Introduction
Sarcomas, including soft tissue sarcomas (STS) and
osteosarcomas, are heterogeneous tumours that arise from
transformed cells of mesenchymal origin, including malignant
tumours made of bone, cartilage, fat, muscle, vascular or
hematopoietic tissues. Patients with a localized sarcoma have a 83%
chance for a 5-year survival, whereas those sarcomas with lymph
node involvement have a reduced prognosis of 54% and the worst
prognosis is 16% for sarcomas that have spread to distant parts of
the body (1). This reflects the
ineffectiveness of current therapy and the importance for the
development of better treatment options to improve outcomes.
The human epidermal growth factor receptor
(EGFR/HER) family of receptor tyrosine kinases, including
EGFR/HER1/ErbB1, HER2/ErbB2, HER3/ErbB3 and HER4/ErbB4, regulates
proliferation, survival, adhesion, and the migration and invasion
of malignant cells (2,3). Following ligand binding to the
extra-celluar domain of HER family members, subsequent signalling
cascades affect gene transcription through three downstream
pathways: ras/raf/mitogen-activated protein kinase (MAPK),
phosphoinositide 3-kinase (PI3K)/AKT and Janus kinase/signal
transducer and activator of transcription (JAK)/signal transducer
and activator of transcription (STAT) (4). Clinical studies have indicated that
the hyperactivation of the HER family is associated with more
aggressive diseases and poor clinical outcomes, so the HER family
has been intensively pursued as therapeutic targets (5 and refs.
therein).
We have previously reported that, in a cohort of 46
consecutive patients with STS, 78% demonstrated a positive
expression of EGFR (6). This
finding is consistent with other series in STS with a mean of 68%
(range 60–77%) (7–11). In a large Japanese study, EGFR
expression was found to be significantly associated with the
histological grade, but was not an independent prognostic factor
for survival (7). Our recent
studies demonstrated that EGFR and downstream signal transducers
were highly expressed and activated in STS cell lines (12), and showed that phosphorylated
(p-)EGFR p-EGFR) and p-Erk are independent prognostic factors for
overall and/or cancer-specific survival in 87 STS samples (13). However, the specific EGFR
inhibitor, gefitinib, was ineffective in terms of preclinical
anti-proliferation, despite the inhibition of pEGFR and signalling
transducers in the EGFR downstream PI3K/AKT and ras/raf/Erk MAPK
pathways (12). Consistently, a
phase II clinical trial demonstrated that single agent gefitinib
was unsatisfactory with low response rates of 21 and 6% at 4 and 6
months, respectively and short disease control in advanced synovial
sarcomas (14). All the above data
suggest that novel approaches targeting this pathway are required
in sarcoma.
Our previous study on gefitinib in sarcoma cell
lines identified the STAT3 escape pathway as a potential resistance
mechanism, due to the increased/unchanged ratio of pSTAT3/pSTAT1
from the JAK/STAT pathway (12).
HER2 overexpression as an upstream regulator has been demonstrated
to activate STAT3 and induce breast cancer growth (15,16).
In addition, HER2 also acts as a transcriptional co-activator of
STAT3 and leads to cyclin D1 promoter activation to enhance tumour
proliferation (17). Furthermore,
blocking HER2 can induce apoptosis (18). These studies provide a rationale
that HER2-induced STAT3 activation may be an escape pathway to
EGFR-specific inhibition and suggest that a panHER inhibitor
blocking both EGFR and HER2 may enhance the therapeutic effect.
Dacomitinib, a panHER inhibitor, has been shown to
exert an irreversible inhibitory effect on the tyrosine kinase
activation of human EGFR/HER1, HER2 and HER4, and is active in both
EGFR-sensitive and EGFR-resistant preclinical models (19–23).
Following initial phase I studies (20,24,25),
in subsequent phase II trials, it was shown to be well-tolerated
and showed encouraging activity (25–27).
In two phase II studies as a first-line therapy, dacomitinib
demonstrated encouraging clinical activity in patients with
recurrent and/or metastatic squamous cell cancer of the head and
neck (SCCHN) and with clinically or molecularly selected advanced
non-small cell lung cancer (NSCLC) (28–30).
Although a randomised phase II trial demonstrated a significant
improvement of progression-free survival (PFS) (31), in two double-blind randomised phase
III trials in patients pretreated with NSCLC, dacomitinib did not
improve PFS/overall survival (OS) compared with the placebo or
erlotinib in an unselected patient population (32,33).
However, a pooled subset analyses demonstrated that dacomitinib
exhibited favourable trends in PFS for the EGFR activation
mutation patient subgroup (34).
Accordingly, dacomitinib is currently in a phase III study of
first-line dacomitinib versus gefitinib in patients with advanced
NSCLC harbouring activating EGFR mutations. Of note, in a
recent interim analysis of the 452 patients in this study,
dacomitinib treatment extended PFS and the duration of response
versus gefitinib (35).
Considering the potential advantages of irreversible
panHER inhibitors over their reversible counterparts, as well as
our previous findings (12) that
sarcoma cells exhibit resistance to gefitinib treatment through the
STAT3 escape pathway, in this study, the panHER inhibitor,
dacomitinib, was examined in sarcoma cell lines. We hypothesized
that the use of dacomitinib would have the potential to increase
the effectiveness of EGFR/HER1-targeted therapy in sarcomas by i)
downregulating STAT3 via the inhibition of HER2 favouring an
increased drive to apoptsis; ii) blocking EGFR/HER1 downstream
ras/raf/MAPK and PI3K/AKT survival signals to further induce
apoptosis; and iii) blocking other HER family member signalling to
overcome resistance to single EGFR/HER1 inhibition. To date, no
panHER inhibitor has been tested in sarcomas. The principal aim of
this study was to investigate the in vitro antitumour effect
and mechanisms of action of dacomitinib mono- and combination
therapy in a panel of 13 human STS and osteosarcoma cell lines.
Materials and methods
Drugs and cell lines
The panHER inhibitor, dacomitinib (PF299804), was
kindly provided by Pfizer USA (New York, NY, USA). The STAT3
inhibitor, S3I-201 (NSC74859), and the EGFR inhibitor, gefitinib,
were purchased from Selleckchem (Scoresby, VIC, Australia) and
Euroasian chemicals (Mumbai, India), respectively. The human
sarcoma cell lines [SW872 (liposarcoma), HT1080 (fibrosarcoma),
SW684 (fibrosarcoma), GCT (undifferentiated pleomorphic sarcoma),
SW982 (synovial sarcoma), A431 (epidermoid carcinoma), SJSA
(osteosarcoma), U2-OS (osteosarcoma), MG63 (osteosarcoma) and
Saos-2 (osteosarcoma)] were purchased from the American Type
Culture Collection (ATTC, Manassas, VA, USA). PC9 (lung
adenocarcinoma) was purchased from Sigma-Aldrich (Castle Hill, NSW,
Australia). The other cell lines [778 (fibrosarcoma), 449B
(liposarcoma; also known as 93T449; STR profile available on ATCC,
cat. no. ATCC CRL-3043), HOS (osteosarcoma) and 143B
(osteosarcoma)] were kindly provided by Professor David Thomas
(Peter MacCallum Cancer Centre, Melbourne, VIC, Australia) and Dr
Florence Pedeatour (Nice University Hospital, Nice, France). The
cell lines were all tested as mycoplasma-free, and were subjected
to identification tests using short tandem repeat (STR) profiling
by CellBank Australia (Westmead, NSW, Australia) and shown to be
consistent with their stated cell lineage.
Cell culture and cell proliferation
assay
All cells were maintained in Rosewell Park Memorial
Institute (RPMI)-1640 medium, supplemented with 10%
heat-inactivated fetal bovine serum (FBS), 2 mM L-glutamine and
antibiotics (50 U/ml penicillin and 50 μg/ml streptomycin)
at 37°C in a humidified 5% CO2 and 95% atmosphere. Cell
culture reagents were purchased from Gibco (Melbourne, VIC,
Australia). Cells were grown as monolayer cultures in
75-cm2 flasks. Once 80–90% confluent, the cells were
detached with 0.05% trypsin-EDTA/PBS (Invitrogen, Carlsbad, CA,
USA) and then cultured in a new flask for use in subsequent
experiments. Cell proliferation assay was carried by crystal violet
colorilmetric assay, as previously described (36). Briefly, 24 h after the cells were
seeded, they were treated with the vehicle [dimethyl sulfoxide
(DMSO), purchased from Amresco (Solon, OH, USA)] or drugs
[including monotherapy with gefitinib, S3I-201, and dacomitinib, as
well as combination therapy (refer to the Results section for the
details of specific mono- or combination treatments, doses and
treatment durations)]. At least duplicate experiments with each
treatment group containing triplicate wells were carried out. After
the required time period, the cells were washed with Dulbecco’s
phosphate-buffered saline (DPBS), stained with 0.5% crystal violet
(Sigma-Aldrich) and incubated with elution solution (0.1 M sodium
citrate and 100% ethanol), followed by light absorbance at 540 nm
on a plate reader (Tecan; Grodig, Salzburg, Austria).
Clonogenic survival assay for adherent
sarcoma cells
After 24 h of seeding, the cells were treated with
the vehicle (DMSO) or drugs (200 nM of dacomitinib, 10 μM of
S3I-201 or 200 nM of dacomitinib plus 10 μM of S3I-201) and
incubated at 37°C. The drugs were present in the medium throughout
the whole incubation period. Once colony-formation (1 colony ≥50
cells, 6–8 days) was observed in the control wells, all related
wells were washed and stained with crystal violet.
Anchorage-independent growth assay
(anti-anoikis assay) by soft agar colony formation assay
The bottom gel was created by mixing 1% agarose with
equivalent volume of 2X RPMI. Logarithmically growing cells were
harvested and suspended in medium containing 0.35% soft agar. The
cells were treated with the vehicle or the drugs in quadruplicate.
Plates were maintained at 37°C in a humidified incubator for 1–2
weeks until colonies were formed. Subsequently, 10% AlamarBlue
(Invitrogen, Carlsbad, CA, USA) was added to each well and
incubated for 4–20 h. The results were evaluated using a
fluorescence microplate reader SpectrMax M3 (Molecular Devices, San
Jose, CA, USA) with an excitation wavelength of 540–570 nm and an
emission wavelength of 580–610 nm.
Western blot analysis
The vehicle-treated and drug-treated cells were
harvested, and total proteins were extracted and measured using
western blot with β-actin as an internal protein loading control,
with our standard procedures (37). Briefly, cells were starved
overnight in RPMI-1640 medium containing 1% FBS and then treated
with the vehicle (DMSO) or drugs (200 nM of dacomitinib and/or 20
μM of S3I-201 for 24 h). The cells were then harvested
following 15 min of incubation with or without 100 ng/ml EGF
stimulation, and total proteins were extracted using RIPA buffer
(both from Sigma-Aldrich) with 1% Protease and Phosphatase
inhibitor cocktails (Merck, Bayswater, VIC, Australia). Protein
concentrations were determined by BCA protein assay (Thermofisher,
Scoresby, VIC, Australia), according to the manufacturer’s
instructions. Subsequently, 50 μg proteins per lane were
separated by 4–20% SDS-PAGE and transferred onto nitrocellulose
membranes (Bio-Rad, Gladesville, NSW, Australia), which were
blocked with 5% skim milk powder in TBS with 01% Tween-20 (TBST)
for 1 h at room temperature, and then probed with primary
antibodies overnight at 4°C. The detection of β-actin was used to
ensure equal loading and proper transfer of the protein.
HRP-conjugated secondary antibodies were detected by
chemiluminescence agent Supersignal Western Dura Extended Duration
(Thermofisher). Membranes were imaged by ImageQuant LAS4000 (GE
Healthcare; Silverwater, NSW, Australia). Densitometric analysis
was performed by ImageQuant TL Software (GE Healthcare) and
presented as ratios of protein expression normalized to relevant
β-actin loading control. All antibodies (STAT antibody kit #9939,
p-STAT antibody kit #9914, HER family antibody kit #8339,
PhosphoPlus AKT activation kit #9280, MAPK family antibody kit
#9926, p-MAPK family antibody kit #9910 and β-actin #4970) were
purchased from Cell Signaling Technology (Arundel, QLD, Australia)
and diluted as per the manufacturer’s instructions.
Statistical analysis
Growth inhibition data were calculated using
GraphPad Prism software, and IC50 curves were fitted
using a non-linear regression model with a sigmoidal dose-response.
Mean percentage growths in different treatment groups from at least
duplicate experiments with triplicate samples against controls were
analysed using ANOVA first. Significant differences obtained from
ANOVA were further analysed by a post-hoc Bonferroni test.
Two-tailed P-values <0.05 were considered to indicate
statistically significant differences.
Results
Positive expression of HER1 and HER2 in
sarcoma cell lines
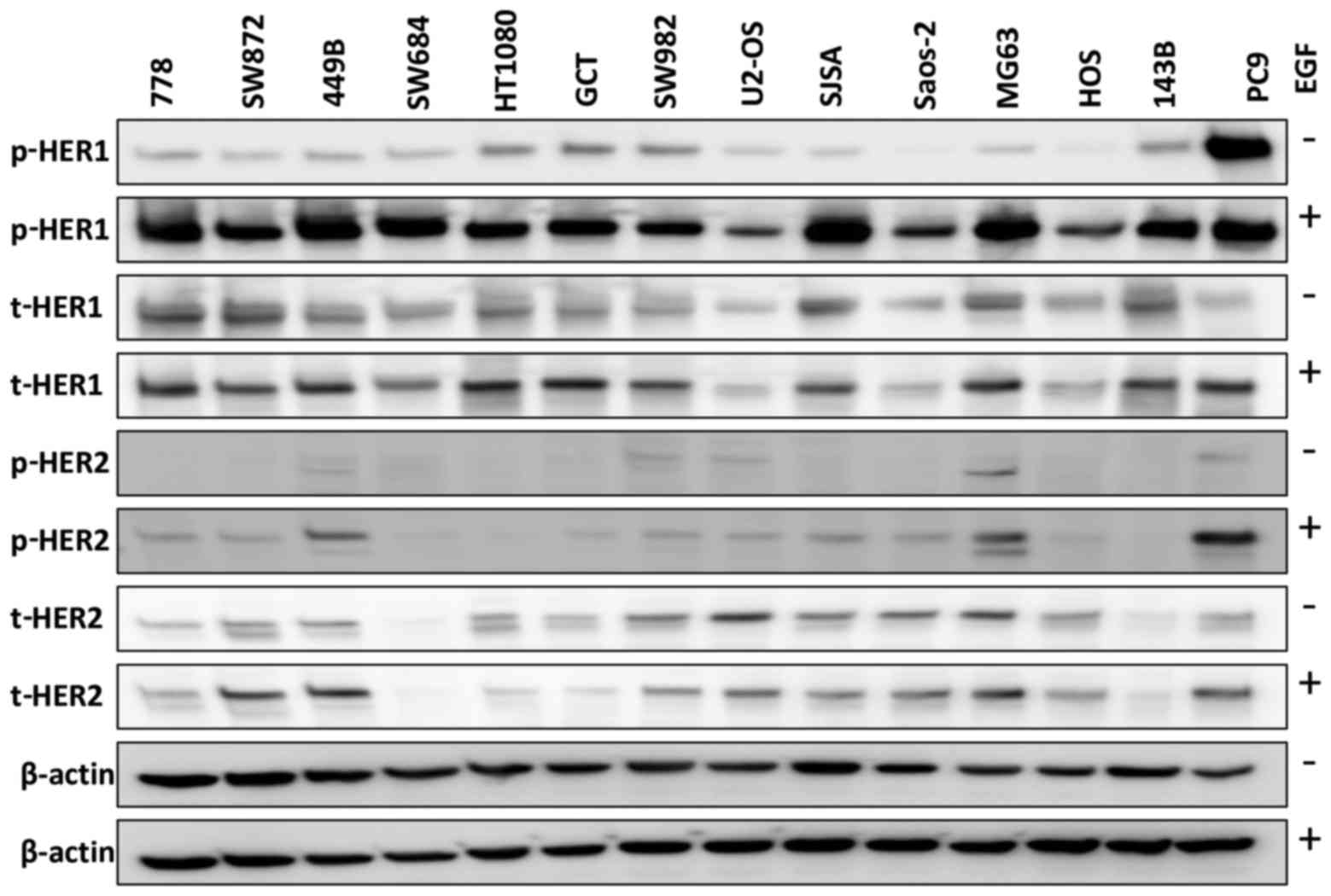
We first examined HER1 expression in 13 sarcoma cell
lines, using the PC9 cells as a positive control [which have been
reported to express abnormally high levels of HER1 (38)]. The sarcoma cell lines expressed
total HER1 (t-HER1) at a similar or higher level to that in the PC9
cells (Fig. 1) in the absence of
ligand EGF stimulation. Phosphorylated HER1 (p-HER1) was
undetectable in the Saos-2 and HOS cells, and was weakly expressed
in the other sarcoma cell lines in the absence of EGF, while the
PC9 cells exhibited a strong p-HER1 expression. We also found that
EGF stimulation (closely mimicking the in vivo setting)
induced p-HER1 expression in all 13 sarcoma cell lines, with the
cells expressing lower (U2-OS, Saos-2 and HOS) or similar levels of
p-HER1 compared with the PC9 cells. Similarly, with ligand EGF
stimulation, 10/13 cell lines exhibited HER2 phosphorylation. In
total, 6/7 STS (86%) and 5/6 osteosarcoma (83%) cell lines
expressed t-HER2 in the absence/presence of EGF.
Inhibition of the phosphorylation of HER
family receptors and signalling factors in PI3K/AKT and
ras/raf/MAPK pathways by dacomitinib monotherapy
Five sarcoma cell lines (3 STS and 2 osteosarcoma)
were treated for 24 h with dacomitinib (PF299804) at its clinically
achievable total plasma concentration (200 nM) (20). As shown in Fig. 2, dacomitinib markedly blocked
EGF-induced p-HER1 expression in all 5 sarcoma cell lines, as well
as that of p-HER2 in 4/5. In addition, dacomitinib decreased the
activation of AKT and SAPK/JNK to baseline levels in all 5 sarcoma
cell lines, and downregulated the the levels of p-Erk and p-p38
MAPK in 4/5 sarcoma cell lines, apart from p-Erk in the 143B cells
and p-p38 in the 449B cells (Fig.
2).
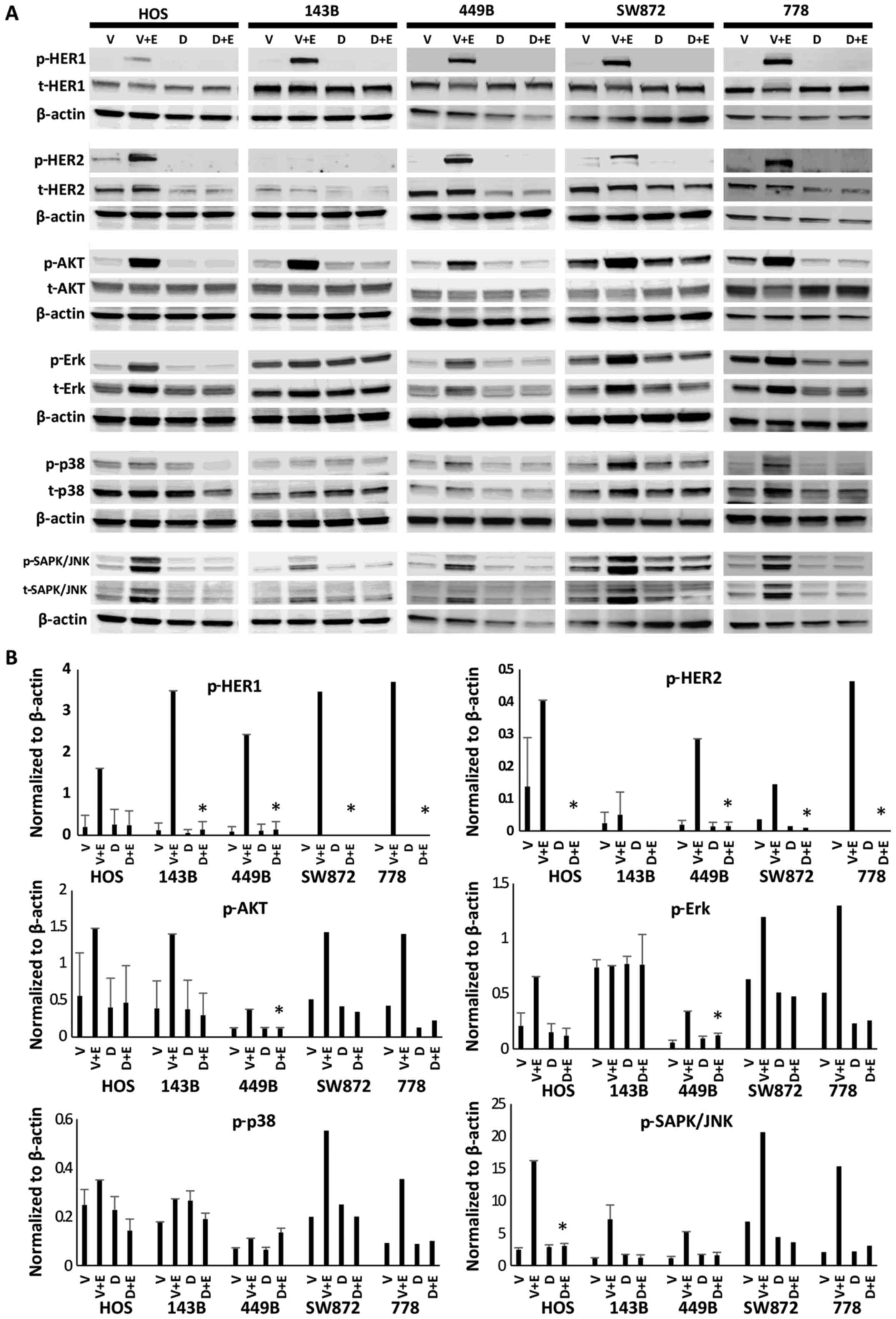 | Figure 2Dacomitinib (PF299804) decreased the
phosphorylation (activity) of the HER family receptors (HER1 and
HER2), as well as signalling factors in the PI3K/AKT and
ras/raf/MAPK downstream pathways (AKT, Erk, p38 MAPK and SAPK/JNK).
A panel of 5 sarcoma cell lines (HOS, 143B, 449B, SW872 and 778)
were treated with 200 nM of dacomitinib for 24 h, followed by
incubation with or without EGF for 15 min, and the the extracted
proteins were then immunoblotted with the indicated
phosphor-specific antibodies. Membranes used for analysis of
phosphorylated proteins were stripped and reblotted with the
respective total antibodies. The expression of β-actin was analysed
from the same cell lines as a loading control. V, vehicle control;
V+E, vehicle control plus EGF stimulation; D, 200 nM dacomitinib
treatment; D+E, 200 nM dacomitinib treatment plus EGF stimulation.
(A) Representative western blot images. (B) Expression levels of
the indicated proteins were quantified by densitometry and
normalized to β-actin loading control. P-values (D+E vs. V+E)
<0.05 were considered statistically significant.
*P<0.05. |
Growth inhibitory effects of dacomitinib
monotherapy on sarcoma cell lines
Following 72 h of treatment with 0.25–15 μM
dacomitinib, the anti-proliferative ability was evaluated. The
IC50 values are summarized in Table I. At the early time point (72 h of
treatment), all the cell lines exhibited a greater sensitivity to
dacomitinib [IC50 values: 1–5 μM for dacomitinib
versus 14–30 μM for gefitinib in our previous study on
gefitinib (12)]. However, the
IC50 values of dacomitinib in the sarcoma cell lines
were still 1,000-fold higher than those in the sensitive control
cell line, PC9 (1 nM). Extending the treatment window for longer
time period of up to 7 days, we found that all the cell lines
exhibited further growth inhibitory effects. The IC50
values for the 778, 449B, Saos-2 and SW684 cells were 3- to 5-fold
lower than those on day 3. In particular, the IC50
values for 6 sarcoma cell lines were scaled down to <1
μM.
 | Table IIC50 values of dacomitinib
in 13 sarcoma cell lines. |
Table I
IC50 values of dacomitinib
in 13 sarcoma cell lines.
| Anti-proliferation
| IC50 of
anti-colony formation (μM) |
|---|
| IC50
value on day 3 (μM) | IC50
value on day 7 (μM) |
|---|
| 449B | 4.4±0.8 | 1.3±0.3 | 0.677±0.057 |
| 778 | 3.1±0.9 | 0.9±0.2 | 1.326±0.032 |
| GCT | 2.2±0.1 | 1.3±0.1 | 1.142±0.027 |
| HT1080 | 2.2±0.5 | 0.9±0.2 | 0.781±0.058 |
| SW684 | 5.6±1.3 | 1.1±0.2 | No colonies
formed |
| SW872 | 3.7±0.6 | 1.6±0.3 | 0.875±0.089 |
| SW982 | 2.0±0.3 | 0.8±0.2 | 0.163±0.055 |
| SJSA | 3.9±1.3 |
1.5±0 | No colonies
formed |
| U2-OS | 3.7±1.5 |
0.5±0 | 0.278±0.083 |
| 143B | 4.6±0.1 | 2.4±0.1 | 0.184 |
| HOS | 3.6±1.0 | 1.3±0.5 | 0.455±0.033 |
| MG63 | 2.3±0.6 |
1.0±0 | No colonies
formed |
| Saos-2 | 1.9±0.3 | 0.6±0.1 | No colonies
formed |
| PC-9 (serving as
sensitive control) | 0.001 | – | – |
| A431 (serving as
sensitive control) | – | – | 0.044 |
In addition, clonogenic survival assay was also
performed to determine the long-term effects of dacomitinib on the
sarcoma cell lines. Dacomitinib at the concentration of <1.4
μM suppressed colony formation by 50% in the sarcoma cell
lines, with IC50 values of 0.163 and 0.184 μM for
the SW982 and 143B cells, respectively, which were higher than
those of the sensitive control cell line, A431 (IC50,
0.044 μM), treated with dacomitinib (Table I).
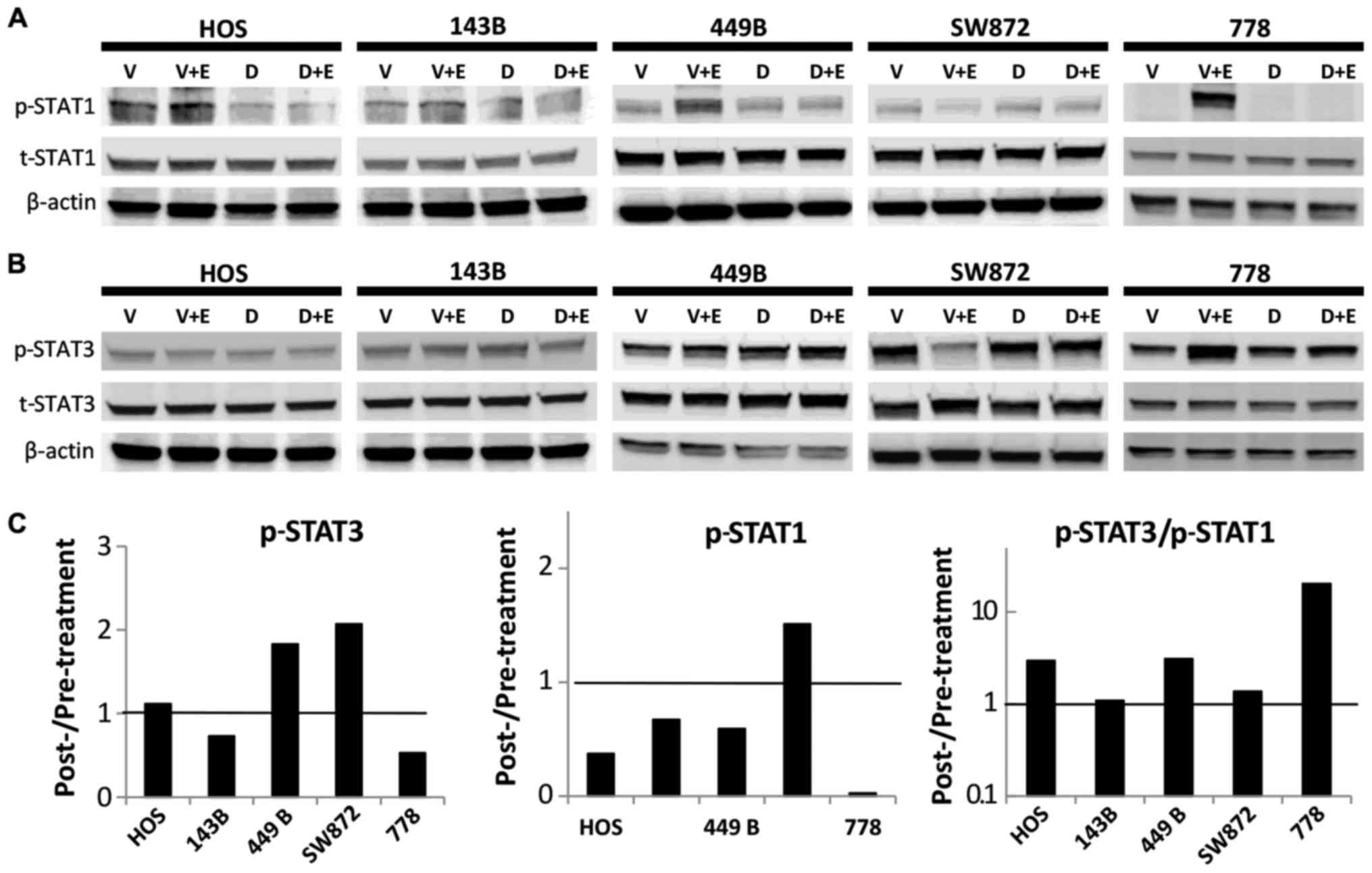
The STAT3 escape pathway and the
resistance of dacomitinib monotherapy in sarcoma
Although dacomitinib markedly inactivated HER family
members and downstream ras/raf/MAPK and PI3K/AKT pathways, it
failed to supress sarcoma cell growth and colony formation at
reasonable IC50 values, compared with the sensitive
control. The increased or unaltered ratio of p-STAT3/p-STAT1 was
found to be associated with the STAT3 escape pathway in our
previous study on the EGFR inhibitor, gefitinib, in sarcoma cell
lines (12). In the present study,
we found that dacomitinib inhibited p-STAT1 expression in 4/5
sarcoma cell lines by 97, 62, 41 and 33 in the 778, HOS, 449B and
143B cells, respectively, compared with the corresponding vehicle
control in the presence of EGF stimulation (Fig. 3A). However, the effects of
dacomitinib on p-STAT3 expression were highly variable: We observed
a decreased p-STAT3 expression in the 143B cells (27%) and 778
cells (47%); however, p-STAT3 expression was unaltered in the HOS
cells and it increased in the 449B cells (1.8-fold) and SW872
(2.1-fold) (Fig. 3B). The ratio of
p-STAT3/p-STAT1 was increased in 3/5 cell lines (HOS, 449B and 778
cells) and unaltered in the 143B and SW872 cells (Fig. 3C), suggesting that the STAT3
relative abundance and activation likely plays an important role in
sarcoma growth, maintenance and resistance mechanisms.
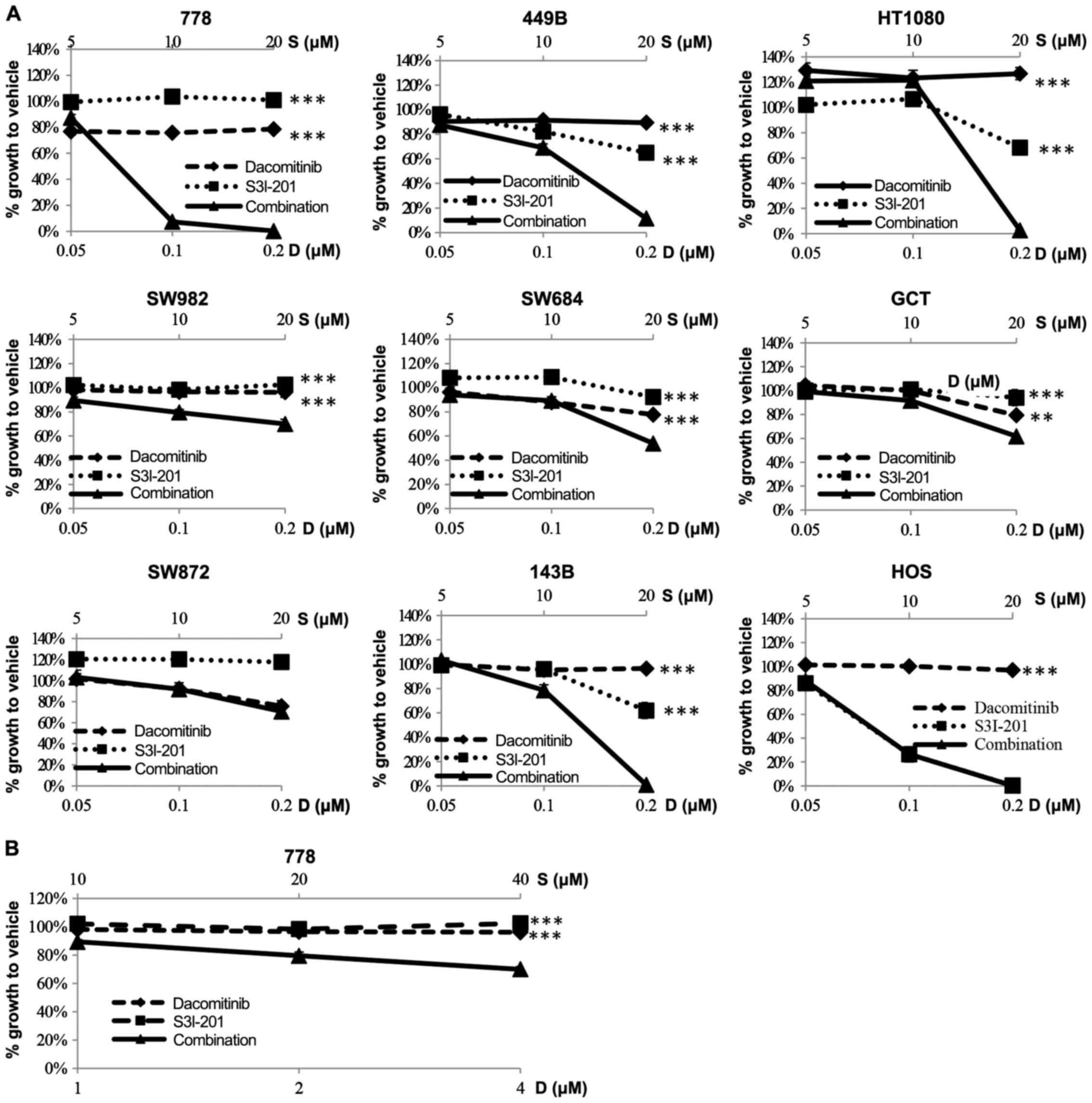
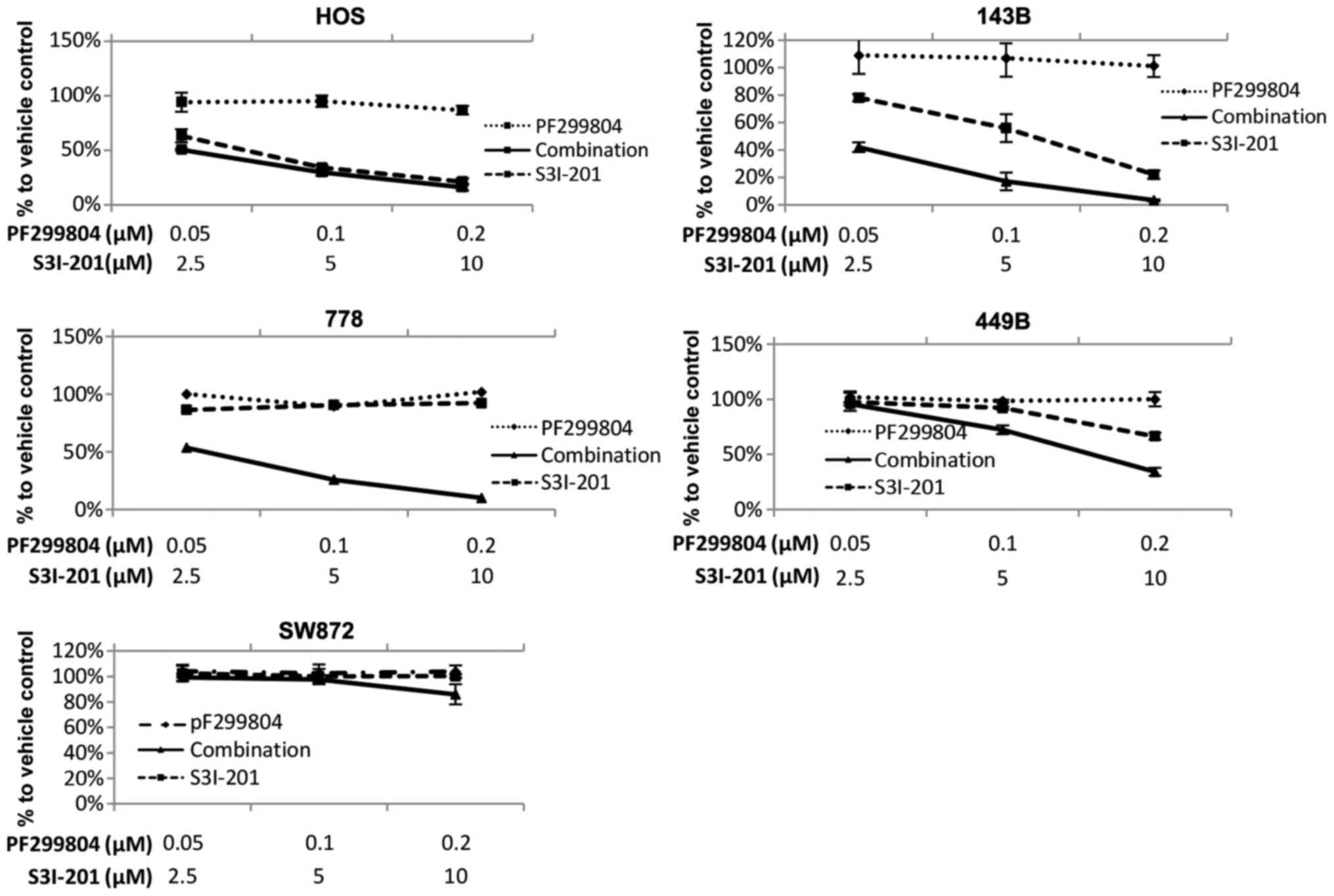
Enhancement of the sensitivity of sarcoma
cells to dacomitinib by the STAT3 inhibitor, S3I-201
A panel of 7 STS and 2 osteosarcoma (HOS and 143B)
cell lines were treated with the vehicle, dacomitinib, or S3I-201,
or a combination of dacomitinib and S3I-201 at 3 concentrations
(lower than the clinically achievable total plasma concentration
200 nM) in the constant ratio (dose ratio of dacomitinib to S3I-201
= 1:100). At 7 days post-administration, in 7/9 cell lines,
combination therapy achieved significantly enhanced
anti-proliferative effects (Fig.
4A). The HOS cells, which were the most sensitive (almost 100%)
to S3I-201 monotherapy, did not exhibit any further growth
inhibition. We also examined combination therapy in the
S3I-201-resistant SW872 cells (36) and found that these cells did not
undergo any further enhancement of the growth inhibitory effects.
Dacomitinib is ~98% bound to plasma proteins in human plasma, as
measured by equilibrium dialysis (kindly suggested by Dr Scott
Weinrich, Director of Early Clinical Development, Pfizer USA). This
indicates that only 2% constitutes the ‘free’ drug in the
circulation of patients; therefore, the 200 nM total plasma
concentration becomes 4 nM ‘free’ drug. Considering that it should
be the ‘free’ drug concentration at the site of action that exerts
the biological activity (39), we
aimed to emulate clinical exposure in vitro by the addition
of up to 4 nM dacomitinib to the most sensitive cell line, 778.
This achieved a significantly enhanced anti-proliferative effect
(combination versus dacomitinib: P=0.003; combination versus
S3I-201: P=0.0002) (Fig. 4B).
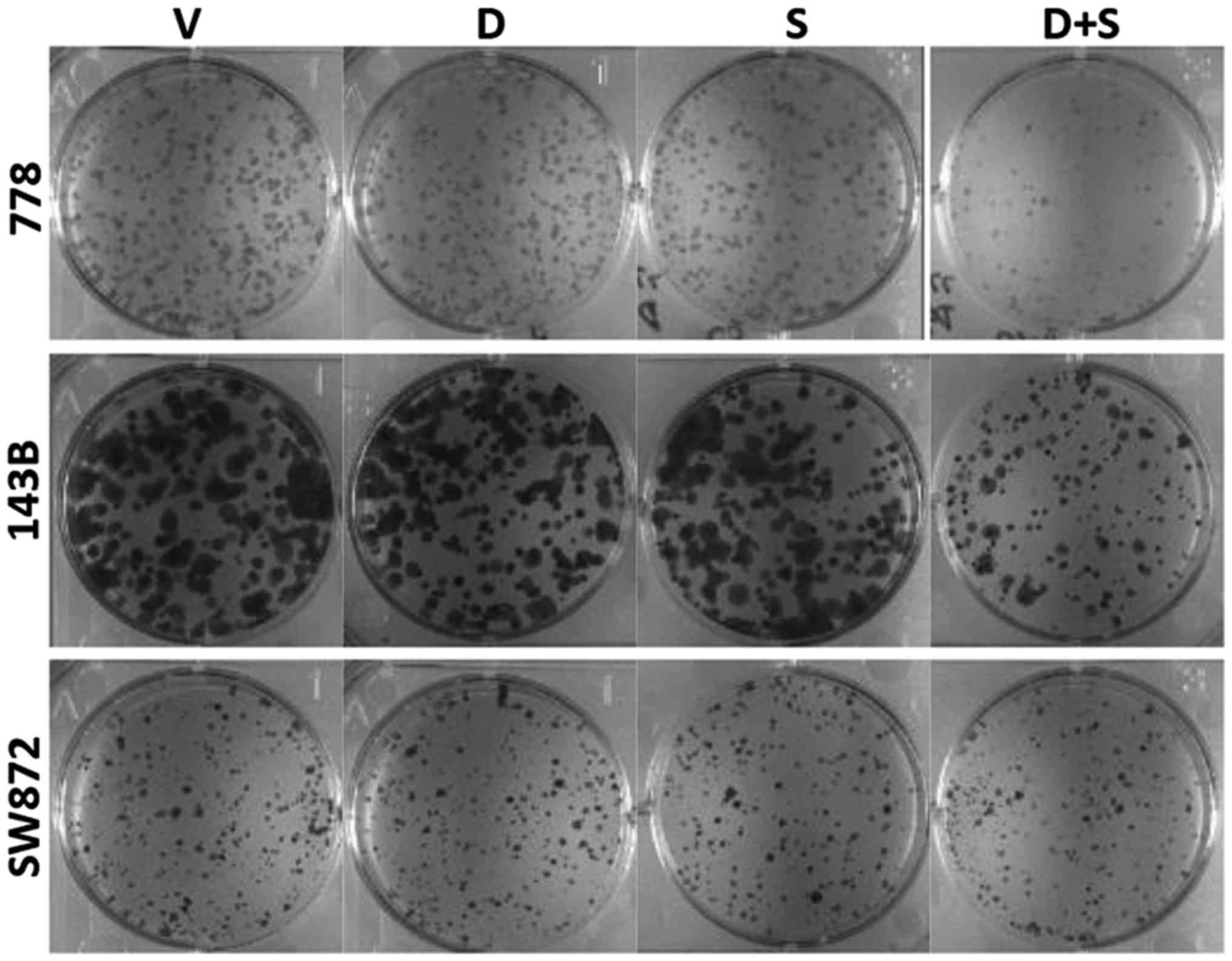
In addition, clonogenic assay was performed on the
778, 143B and SW872 cells, representing a sensitive STS cell line,
a sensitive osteosarcoma cell line and a resistant cell line,
respectively, to further examine the cell responses to the
combination therapy. Consistently, the 778 and 143B cells exhibited
an enhanced inhibition of their colony-forming ability (Fig. 5), whereas the SW872 cells were
still resistant to both monotherapies and combination therapy.
Enhancement of anoikis by dacomitinib and
S3I-201 in sarcoma cell lines
Anchorage-independent growth (the ability to evade
anoikis) using the 3D soft agar colony formation assay was then
applied to assess cancer metastatic (anoikis-resistance) ability in
sarcoma cell lines following treatment with dacomitinib and S3I-201
monotherapy or their combination. The results shown in Fig. 6 confirmed that combination therapy
markedly enhanced anoikis (apoptosis occurred when the cells
detached to the extracellular matrix) in the 778, 449B and 143B
cells. However, this enhancement was not observed in the HOS cells,
in which the combination therapy did not further enhance anoikis
compared to treatment with S3I-201 alone treatment, no in the SW872
cells, in which all treatments (both drug mono- and
combination-therapies) did not restore anoikis.
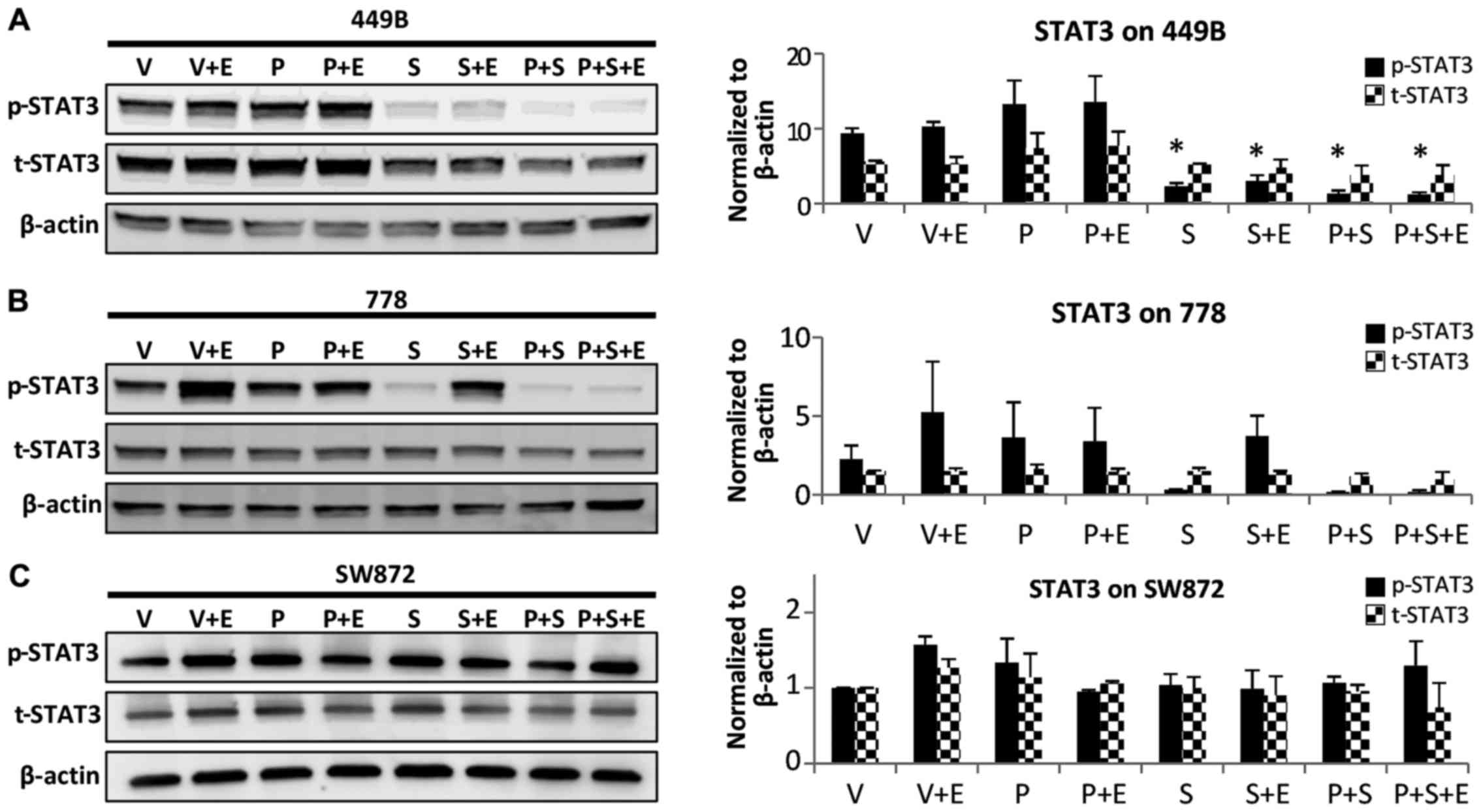
Contribution of the downregulation of
STAT3 phosphorylation to the enhanced effects observed with
combination therapy
Using different methods (crystal violet
colorimetric, clonogenic and anoikis assays), we demonstrated that
combination treatment with S3I-201 enhanced the sensitivity to
dacomitinib in the sarcoma cell lines. As a preliminary
investigation of the potential mechanisms of action behind the
enhancement, western blot analysis was performed on 3 sarcoma cell
lines, representing a strong enhancement (778 cells), moderate
enhancement (449B cells) and resistance (SW872 cells). In the 778
cells, treatment with S3I-201 alone downregulated constitutive
STAT3 phosphorylation; however, the blockage of p-STAT3 was
partially recovered by EGF stimulation. As expected, the
combination therapy induced almost complete (94%) inhibition of
STAT3 phosphorylation, even with EGF stimulation (Fig. 7). In the 449B cells, the addition
of 200 nM dacomitinib also led to the further blockage of STAT3
phosphorylation compared to treatment with S3I-201 alone (from 77
to 90%). However, in the SW872 cells, which exhibited resistance to
both monotherapies and combination treatment in our
anti-proliferation assay, combination therapy did not effectively
inhibit STAT3 phosphorylation, as shown by the results of western
blot analysis (Fig. 7).
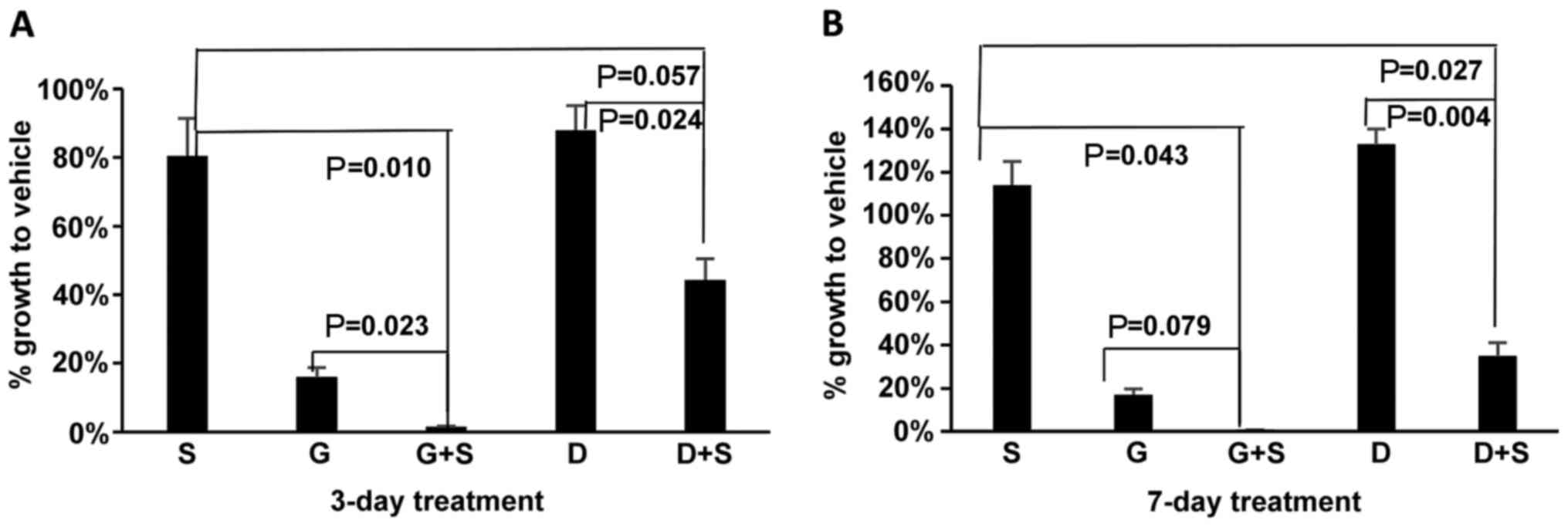
Comparison of panHER inhibition with HER1
in combination with STAT3 inhibition
Previously, we reported that the addition of the
STAT3 inhibitor, S3I-201, to the EGFR inhibitor, gefitinib,
achieved synergistic anti-proliferative and pro-apoptotic effects
in sarcoma cell lines (12). In
this study, to compare the potential effects of concurrent
inhibition using dacomitinib-S3I-201 with previous
gefitinib-S3I-201, we examined the growth-inhibiting ability of the
combination therapies at the clinically achievable concentrations
for each drug (10 μM for gefitinib and 200 nM for
dacomitinib) at the same time (Fig.
8). After 24 h of seeding, the HT1080 cells were treated with
the vehicle (DMSO), S3I-201, gefitinib, dacomitinib, gefitinib plus
S3I-201 or dacomitinib plus S3I-201 for 3 or 7 days. In terms of
combination versus monotherapy with dacomitinib or gefitinib,
treatment with dacomitinib led to a similar P-value (0.024) as
gefitinib (0.023) after 3 days of treatment, and to an even lower
P-value (0.004) than gefitinib (0.079) after 7 days of treatment.
The combination treatment with dacomitinib and S3I-201 (56 and 65%
growth inhibition on days 3 and 7, respectively) significantly
enhanced the anti-proliferative effects compared to dacomitinib
monotherapy; however, it exerted less potent inhibitory effects
than gefitinib-S3I-201 (98 and 99% inhibition) on sarcoma cell
growth.
Discussion
In this study, we discovered that apart from
EGFR/HER1, another HER family receptor, HER2, was also
overexpressed in human sarcoma cell lines. The panHER inhibitor,
dacomitinib, successfully inhibited the activation of HER family
members (p-EGFR and p-HER2), as well as HER downstream pathway
signalling transducers (p-AKT, p-Erk, p-p38 MAPK and p-SAPK/JNK).
Despite the suppression of these pathways, the results of cell
proliferation and colony formation assay revealed that all 13
sarcoma cell lines were insensitive to dacomitinib. We demonstrated
that dacomitinib increased or did not significantly alter the ratio
of oncogene p-STAT3 versus tumour suppressor p-STAT1, indicating
that STAT3 may represent an escape pathway that correlates with the
resistance of sarcoma cell lines to dacomitinib. To conclusively
determine causation will require additional studies with siRNA
and/or transgenic mouse models in the future. This mechanism (the
STAT3 escape pathway), if further validated, may prove to be a
targe-table mechanism of resistance to EGFR blockade in
sarcoma.
Even though the overexpression of p-EGFR and its
downstream signal transducers is noted in sarcoma tissues and cell
lines and is negatively associated with sarcoma outcomes, we have
shown a limited inhibitory effect of EGFR pathway blockade using
the specific EGFR inhibitor, gefitinib, due to the relative
activation of STAT3 (12). The
observation of HER2-positive expression in sarcoma cell lines
indicated that the blockade of a single receptor of the HER family
may be compromised by signalling through other members. HER2
overexpression has been demonstrated to activate STAT3 and to act
as a transcriptional co-activator of STAT3 and contribute to tumour
initiation and growth (15–17).
The activation of HER2 signalling has been reported to be
associated with the primary resistance of metastatic colorectal
carcinoma to EGFR-targeted therapy (40). In addition, MET
amplification/overexpression has also been reported to promote
resistance to gefitinib by driving the HER3-dependent activation of
PI3K in a gefitinib-sensitive lung cancer cell line (HCC827)
(41,42). Therefore, we hypothesized that
targeting all HER family members using a panHER inhibitor may
enhance the inhibitory effects of targeted therapy in sarcoma.
Dacomitinib is a highly effective panHER inhibitor both in
vitro and in vivo in a broad range of human cancer cell
lines (21–23,43–45).
In this study, we demonstrated that dacomitinib
markedly suppressed the activation of both EGFR/HER1 and HER2, as
well as their representative downstream signalling factors in
ras/raf/MAPK and PI3K/AKT pathways, including p-AKT, p-Erk, p-p38
and p-SAPK/JNK. This was consistent with the findings of other
studies on breast cancer and head and neck squamous cell carcinoma
cell lines (44,45). Despite this, the anti-proliferative
effect of dacomitinib in our study indicated that all 13 sarcoma
cell lines were resistant to the drug with IC50 values
of ~1 μM, greater than that of our sensitive control lung
cancer cell line, PC9 (1 nM), which is similar to previous reports
on non-sarcoma cell lines (21,46).
The results of western blot analysis revealed that dacomitinib did
not decrease the ratio of p-STAT3/p-STAT1. Dacomitinib, despite its
additional blockade of HER2 compared with gefitinib blocking HER1
only, showed lower IC50 values, but no improvement in
biologically and potentially clinically meaningful antitumour
effects. Rather than the EGF pathway not being relevant to tumour
promotion in sarcoma, this absence of benefit appears to be a
result of failure to overcome the STAT3 escape pathway, which may
be regulated by HER-independent upstream mediators. In head and
neck squamous cell carcinoma and lung adeno-carcinomas, STAT3
constitutive activation has been reported to use an
autocrine/paracrine-activating loop through alternative pathways,
such as IL-6/gp130 (47,48). HER-independent JAK/STAT3 (such as
IL-6/JAK/STAT3) leads to the ineffectiveness of targeting HER
family pathway. STAT3 is a more downstream point of convergence in
many ligand/receptor pathways (such as growth factor and cytokine
receptors) and non-receptor tyrosine kinase pathways (such as Src)
and consequently cross-talk among these signalling pathways may
contribute to resistance to EGFR/panHER inhibitors (49).
In this study, we found that the addition of S3I-201
to dacomitinib induced significant enhancement of the
anti-proliferative effects (7/9 cell lines), further inhibited
colony formation (2/3 cell lines) and enhanced anoikis (3/5 of
sarcoma cell lines). Our preliminary STAT3 expression and
regulation analysis revealed the additional inactivation of STAT3
by the combination treatment in sensitive sarcoma cell lines. By
contrast, in the SW872 liposarcoma cell line, which exhibited
resistance to the combination therapy in the anti-proliferation
assay, STAT3 activation was not inhibited by the combination
treatment. Comparing the current dacomitinib-S3I-201 combination
with the one in our previous study (gefitinib-S3I-201) (13), we found that dacomitinib-S3I-201
did not demonstrate superior anti-proliferative activities.
Consistent with our findings, the activation of
JAK/STAT3 signalling pathway mediated by autocrine and paracrine
IL-6R has been found to be associated with the development of drug
resistance to the irreversible panHER inhibitors, dacomitinib and
afatinib in non-small cell lung cancer (50). The blockade of the IL-6R/JAK/STAT3
signalling pathway significantly enhanced the sensitivity to these
irreversible inhibitors in both in vitro and in vivo
models. Taken together, these results of STAT inhibition used to
overcome primary resistance to EGFR blockade encourage the further
exploration of this approach. Further experiments will require
optimisation through next-generation inhibitors, different drug
ratios, and the sequence of drug administration in vitro,
followed by assessing the effectiveness and safety of combination
therapy using both panHER and STAT3 inhibitors in vivo in
sarcoma animal models.
We thus conclude that neither the first-generation
reversible specific EGFR inhibitor, gefitinib, nor the
second-generation irreversible panHER inhibitor, dacomitinib, as
single agents are likely to have clinical utility in sarcoma. The
relative abundance and activation of STAT3 may be involved in the
resistance mechanism in both EGFR- and panHR-targeted therapies. To
the best of our knowledge, our results are the first to demonstrate
the significantly and highly anti-proliferative effects of the
combination of the irreversible panHER inhibitor, dacomitinib, and
the STAT3 inhibitior, S3I-201, in sarcoma cell lines. These results
provide a rationale for further in vitro and in vivo
studies inhibiting both EGFR/panHER and STAT3 in combination for
the treatment of sarcoma.
Acknowledgments
Not applicable.
Funding
This study was supported by the Pfizer USA (grant
no. IIR #WS1914693). XW was awarded Rainbows for Kate PhD
Scholarship from the Australasian Sarcoma Study Group.
Availability of data and materials
The analysed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors’ contributions
XW contributed to performing the experiments, data
acquisition and analysis and manuscript drafting. JLY contributed
to Pfizer IIR grant application, project design, supervision, grant
management, statistical analysis and manuscript revision. DG and
PJC assisted in the study design and conception and revised the
manuscript. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sarcoma, Soft Tissue: Statistics. ASCO,
Cancer.Net Editorial Board; 2016, http://www.cancer.net/cancer-types/sarcoma-soft-tissue/statistics.
|
|
2
|
Baselga J and Arteaga CL: Critical update
and emerging trends in epidermal growth factor receptor targeting
in cancer. J Clin Oncol. 23:2445–2459. 2005. View Article : Google Scholar
|
|
3
|
Hynes NE and Lane HA: ERBB receptors and
cancer: The complexity of targeted inhibitors. Nat Rev Cancer.
5:341–354. 2005. View
Article : Google Scholar
|
|
4
|
Wang X, Batty KM, Crowe PJ, Goldstein D
and Yang JL: The potential of panHER inhibition in cancer. Front
Oncol. 5:22015. View Article : Google Scholar
|
|
5
|
Holbro T and Hynes NE: ErbB receptors:
Directing key signaling networks throughout life. Annu Rev
Pharmacol Toxicol. 44:195–217. 2004. View Article : Google Scholar
|
|
6
|
Yang JL, Hannan MT, Russell PJ and Crowe
PJ: Expression of HER1/EGFR protein in human soft tissue sarcomas.
Eur J Surg Oncol. 32:466–468. 2006. View Article : Google Scholar
|
|
7
|
Sato O, Wada T, Kawai A, Yamaguchi U,
Makimoto A, Kokai Y, Yamashita T, Chuman H, Beppu Y, Tani Y, et al:
Expression of epidermal growth factor receptor, ERBB2 and KIT in
adult soft tissue sarcomas: A clinicopathologic study of 281 cases.
Cancer. 103:1881–1890. 2005. View Article : Google Scholar
|
|
8
|
Teng HW, Wang HW, Chen WM, Chao TC, Hsieh
YY, Hsih CH, Tzeng CH, Chen PC and Yen CC: Prevalence and
prognostic influence of genomic changes of EGFR pathway markers in
synovial sarcoma. J Surg Oncol. 103:773–781. 2011. View Article : Google Scholar
|
|
9
|
Cascio MJ, O’Donnell RJ and Horvai AE:
Epithelioid sarcoma expresses epidermal growth factor receptor but
gene amplification and kinase domain mutations are rare. Mod
Pathol. 23:574–580. 2010. View Article : Google Scholar
|
|
10
|
Keizman D, Issakov J, Meller I, Maimon N,
Ish-Shalom M, Sher O and Merimsky O: Expression and significance of
EGFR in malignant peripheral nerve sheath tumor. J Neurooncol.
94:383–388. 2009. View Article : Google Scholar
|
|
11
|
Biscuola M, Van de Vijver K, Castilla MA,
Romero-Pérez L, López-García MÁ, Díaz-Martín J, Matias-Guiu X,
Oliva E and Palacios Calvo J: Oncogene alterations in endometrial
carcinosarcomas. Hum Pathol. 44:852–859. 2013. View Article : Google Scholar
|
|
12
|
Wang X, Goldstein D, Crowe PJ, Yang M,
Garrett K, Zeps N and Yang JL: Overcoming resistance of targeted
EGFR monotherapy by inhibition of STAT3 escape pathway in soft
tissue sarcoma. Oncotarget. 7:21496–21509. 2016.
|
|
13
|
Yang JL, Gupta RD, Goldstein D and Crowe
PJ: Significance of phosphorylated epidermal growth factor receptor
and its signal transducers in human soft tissue sarcoma. Int J Mol
Sci. 18:182017. View Article : Google Scholar
|
|
14
|
Ray-Coquard I, Le Cesne A, Whelan JS,
Schoffski P, Bui BN, Verweij J, Marreaud S, van Glabbeke M,
Hogendoorn P and Blay JY: A phase II study of gefitinib for
patients with advanced HER-1 expressing synovial sarcoma refractory
to doxorubicin-containing regimens. Oncologist. 13:467–473. 2008.
View Article : Google Scholar
|
|
15
|
Hawthorne VS, Huang WC, Neal CL, Tseng LM,
Hung MC and Yu D: ErbB2-mediated Src and signal transducer and
activator of transcription 3 activation leads to transcriptional
up-regulation of p21Cip1 and chemoresistance in breast cancer
cells. Mol Cancer Res. 7:592–600. 2009. View Article : Google Scholar
|
|
16
|
Proietti CJ, Rosemblit C, Beguelin W,
Rivas MA, Díaz Flaqué MC, Charreau EH, Schillaci R and Elizalde PV:
Activation of Stat3 by heregulin/ErbB-2 through the co-option of
progesterone receptor signaling drives breast cancer growth. Mol
Cell Biol. 29:1249–1265. 2009. View Article : Google Scholar
|
|
17
|
Béguelin W, Díaz Flaqué MC, Proietti CJ,
Cayrol F, Rivas MA, Tkach M, Rosemblit C, Tocci JM, Charreau EH,
Schillaci R, et al: Progesterone receptor induces ErbB-2 nuclear
translocation to promote breast cancer growth via a novel
transcriptional effect: ErbB-2 function as a coactivator of Stat3.
Mol Cell Biol. 30:5456–5472. 2010. View Article : Google Scholar
|
|
18
|
Gao L, Li F, Dong B, Zhang J, Rao Y, Cong
Y, Mao B and Chen X: Inhibition of STAT3 and ErbB2 suppresses tumor
growth, enhances radiosensitivity, and induces
mitochondria-dependent apoptosis in glioma cells. Int J Radiat
Oncol Biol Phys. 77:1223–1231. 2010. View Article : Google Scholar
|
|
19
|
Boyer MJ, Blackhall FH, Park K, Barrios
CH, Krzakowski MJ, Taylor I, Liang JQ, Denis LJ, O’Connell JP and
Ramalingam SS: Efficacy and safety of PF299804 versus erlotinib
(E): A glolbal, randomized phase II trial in patients (pts) with
advanced non-small cell lung cancer (NSCLC) after failure of
chemotherapy (CT). In: ASCO Annual Meeting 2010; J Clin Oncol;
Chicago, IL; 2010
|
|
20
|
Jänne PA, Boss DS, Camidge DR, Britten CD,
Engelman JA, Garon EB, Guo F, Wong S, Liang J, Letrent S, et al:
Phase I dose-escalation study of the pan-HER inhibitor, PF299804,
in patients with advanced malignant solid tumors. Clin Cancer Res.
17:1131–1139. 2011. View Article : Google Scholar
|
|
21
|
Engelman JA, Zejnullahu K, Gale C-M,
Lifshits E, Gonzales AJ, Shimamura T, Zhao F, Vincent PW, Naumov
GN, Bradner JE, et al: PF00299804, an irreversible pan-ERBB
inhibitor, is effective in lung cancer models with EGFR and ERBB2
mutations that are resistant to gefitinib. Cancer Res.
67:11924–11932. 2007. View Article : Google Scholar
|
|
22
|
Gonzales AJ, Hook KE, Althaus IW, Ellis
PA, Trachet E, Delaney AM, Harvey PJ, Ellis TA, Amato DM, Nelson
JM, et al: Antitumor activity and pharmacokinetic properties of
PF-00299804, a second-generation irreversible pan-erbB receptor
tyrosine kinase inhibitor. Mol Cancer Ther. 7:1880–1889. 2008.
View Article : Google Scholar
|
|
23
|
Nam H-J, Ching KA, Kan J, Kim HP, Han SW,
Im SA, Kim TY, Christensen JG, Oh DY and Bang YJ: Evaluation of the
antitumor effects and mechanisms of PF00299804, a pan-HER
inhibitor, alone or in combination with chemotherapy or targeted
agents in gastric cancer. Mol Cancer Ther. 11:439–451. 2012.
View Article : Google Scholar
|
|
24
|
Takahashi T, Boku N, Murakami H, Naito T,
Tsuya A, Nakamura Y, Ono A, Machida N, Yamazaki K, Watanabe J, et
al: Phase I and pharmacokinetic study of dacomitinib (PF-00299804),
an oral irreversible, small molecule inhibitor of human epidermal
growth factor receptor-1, -2, and -4 tyrosine kinases, in Japanese
patients with advanced solid tumors. Invest New Drugs.
30:2352–2363. 2012. View Article : Google Scholar
|
|
25
|
Park K, Cho BC, Kim DW, Ahn MJ, Lee SY,
Gernhardt D, Taylor I, Campbell AK, Zhang H, Giri N, et al: Safety
and efficacy of dacomitinib in korean patients with KRAS wild-type
advanced non-small-cell lung cancer refractory to chemotherapy and
erlotinib or gefitinib: A phase I/II trial. J Thorac Oncol.
9:1523–1531. 2014. View Article : Google Scholar
|
|
26
|
Reckamp KL, Giaccone G, Camidge DR,
Gadgeel SM, Khuri FR, Engelman JA, Koczywas M, Rajan A, Campbell
AK, Gernhardt D, et al: A phase 2 trial of dacomitinib
(PF-00299804), an oral, irreversible pan-HER (human epidermal
growth factor receptor) inhibitor, in patients with advanced
non-small cell lung cancer after failure of prior chemotherapy and
erlotinib. Cancer. 120:1145–1154. 2014. View Article : Google Scholar
|
|
27
|
Oh DY, Lee KW, Cho JY, Kang WK, Im SA, Kim
JW and Bang YJ: Phase II trial of dacomitinib in patients with
HER2-positive gastric cancer. Gastric Cancer. 19:1095–1103. 2016.
View Article : Google Scholar
|
|
28
|
Abdul Razak AR, Soulières D, Laurie SA,
Hotte SJ, Singh S, Winquist E, Chia S, Le Tourneau C, Nguyen-Tan
PF, Chen EX, et al: A phase II trial of dacomitinib, an oral
pan-human EGF receptor (HER) inhibitor, as first-line treatment in
recurrent and/or metastatic squamous-cell carcinoma of the head and
neck. Ann Oncol. 24:761–769. 2013. View Article : Google Scholar
|
|
29
|
Jänne PA, Ou SH, Kim DW, Oxnard GR,
Martins R, Kris MG, Dunphy F, Nishio M, O’Connell J, Paweletz C, et
al: Dacomitinib as first-line treatment in patients with clinically
or molecularly selected advanced non-small-cell lung cancer: A
multicentre, open-label, phase 2 trial. Lancet Oncol. 15:1433–1441.
2014. View Article : Google Scholar
|
|
30
|
Kris MG, Camidge DR, Giaccone G, Hida T,
Li BT, O’Connell J, Taylor I, Zhang H, Arcila ME, Goldberg Z, et
al: Targeting HER2 aberrations as actionable drivers in lung
cancers: Phase II trial of the pan-HER tyrosine kinase inhibitor
dacomitinib in patients with HER2-mutant or amplified tumors. Ann
Oncol. 26:1421–1427. 2015. View Article : Google Scholar
|
|
31
|
Ramalingam SS, Blackhall F, Krzakowski M,
Barrios CH, Park K, Bover I, Seog Heo D, Rosell R, Talbot DC, Frank
R, et al: Randomized phase II study of dacomitinib (PF-00299804),
an irreversible pan-human epidermal growth factor receptor
inhibitor, versus erlotinib in patients with advanced
non-small-cell lung cancer. J Clin Oncol. 30:3337–3344. 2012.
View Article : Google Scholar
|
|
32
|
Ellis PM, Shepherd FA, Millward M, Perrone
F, Seymour L, Liu G, Sun S, Cho BC, Morabito A, Leighl NB, et al
NCIC CTG; Australasian Lung Cancer Trials Group; NCI Naples
Clinical Trials Unit: Dacomitinib compared with placebo in
pretreated patients with advanced or metastatic non-small-cell lung
cancer (NCIC CTG BR.26): A double-blind, randomised, phase 3 trial.
Lancet Oncol. 15:1379–1388. 2014. View Article : Google Scholar
|
|
33
|
Ramalingam SS, Jänne PA, Mok T, O’Byrne K,
Boyer MJ, Von Pawel J, Pluzanski A, Shtivelband M, Docampo LI,
Bennouna J, et al: Dacomitinib versus erlotinib in patients with
advanced-stage, previously treated non-small-cell lung cancer
(ARCHER 1009): A randomised, double-blind, phase 3 trial. Lancet
Oncol. 15:1369–1378. 2014. View Article : Google Scholar
|
|
34
|
Ramalingam SS, O’Byrne K, Boyer M, Mok T,
Jänne PA, Zhang H, Liang J, Taylor I, Sbar EI and Paz-Ares L:
Dacomitinib versus erlotinib in patients with EGFR-mutated advanced
nonsmall-cell lung cancer (NSCLC): Pooled subset analyses from two
randomized trials. Ann Oncol. 27:423–429. 2016. View Article : Google Scholar
|
|
35
|
Wu YL, Cheng Y, Zhou X, Lee KH, Nakagawa
K, Niho S, Tsuji F, Linke R, Rosell R, Corral J, et al: Dacomitinib
versus gefitinib for the first-line treatment of advanced EGFR
mutation positive non-small cell lung cancer (ARCHER 1050): A
randomized, open-label phase III trial. Lancet Oncol. 18:1454–1466.
2017. View Article : Google Scholar
|
|
36
|
Wang X, Goldstein D, Crowe PJ and Yang
J-L: S3I-201, a Novel STAT3 Inhibitor, Inhibits Growth of Human
Soft Tissue Sarcoma Cell Lines. World J Cancer Res. 1:61–68. 2013.
View Article : Google Scholar
|
|
37
|
Wang X, Goldstein D, Crowe PJ and Yang JL:
Impact of STAT3 inhibition on survival of osteosarcoma cell lines.
Anticancer Res. 34:6537–6545. 2014.
|
|
38
|
Koizumi F, Shimoyama T, Taguchi F, Saijo N
and Nishio K: Establishment of a human non-small cell lung cancer
cell line resistant to gefitinib. Int J Cancer. 116:36–44. 2005.
View Article : Google Scholar
|
|
39
|
Muller PY and Milton MN: The determination
and interpretation of the therapeutic index in drug development.
Nat Rev Drug Discov. 11:751–761. 2012. View Article : Google Scholar
|
|
40
|
Ciardiello F and Normanno N: HER2
signaling and resistance to the anti-EGFR monoclonal antibody
cetuximab: A further step toward personalized medicine for patients
with colorectal cancer. Cancer Discov. 1:472–474. 2011. View Article : Google Scholar
|
|
41
|
Engelman JA, Zejnullahu K, Mitsudomi T,
Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen
J, et al: MET amplification leads to gefitinib resistance in lung
cancer by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar
|
|
42
|
Bean J, Brennan C, Shih JY, Riely G, Viale
A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, et al: MET
amplification occurs with or without T790M mutations in EGFR mutant
lung tumors with acquired resistance to gefitinib or erlotinib.
Proc Natl Acad Sci USA. 104:20932–20937. 2007. View Article : Google Scholar
|
|
43
|
Nam H-J, Kim H-P, Yoon Y-K, Song SH, Min
AR, Han SW, Im SA, Kim TY, Oh DY and Bang YJ: The irreversible
pan-HER inhibitor PF00299804 alone or combined with gemcitabine has
an antitumor effect in biliary tract cancer cell lines. Invest New
Drugs. 30:2148–2160. 2012. View Article : Google Scholar
|
|
44
|
Kalous O, Conklin D, Desai AJ, O’Brien NA,
Ginther C, Anderson L, Cohen DJ, Britten CD, Taylor I, Christensen
JG, et al: Dacomitinib (PF-00299804), an irreversible Pan-HER
inhibitor, inhibits proliferation of HER2-amplified breast cancer
cell lines resistant to trastuzumab and lapatinib. Mol Cancer Ther.
11:1978–1987. 2012. View Article : Google Scholar
|
|
45
|
Ather F, Hamidi H, Fejzo MS, Letrent S,
Finn RS, Kabbinavar F, Head C and Wong SG: Dacomitinib, an
irreversible Pan-ErbB inhibitor significantly abrogates growth in
head and neck cancer models that exhibit low response to cetuximab.
PLoS One. 8:e561122013. View Article : Google Scholar
|
|
46
|
Ercan D, Zejnullahu K, Yonesaka K, Xiao Y,
Capelletti M, Rogers A, Lifshits E, Brown A, Lee C, Christensen JG,
et al: Amplification of EGFR T790M causes resistance to an
irreversible EGFR inhibitor. Oncogene. 29:2346–2356. 2010.
View Article : Google Scholar
|
|
47
|
Sriuranpong V, Park JI, Amornphimoltham P,
Patel V, Nelkin BD and Gutkind JS: Epidermal growth factor
receptor-independent constitutive activation of STAT3 in head and
neck squamous cell carcinoma is mediated by the autocrine/paracrine
stimulation of the interleukin 6/gp130 cytokine system. Cancer Res.
63:2948–2956. 2003.
|
|
48
|
Gao SP, Mark KG, Leslie K, Pao W, Motoi N,
Gerald WL, Travis WD, Bornmann W, Veach D, Clarkson B, et al:
Mutations in the EGFR kinase domain mediate STAT3 activation via
IL-6 production in human lung adenocarcinomas. J Clin Invest.
117:3846–3856. 2007. View Article : Google Scholar
|
|
49
|
Wang X, Crowe PJ, Goldstein D and Yang JL:
STAT3 inhibition, a novel approach to enhancing targeted therapy in
human cancers (Review). Int J Oncol. 41:1181–1191. 2012. View Article : Google Scholar
|
|
50
|
Kim SM, Kwon O-J, Hong YK, Kim JH, Solca
F, Ha SJ, Soo RA, Christensen JG, Lee JH and Cho BC: Activation of
IL-6R/JAK1/STAT3 signaling induces de novo resistance to
irreversible EGFR inhibitors in non-small cell lung cancer with
T790M resistance mutation. Mol Cancer Ther. 11:2254–2264. 2012.
View Article : Google Scholar
|