Introduction
Urinary bladder cancer has become one of the most
common urological malignancies, and its incidence ranks eighth
among all malignant types of cancer. It is estimated that 79,030
new cases of bladder cancer and 16,870 associated mortalities
occurred in 2017 (1).
Muscle-invasive bladder cancer patients are at a great risk of
recurrence and metastasis subsequent to radical cystectomy.
Furthermore, the 5-year survival of these patients is only 30–50%
(2,3). Chemotherapy is a beneficial treatment
for prolonging the survival time of patients at an advanced tumor
stage. Gemcitabine, a cytosine analogue that inhibits DNA
synthesis, has been widely used to treat bladder cancer, and
gemcitabine in combination with other chemotherapeutic drugs has
become the preferred standard treatment for advanced urothelial
carcinoma (4–6). However, drug resistance leads to
treatment failure in bladder cancer. Therefore, it is important to
explore the underlying mechanism of gemcitabine resistance.
It has been reported that the tumor microenvironment
is involved in the development of chemoresistance (7). Solid tumors are commonly exposed to a
hypoxic environment due to their abnormal vascular system,
treatment- or malignancy-associated anemia, and low levels of
intra-tumor infusion. The resulting hypoxia becomes an important
feature of the tumor microenvironment (8). The adaptive response of cells to
hypoxia is achieved by altering the transcription of certain genes.
Hypoxia-inducible factor 1 (HIF-1) is an important nuclear
transcription factor that regulates hypoxia responses in cells.
HIF-1 controls the expression of several downstream genes, which
affect energy metabolism, glycolysis, angiogenesis, DNA repair, and
the cell cycle progression. These genes not only control tumor
proliferation and metastasis, but also promote chemotherapy
resistance (9–11).
In hepatocellular carcinoma cells, autophagy is a
protective mechanism involved in the resistance to chemotherapy
under hypoxic conditions (12).
Cell autophagy is a complex process involving lysosomal degradation
of intracellularly damaged organelles and long-lived proteins,
providing for the reuse of raw materials for cell survival under
external conditions, such as hypoxia, a lack of growth factors or
nutrient starvation conditions (13). It has been demonstrated that HIF-1
is involved in autophagy regulation (14). The downregulation of HIF-1α
suppresses autophagy and promotes anticancer agent-induced
apoptosis (15).
Therefore, in order to validate the hypothesis that
HIF-1α mediates hypoxia-induced protective autophagy associated
with gemcitabine resistance, the present study investigated the
impact of hypoxia on cells treated with gemcitabine. Next, the
study examined whether autophagy inhibition reversed the
cytotoxicity of bladder cancer cells under hypoxia, and
investigated the underlying mechanism of hypoxia-induced autophagy
leading to gemcitabine resistance.
Materials and methods
Reagents and antibodies
The anti-light chain 3B (LC3B) antibody (1:1,000;
Cat. no. L7543) was purchased from Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany), the anti-p62 antibody (1:2,000; Cat. no.
610497) was obtained from BD Biosciences (New York, NY, USA), and
the anti-HIF-1α antibody (1:1,000; Cat. no. 113642) was purchased
from Abcam (Cambridge, MA, USA). Anti-poly(ADP-ribose) polymerase
(PARP; 1:1,000; Cat. no. 95325), anti-cleaved caspase-3 (1:1, 000;
Cat. no. 9664S) and anti-Beclin1 antibodies (1:1,000; Cat. no.
3495S) were obtained from Cell Signaling Technology, Inc. (Danvers,
MA, USA). An antibody against BCL2/adenovirus E1B 19 kDa
protein-interacting protein 3 (BNIP3;1:500; Cat. no. sc-56167) was
obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
Anti-β-actin antibody (1:1,000; Cat. no. Abm-0001) was from Zoonbio
Biotechnology Co., Ltd. (Nanjing, China), while anti-rabbit
(1:5,000; Cat. no. ASS1006)/mouse (1:5,000; Cat. no. ASS1021)
secondary antibodies were purchased from Abgent (San Diego, CA,
USA). 3-Methyladenine (3MA; 5 mM), chloroquine (CQ; 10 µM),
gemcitabine and cell counting kit-8 (CCK-8) reagents were obtained
from Selleck Chemicals (Houston, TX, USA). 3MA and CQ were both
formulated in triple-distilled water. The bladder cancer cells were
treated with 3MA or CQ for 1 h at room temperature prior to
stimulation with hypoxia and GEM. RPMI-1640 medium was obtained
from HyClone (GE Healthcare Life Sciences, Logan, UT, USA). Fetal
bovine serum (FBS) and trypsin were purchased from Thermo Fisher
Scientific, Inc. (Gibco, Waltham, MA, USA). The green fluorescent
protein (GFP)-LC3 adenoviral vectors were acquired from Hanbio
Biotechnology Co., Ltd. (Shanghai, China).
Tissue sample collection
A total of 20 paired specimens of bladder cancer and
matched adjacent tissues were collected from the Department of
Pathology, The First Affiliated Hospital of Chongqing Medical
University. All specimens were histopathologically diagnosed. Among
the patients, there were 16 males and 4 females, aged 50–80 years
with a mean age of 66.5 years. They were admitted from January,
2013 to October, 2017. No patient received chemotherapy or
radiotherapy prior to surgery. In total, 6 patients underwent
transurethral resection of the bladder tumor (TURBT) and 14
patients underwent radical curative resection the bladder tumor
(RC). Pathological grading and staging were determined according to
the WHO 1973 criteria for grade and the 2002 TNM classification
system (16,17). The study was approved by the Ethics
Committee of the First Affiliated Hospital of Chongqing Medical
University, and written informed consent was obtained from the
patients or the patients' families.
Cell lines and culture conditions
The human bladder cancer T24 cells were provided by
the Chongqing Key Laboratory of Molecular Oncology and Epigenetics
at the First Affiliated Hospital of Chongqing Medical University
[Chongqing, China; the cells were originally obtained from the
American Type Culture Collection (ATCC), Manassas, VA, USA]. The
cells were maintained in RPMI-1640 medium supplemented with 10%
FBS, 0.1 mg/ml penicillin and 0.1 mg/ml streptomycin at 37°C. In
order to simulate the hypoxic environment, a hypoxia incubator
(Sanyo Electric Co., Ltd., Osaka, Japan) with 1% O2 at
37°C was used for cell cultures under hypoxic conditions, as
previously described (18).
Cell viability assay
Cells were seeded in 96-well plates at
5×103 cells/well with 200 µl medium for 24 h and
then treated with gemcitabine at the indicated concentrations (0,
5, 10, 20, 30, 40, 50 and 60 µM) under hypoxic or normoxic
conditions for a further 24 h. In order to simulate a hypoxic
environment, a hypoxic incubator (Sanyo Electric Co., Ltd.) with 1%
O2 at 37°C was used to expose the cells to hypoxia.
Following incubation, 10 µl CCK-8 and 100 µl medium
were added into each well, and incubated for 1 h. The absorbance
(450 nm) was measured by the Tecan Infinite F200/M200 type
multifunction microplate reader (Tecan Group, Ltd., Männedorf,
Switzerland). The viability percentage of the cells based on the
optical density (OD) was calculated as follows: Cell viability (%)
= (Average OD value of the experimental group − average OD value of
blank group) / (average OD value of control group − average OD
value of blank group) × 100%. All experiments were conducted in
triplicate. The half maximal inhibitory concentration
(IC50) was calculated using SPSS software (version 17.0;
SPSS, Inc., Chicago, IL, USA).
Annexin V/propidium iodide (PI)
assay
The cells were treated as follows: i) When the cell
density reached 70–80%, the cells were pre-treated with 3MA (5 mM)
for 1 h, and then cells were incubated under hypoxic or normoxic
conditions for 24 h with or without gemcitabine (5 µM)
treatment, respectively. The specific groups were as follows: The
normoxic control group, hypoxic group, hypoxia plus 3MA group, GEM
group, GEM group under hypoxic conditions and gemcitabine combined
with the 3MA group under hypoxic conditions. ii) When the cell
density reached 40–50%, HIF-1α siRNA and NC siRNA were transfected
into T24 cells for 48 h, and the cells were then incubated under
hypoxic or normoxic conditions for 24 h with or without gemcitabine
(5 µM) treatment, respectively. The specific groups were as
follows: The normoxic control group, hypoxia group, GEM group, GEM
group under hypoxic conditions, GEM with NC siRNA group under
hypoxic conditions and the GEM with siHIF-1α group under hypoxic
conditions. The cells were then trypsinized, washed with cold PBS 3
times, collected and then resuspended in 200 µl Annexin
V-FITC binding buffer, which included 5 µl Annexin V-FITC
and 10 µl PI for 10 min in the dark. For this, the Annexin
V-FITC and PI kit (Beyotime Institute of Biotechnology, Haimen,
China) was used. Subsequently, all cells were stained for 10 min in
the dark. Apoptotic cells were measured by flow cytometry (BD
Biosciences, San Jose, CA, USA).
Electron microscopy
Subsequent to the indicated treatments, the cells
were digested by trypsin and collected by centrifugation at 1,200 ×
g for 10 min at room temperature. The cells were sequentially fixed
with 2.5% glutaraldehyde for 2 h and 1% osmium tetroxide for 1 h,
and then dehydrated with acetone and embedded in Epon resin
(Sigma-Aldrich/Merck KGaA, Darmstadt, Germany) and the embedded
material was then cut into 50-nm-thick sections using an
ultramicrotome. Ultra-thin (50 nm) slices were double-stained with
uranyl acetate and lead citrate (both from Sigma-Aldrich/Merck
KGaA) for 15 min at room temperature. Subsequently, a transmission
electron microscope (Hitachi-HTT7700; Hitachi, Ltd., Tokyo, Japan)
was used to observe the autophagosomes.
Confocal microscopy
Bladder cancer cells were seeded
(105/well) into confocal culture dishes (20 mm; NEST
Biotechnology Co., Ltd., Wuxi, China) for 24 h, and an adenovirus
vector carrying GFP-LC3 (acquired from Hanbio Biotechnology Co.,
Ltd.) was transfected into the cells for 24 h according to the
manufacturer's instructions. Subsequent to the indicated
treatments, the cells were fixed in 4% formaldehyde at room
temperature for 10 min and then washed with phosphate-buffered
saline (PBS). The samples were observed under a confocal laser
scanning microscope (Carl Zeiss-LSM700; Zeiss AG, Jena, Germany),
and the numbers of GFP-LC3-positive puncta in each cell were
counted by ImageJ software version 1.48 (National Institutes of
Health, Bethesda, MD, USA).
Small interfering RNA (siRNA)
transfection
The bladder cancer cells were transfected with the
following: HIF-1α siRNA, 5′-GCUGAUUUGUGAACCCAUUTT-3′ (sense) and
5′-AAUGGGUUCACAAAUCAGCTT-3′ (antisense); control siRNA,
5′-UUCUCCGAACGUGUCACGUTT-3′ (sense) and 5′-ACGUGACACGUUCGGAGAATT-3′
(anti-sense). Transfection reagents (siRNA-Mate; GenePharma Co.,
Ltd., Shanghai, China) and Opti-MEM (Invitrogen; Thermo Fisher
Scientific, Inc.) were used based according to the manufacturer's
instructions. Transfection was performed at room temperature for 10
min. The knockdown efficiency was then measured by
immunoblotting.
Immunoblotting
In order to extract total protein lysates, treated
cells were lysed in radioimmunoprecipitation assay lysis buffer and
1% phenylmethanesulfonyl fluoride (both from Beyotime Institute of
Biotechnology) and centrifuged at 12,000 × g for 15 min at 4°C. A
bicinchoninic acid kit (Beyotime Institute of Biotechnology) was
then used to assess the protein concentration. Identical amounts of
protein (40 µg) in each lane were separated with SDS-PAGE
(12% gels) and transferred onto polyvinylidene difluoride
membranes. After blocking in 5% fat-free milk in Tris-buffered
saline/Tween-20 (TBST) for 1 h, the bands were probed with various
antibodies (mentioned above) at 4°C overnight. On the following
day, the bands were washed three times with TBST for 10 min, then
incubated with the goat anti-rabbit/mouse secondary antibody for 1
h, and washed again three times with TBST. Finally, the bands were
exposed to an enhanced chemiluminescence detection reagent (EMD
Millipore, Burlington, MA, USA) and images were captured using the
Fusion Chemical Imaging System (Vilber Lourmat, Paris, France). The
membranes were exposed to a gel imager and the expression of the
target protein was visualized by detecting specific bands. ImageJ
software version 1.48 was used to quantify each specific blot
band.
Immunohistochemistry
Formalin-fixed and paraffin-embedded tissue sections
were dehydrated with xylene, hydrated with ethanol, and then soaked
in 0.01 mol/l citrate buffer for 30 min at 96°C to conduct antigen
repair. Next, 3% hydrogen peroxide was used to eliminate the
endogenous peroxidase. The sections were then blocked with 10% goat
serum at 37°C for 30 min and incubated with the anti-HIF-1α primary
antibody overnight at 4°C. On the following day, the slides were
incubated at 37°C for 30 min, washed with PBS, and incubated with
the horseradish peroxidase-conjugated secondary antibody (OriGene
Technologies, Inc., Beijing, China) for 30 min. The slides were
then stain with DAB (OriGene Technologies, Inc.) for ~1 min, and
the cell nucleus was stained with hematoxylin for 10 sec. PBS
instead of the primary antibody was used as a negative control.
Differential expression of HIF-1α staining was scored using a
semiquantitative grading system based on the staining intensity and
extent. The HIF-1 staining extent was scored according to the
percentage of positive staining cells as follows: 0, positive cells
of ≤5%; 1, 10–25% positive cells; 2, 26–50% positive cells; 3,
51–75% positive cells; and 4, >75% positive cells. Similarly,
the HIF-1 staining intensity was scored as follows: 0, no staining;
1, low staining; 2, mild staining; and 3, high staining. The final
score was the sum of the two indicators, and was between 0 and 7. A
score of ≥4 was considered to indicate high HIF-1α expression,
while a score of <4 was defined as low HIF-1α expression
(19). The immunohistochemical
staining level was assessed and scored by two independent
pathologists, who were blinded to the origin of the tumor and
paracarcinoma tissues.
Statistical analysis
At least three independent experiments were utilized
to collect the quantitative data, and results are presented as the
mean ± standard deviation. SPSS software (version 17.0; SPSS, Inc.)
was used for the statistical analyses. One-way analysis of variance
with Fisher's Least Significant Difference post hoc test was
applied to analyze the statistical differences among different
treatment groups. The statistical difference between two groups was
analyzed by the least significant difference t-test. P<0.05 was
considered to be an indicator of a statistically significant
difference.
Results
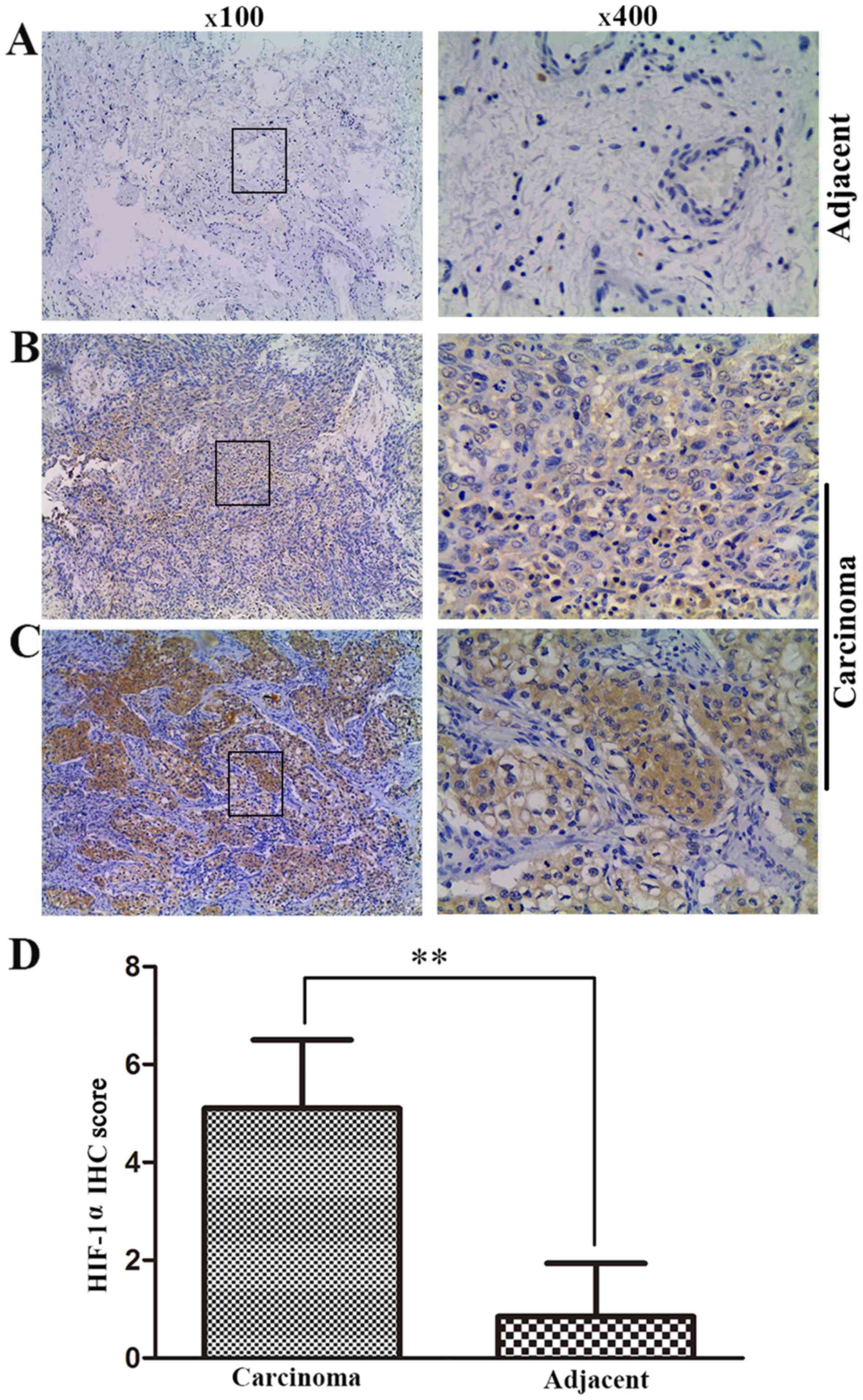
Overexpression of HIF-1α in tumor tissues
of bladder cancer patients
To determine whether hypoxia was a general
characteristic in tumor tissues, the present study tested the
expression of the hypoxia-specific indicator HIF-1α in 20 paired
bladder cancer and adjacent non-cancerous tissues using
immunohistochemistry. HIF-1α was mostly detected in the cytoplasm,
and the majority of the tumor samples had higher HIF-1α expression
as compared with that in the matched paracarcinoma tissues
(Fig. 1A–C). The quantitative
scores of HIF-1α expression in tumor tissues were almost 5-fold
greater compared with those of the paracarcinoma tissues (5.15 vs.
0.71, respectively; P<0.01; Fig.
1D). These results revealed that hypoxia was a general
characteristic in bladder cancer.
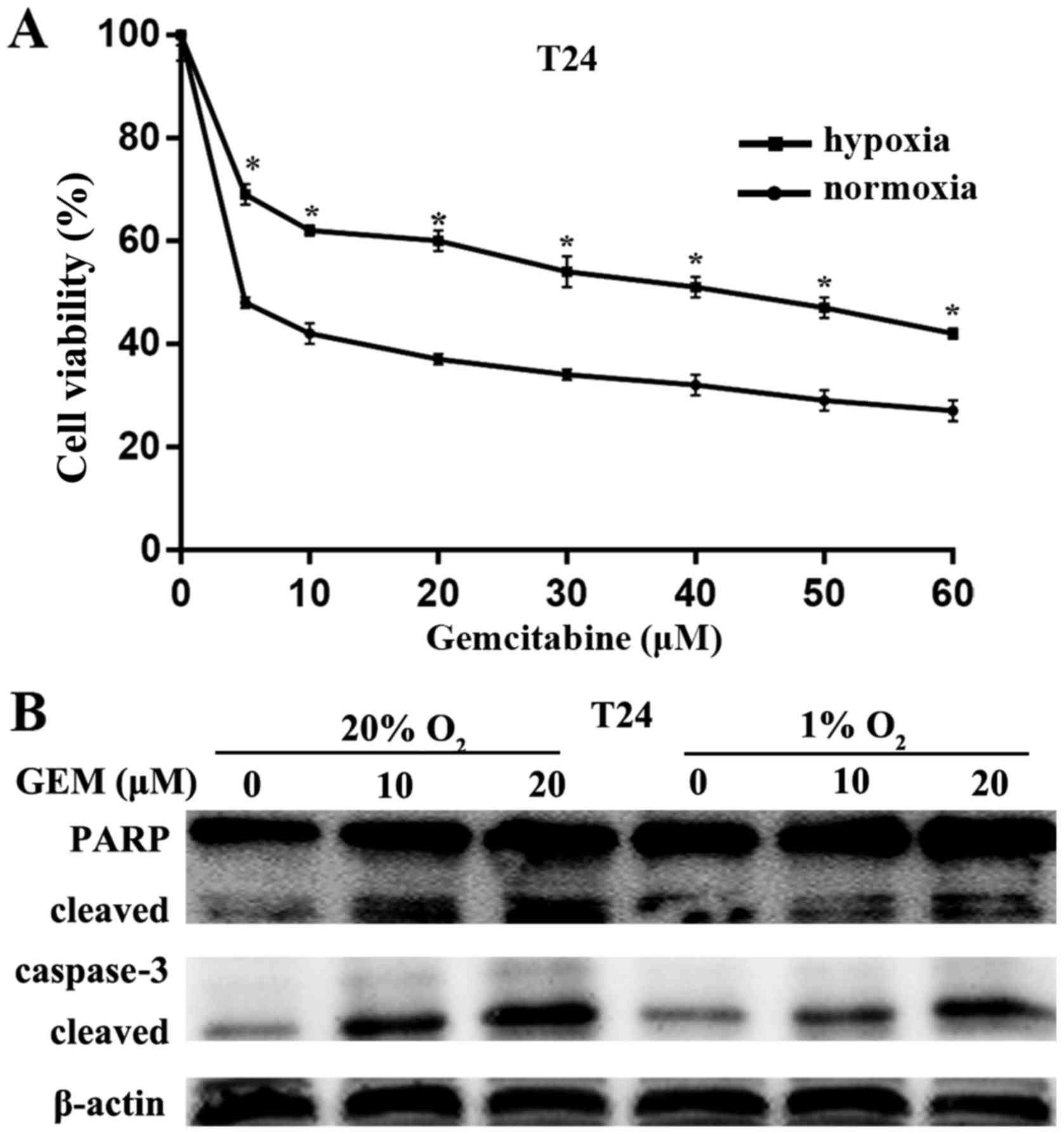
Hypoxia induces gemcitabine resistance in
bladder cancer cells
To detect the impact of hypoxia on gemcitabine
resistance, the chemosensitivity of the T24 cells was examined
under hypoxic or normoxic conditions. The cells were exposed to a
series of concentrations of gemcitabine ranging between 0 and 60
µM for 24 h under normoxic conditions (20% O2) or
hypoxia (1% O2). Subsequently, the CCK-8 assay was used
to evaluate the cell viability (Fig.
2A). The cell viability decreased in a dose-dependent manner
and was higher conditions as compared with that under normoxic
conditions. The half maximal inhibitory concentration
(IC50) value for T24 cells under normoxic conditions was
4.104±0.6132 µM, while the IC50 value of T24
cells conditions was 38.40±1.001 µM. Therefore, the
gemcitabine concentration of 5 µM was utilized for the
treatment of T24 cells in subsequent experiments. Furthermore,
gemcitabine caused the cleaved caspase-3 and cleavage of PARP in a
dose-dependent manner (Fig. 2B).
Under normoxic conditions, gemcitabine caused more significant
cleaved caspase-3 and cleavage of PARP as compared with that under
hypoxia (Fig. 2B). These data
suggested that hypoxia reduced the chemosensitivity of bladder
cancer cells to gemcitabine.
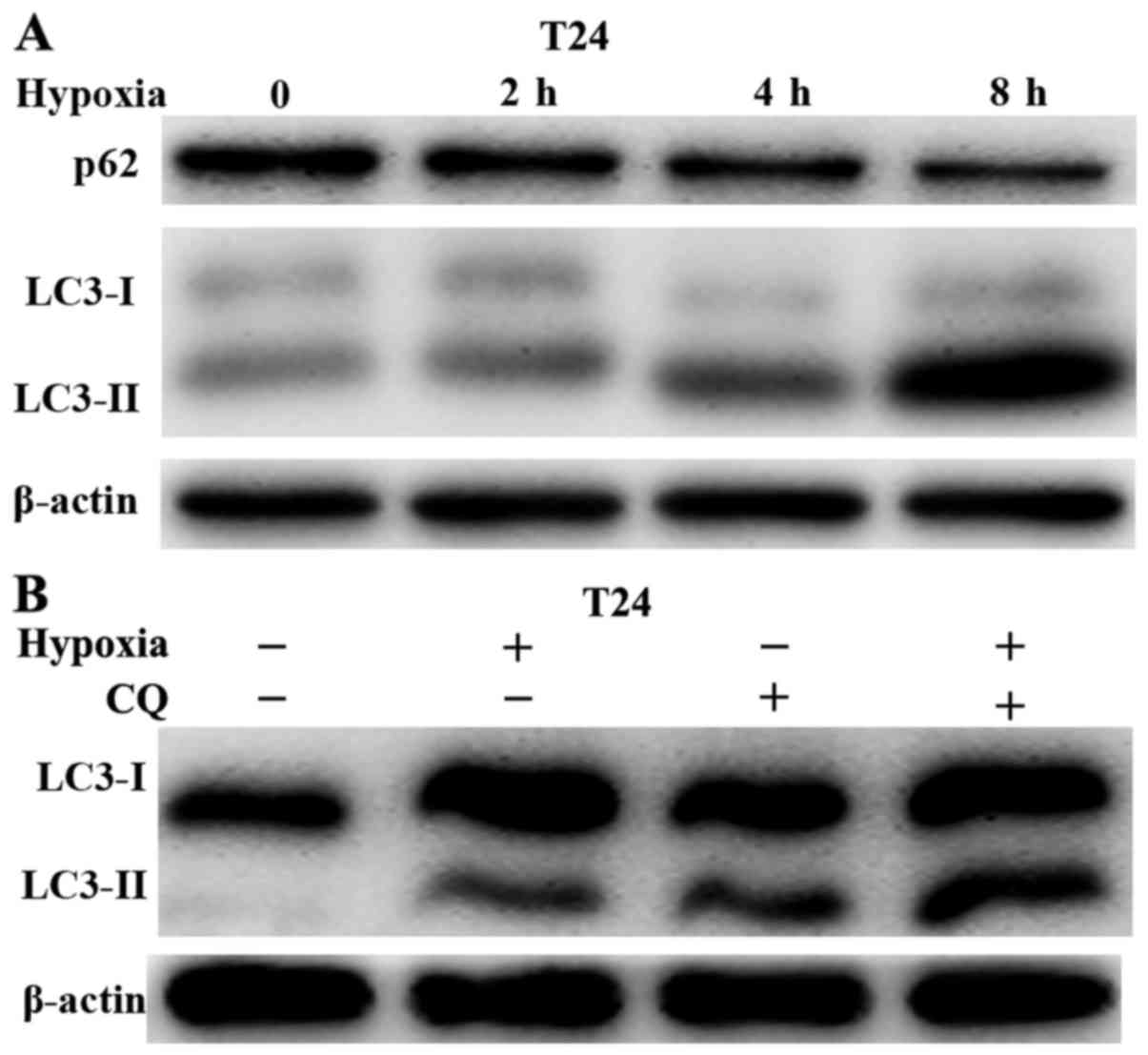
Hypoxia induces autophagy in bladder
cancer cells
Previous studies (20,21)
have demonstrated that hypoxia leads to resistance to gemcitabine,
and it was further demonstrated that autophagy was induced during
hypoxia and nutritional deficiency. Thus, the current study
investigated whether hypoxia induced autophagy in bladder cancer
cells. First, immunoblotting was used to confirm the occurrence of
autophagy under hypoxic conditions. Hypoxia improved the expression
of LC3-II, with p62 degradation occurring in a time-dependent
manner (Fig. 3A). In addition,
autophagy flux was examined following co-treatment with hypoxia and
a lysosomal inhibitor, CQ (10 µM). Although hypoxia or CQ
alone increased LC3-II levels, the combination of hypoxia with CQ
resulted in a marked increase in LC3-II levels (Fig. 3B). Taken together, these data
revealed that hypoxia activated autophagy in bladder cancer
cells.
Hypoxia elevates gemcitabine-induced
autophagy in bladder cancer cells
The current study next examined the occurrence of
autophagy in bladder cancer cells treated with gemcitabine under
hypoxic conditions. T24 cells were incubated in complete medium
under hypoxic or normoxic conditions, with or without gemcitabine
for 8 h. Exposure to hypoxia and treatment with gemcitabine
increased LC3-II levels compared with the levels upon exposure to
hypoxia or treatment with gemcitabine alone. In addition, the LC3-I
to LC3-II conversion was almost completely abolished by the
addition of 3MA (5 mM) in the gemcitabine-treated cells under
hypoxic conditions or in cells exposed to hypoxia alone (Fig. 4A).
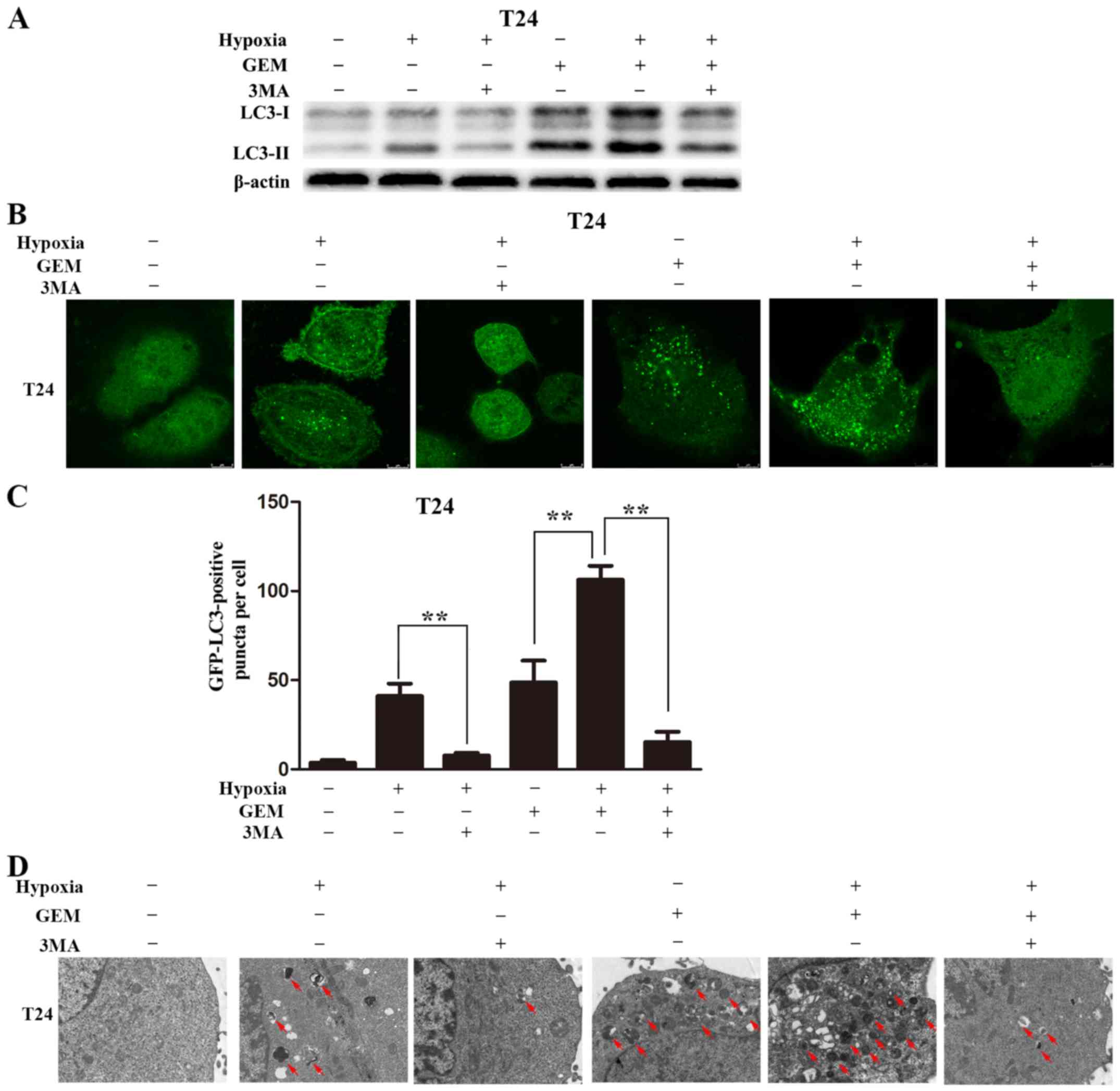 | Figure 4Hypoxia augmented GEM-induced
autophagy in bladder cancer cells. (A) T24 cells were pretreated
with 3MA (5 mM) for 1 h, and then incubated in complete medium
under hypoxia or normoxia with or without GEM (5 µM) for an
additional 8 h. LC3 was detected by immunoblotting. (B) T24 cells
were transfected with GFP-LC3 adenovirus for 24 h, pretreated with
3MA (5 mM) for 1 h, and then incubated in complete medium under
hypoxia or normoxia with or without GEM (5 µM) for an
additional 8 h. GFP-LC3 puncta were detected by confocal microscopy
(scale bar, 8 µm). (C) Number of GFP-LC3 puncta per cell
(>10) were calculated, and are displayed in the histogram. (D)
Cells were pretreated with 3MA (5 mM) for 1 h, and then incubated
in complete medium under hypoxia or normoxia with or without GEM (5
µM) for an additional 8 h. Autophagosomes were detected by
transmission electron microscopy (scale bar, 2 µm). The red
arrows designate the autophagosomes. Data represent the mean ±
standard deviation from three independent experiments.
**P<0.01. GEM, gemcitabine; 3MA, 3-methyladenine;
LC3, light chain 3. |
Autophagy was also observed by monitoring
the formation of GFP spots using confocal microscopy
Following transfection using a GFP-LC3 adenovirus
for 24 h, the cells were maintained in complete medium under
normoxic or hypoxic conditions, with or without gemcitabine for 8
h. The number of GFP-LC3 puncta in the gemcitabine-treated cells
under hypoxic conditions was significantly increased in comparison
with that in the hypoxia or gemcitabine-treated cells alone.
Furthermore, following pretreatment with 3MA, GFP-LC3 puncta
accumulation significantly decreased in the gemcitabine-treated
cells under hypoxic conditions or in cells cultured under hypoxic
conditions alone (Fig. 4B and
C).
Transmission electron microscopy was also used to
observe autophagosomes. Autophagic vacuoles were detected following
exposure to hypoxia or gemcitabine treatment alone, and the amount
of autophagic vacuoles was augmented when cells were exposed to
gemcitabine and hypoxia. However, following pretreatment with 3MA,
the autophagic vacuoles decreased (Fig. 4D). In conclusion, these results
indicated that hypoxia augmented gemcitabine-induced autophagy in
bladder cancer cell lines.
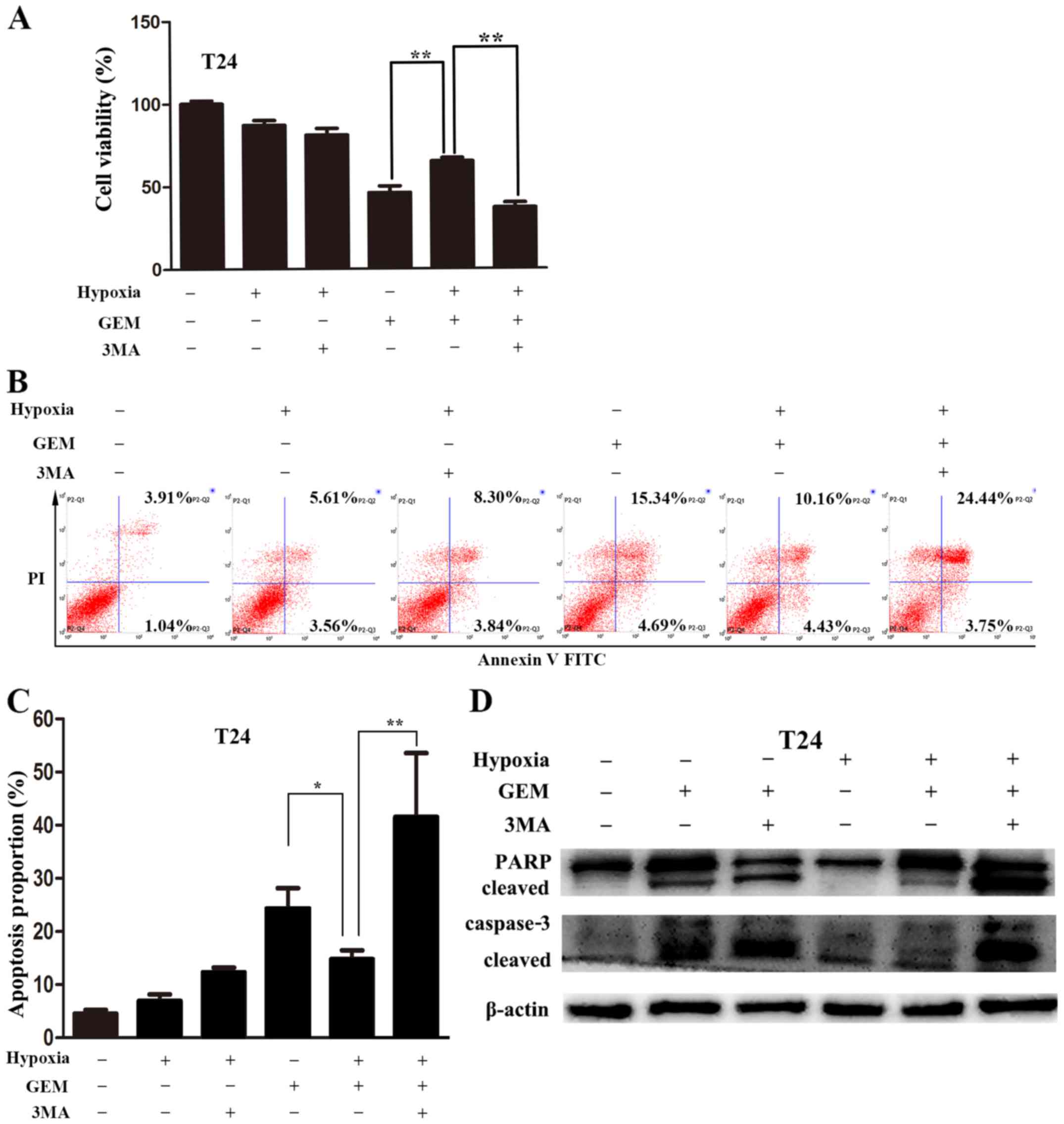
Blocking autophagy enhances bladder
cancer cell sensitivity to gemcitabine under hypoxic
conditions
To verify that autophagy had a protective effect in
bladder cancer cells obtaining resistance to gemcitabine under
hypoxic conditions, 3MA, which is an inhibitor of
phosphatidylinositol-3 kinase (PI3K), was utilized to inhibit
autophagy. The CCK-8 assay revealed that 3MA (5 mM) strongly
enhanced the inhibitory effect of gemcitabine on cell viability
under hypoxic conditions (Fig.
5A). Annexin V-FITC/PI staining followed by flow cytometry was
subsequently used to analyze apoptosis. The apoptotic percentages
of T24 cells treated with gemcitabine under normoxic conditions
were higher than those observed under hypoxic conditions. However,
the apoptotic percentages of T24 cells treated with gemcitabine
under hypoxic conditions was significantly augmented following the
suppression of autophagy with 3MA (Fig. 5B and C). A similar apoptosis
enhancement was also detected using immunoblotting. Increased
cleaved caspase-3 and cleavage of PARP were noted when cells were
co-treated with gemcitabine and 3MA under hypoxic conditions
(Fig. 5D). These results suggested
that the resistance of bladder cancer cells to gemcitabine may be
attributed to hypoxia-mediated protective autophagy. Decreasing
autophagy may therefore enhance gemcitabine-induced apoptosis.
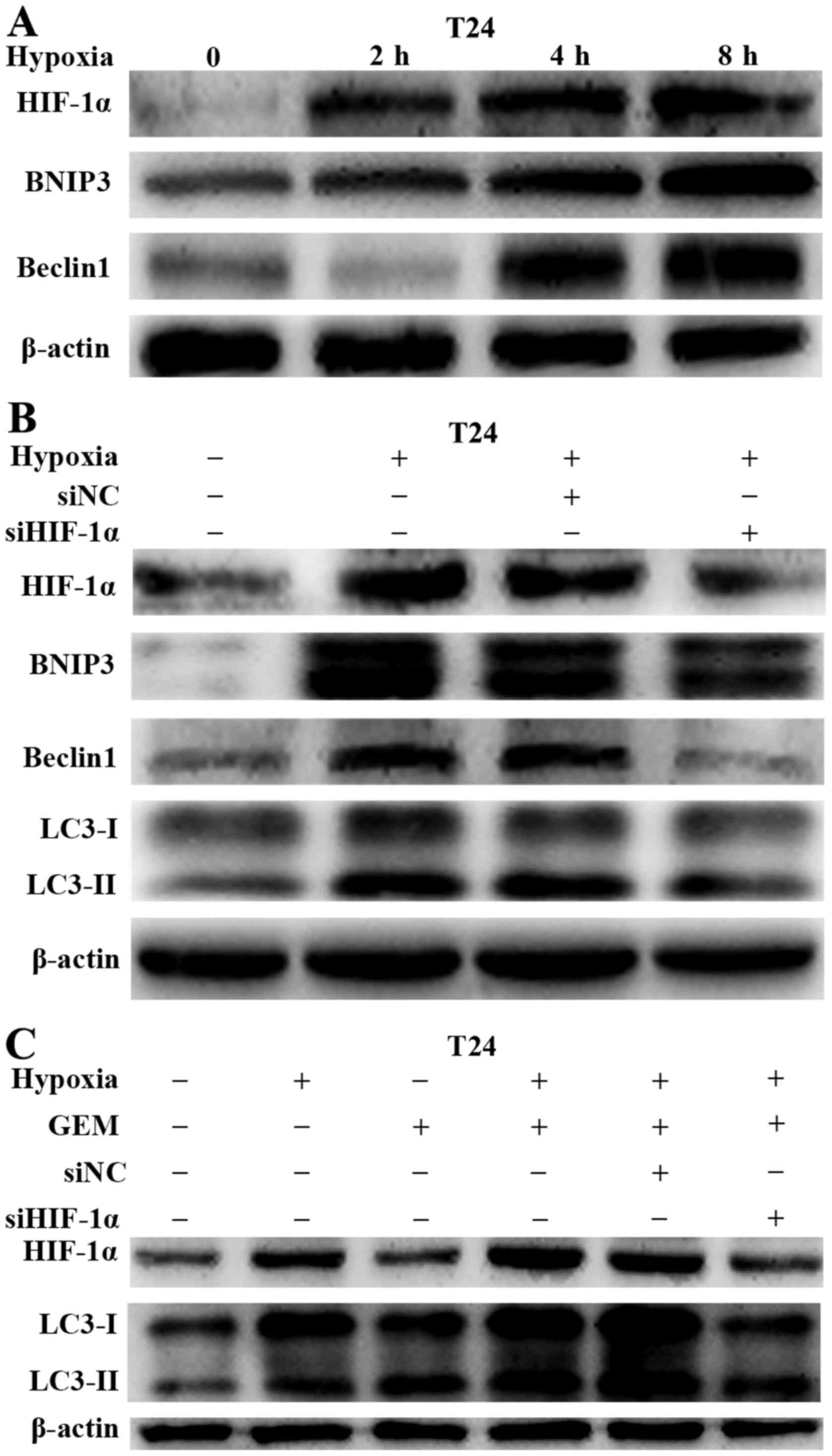
HIF-1α/BNIP3/Beclin1 pathway mediates
hypoxia-activated autophagy in bladder cancer cells
To investigate the mechanism through which hypoxia
induced autophagy in bladder cancer cells, the study determined
whether HIF-1α was involved in the process of hypoxia-induced
autophagy. To verify the hypothesis, the activation of HIF-1α and
its downstream target proteins (BNIP3 and Beclin1) was first
examined in bladder cancer cells under hypoxic conditions. HIF-1α,
BNIP3 and Beclin1 levels were increased in a time-dependent manner
under hypoxic conditions in T24 cells (Fig. 6A). A specific siRNA targeting
HIF-1α was then transfected into bladder cancer cell lines to
knockdown HIF-1α expression. As expected, the inhibition of HIF-1α
expression decreased the levels of the HIF-1α downstream target
proteins BNIP3 and Beclin1, and also reduced hypoxia-induced
autophagy (Fig. 6B). The results
mentioned above suggested that hypoxia promoted gemcitabine-induced
autophagy. Therefore, the present study further investigated
whether HIF-1α was involved in the gemcitabine-induced autophagy
increase in bladder cancer cells. Following the downregulation of
HIF-1α, gemcitabine failed to induce more LC3-II accumulation under
hypoxic conditions (Fig. 6C). This
demonstrated that HIF-1α was a main regulator of hypoxia-induced
autophagy.
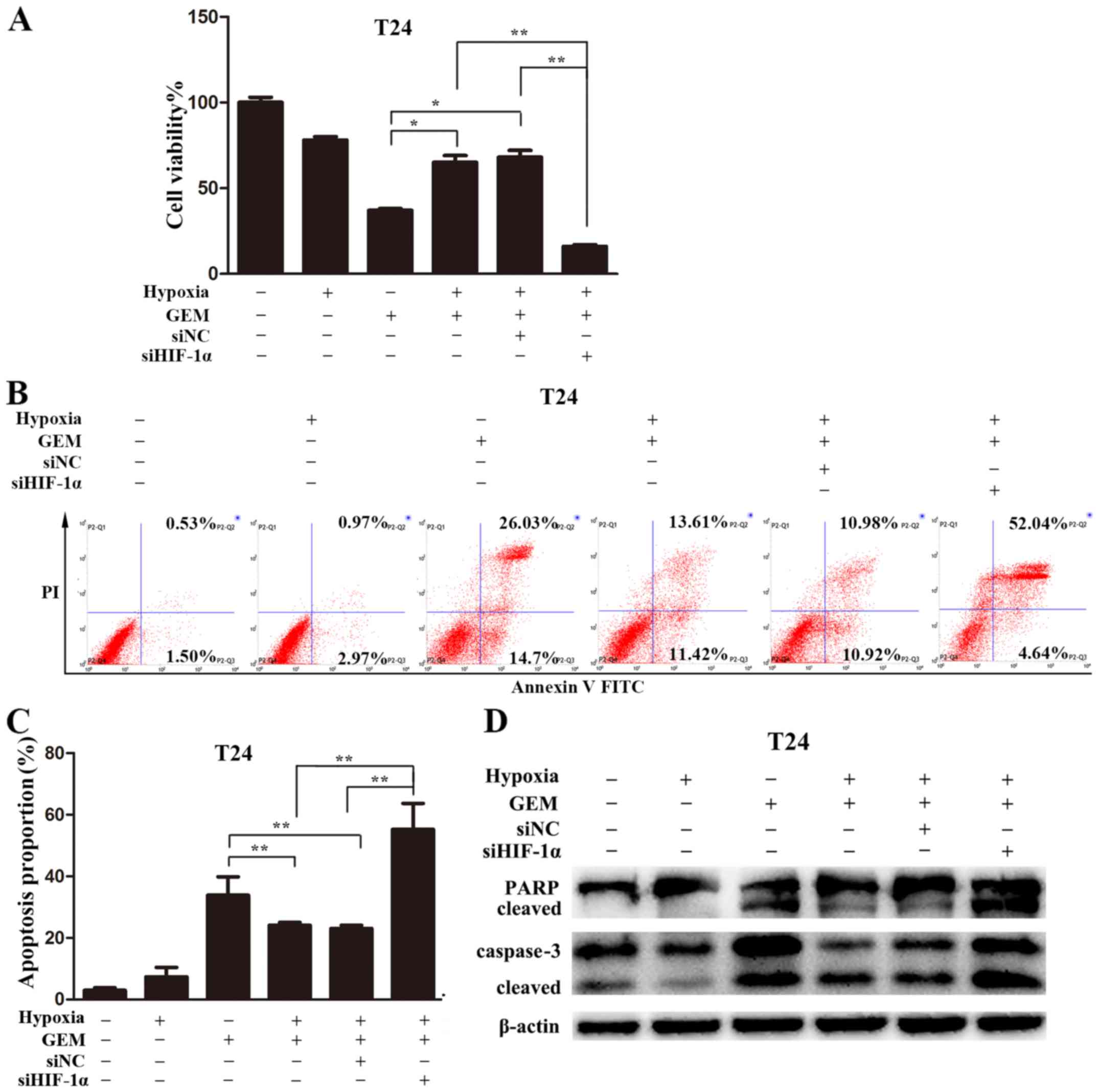
HIF-1α knockdown promoted
gemcitabine-induced apoptosis in bladder cancer under hypoxic
conditions
To further investigate the role of HIF-1α in
gemcitabine resistance, the effects of HIF-1α knockdown on
gemcitabine-induced cytotoxicity were examined. The knockdown of
HIF-1α almost completely eliminated the resistance of bladder
cancer cells to gemcitabine under hypoxic conditions. In addition,
HIF-1α knockdown strongly inhibited cell viability compared with
that in cells treated with gemcitabine under hypoxic conditions
alone (Fig. 7A). Annexin V-FITC/PI
staining followed by flow cytometry was used to measure apoptosis
levels. The data revealed that gemcitabine-induced apoptosis
increased significantly compared with that in the hypoxia group
(Fig. 7B and C). However,
gemcitabine-induced apoptosis of bladder cancer cells was reversed
following the inhibition of HIF-1α expression. Consistently,
immunoblotting demonstrated the same apoptotic changes. Knockdown
of HIF-1α was observed to enhance the activation of caspase-3 and
PARP cleavage (Fig. 7D). These
results suggested that hypoxia-induced protective autophagy
mediated by HIF-1α may lead to bladder cancer cell resistance to
gemcitabine.
Discussion
The current study provided evidence that
HIF-1α-mediated hypoxia-induced autophagy was critical to
gemcitabine resistance. Autophagy was evidently induced when T24
cells were treated with hypoxia or gemcitabine alone, and it was
noted that hypoxia significantly enhanced autophagy when bladder
cancer cells were exposed to gemcitabine. In addition, HIF-1α
expression was increased in bladder cancer tissues and bladder
cancer cells treated with hypoxia. HIF-1α siRNA or treatment with
the autophagy inhibitor 3MA inhibited the hypoxia-activated
autophagy and augmented the gemcitabine-induced apoptosis.
Furthermore, HIF-1α strongly elevated BNIP3 and Beclin1 expression
levels. Suppression of HIF-1α was observed to downregulate the
expression levels of BNIP3 and Beclin1, suggesting that the
HIF-1α/BNIP3/Beclin1 signaling cascade pathway was involved in
hypoxia-induced autophagy. Taken together, the present study
results suggested that targeting autophagy or HIF-1α increased the
chemosensitivity of bladder cancer cells to gemcitabine.
Chemotherapy is an important treatment for advanced
bladder cancer; however, resistance to anti-cancer drugs has been a
key obstacle to further extending the survival of patients. Recent
studies have suggested that the local microenvironment of the tumor
(particularly hypoxia) serves a significant part in the induction
of tumor resistance (21,22). Consistent with the findings of
previous studies (20,21,23),
the present study found that T24 cells treated with gemcitabine
under hypoxic conditions exhibited significant treatment resistance
compared with that observed under normoxic conditions. The
underlying mechanisms of hypoxia-induced chemoresistance are
complex, and include multiple drug-resistant gene expression,
reduced reactive oxygen species (ROS) levels, cell cycle arrest,
gene mutations and drug concentration decreases (8,24–27).
Furthermore, autophagy activation is another possible mechanism
protecting cells from chemotherapeutic drug-induced apoptosis. It
is generally accepted that autophagy is a mechanism of cellular
defense and stress regulation. In the absence of oxygen or
nutritional deficiencies, autophagy selectively removes some of the
damaged organelles, including mitochondria, endoplasmic reticulum
and peroxisomes, to maintain cell survival (28,29).
In recent years, it was demonstrated that autophagy was also a
protective mechanism in the process of chemoresistance under
hypoxic conditions in a variety of tumor cells, such as in liver,
lung and colon cancer (30–32).
However, whether autophagy is involved in the process of bladder
cancer cell resistance to chemotherapy under a hypoxic
microenvironment is not clear.
In the present study, similarly to previous studies
(6,18,33),
it was observed that autophagy was activated when bladder cancer
cells were exposed to hypoxia, as well as to gemcitabine. Notably,
autophagy was enhanced when the cells were exposed to hypoxia and
gemcitabine simultaneously, including increased GFP-LC3 puncta,
enhanced formation of autophagic vacuoles and upregulation of
LC3-II levels. However, when autophagy was blocked by treatment
with 3MA, a commonly used inhibitor of autophagy that inhibits the
conversion of LC3-I to LC3-II by inhibiting PI3K (34), the hypoxia-induced autophagy was
significantly reduced. Furthermore, it has been demonstrated that
3MA combined with chemotherapy drugs under hypoxic conditions can
effectively promote tumor cell apoptosis (14,15).
As expected, 3MA reversed the chemosensitivity of bladder cancer
cells to gemcitabine under hypoxic conditions, which was
demonstrated by the enhanced cell proliferation inhibition,
increased apoptotic cell percentage, and increased caspase-3
activation and PARP cleavage.
The adaptation of cells to hypoxia mainly depends on
changes in gene transcription levels (35). HIF-1α is an oxygen-regulating
subunit of HIF-1, a hypoxia-dependent protein, that serves a major
hypoxic regulatory role and determines HIF-1 activity. The α
subunit of HIF-1α is easily degraded at high concentrations of
oxygen. After stably and rapidly accumulating in the nucleus under
hypoxic conditions, HIF-1α binds with HIF-1β to activate the HIF-1
heterodimeric complex, which promotes the expression of hypoxia
target genes through combining with the hypoxia response element
(33,36,37).
Studies have revealed that the associations between hypoxia and
autophagy are mainly through HIF-1-regulated expression of
downstream target proteins BNIP3 and BNIP3L, which release Beclin1
to activate autophagy by disrupting the interaction between Beclin1
and Bcl-2/Bcl-xL complexes (33,38–40).
Another study has suggested that hypoxia-induced cancer cell
autophagy was an HIF-1 independent pathway. Hypoxia activated
autophagy through activating AMPK-mTOR signaling (41–43).
In addition, the unfolded protein region generated by the
endoplasmic reticulum under hypoxic stimulation activated
transcription factor 4 to induce autophagy (44–46).
The present study demonstrated that the expression of HIF-1α in
bladder cancer cells under hypoxic conditions was accompanied with
the activation of the downstream proteins BNIP3 and Beclin1.
Suppressing HIF-1α reduced the expression of BNIP3 and Beclin1, and
the autophagy activation, suggesting that the HIF-1α/BNIP3/Beclin1
signaling cascade was involved in hypoxia-induced autophagy.
Furthermore, when HIF-1α was upregulated under hypoxic conditions,
the gemcitabine-induced apoptosis was significantly reduced.
However, when HIF-1α was suppressed, gemcitabine-induced apoptosis
was significantly increased. These results indicated that HIF-1α
inhibited the gemcitabine apoptotic cytotoxicity in bladder cancer
cells by mediating autophagy.
In conclusion, the results of the current study
revealed that resistance to gemcitabine in bladder cancer cells
under hypoxic conditions was due to elevated levels of autophagy,
which was regulated by HIF-1α-associated signaling pathways.
Therefore, the hypoxia-autophagy pathway may be a target for
enhancing the effectiveness of gemcitabine chemotherapy in bladder
cancer patients.
Acknowledgments
Not applicable.
Funding
This study was supported by funding from Natural
Science Foundation of China (grant no. 81372758), the Natural
Science Fund Project of Chongqing (grant no. cstc2013jcyjA10058),
and the Innovative Project of Science Research for Postgraduate of
Chongqing Municipal Education Committee (CYS15141).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WH designed the research; XY performed the
experiments; XY, HY, YZ, HT, QW, XL and YZ analyzed the data; XY
wrote the manuscript. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
the First Affiliated Hospital of Chongqing Medical University, and
written informed consent was obtained from the patients or the
patients' families.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
Statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ghoneim MA, Abdel-Latif M, el-Mekresh M,
Abol-Enein H, Mosbah A, Ashamallah A and el-Baz MA: Radical
cystectomy for carcinoma of the bladder: 2,720 consecutive cases 5
years later. J Urol. 180:121–127. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Park JC, Citrin DE, Agarwal PK and Apolo
AB: Multimodal management of muscle-invasive bladder cancer. Curr
Probl Cancer. 38:80–108. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Geldart T, Chester J, Casbard A, Crabb S,
Elliott T, Protheroe A, Huddart RA, Mead G, Barber J, Jones RJ, et
al: SUCCINCT: An open-label, single-arm, non-randomised, phase 2
trial of gemcitabine and cisplatin chemotherapy in combination with
sunitinib as first-line treatment for patients with advanced
urothelial carcinoma. Eur Urol. 67:599–602. 2015. View Article : Google Scholar :
|
|
5
|
Toschi L, Finocchiaro G, Bartolini S,
Gioia V and Cappuzzo F: Role of gemcitabine in cancer therapy.
Future Oncol. 1:7–17. 2005. View Article : Google Scholar
|
|
6
|
Huang XL, Zhang H, Yang XY, Dong XY, Xie
XY, Yin HB, Gou X, Lin Y and He WY: Activation of a c-Jun
N-terminal kinase-mediated autophagy pathway attenuates the
anticancer activity of gemcitabine in human bladder cancer cells.
Anticancer Drugs. 28:596–602. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dauer P, Nomura A, Saluja A and Banerjee
S: Microenvironment in determining chemo-resistance in pancreatic
cancer: Neighborhood matters. Pancreatology. 17:7–12. 2017.
View Article : Google Scholar :
|
|
8
|
Yasuda H: Solid tumor physiology and
hypoxia-induced chemo/radio-resistance: Novel strategy for cancer
therapy: Nitric oxide donor as a therapeutic enhancer. Nitric
Oxide. 19:205–216. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Frolova O, Samudio I, Benito JM, Jacamo R,
Kornblau SM, Markovic A, Schober W, Lu H, Qiu YH, Buglio D, et al:
Regulation of HIF-1α signaling and chemoresistance in acute
lymphocytic leukemia under hypoxic conditions of the bone marrow
microenvironment. Cancer Biol Ther. 13:858–870. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Qiu Y, Li P and Ji C: Cell death
conversion under hypoxic condition in tumor development and
therapy. Int J Mol Sci. 16:25536–25551. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schito L and Semenza GL: Hypoxia-inducible
factors: Master regulators of cancer progression. Trends Cancer.
2:758–770. 2016. View Article : Google Scholar
|
|
12
|
Song J, Qu Z, Guo X, Zhao Q, Zhao X, Gao
L, Sun K, Shen F, Wu M and Wei L: Hypoxia-induced autophagy
contributes to the chemoresistance of hepatocellular carcinoma
cells. Autophagy. 5:1131–1144. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mizushima N: Autophagy: Process and
function. Genes Dev. 21:2861–2873. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu HM, Jiang ZF, Ding PS, Shao LJ and Liu
RY: Hypoxia-induced autophagy mediates cisplatin resistance in lung
cancer cells. Sci Rep. 5:122912015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu XW, Su Y, Zhu H, Cao J, Ding WJ, Zhao
YC, He QJ and Yang B: HIF-1α-dependent autophagy protects HeLa
cells from fenretinide (4-HPR)-induced apoptosis in hypoxia.
Pharmacol Res. 62:416–425. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Helpap B: New WHO classification of
urothelial carcinoma of the urinary bladder. Verh Dtsch Ges Pathol.
86:57–66. 2002.In German.
|
|
17
|
Compérat EM, Burger M, Gontero P, Mostafid
AH, Palou J, Rouprêt M, van Rhijn BWG, Shariat SF, Sylvester RJ,
Zigeuner R, et al: Grading of urothelial carcinoma and the new
'World Health Organisation Classification of Tumours of the Urinary
System and Male Genital Organs 2016'. Eur Urol Focus.
S2405-4569(18)30004-X. 2018. View Article : Google Scholar
|
|
18
|
Lee JG, Shin JH, Shim HS, Lee CY, Kim DJ,
Kim YS and Chung KY: Autophagy contributes to the chemo-resistance
of non-small cell lung cancer in hypoxic conditions. Respir Res.
16:1382015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huang Z, Zhong Z, Zhang L, Wang X, Xu R,
Zhu L, Wang Z, Hu S and Zhao X: Down-regulation of HMGB1 expression
by shRNA constructs inhibits the bioactivity of urothelial
carcinoma cell lines via the NF-κB pathway. Sci Rep. 5:128072015.
View Article : Google Scholar
|
|
20
|
He X, Wang J, Wei W, Shi M, Xin B, Zhang T
and Shen X: Hypoxia regulates ABCG2 activity through the
activivation of ERK1/2/HIF-1α and contributes to chemoresistance in
pancreatic cancer cells. Cancer Biol Ther. 17:188–198. 2016.
View Article : Google Scholar :
|
|
21
|
Ko YH, Cho Y-S, Won HS, Jeon EK and Hong
YS: Possible role of autophagy inhibition in hypoxia-induced
chemoresistance of pancreatic cancer cells. J Clin Oncol. 30(Suppl
4): 2242012. View Article : Google Scholar
|
|
22
|
Ma Q, Zhang Y, Liu T, Jiang K, Wen Y, Fan
Q and Qiu X: Hypoxia promotes chemotherapy resistance by
down-regulating SKA1 gene expression in human osteosarcoma. Cancer
Biol Ther. 18:177–185. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guo Q and Qin W: DKK3 blocked
translocation of β-catenin/EMT induced by hypoxia and improved
gemcitabine therapeutic effect in pancreatic cancer Bxpc-3 cell. J
Cell Mol Med. 19:2832–2841. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Guo XL, Li D, Sun K, Wang J, Liu Y, Song
JR, Zhao QD, Zhang SS, Deng WJ, Zhao X, et al: Inhibition of
autophagy enhances anticancer effects of bevacizumab in
hepatocarcinoma. J Mol Med (Berl). 91:473–483. 2013. View Article : Google Scholar
|
|
25
|
Wang Q, Yang Y, Wang L, Zhang PZ and Yu L:
Acidic domain is indispensable for MDM2 to negatively regulate the
acetylation of p53. Biochem Biophys Res Commun. 374:437–441. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wartenberg M, Hoffmann E, Schwindt H,
Grünheck F, Petros J, Arnold JR, Hescheler J and Sauer H: Reactive
oxygen species-linked regulation of the multidrug resistance
transporter P-glycoprotein in Nox-1 overexpressing prostate tumor
spheroids. FEBS Lett. 579:4541–4549. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen WL, Wang CC, Lin YJ, Wu CP and Hsieh
CH: Cycling hypoxia induces chemoresistance through the activation
of reactive oxygen species-mediated B-cell lymphoma extra-long
pathway in glioblastoma multiforme. J Transl Med. 13:3892015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ravanan P, Srikumar IF and Talwar P:
Autophagy: The spotlight for cellular stress responses. Life Sci.
188:53–67. 2017. View Article : Google Scholar
|
|
29
|
Yin H, Yang X, Gu W, Liu Y, Li X, Huang X,
Zhu X, Tao Y, Gou X and He W: HMGB1-mediated autophagy attenuates
gemcitabine-induced apoptosis in bladder cancer cells involving JNK
and ERK activation. Oncotarget. 8:71642–71656. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kabir N and Yong Y: Hypoxia-induced
autophagy in hepatocellular carcinoma and anticancer therapy. Natl
J Physiol Pharm Pharmacol. 7:771–780. 2017.
|
|
31
|
Notte A, Ninane N, Arnould T and Michiels
C: Hypoxia counteracts taxol-induced apoptosis in MDA-MB-231 breast
cancer cells: Role of autophagy and JNK activation. Cell Death Dis.
4:e6382013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Selvakumaran M, Amaravadi RK, Vasilevskaya
IA and O'Dwyer PJ: Autophagy inhibition sensitizes colon cancer
cells to antiangiogenic and cytotoxic therapy. Clin Cancer Res.
19:2995–3007. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zou YM, Hu GY, Zhao XQ, Lu T, Zhu F, Yu SY
and Xiong H: Hypoxia-induced autophagy contributes to
radioresistance via c-Jun-mediated Beclin1 expression in lung
cancer cells. J Huazhong Univ Sci Technolog Med Sci. 34:761–767.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vergne I, Roberts E, Elmaoued RA, Tosch V,
Delgado MA, Proikas-Cezanne T, Laporte J and Deretic V: Control of
autophagy initiation by phosphoinositide 3-phosphatase Jumpy. EMBO
J. 28:2244–2258. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Adams JM, Difazio LT, Rolandelli RH, Luján
JJ, Haskó G, Csóka B, Selmeczy Z and Németh ZH: HIF-1: A key
mediator in hypoxia. Acta Physiol Hung. 96:19–28. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li YN, Hu JA and Wang HM: Inhibition of
HIF-1α affects autophagy mediated glycosylation in oral squamous
cell carcinoma cells. Dis Markers. 2015:2394792015. View Article : Google Scholar
|
|
37
|
Harada H, Itasaka S, Kizaka-Kondoh S,
Shibuya K, Morinibu A, Shinomiya K and Hiraoka M: The Akt/mTOR
pathway assures the synthesis of HIF-1alpha protein in a glucose-
and reoxygenation-dependent manner in irradiated tumors. J Biol
Chem. 284:5332–5342. 2009. View Article : Google Scholar
|
|
38
|
Hu YL, DeLay M, Jahangiri A, Molinaro AM,
Rose SD, Carbonell WS and Aghi MK: Hypoxia-induced autophagy
promotes tumor cell survival and adaptation to antiangiogenic
treatment in glioblastoma. Cancer Res. 72:1773–1783. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bellot G, Garcia-Medina R, Gounon P,
Chiche J, Roux D, Pouysségur J and Mazure NM: Hypoxia-induced
autophagy is mediated through hypoxia-inducible factor induction of
BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol.
29:2570–2581. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Huang HY, Wang WC, Lin PY, Huang CP, Chen
CY and Chen YK: The roles of autophagy and hypoxia in human
inflammatory periapical lesions. Int Endod J. 51(Suppl 2):
e125–e145. 2018. View Article : Google Scholar
|
|
41
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jin Y, Bai Y, Ni H, Qiang L, Ye L, Shan Y
and Zhou M: Activation of autophagy through calcium-dependent
AMPK/mTOR and PKCθ pathway causes activation of rat hepatic
stellate cells under hypoxic stress. FEBS Lett. 590:672–682. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu H, Qiu H, Xiao Q and Le W: Chronic
hypoxia-induced autophagy aggravates the neuropathology of
Alzheimer's disease through AMPK-mTOR signaling in the
APPSwe/PS1dE9 mouse model. J Alzheimers Dis. 48:1019–1032. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fang Y, Tan J and Zhang Q: Signaling
pathways and mechanisms of hypoxia-induced autophagy in the animal
cells. Cell Biol Int. 39:891–898. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pike LR, Singleton DC, Buffa F, Abramczyk
O, Phadwal K, Li JL, Simon AK, Murray JT and Harris AL:
Transcriptional up-regulation of ULK1 by ATF4 contributes to cancer
cell survival. Biochem J. 449:389–400. 2013. View Article : Google Scholar
|
|
46
|
Rzymski T, Milani M, Pike L, Buffa F,
Mellor HR, Winchester L, Pires I, Hammond E, Ragoussis I and Harris
AL: Regulation of autophagy by ATF4 in response to severe hypoxia.
Oncogene. 29:4424–4435. 2010. View Article : Google Scholar : PubMed/NCBI
|