cell motility and spread into the surrounding
tissues; in this manner, RhoA has a pivotal role in the modulation
of metastatic spread.
Simvastatin-mediated blocking of the prenylation and
geranylgeranylation of Rac1, Cd42 and RhoA inhibits the three
principal pathways of actin polymerization and filopodia,
lamellipodia and invadopodia formation; in this manner, simvastatin
may decrease the capability of cancer cells to invade the
extracellular matrix, and thus may have a potential role in the
control of tumor progression.
In the present study, we demonstrated that
simvastatin can inhibit normal actin polymerization (which is
fundamental for cytoskeletal formation and maintenance of normal
cytoarchitecture), thereby inducing the loss of actin cytoskeleton
integrity and the relative inhibition of invadopodia formation,
which can decrease cell motility and spreading in the extracellular
matrix or other surrounding tissues; by these mechanisms,
simvastatin may contribute to the reduction of metastatic spread.
Furthermore, in NTERA-2 teratocarcinoma cells, HepG2 hepatoblastoma
cells and MCF7 breast cancer cells, we demonstrated that
simvastatin induces a strong down-regulation of the three principal
stemness genes, Oct4, Sox2, and Nanog, and inhibits the expression
of β-catenin, which has a pivotal role in EMT.
Collectively, these data demonstrate that
simvastatin, through inhibition of the mevalonate pathway and the
associated inhibition of the farnesylation and geranylgeranylation
processes, induces a destructuration of the cytoskeleton, and that
it is the destabilized cytoskeleton that results in the inhibition
of stemness gene expression and lamellipodia and filopodia
formation. In this manner, simvastatin can modulate the equilibrium
between proliferation, differentiation and metastatic spreading of
cancer cells. On the basis of our experimental results, we believe
that simvastatin, used at an appropriate dosage and in combination
with conventional chemotherapy, may have a potential utility in the
therapy or prevention of various cancer types.
Materials and methods
Cell culture
The NTERA-2 pluripotent human embryonic carcinoma
cells, HepG2 human hepatoblastoma cells and MCF7 human breast
cancer cells were purchased from the American Type Culture
Collection (ATCC; Manassas, VA, USA) and cultured in Dulbecco's
modified Eagle's medium (DMEM) supplemented with L-glutamine, 100
U/ml penicillin, 10 μg/ml streptomycin and 10% fetal bovine
serum (FBS), in a 5% CO2 incubator at 37°C.
Simvastatin and cytochalasin
treatment
Simvastatin (carboxylate form; Calbiochem-Merck Co.,
Darmstadt, Germany) was dissolved in dimethyl sulfoxide (DMSO) to
produce a 20-mM stock solution and stored at −20°C. In all
experiments, plated cells were treated with 20 μM
simvastatin for 24 h in normal culture conditions. Cytochalasin D
(C2618; Sigma-Aldrich, Hamburg, Germany) was prepared as 10-mM
stock solution in DMSO and used at concentration of 5 μM in
cell cultures for 6 h. The control cells were treated with DMSO
only.
RNA extraction and cDNA synthesis
Total RNA was extracted from cells untreated or
treated with simvastatin by scraping in TRIzol® reagent
(Invitrogen, Carlsbad, CA, USA). Total RNA was purified according
to the manufacturer's protocol and the purified RNA integrity was
verified by electrophoresis on 1% agarose gel in 1X TBE. cDNA
synthesis was carried out with an EasyScript™ cDNA Synthesis kit
(Applied Biological Materials Inc., Richmond, BC, Canada) according
to the manufacturer's protocol, starting from 1 μg of total
RNA. cDNA synthesis was verified by amplification of a housekeeping
gene, such as β-actin or GAPDH (primers listed below), using the
SapphireAmp® Fast PCR Master Mix, in an Applied
Biosystems GeneAmp PCR System 9700 thermocycler.
Real-time quantitative PCR (qPCR)
The expression levels of stemness genes were
quantified by qPCR. For each sample, a mixture consisting of 10
μl of SYBR-Green (Thermo Scientific Maxima
SYBR-Green/Fluorescein qPCR Master Mix 2X), 1 μl reverse and
forward primers (10 μM), and 5 μl cDNA was prepared
in a 20 μl of total volume, according to the manufacturer's
recommendations, and analyzed in a Bio-Rad Thermal Cycler. The Ct
values of the genes of interest were normalized with respect to the
β-actin or GAPDH housekeeping gene. The PCR conditions was: 10 min
95°C for Taq activation, followed by 40 cycles of 15 sec at 95°C
for denaturation and 60 sec at 60°C for annealing/extension.
The sequences of primers used were as follows:
h-β-actin forward, GTGGCCGAGGACTTTGATTG and h-β-actin reverse,
GGACTGGGCCATTCTCCTTA; GAPDH forward, AGTCAGCCGCATCTTCTTTT and GAPDH
reverse, GTGAAGCGCCAGTGGACTC; h-OCT4 forward, ACATCAAAG C
TCTGCAGAAAGAACT and h-OCT4 reverse, CTGAATACCTTCCCAAATAGAACCC;
h-SOX2 forward, TACAGCATGTCCTACTCGA and h-SOX2 reverse,
TGGAGTGGGAGGAAGAGGTA; h-NANOG forward, CAGCCAAATTCTCCTGCCAG and
h-NANOG reverse, CACGTCTTCAGGTTGATGT; h-β-catenin forward,
AGCTTCCAGACACGCTATCA and h-β-catenin reverse, CCAGTAAGCCCTCACGATGA.
All primers were purchased from Metabion International
(Steinkirchen, Germany.
Immunofluorescence analysis
Untreated or simvastatin-treated cells were grown on
coverslips in 24-well cell culture plates to ~80% confluence. In
preparation for immunofluorescence, cells were rinsed with
phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde for
30 min at room temperature and permeabilized with 0.1% Triton X-100
for 10 min at room temperature. The following primary antibodies
were used: OCT-3/4 (C-10; #sc-5279; 1:500; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA); Sox2 (L1D6A2, cat. no.
4900; 1:500; Cell Signaling Technology, Danvers, MA, USA); Nanog
(1E6C4, cat. no. 4893; 1:500; Cell Signaling Technology); and
β-catenin (H-102; #sc-7199; 1:500; Santa Cruz Biotechnology). After
washing three times in PBS, cells were incubated with secondary
antibodies (1:200; Alexa-Fluor; Molecular Probes/Thermo Fisher
Scientific, Inc., Waltham, MA, USA). For actin detection, cells
were stained with Alexa Fluor 488 Phalloidin (#A12379); for the
detection of cell membranes, cells were stained with the membrane
stain wheat germ agglutinin-Alexa Fluor 488 conjugate (Invitrogen,
Carlsbad, CA, USA) following the manufacturer's instructions. Cell
nuclei were counterstained with DAPI (Invitrogen). The coverslips
were mounted with a drop of Mowiol® mounting medium and
observed with an AR1 confocal laser scanning microscope (Nikon,
Tokyo, Japan) using the NIS elements software.
Wound-healing assay
In order to understand the effect of simvastatin on
the progression and invasion of the cancer cells utilized in our
experiments, we performed a wound-healing assay. In brief, cells
were seeded into 60-mm dishes at 5×105 cells/plate. When
the confluence reached ~90%, a single scratch wound was created on
the cell layer with a 200 μl pipette tip. After 24 h, in
untreated cells and in 20 μM simvastatin-treated cells, the
number of cells that had grown into the scratch area was observed
by inverted microscopy (DMI6000 Leica microscope; Leica
Microsystems, Wetzlar, Germany). The cells were quantified by
counting.
Statistical analysis
Each experiment was repeated three times. Results
are expressed as the mean ± SEM, and the effects were compared with
untreated control cells. Paired t-tests were used to analyze the
effects of simvastatin and cytochalasin. P<0.05 was considered
to indicate statistical significance (P<0.05; P<0.01;
P<0.001).
Results
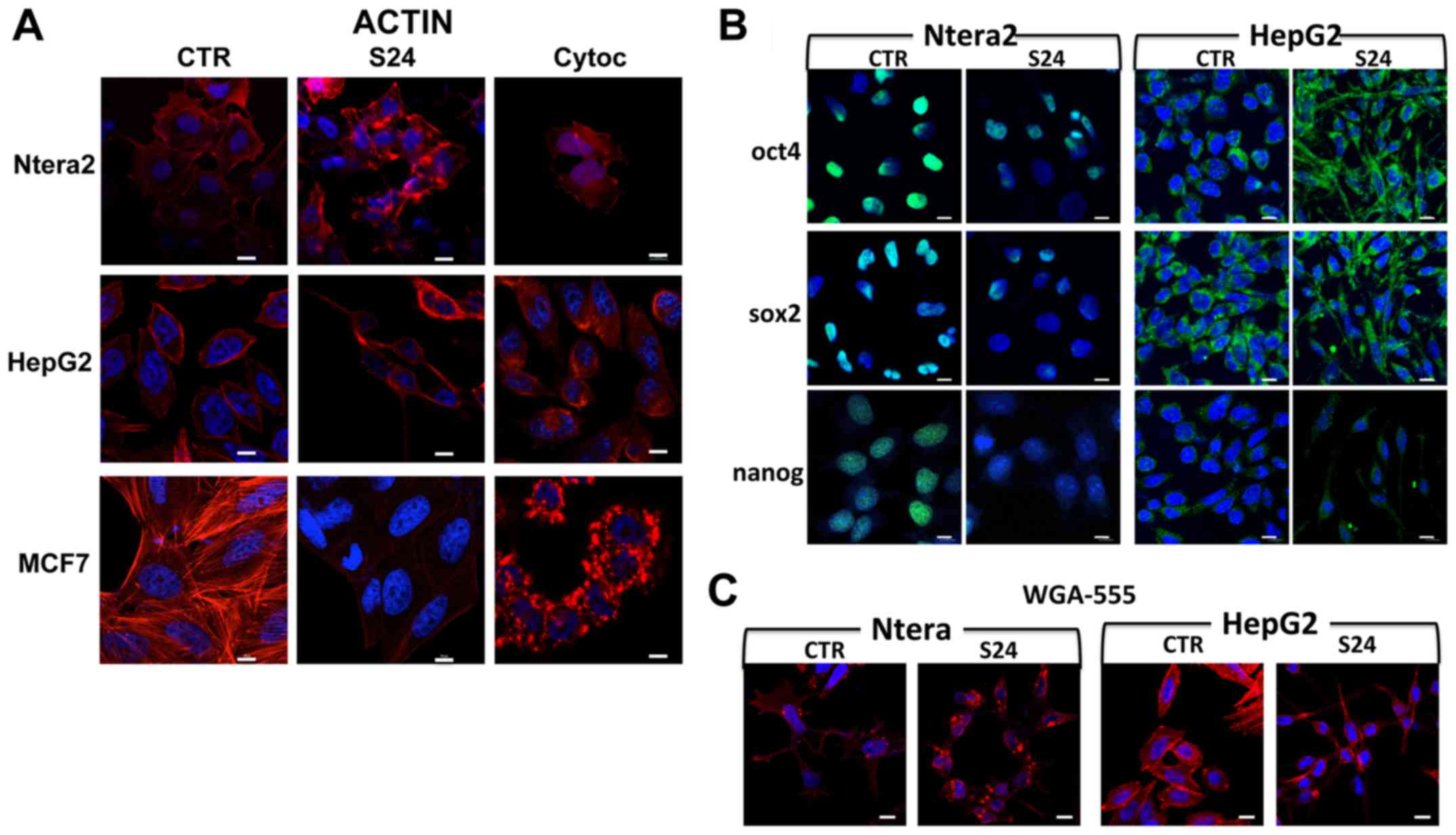
Immunofluorescence analysis of the actin
cytoskeleton
Treatment of NTERA-2, HepG2 and MCF7 human cancer
cells with 20 μM simvastatin for 24 h induced strong
modifications in actin structure and cytoskeletal conformation
(Fig. 2A). These modifications
were different in the various cell lines, indicating a different
sensitivity of each cell type to the drug. Moreover, simvastatin
induced actin destructuration, which differed markedly from that
observed following cytochalasin treatment (Fig. 2A).
In NTERA-2 cells, the cytoskeleton structural
modification was obvious; in the untreated control cells, a
well-structured frame of fibers occupied the entire cytoplasm and
there were projections from the membrane cytoskeleton to the
nucleus, whereas this organization had completely disappeared in
the simvastatin-treated cells. The same result was obtained with
the cytochalasin treatment. In both treatment groups, the actin
molecules accumulated in a disorganized manner in parts of the
cytoplasm.
In untreated HepG2 cells, the actin fibers were
localized mainly at the periphery, inside the membrane, with
well-defined projections to the nuclei. After the simvastatin
treatment (Fig. 2A), this
organization disappeared completely, and the actin remained
localized at the extremities of the cell, and appeared compressed,
assuming a polarized form. In presence of the cytochalasin, none of
this structure was maintained, and the fibers were not visible, but
the actin appeared to accumulate in a disorganized manner in the
cytoplasm.
In untreated MCF7 breast cancer cells, the structure
of the actin fibers was evident, with clear projections from the
membrane to the nuclei. With simvastatin, this structure was
gradually lost and the fiber bundles were less evident. The
addition of cytochalasin induced a complete loss of the fiber
conformation, and the actin appeared fragmented in the cytoplasm
(Fig. 2A).
These results suggest that simvastatin induces
cytoskeletal conformational changes, presumably related to the loss
of post-translational modification of proteins involved in actin
cytoskeleton formation, such as RhoA, Cd42 or ROCK GTPases. These
proteins, to be functionally active, must be prenylated or
geranylated, and simvastatin specifically inhibits these
modifications.
Simvastatin induces changes in the
localization of stemness-related proteins
The expression of the selected stemness-related
proteins was studied by immunofluorescence analysis following
simvastatin treatment.
As shown in Fig.
2B, stronger expression of the stemness-related proteins was
observed in the NTERA-2 cells and, specifically in this cell
population, the changes in the stemness protein localization in the
presence of simvastatin were more clearly observable. In untreated
NTERA-2 cells, the expression of Oct4 was very strong, and the
protein was localized only in the nuclei. Simvastatin treatments
significantly reduced the Oct4 protein expression in the nuclei
(Fig. 2B). Similar results were
obtained with Sox2: the expression of the protein was very strong,
with the protein concentrated principally in the nuclei, and the
simvastatin treatment induced an evident decrease in the expression
of Sox2, with only a minority of nuclei showing expression of the
protein. Similarly, the expression of Nanog was evident in the
nuclei in untreated cells; by contrast, in cells treated with
simvastatin, the protein expression was strongly reduced and almost
disappeared from the nuclei (Fig.
2B).
Less evident were the localization differences
obtained in untreated HepG2 cells. In these cells, the
morphological changes induced by the simvastatin were very strong,
and the cytoplasm was compacted at the two cell poles; for this
reason, the Oct4 and Sox2 proteins, which in untreated cells
appeared uniformly localized around the nucleus, were confined at
the extremities of the cell. This particular form does allow
individuation of the specific changes in stemness protein
localization (Fig. 2B). Nanog is
expressed at a low level in HepG2 cells, and the localization
differences after simvastatin treatment were limited (Fig. 2B). These findings suggest that, in
some cells, entry of the protein into the nucleus is impaired, and
thus the protein remains accumulated in the cytoplasm.
In MCF7 cells, the levels of the stemness-related
proteins were very low, and it was not possible to observe
differences in expression and localization between the untreated
and simvastatin-treated cells (data not shown).
As shown in Fig.
2C, in NTERA-2 and HepG2 cells, simvastatin induced
morphological modifications; immunofluorescence analysis using the
membrane marker WGA-488 showed the effects of simvastatin on cell
morphology, as previously shown in Fig. 2A, wherein cells were marked with
phalloidin to reveal actin structure.
Stemness-related gene expression in
simvastatin- and cytochalasin-treated cells
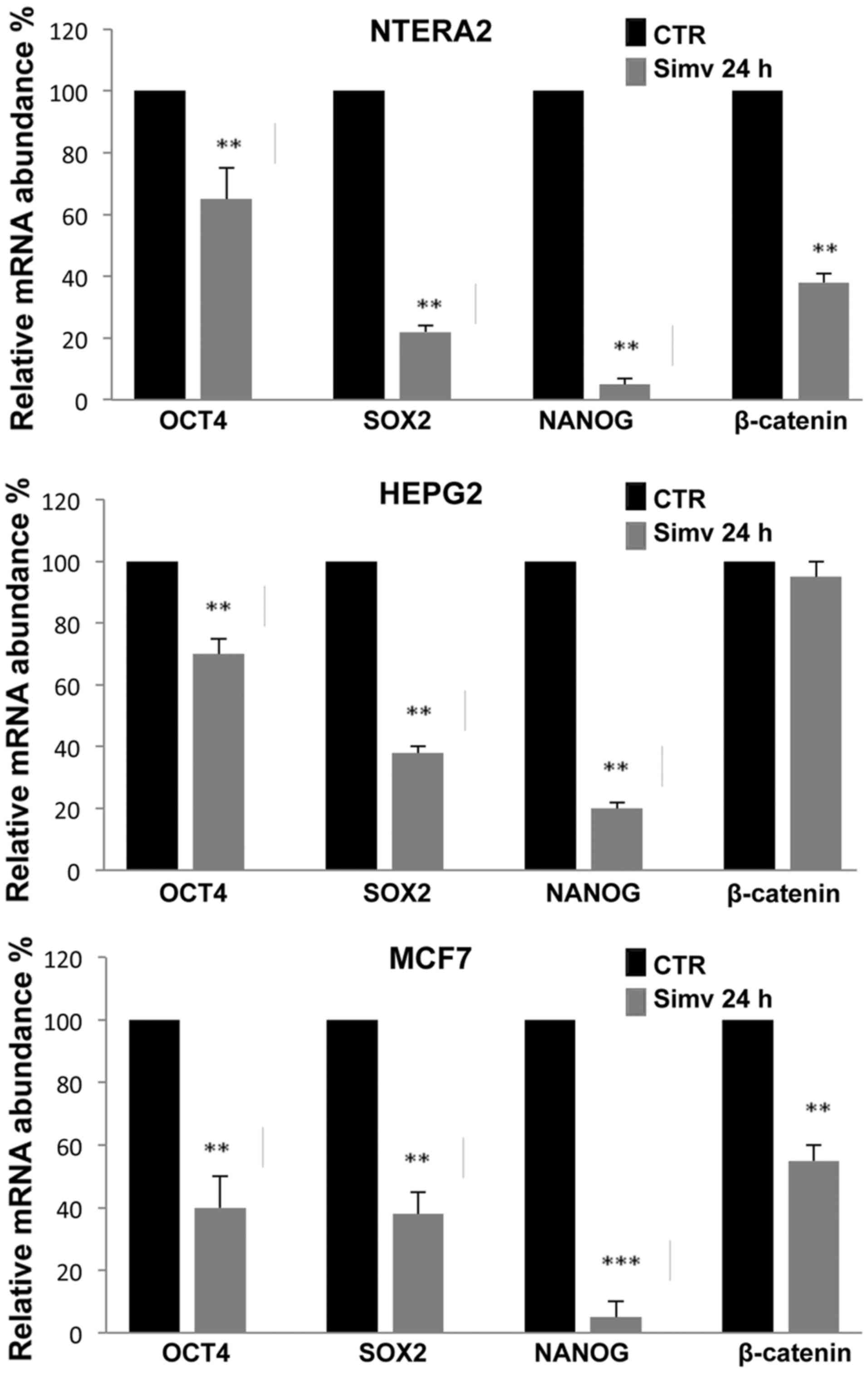
As shown in Fig. 3,
RT-qPCR analysis revealed that 20 μM simvastatin treatment
for 24 h strongly inhibited the expression of the stemness-related
genes Oct4, Sox2 and Nanog in the three human cancer cell lines
investigated (NTERA-2, HepG2 and MCF7). In all three cell types
analyzed, the effects of simvastatin treatment were most evident
with Nanog expression, which in NTERA-2 and MCF7 reached an
inhibition rate of 90%. More limited was the expression reduction
observed for Oct4, which was ~60%, in all the cells analyzed while
for Sox2 the greatest inhibition rate was observed in NTERA-2
cells. The reduction of β-catenin expression induced by simvastatin
was evident only in NTERA-2 and MCF7 cells, while no substantial
inhibition was observed in HepG2 cells (Fig. 3).
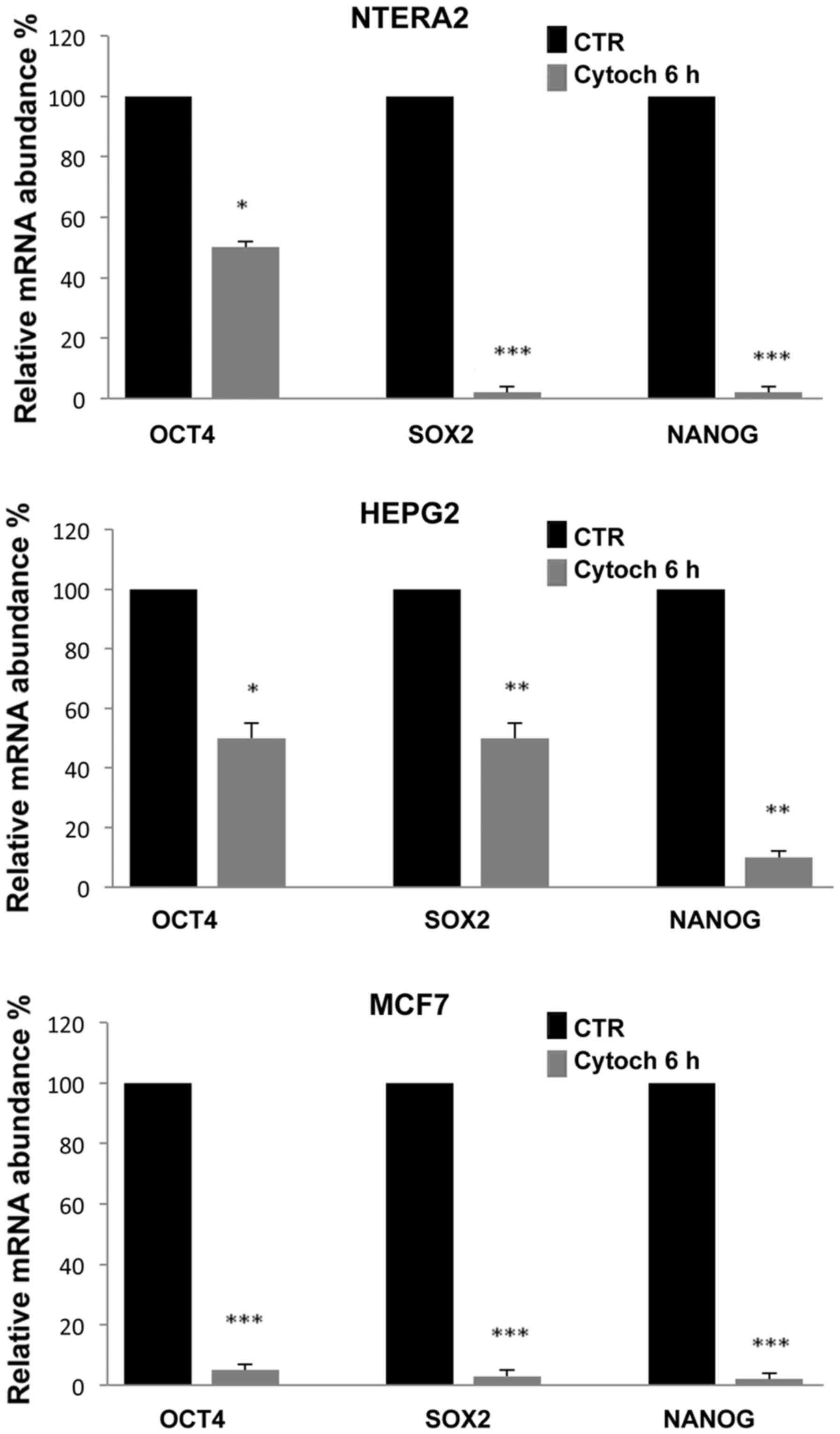
To evaluate whether the inhibition of
stemness-related gene expression was correlated with the
destructuration of the cytoskeleton, we cultured cells in the
presence of 5 μM cytochalasin, a well known inhibitor of
actin polymerization. Notably, after 6 h of culture, the
cytochalasin induced a significant reduction in the expression of
all three stemness-related genes analyzed, which, as shown in
Fig. 4, was comparable to the
inhibition levels induced by simvastatin. The most evident
reduction was observed in NTERA-2 and MCF7 cells, and the gene most
sensitive to cytochalasin treatment was Nanog.
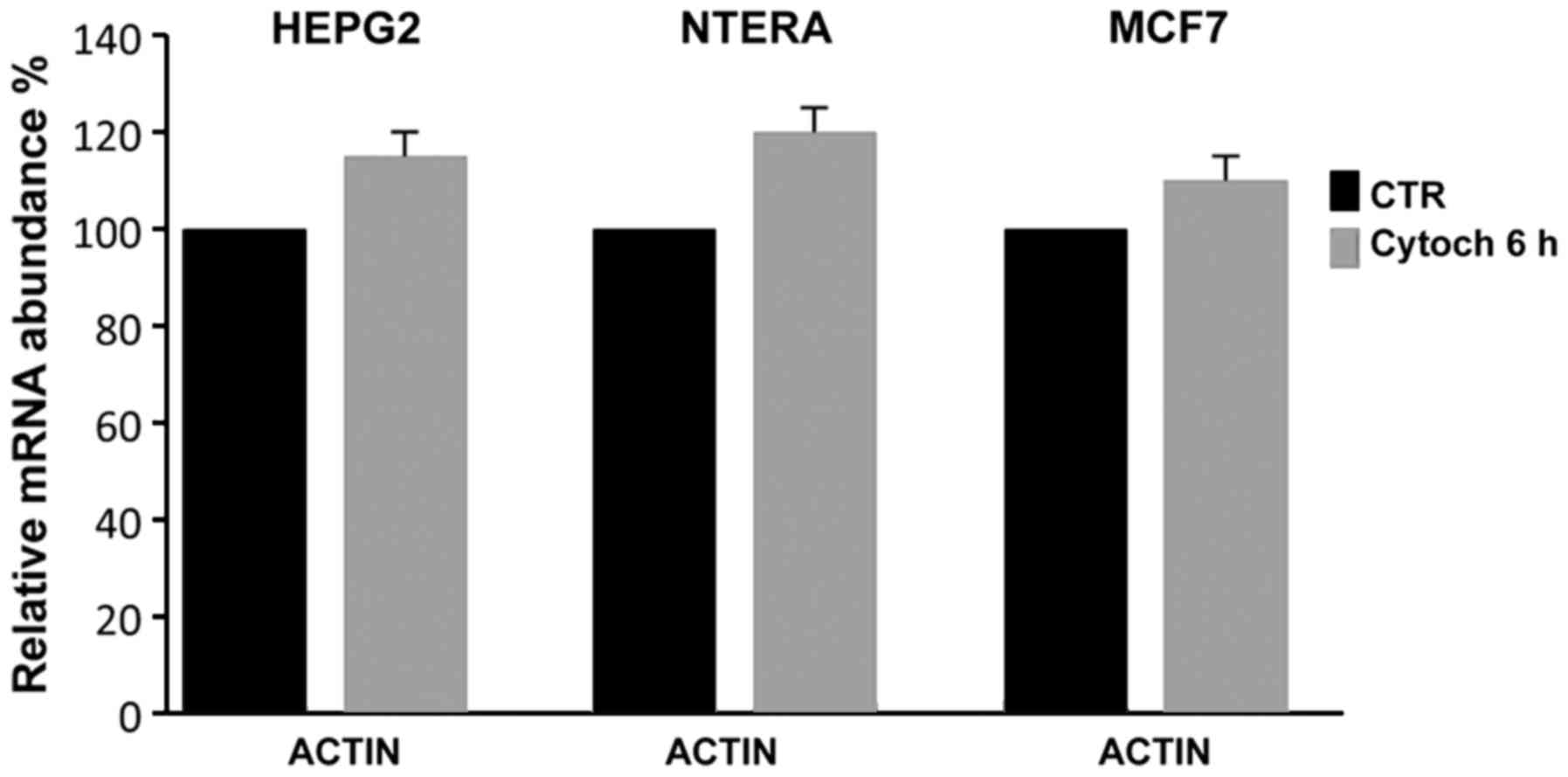
To verify whether the cytochalasin treatment
affected the expression of actin, which is used in RT-qPCR
expression analyses as an internal standard, we controlled its
expression relative to another house-keeping gene, GAPDH; as shown
in Fig. 5, even when actin
polymerization and cell cytoskeletal structure were evidently
inhibited, cytochalasin did not inhibit the actin expression,
demonstrating that it could be used as an internal control in
RT-qPCR expression analyses even in the presence of
cytochalasin.
We hypothesized that alteration of the actin
cytoskeleton structure may induce modification of the projections
of actin fibers to the nucleoskeleton, and that modification of
this strain transfer may have a regulatory action on certain
stemness-related genes. RT-qPCR demonstrated that simvastatin and
cytochalasin could downregulate the transcription of the analyzed
stemness genes, and simvastatin treatment was found to induce a
change in the expression and localization of the respective
proteins.
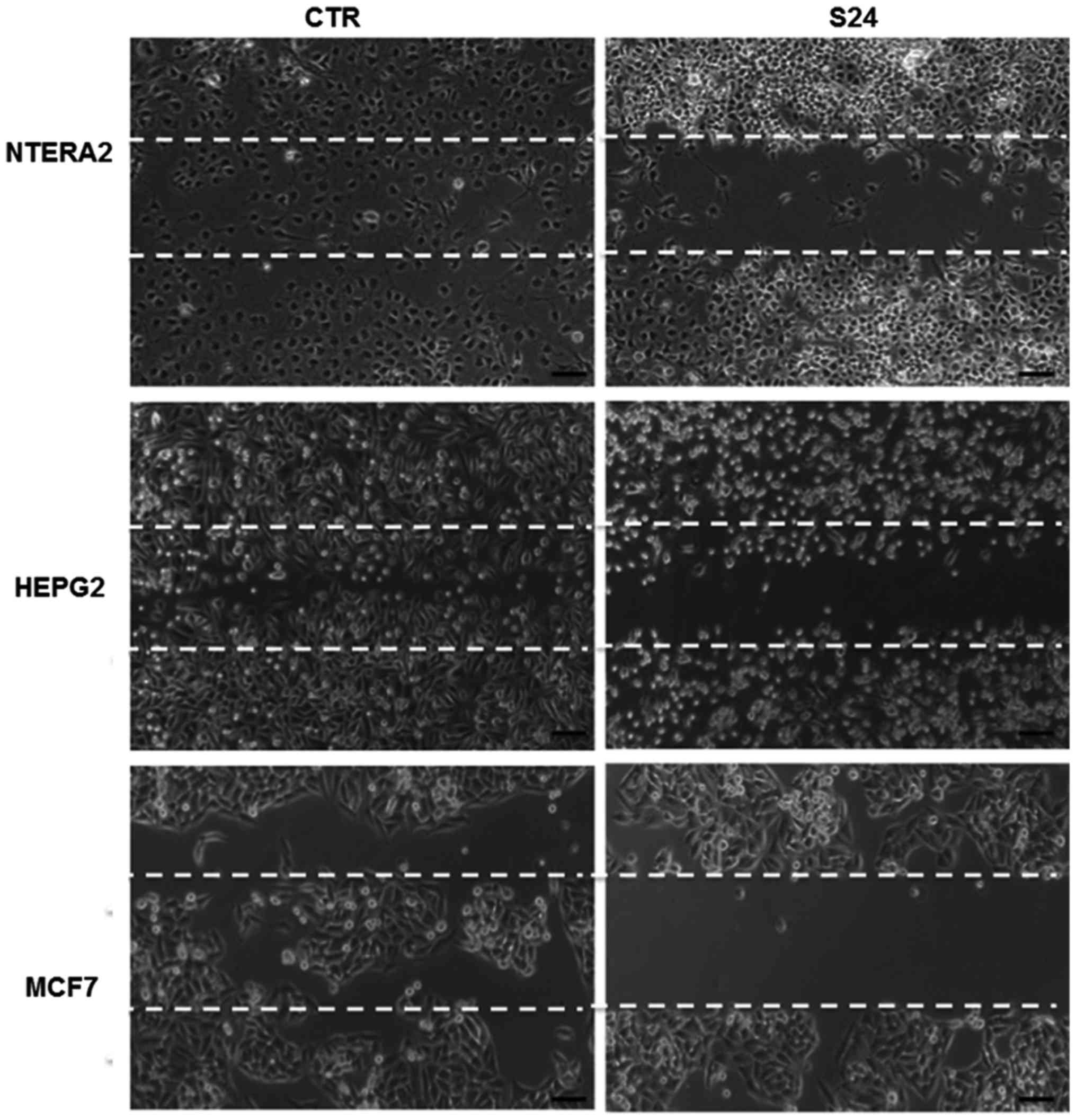
Wound healing assay
To explore whether simvastatin treatment could
affect cell migration, we performed a scratch-wound assay in
NTERA-2, HepG2 and MCF7 cells. As shown in Fig. 6, in the untreated cells, the
scratched area was nearly completely repopulated after 24 h; by
contrast, in the cells treated with 20 μM simvastatin, the
cell growth was inhibited and the scratched area remained
unpopulated after 24 h, demonstrating that simvastatin may inhibit
cancer cell growth and migration. This experiment was replicated
three times, confirming the results obtained.
Discussion
In the present study, we demonstrated that
simvastatin, by blocking the mevalonate pathway and the associated
processes of farnesylation and geranylgeranylation, induces
inhibition of the normal actin polymerization that is fundamental
for cytoskeletal formation and maintenance of normal
cytoarchitecture; the resultant destabilization of the cytoskeleton
seems to inhibit the expression of stemness-related genes, which
likely modulates the equilibrium between proliferation and
differentiation.
Moreover, loss of actin cytoskeleton integrity
inhibits lamellipodia and invadopodia formation, decreasing cell
motility and invasion of the cancer cells into the extracellular
matrix or surrounding tissues, thereby reducing metastatic spread.
Inhibition of cell spreading by simvastatin was also demonstrated
by wound healing experiments, in which simvastatin appeared to
reduce cell migration.
From a more general perspective, we could
hypothesize that any condition or drug that affects the structure
of the cytoskeleton may influence the formation of actin fiber
projections from the cytoskeleton to the nucleoskeleton. If actin
projections to the nucleoskeleton exert a strain transfer and, as
observed, this strain has regulatory action on certain genes, we
can hypothesize the presence of mechanosensitive molecules at or
within cell membranes confers to the cytoskeleton a more important
function beyond simply its structural functions. In this scenario,
active communication between the membrane cytoskeleton and the
nucleoskeleton may occur, with regulatory message transfer, and
thus, we may hypothesize the existence of a functional
mechanotransduction pathway that depends specifically on nuclear
strain transfer and nuclear deformation.
These findings may lead to a more dynamic
perspective on cell-cell interactions within a tissue or an organ.
Each individual cell, through partial perturbation of the membrane
cytoskeleton, could be affected by the presence of neighboring
cells, and changes in the conformational state, induced by changes
in cell morphology, could lead to cryptic binding sites being
exposed, thereby recruiting or activating molecules involved in
downstream signaling (56).
In this complex interaction system, any perturbation
at the membrane level could be transferred through the cytoskeleton
to the nucleoskeleton of the neighboring cells. This strain
transfer could exert regulatory pressure on target genes.
Collectively, these aspects could produce a
directional flow of information by which, in different moments,
different genes could be overexpressed or underexpressed in the
three spatial directions. If the cytoskeleton exercises a function
so important, it is easy to imagine that any destruction of the
actin framework may induce a silencing or a strong downregulation
of all the genes controlled by the transfer of the actomyosin fiber
strain.
In our experiments, we demonstrated that simvastatin
has the capability to strongly influence cytoskeletal conformation.
It has been well-documented that simvastatin blocks the
isoprenylation and geranylgeranylation of RhoA, which in turn
blocks the translocation of RhoA from the cytoplasm to the
membrane, where it can bind to a set of effector molecules that are
important for maintaining the undifferentiated status of the cell.
On this basis, we may also speculate that the simvastatin-induced
inhibition of RhoA functioning may indirectly induce
differentiation-related processes. Moreover, the inhibition of RhoA
geranylation may account for the inhibition of ROCK activity, which
has an important role in mediating changes in the structure of the
actin cytoskeleton. Thus, simvastatin, by directly regulating actin
polymerization and de-polymerization, can decrease strain transfer
to the nucleus and induce loss of the
cytoskeletal-to-nucleoskeletal connectivity; this connectivity is
required for the transmission of regulatory signals that activate
kinase cascades or transcriptional factors controlling the
activation of proliferative genes or inactivation of
differentiation genes. The loss of these signals may induce the
downregulation of certain stemness-related genes which, on the
basis of these considerations, could be considered
nucleoskeleton-associated and mechanically regulated genes.
However, we think that is not easy to define if the
de-structuration of the cytoskeleton is a secondary effect of
simvastatin treatment or the inhibition of post-translational
protein modification, have a precise role in the structuration of
actin cytoskeleton, according to our hypothesis more than
alteration of cytoskeleton is the alteration of the strain transfer
to have a direct role in the transmission of regulatory signal on
genes expression. We want, however, to underline that ours is not a
conclusion, but this function of simvastatin is at the moment only
a hypothesis and more experiments are necessary to confirm this
possible regulatory mechanism.
If the target genes of these pathways are
proliferative genes, as the stemness genes typically are, this
regulatory action could lead to different cell proliferation rates
in different spatial directions, and this could result in
preferential localized cell growth, whereby exercising additional
directional pressure could induce further directional growth, with
possible feed-back regulation.
On another more general point of view, it is easy to
consider that differences in cell proliferation in the three
spatial directions, in a determined time interval, form the basis
of the determination of body shape. When this occurs during normal
development, in the absence of a pathological condition, normal
morphological differences result. Outside normal development, a
directional or localized activation of cell proliferation can
result in a pathological state and, if this hyper-proliferation
derives from the activation of proliferative genes and the
inhibition of differentiation genes, a tumor phenotype may
occur.
Thus, it appears likely that perturbation or
destruction of the cytoskeleton by simvastatin, resulting in
inhibition of strain transfer, may have important effects on the
control of pathological cell growth. We may also hypothesize that
the cell membrane and cytoskeleton serve a much more general role,
with a sort of additional code for the control of the gene
activity. In this context, the membrane proteins could control the
strain transfer from cytoskeleton to nucleus, which could have a
role in the transcriptional control of stemness- and/or
differentiation-related genes.
In the present study, we have described, in several
cancer cell types, the role of simvastatin in inducing the
destruction of the cytoskeleton and the resultant transcriptional
inhibition of three well-studied stemness genes, Oct4, Sox2 and
Nanog. We obtained the same results, including the significant
inhibition of expression of Oct4, Sox2 and Nanog, with the use of
cytochalasin D, a drug usually used to inhibit cytoskeleton
formation.
On the basis of these results, we can speculate that
the downregulation of stemness-related gene expression may be
determined by the loss of appropriate strain transfer from
cytoskeleton to nucleus, which does not allow the exposure of
cryptic binding sites and recruitment of the molecules normally
involved in the downstream regulatory cascade.
As mentioned previously, the effect of simvastatin
derives from the inhibition of RhoA and ROCK GTPase activity, as
consequence of the inhibition of the prenylation and
geranylgeranylation of these proteins. These GTPases have an
important effect on the regulation of the translation rate and
exact localization of the protein within the cell.
We have observed, principally in NTERA-2 cells, in
which Oct4, Sox2 and Nanog genes are strongly expressed, a
reduction in the nuclear localization of all three stemness-related
proteins. The evident loss of localization of these proteins in the
nuclei, where they function as important transcription factors,
leads us to believe that simvastatin could have a possible role in
the inactivation of the function of stemness genes, which may
result in the blocking of proliferation and induction of
differentiation. In the other analyzed cancer cell lines, the
effect of simvastatin was less clear than in NTERA-2 cells,
suggesting that the inhibitory effect of simvastatin predominantly
occurs in cells in which stemness gene expression is strong.
Moreover, by blocking the prenylation and
geranylgeranylation of Rac1 and Cd42, simvastatin inhibits the
three principal pathways of actin polymerization that occur during
the formation of protrusions during cell movement, as well as the
translocation and retraction steps involved in filopodia,
lamellipodia and invadopodia formation. In this manner, this drug
may decrease the ability of cancer cells to invade the
extracellular matrix. The capacity of cancer cells to move through
the extracellular matrix and into neighboring tissues is the basis
of the process of metastasis formation. We suggest that
simvastatin, through inhibition of appropriate cytoskeleton
formation, may suppress the formation of invadopodia complexes
necessary for cancer cell invasion. On the basis of these
considerations, we hypothesize that simvastatin, in combination
with conventional therapy, may serve a possible role in the control
of tumor progression.
Acknowledgments
The authors are grateful to Mr. F. Moscatiello and
Mr. S. Arbucci for their skillful technical assistance. The present
study was financially supported by the Institute of Genetics and
Biophysics A. Buzzati Traverso CNR.
References
|
1
|
Spampanato C, De Maria S, Sarnataro M,
Giordano E, Zanfardino M, Baiano S, Cartenì M and Morelli F:
Simvastatin inhibits cancer cell growth by inducing apoptosis
correlated to activation of Bax and down-regulation of BCL-2 gene
expression. Int J Oncol. 40:935–941. 2012. View Article : Google Scholar
|
|
2
|
Zanfardino M, Spampanato C, De Cicco R,
Buommino E, De Filippis A, Baiano S, Barra A and Morelli F:
Simvastatin reduces melanoma progression in a murine model. Int J
Oncol. 43:1763–1770. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liao JK and Laufs U: Pleiotropic effects
of statins. Annu Rev Pharmacol Toxicol. 45:89–118. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Goldstein JL and Brown MS: Regulation of
the mevalonate pathway. Nature. 343:425–430. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee MH, Cho YS and Han YM: Simvastatin
suppresses self-renewal of mouse embryonic stem cells by inhibiting
RhoA geranylgeranylation. Stem Cells. 25:1654–1663. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sorrentino G, Ruggeri N, Specchia V,
Cordenonsi M, Mano M, Dupont S, Manfrin A, Ingallina E, Sommaggio
R, Piazza S, et al: Metabolic control of YAP and TAZ by the
mevalonate pathway. Nat Cell Biol. 16:357–366. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang S and Cui W: Sox2, a key factor in
the regulation of pluripotency and neural differentiation. World J
Stem Cells. 6:305–311. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Boyer LA, Lee TI, Cole MF, Johnstone SE,
Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG,
et al: Core transcriptional regulatory circuitry in human embryonic
stem cells. Cell. 122:947–956. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen X, Xu H, Yuan P, Fang F, Huss M, Vega
VB, Wong E, Orlov YL, Zhang W, Jiang J, et al: Integration of
external signaling pathways with the core transcriptional network
in embryonic stem cells. Cell. 133:1106–1117. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Loh KM and Lim B: A precarious balance:
Pluripotency factors as lineage specifiers. Cell Stem Cell.
8:363–369. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Thomson M, Liu SJ, Zou LN, Smith Z,
Meissner A and Ramanathan S: Pluripotency factors in embryonic stem
cells regulate differentiation into germ layers. Cell. 145:875–889.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Askarian-Amiri ME, Seyfoddin V, Smart CE,
Wang J, Kim JE, Hansji H, Baguley BC, Finlay GJ and Leung EY:
Emerging role of long non-coding RNA SOX2OT in SOX2 regulation in
breast cancer. PLoS One. 9:e1021402014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bowles J, Schepers G and Koopman P:
Phylogeny of the SOX family of developmental transcription factors
based on sequence and structural indicators. Dev Biol. 227:239–255.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wilson M and Koopman P: Matching SOX:
Partner proteins and co-factors of the SOX family of
transcriptional regulators. Curr Opin Genet Dev. 12:441–446. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kamachi Y, Uchikawa M and Kondoh H:
Pairing SOX off: With partners in the regulation of embryonic
development. Trends Genet. 16:182–187. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ambrosetti DC, Schöler HR, Dailey L and
Basilico C: Modulation of the activity of multiple transcriptional
activation domains by the DNA binding domains mediates the
synergistic action of Sox2 and Oct-3 on the fibroblast growth
factor-4 enhancer. J Biol Chem. 275:23387–23397. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Loh YH, Wu Q, Chew JL, Vega VB, Zhang W,
Chen X, Bourque G, George J, Leong B, Liu J, et al: The Oct4 and
Nanog transcription network regulates pluripotency in mouse
embryonic stem cells. Nat Genet. 38:431–440. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tomioka M, Nishimoto M, Miyagi S,
Katayanagi T, Fukui N, Niwa H, Muramatsu M and Okuda A:
Identification of Sox-2 regulatory region which is under the
control of Oct-3/4-Sox-2 complex. Nucleic Acids Res. 30:3202–3213.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jauch R, Aksoy I, Hutchins AP, Ng CK, Tian
XF, Chen J, Palasingam P, Robson P, Stanton LW and Kolatkar PR:
Conversion of Sox17 into a pluripotency reprogramming factor by
reengineering its association with Oct4 on DNA. Stem Cells.
29:940–951. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Loh KM and Lim B: A precarious balance:
Pluripotency factors as lineage specifiers. Cell Stem Cell.
8:363–369. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang Z, Oron E, Nelson B, Razis S and
Ivanova N: Distinct lineage specification roles for NANOG, OCT4,
and SOX2 in human embryonic stem cells. Cell Stem Cell. 10:440–454.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Takahashi K and Yamanaka S: Induction of
pluripotent stem cells from mouse embryonic and adult fibroblast
cultures by defined factors. Cell. 126:663–676. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nakagawa M, Koyanagi M, Tanabe K,
Takahashi K, Ichisaka T, Aoi T, Okita K, Mochiduki Y, Takizawa N
and Yamanaka S: Generation of induced pluripotent stem cells
without Myc from mouse and human fibroblasts. Nat Biotechnol.
26:101–106. 2008. View
Article : Google Scholar
|
|
24
|
Huangfu D, Osafune K, Maehr R, Guo W,
Eijkelenboom A, Chen S, Muhlestein W and Melton DA: Induction of
pluripotent stem cells from primary human fibroblasts with only
Oct4 and Sox2. Nat Biotechnol. 26:1269–1275. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Okita K, Ichisaka T and Yamanaka S:
Generation of germline-competent induced pluripotent stem cells.
Nature. 448:313–317. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Alonso MM, Diez-Valle R, Manterola L,
Rubio A, Liu D, Cortes-Santiago N, Urquiza L, Jauregi P, Lopez de
Munain A, Sampron N, et al: Genetic and epigenetic modifications of
Sox2 contribute to the invasive phenotype of malignant gliomas.
PLoS One. 6:e267402011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Basu-Roy U, Seo E, Ramanathapuram L, Rapp
TB, Perry JA, Orkin SH, Mansukhani A and Basilico C: Sox2 maintains
self renewal of tumor-initiating cells in osteosarcomas. Oncogene.
31:2270–2282. 2012. View Article : Google Scholar
|
|
28
|
Chen Y, Shi L, Zhang L, Li R, Liang J, Yu
W, Sun L, Yang X, Wang Y, Zhang Y, et al: The molecular mechanism
governing the oncogenic potential of SOX2 in breast cancer. J Biol
Chem. 283:17969–17978. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cox JL, Wilder PJ, Desler M and Rizzino A:
Elevating SOX2 levels deleteriously affects the growth of
medulloblastoma and glioblastoma cells. PLoS One. 7:e440872012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Girouard SD, Laga AC, Mihm MC, Scolyer RA,
Thompson JF, Zhan Q, Widlund HR, Lee CW and Murphy GF: SOX2
contributes to melanoma cell invasion. Lab Invest. 92:362–370.
2012. View Article : Google Scholar
|
|
31
|
Leis O, Eguiara A, Lopez-Arribillaga E,
Alberdi MJ, Hernandez-Garcia S, Elorriaga K, Pandiella A, Rezola R
and Martin AG: Sox2 expression in breast tumours and activation in
breast cancer stem cells. Oncogene. 31:1354–1365. 2012. View Article : Google Scholar
|
|
32
|
Lengerke C, Fehm T, Kurth R, Neubauer H,
Scheble V, Müller F, Schneider F, Petersen K, Wallwiener D, Kanz L,
et al: Expression of the embryonic stem cell marker SOX2 in
early-stage breast carcinoma. BMC Cancer. 11:42–52. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li XL, Eishi Y, Bai YQ, Sakai H, Akiyama
Y, Tani M, Takizawa T, Koike M and Yuasa Y: Expression of the
SRY-related HMG box protein SOX2 in human gastric carcinoma. Int J
Oncol. 24:257–263. 2004.PubMed/NCBI
|
|
34
|
Sanada Y, Yoshida K, Ohara M, Oeda M,
Konishi K and Tsutani Y: Histopathologic evaluation of stepwise
progression of pancreatic carcinoma with immunohistochemical
analysis of gastric epithelial transcription factor SOX2:
Comparison of expression patterns between invasive components and
cancerous or nonneoplastic intraductal components. Pancreas.
32:164–170. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sattler HP, Lensch R, Rohde V, Zimmer E,
Meese E, Bonkhoff H, Retz M, Zwergel T, Bex A, Stoeckle M, et al:
Novel amplification unit at chromosome 3q25-q27 in human prostate
cancer. Prostate. 45:207–215. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Seo E, Basu-Roy U, Zavadil J, Basilico C
and Mansukhani A: Distinct functions of Sox2 control self-renewal
and differentiation in the osteoblast lineage. Mol Cell Biol.
31:4593–4608. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ben-Porath I, Thomson MW, Carey VJ, Ge R,
Bell GW, Regev A and Weinberg RA: An embryonic stem cell-like gene
expression signature in poorly differentiated aggressive human
tumors. Nat Genet. 40:499–507. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rodriguez-Pinilla SM, Sarrio D,
Moreno-Bueno G, Rodriguez-Gil Y, Martinez MA, Hernandez L,
Hardisson D, Reis-Filho JS and Palacios J: Sox2: A possible driver
of the basal-like phenotype in sporadic breast cancer. Mod Pathol.
20:474–481. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lengerke C, Fehm T, Kurth R, Neubauer H,
Scheble V, Müller F, Schneider F, Petersen K, Wallwiener D, Kanz L,
et al: Expression of the embryonic stem cell marker SOX2 in
early-stage breast carcinoma. BMC Cancer. 11:42–52. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Leis O, Eguiara A, Lopez-Arribillaga E,
Alberdi MJ, Hernandez-Garcia S, Elorriaga K, Pandiella A, Rezola R
and Martin AG: Sox2 expression in breast tumours and activation in
breast cancer stem cells. Oncogene. 31:1354–1365. 2012. View Article : Google Scholar
|
|
41
|
Basu-Roy U, Bayin NS, Rattanakorn K, Han
E, Placantonakis DG, Mansukhani A and Basilico C: Sox2 antagonizes
the Hippo pathway to maintain stemness in cancer cells. Nat Commun.
6:1–11. 2015. View Article : Google Scholar
|
|
42
|
Li X, Xu Y, Chen Y, Chen S, Jia X, Sun T,
Liu Y, Li X, Xiang R and Li N: SOX2 promotes tumor metastasis by
stimulating epithelial-to-mesenchymal transition via regulation of
WNT/β-catenin signal network. Cancer Lett. 336:379–389. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chen Y, Shi L, Zhang L, Li R, Liang J, Yu
W, Sun L, Yang X, Wang Y, Zhang Y, et al: The molecular mechanism
governing the oncogenic potential of SOX2 in breast cancer. J Biol
Chem. 283:17969–17978. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Saigusa S, Tanaka K, Toiyama Y, Yokoe T,
Okugawa Y, Ioue Y, Miki C and Kusunoki M: Correlation of CD133,
OCT4, and SOX2 in rectal cancer and their association with distant
recurrence after chemoradiotherapy. Ann Surg Oncol. 16:3488–3498.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jia X, Li X, Xu Y, Zhang S, Mou W, Liu Y,
Liu Y, Lv D, Liu CH, Tan X, et al: SOX2 promotes tumorigenesis and
increases the anti-apoptotic property of human prostate cancer
cell. J Mol Cell Biol. 3:230–238. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Polyak K and Weinberg RA: Transitions
between epithelial and mesenchymal states: Acquisition of malignant
and stem cell traits. Nat Rev Cancer. 9:265–273. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Xu J, Lamouille S and Derynck R:
TGF-beta-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Bryant DM and Stow JL: The ins and outs of
E-cadherin trafficking. Trends Cell Biol. 14:427–434. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ikenouchi J, Matsuda M, Furuse M and
Tsukita S: Regulation of tight junctions during the
epithelium-mesenchyme transition: Direct repression of the gene
expression of claudins/occludin by Snail. J Cell Sci.
116:1959–1967. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Strauss R, Li ZY, Liu Y, Beyer I, Persson
J, Sova P, Möller T, Pesonen S, Hemminki A, Hamerlik P, et al:
Analysis of epithelial and mesenchymal markers in ovarian cancer
reveals phenotypic heterogeneity and plasticity. PLoS One.
6:e161862011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Jiang YG, Luo Y, He DL, Li X, Zhang LL,
Peng T, Li MC and Lin YH: Role of Wnt/beta-catenin signaling
pathway in epithelial-mesenchymal transition of human prostate
cancer induced by hypoxia-inducible factor-1alpha. Int J Urol.
14:1034–1039. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Beavon IR: The E-cadherin-catenin complex
in tumour metastasis: Structure, function and regulation. Eur J
Cancer. 36:1607–1620. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Brabletz T, Jung A, Spaderna S, Hlubek F
and Kirchner T: Opinion: Migrating cancer stem cells - an
integrated concept of malignant tumor progression. Nat Rev Cancer.
5:744–749. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Driscoll TP, Cosgrove BD, Heo SJ, Shurden
ZE and Mauck RL: Cytoskeletal to nuclear strain transfer regulates
YAP signaling in mesenchymal stem cells. Biophys J. 108:2783–2793.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Artman L, Dormoy-Raclet V, von Roretz C
and Gallouzi IE: Planning your every move: The role of β-actin and
its post-transcriptional regulation in cell motility. Semin Cell
Dev Biol. 34:33–43. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Driscoll TP, Cosgrove BD, Heo SJ, Shurden
ZE and Mauck RL: Cytoskeletal to nuclear transfer regulate YAP
signaling in mesenchymal stem cells. Biophys J. 108:2783–2793.
2015. View Article : Google Scholar : PubMed/NCBI
|