1. Introduction
Leukemia, a common hematological malignancy,
originates from clonal leukemia cells with uncontrolled
proliferation, blocked cell apoptosis and differentiation
disorders. Leukemia is characterized by accumulations of immature
leukemic blasts in the bone marrow and hematopoietic tissues,
infiltration of non-hematopoietic tissues and organs, and
inhibition of normal hematopoietic function. The incidence of
leukemia is increasing at an average rate of 0.7% annually in
children and adolescents of the United States; in China the
incidence of leukemia is approximately 5.17/100,000 individuals and
the mortality rate is 3.94/100,000 individuals, which severely
endangers the lives of patients (1,2).
There are several types of leukemia, and these can be divided into
four common types according to cell morphology and biochemical
characteristics as follows: Acute lymphoblastic leukemia (ALL),
chronic lymphoblastic leukemia (CLL), acute myeloid leukemia (AML)
and chronic myeloid leukemia (CML) (3). The pathogenesis and clinical
treatment for leukemias are 'hot' and difficult research directions
in the field of cancer. STAT proteins (STATs) belong to a family of
transcription factors that are activated by polypeptide ligands,
such as cytokines and growth factors. STATs comprise seven members:
STAT1, STAT2, STAT3, STAT4, STAT5 (a/b) and STAT6 (4). STAT3 is a crucial member of the STAT
family, which forms a dimer following activation, enters the
nucleus, and regulates the transcription of diverse target genes
(5,6). Since it is closely associated with
cell proliferation, differentiation and apoptosis, its abnormal
expression and activity are involved in the development of certain
diseases, such as hyper-IgE syndrome and developmental
abnormalities (7). An increasing
number of studies have found that STAT3 abnormal expression and
activation are accompanied by leukemia development, which indicates
the potential role of STAT3 in the pathogenesis of leukemia
(8–11). The present review focuses on the
roles of STAT3 in the pathogenesis, diagnosis, treatment and
prognosis of different types of leukemia.
2. The structure and regulation of the
activity of STAT3
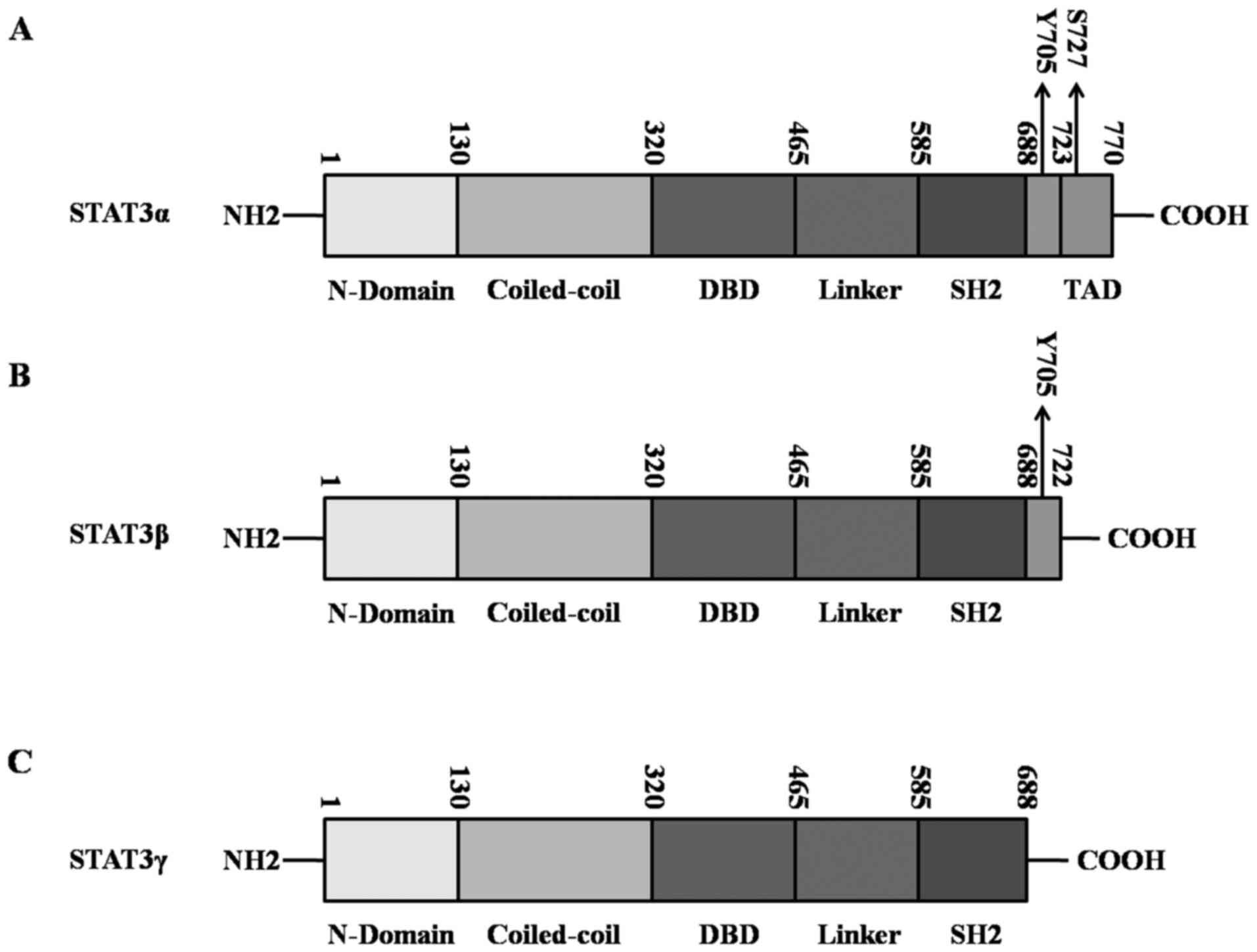
The structure of STAT3
The STAT3 encoding gene is located on the human
genome chromosome 17 (17q21.1) and the protein contains six
classical functional segments of the STAT family: i) N-terminal
domain (ND), which is able to stabilize the dimerized STAT3 and
promote the formation of tetramers of two STAT3 dimers to make it
more stable with DNA (12); ii)
coiled-coil domain (CCD), which mediates STAT3 direct binding to
the receptor and facilitates STAT3 phosphorylation on 705-tyrosine
site (Y705) (13); iii) DNA
binding domain (DBD), which, by recognizing the γ-interferon
activating sequence (GAS), will direct STAT3 to the promoters of
target genes, and initiate transcriptional activation of the target
genes (14); iv) the linker
region, the function of which unknown at present; v) Src homology 2
(SH2) domain, the most conserved part of STAT3, which shares the
same core sequence 'GTFLLRFSS' with the SH2 domain of tyrosine
kinase Src, and the phosphorylation and subsequent dimerization of
SH2 domain and plays a critical role in the process of signal
transduction (15); vi) C-terminal
transcriptional activation domain (TAD), in which there are several
important tyrosines/serines located or near the region, and the
phosphorylation of specific residues are important for STAT3
function. For example, Y705 is located between SH2 and TAD, which
is crucial for STAT3 activation and dimerization, and the
727-serine site (S727) is located in TAD, which is thought to
enhance the transcriptional activity of STAT3 (16–18)
(Fig. 1A).
STAT3 has four isoforms: STAT3α, STAT3β, STAT3γ and
STAT3δ. Among the structures of STAT3 isoforms, STAT3α is the most
common structure. It consists of the ND, CCD, DBD, Linker, SH2 and
TAD domains, with a molecular weight of approximately 92 kDa, and
is mainly associated with cell proliferation and transformation
(19,20) (Fig.
1A). STAT3β originates from the alternative splicing of the
STAT3 gene transcript, which results in a 55 amino acid deletion at
the 3' end of the open reading frame of STAT3α with a molecular
weight of approximately 83 kDa; STAT3β lacks the C-terminal
transactivation domain and S727, and acts as a dominant
transcriptional inhibitory factor that is particularly crucial for
granulocyte colony-stimulating factor (G-CSF)-mediated cell
differentiation (21–23) (Fig.
1B). STAT3γ, with molecular weight of 72 kDa, acts as a
dominant negative form of STAT3α. It lacks the C-terminal
transactivating portion, and due to controlled proteolysis, the
corresponding residues of S727 and Y705 of STAT3α are absent in
STAT3γ, which retains the SH2 domain and can be recruited to
tyrosine-phosphorylated receptor proteins through the SH2 domain
(Fig. 1C). STAT3γ is predominantly
activated in terminal differentiated neutrophils, and is involved
in the regulation of cell proliferation (19,24).
STAT3δ, a putative isoform of STAT3 with a yet unknown structure,
has been found to be expressed during the early stage of
granulocytic differentiation (25).
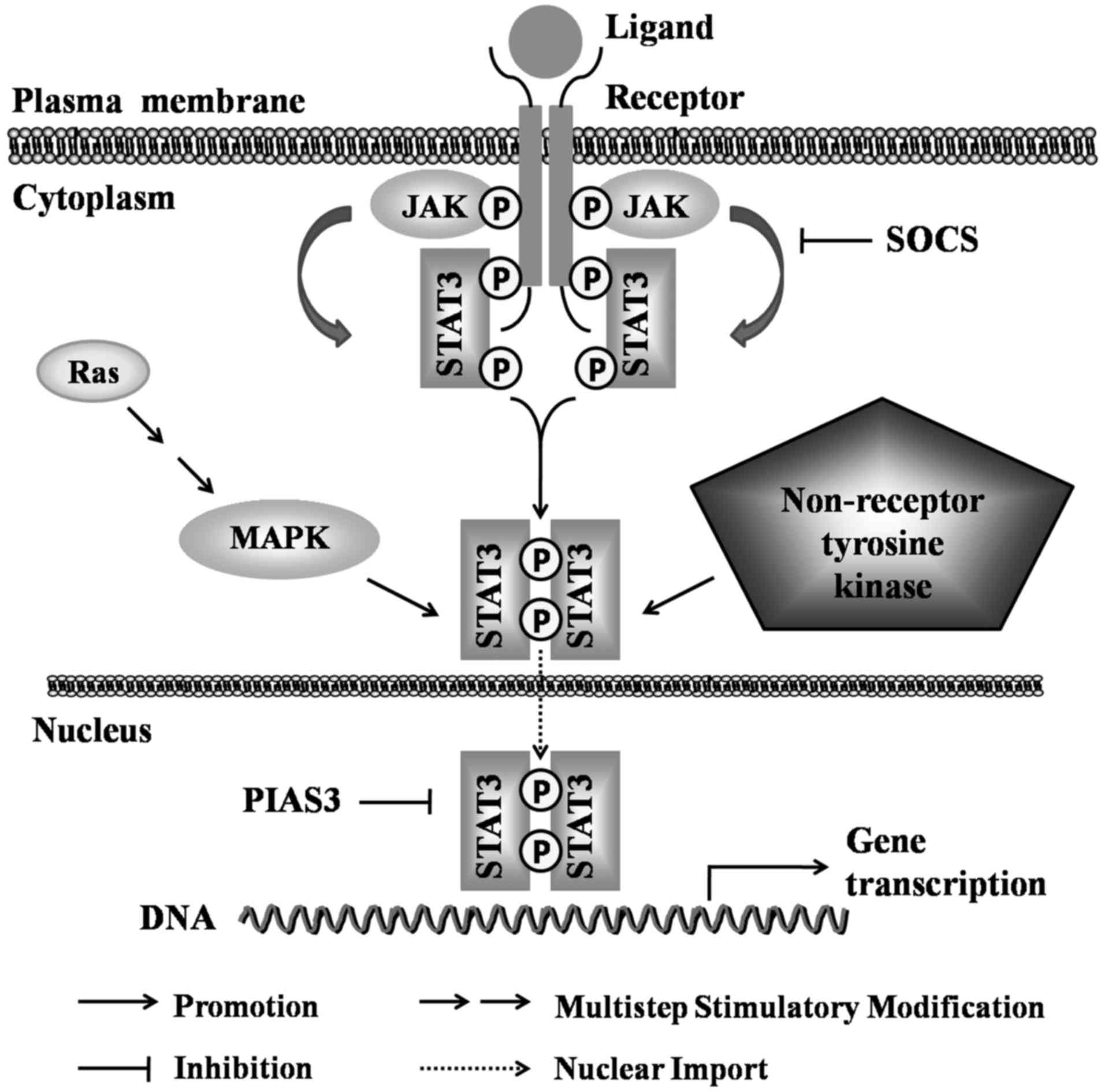
The regulation of STAT3 activity
STAT3 can be activated through a number of
mechanisms, including through the Janus kinase (JAK)/STAT3,
Ras/mitogen-activated protein kinase (MAPK) and non-receptor
tyrosine kinase signaling pathways (26,27).
STAT3 is negatively regulated by two types of regulating factors,
including the suppressor of cytokine signaling (SOCS) and the
protein inhibitor of activated STAT (PIAS), which regulate the
active status of STAT3 through different mechanisms (28). These activation pathways and
regulatory factors regulate STAT3 activity and function
synergistically, and play essential roles in physiological and
pathological processes (29–31)
(Fig. 2). A description of these
activation pathways and regulatory factors is provided as
follows:
a) The JAK/STAT3 pathway
The Janus kinase (JAK) family consists of four
non-receptor tyrosine kinases, JAK1, JAK2, JAK3 and tyrosine kinase
2 (TYK2), which not only phosphorylate the bound cytokine
receptors, but also a number of signaling molecules, which contain
specific SH2 domains (32).
Different cytokine receptors on the cell membrane bind the
corresponding ligands to form homologous or heterodimers, which
drive the mutual phosphorylation of the JAKs in proximity and
facilitate the activation of STAT3 (33–38)
(Table I). Mechanistically, the
activated JAKs will phosphorylate tyrosine residues on the
receptor, which provides a 'docking site' with the surrounding
amino acid sequence for STAT3 protein recruitment by SH2 domain.
Subsequently, STAT3 protein is phosphorylated by JAKs predominantly
at the Y705 site, leading to the activation and dimerization of
STAT3, which will rapidly enter the nucleus, specifically bind to
the GAS sequence (TTC/ANNNG/TAA) or the interferon-stimulated
response element sequence (AGTTTCNNTTCNC/T), and initiate the
activation and transcription of target genes (13,39,40).
In addition, the activity of STAT3 in the nucleus needs to be
tightly controlled. For example, STAT3 activity can be 'shut down'
by the dephosphorylation effects of tyrosine phosphatase in the
nucleus, or by proteolytic enzyme degradation of STAT3 protein.
 | Table IReceptor-specific JAKs in the
JAK/STAT3 pathway. |
Table I
Receptor-specific JAKs in the
JAK/STAT3 pathway.
| Receptors | JAKs | (Refs.) |
|---|
| Type II cytokine
receptor | | |
| Interleukin-10
receptor | JAK1, TYK2 | (33) |
| Receptor
tyrosine kinase family | | |
| Epidermal growth
factor receptor | JAK1, JAK2 | (34) |
| Platelet-derived
growth factor receptor | JAK1, JAK2,
TYK2 | (34) |
| Colony stimulating
factor 1 receptor | JAK1, TYK2 | (35) |
| Vascular
endothelial growth factor receptor | JAK2 | (36) |
| Receptors with
gp130 domain | | |
| Interleukin-6
receptor | JAK1, JAK2,
TYK2 | (33) |
| Interleukin-11
receptor | JAK1, JAK2,
TYK2 | (33) |
| Oncostatin M
receptor | JAK1, JAK2,
TYK2 | (37) |
| Leukemia
inhibitory factor receptor | JAK1, JAK2,
TYK2 | (37) |
| Ciliary
neurotrophic factor receptor | JAK1, JAK2 | (35) |
| Granulocyte
colony-stimulating factor receptor | JAK1, JAK2,
TYK2 | (38) |
| Cardiotrophin-1
receptor | JAK1, JAK2 | (35) |
| Receptors with
γC domain | | |
| Interleukin-2
receptor | JAK1, JAK3 | (33) |
| Interleukin-7
receptor | JAK1, JAK3 | (35) |
| Interleukin-9
receptor | JAK1, JAK3 | (33) |
| Interleukin-15
receptor | JAK1, JAK3 | (35) |
b) The Ras-MAPK activation
pathway
The role of Ras signaling in STAT3 activation has
been demonstrated by numerous studies, whereby the Ras-induced
activation of MAPKs and the subsequent MAPK-mediated
phosphorylation of STAT3 may be required for STAT3 activity
(41). The MAPK family includes
extracellular signal-regulated kinases (ERKs), c-Jun N-terminal
kinase (JNK) and p38 MAPK (p38), which act as components in the Ras
signaling pathway with serine/threonine protein kinase activity
(27). STAT3 is one of the MAPK
substrates, and the MAPK-mediated phosphorylation of STAT3 on S727
will lead to STAT3 dimerization, its entry into the nucleus, its
binding to specific DNA sequences in the promoters of genes, and
the initiation of the activation and transcription of target genes;
when MAPK is blocked, the promoting effects of STAT3 on target gene
transcription are significantly decreased (42).
c) Non-receptor tyrosine kinase
signaling pathways
In addition to JAK/STAT3 and Ras/MAPK pathways,
non-receptor tyrosine kinases, such as activated Src kinase and
Abelson leukemia protein (Abl) can also directly phosphorylate
STAT3 protein independently of ligand-induced receptor pathways.
The activation of non-receptor tyrosine kinase signaling pathways
also causes the simultaneous activation of MAPK family members,
including p38, ERK and JNK, which phosphorylate STAT3 on S727 in
the C-terminal transactivation domain. Following entry into the
nucleus, the activated STAT3 proteins bind to specific DNA-response
elements in the promoters of genes and initiate the expression of
unique genes through interactions with other transcriptional
regulatory components (27,41).
d) SOCS and PIAS regulatory
pathways
STAT3 activity is negatively regulated by two major
regulators, SOCS and PIAS (28).
The SOCS family consists of eight members: SOCS1-7 and
cytokine-inducible SH2-containing protein (CISH), and they
negatively regulate the JAK/STAT3 signaling pathway via several
mechanisms as follows: i) All SOCSs bind to receptor complexes or
associated JAKs through their SH2 domains, recruit Elongin B/C
heterodimers, Cullin5 and other components of a E3 ubiquitination
complex, and lead to the degradation of receptors or associated
JAKs via the proteasomal pathway; ii) SOCS1 and SOCS3 directly
inhibit JAKs tyrosine activity via their kinase inhibitory region
by binding to the JAK activation loop; iii) CISH, SOCS2 and SOCS3
can inhibit signaling transduction via their ability to bind to
phosphotyrosine residues on receptors, thereby blocking the binding
of other SH2-containing signaling molecules (JAKs/STAT3) to the
receptors; iv) SOCS7 can prevent the nuclear translocation of
p-STAT3, and inhibit the transcription of target genes (43,44).
PIAS3 is currently known to be associated with STAT3
activity in the PIAS family, which affects STAT3 activation via
several molecular mechanisms as follows: i) PIAS3 may inhibit
transcription by blocking the DNA-binding activity of p-STAT3; ii)
PIAS3 may suppress transcription by recruiting other co-regulators,
such as histone deacetylases; iii) PIAS3 may regulate transcription
by promoting the sumoylation of p-STAT3; iv) PIAS3 may suppress
transcription by sequestering p-STAT3 to certain subnuclear
structures where co-repressor complexes are enriched (45).
3. The role of STAT3 in the pathogenesis of
leukemia
Leukemia cells and normal hematopoietic cells in the
same bone marrow microenvironment have entirely different
biological characteristics; normal hematopoietic cells, under the
stimulation of cytokines, will continue to differentiate and
mature; however, leukemia cells are characterized by
differentiation failure and infinite proliferation. In normal
cells, the activation of STAT3 is rapid and transient; however, in
leukemia cells, abnormal STAT3 expression and activation always
occur, which accelerate leukemia cell proliferation, block leukemia
cell differentiation and inhibit leukemia cell apoptosis, leading
to the occurrence and development of leukemia (46).
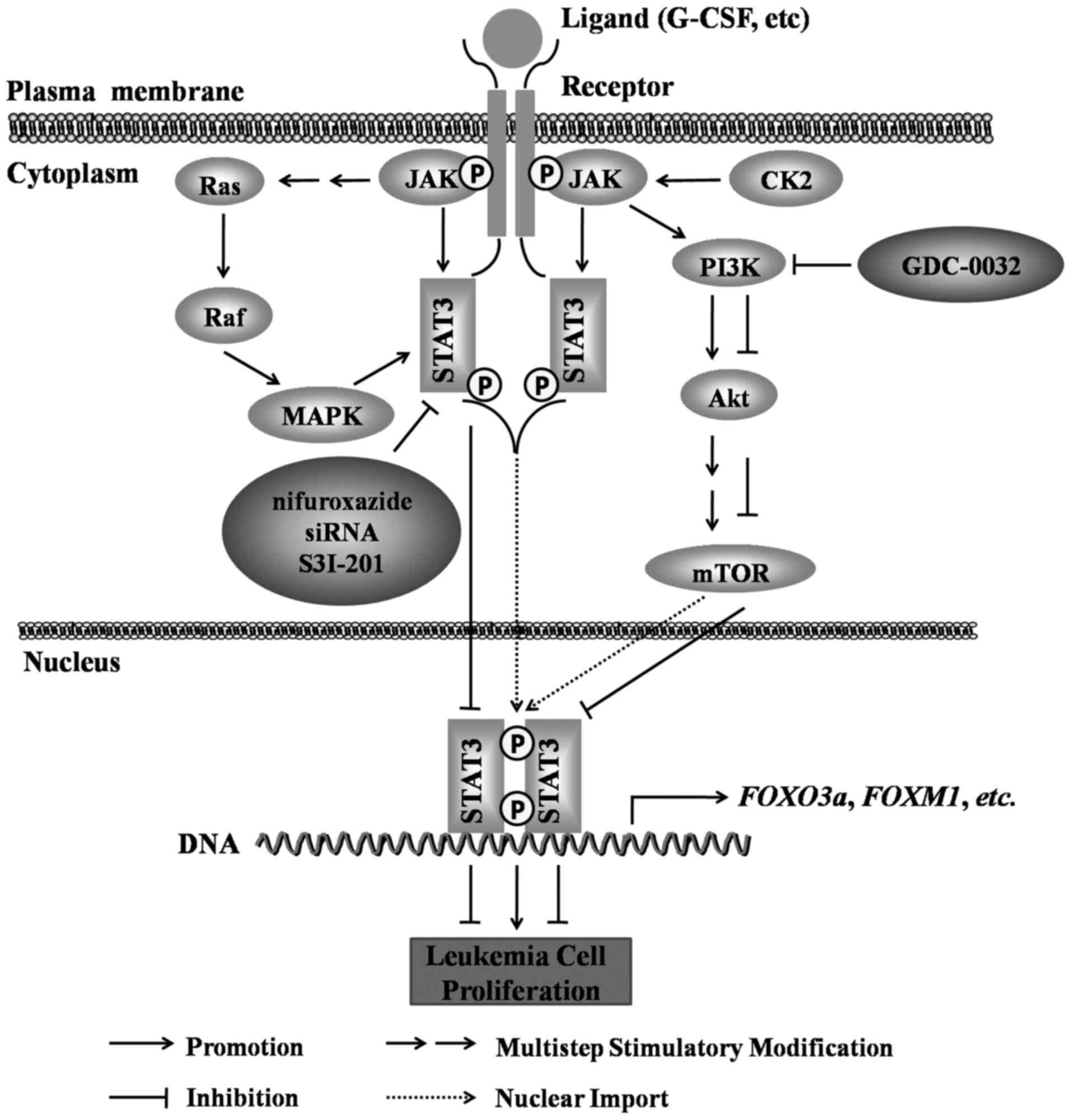
STAT3 and leukemia cell
proliferation
In the process of leukemia cell proliferation, STAT3
can promote the proliferation of leukemia cells through JAK/STAT3,
Ras/Raf/MAPK, PI3K/AKT/mammalian target of rapamycin (mTOR) and
other signaling pathways. For example, G-CSF stimulates the
activation of STAT3α to promote the proliferation of AML cell lines
(47), and protein kinase CK2
regulates the transcription of the Forkhead box O3 (FOXO3a)
gene by activating STAT3 through JAK/STAT3 and PI3K/AKT/mTOR, and
promotes the proliferation of leukemia stem cells (48). Moreover, STAT3 can also be
activated via the JAK/STAT3 and Ras/Raf/MAPK pathways, which
initiates the transcription of the Forkhead box M1 (FOXM1)
gene and promotes the proliferation of the CML cell line, K562
(49), wherease the inhibition of
STAT3 activity can effectively reduce the proliferation and
survival of leukemia cells (50–52).
For example, the STAT3 inhibitor, nifuroxazide, and the PI3K
inhibitor, GDC-0032, alone or in combination, have been shown to
effectively reduce the proliferation of human ALL CCRF-CEM cells
(50). In CD34 antigen-positive
AML (AML CD34+) cells, the siRNA-mediated knockdown of
STAT3 has been shown to significantly inhibit the growth and
survival of AML CD34+ cells (51). In addition, it has been
demonstrated that the combined use of the survivin gene
inhibitor, YM155, with the STAT3 inhibitor, S3I-201, significantly
inhibit the proliferation of YM155-tolerant human T lymphocytic
leukemia MT-2 cells (52). These
studies collectively suggest that STAT3 overexpression can promote
leukemia cell proliferation and that the inhibition of STAT3
activity can reduce leukemia cell survival and proliferation
(Fig. 3).
STAT3 and leukemia cell
differentiation
Hematopoietic progenitor cells can be divided into
myeloid and lymphoid progenitor cells. Myeloid progenitor cells
have the potential to multi-differentiate into myeloid cells
(erythrocytosis, granulocytosis, thrombocytosis, etc.), while
lymphoid progenitor cells have the potential to differentiate into
lymphatic subfamilies. Leukemia cells, due to differentiation and
maturity disorders, are always accompanied by abnormal hyperplasia,
and are stagnated at different stages of cell development. For
example, acute leukemia (AL) cell differentiation is stagnated at
the relatively earlier stages, and the majority of AL cells are
progenitor cells and early immature cells; chronic leukemia (CL)
cell differentiation is stagnated at the later stages, and the
majority of CL cells are mature naive cells or mature cells. We
hereby discuss the roles of STAT3 in the differentiation of
leukemia cells as follows:
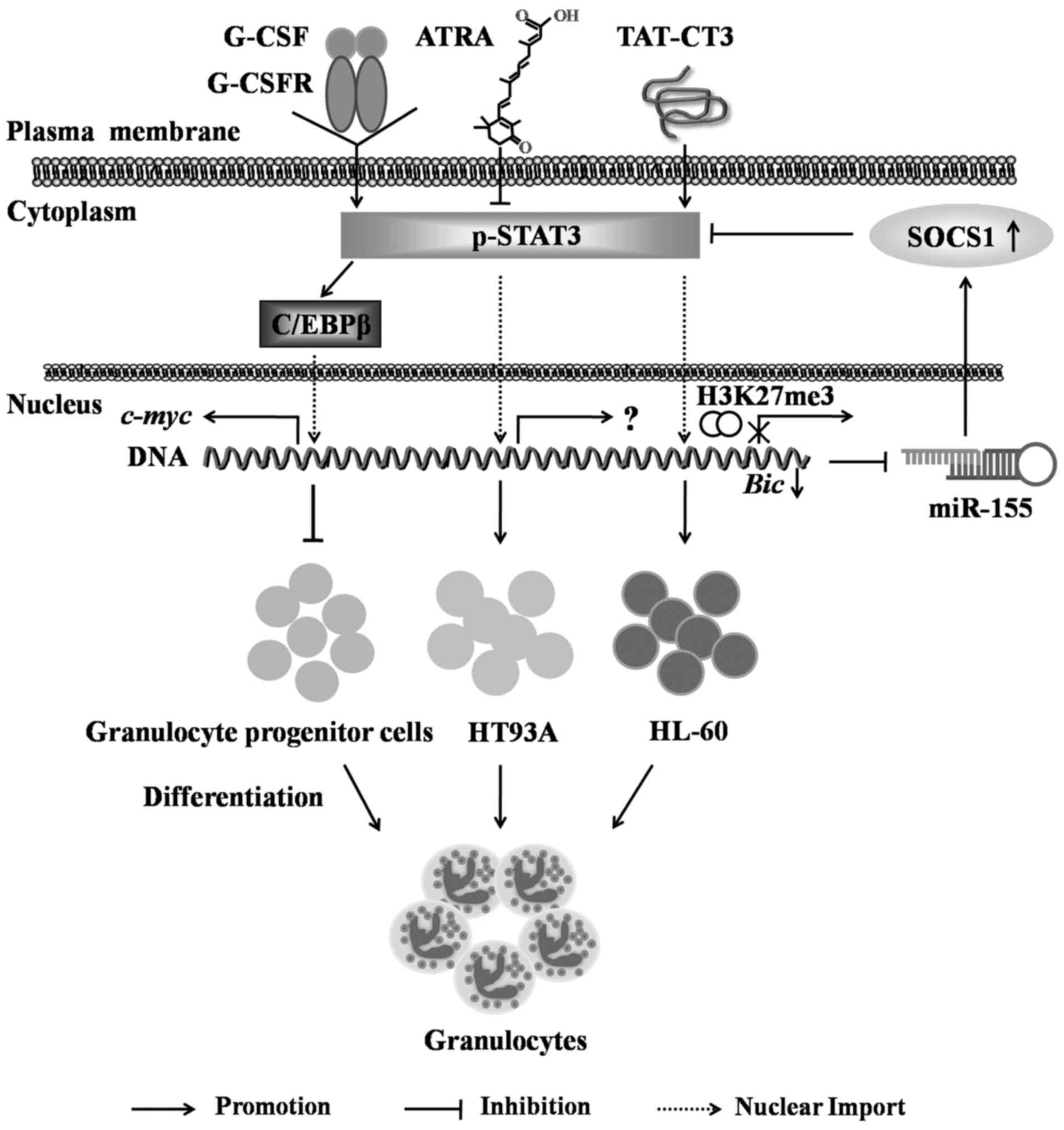
a) STAT3 in granulocyte
differentiation and its role in leukemia
Granulocyte differentiation involves a variety of
cytokines and transcription factors, of which G-CSF plays an
essential role during the process. The binding of G-CSF to the
G-CSF receptor (G-CSFR) causes the intracellular phosphorylation of
the Y705 domain of STAT3 and the activation of multiple signal
transduction pathways, including Ras/Raf/MAPK, PI3K and JAK/STAT
cascades (53). In signaling
transduction, the STAT3 protein regulates G-CSFR-mediated signaling
pathways and inhibits granulocyte differentiation (54). Previous studies have suggested that
STAT3 exerts inhibitory effects on granulocyte differentiation and
maturation, since STAT3-deficient mice exhibit an increased ratio
of mature to immature neutrophils in their bone marrow, peripheral
blood and spleen following sustained exposure to G-CSF in
vivo (55). Previous studies
have reported that during granulopoiesis, the binding of G-CSF to
G-CSFR activates STAT3, which subsequently stimulates the
expression of CCAAT enhancer binding protein (C/EBPβ),
promoting c-myc gene transcription, and thus inhibiting the
differentiation of bone marrow granulocytic progenitors into
granulocytes (54,56,57)
(Fig. 4). However, it should be
noted that isoforms of STAT3 may function differently during
G-CSF-directed granulocyte differentiation. It has been reported
that G-CSF activates STAT3α in three of six uncultured AML patient
samples and in all examined AML cell lines, apart from HL-60. G-CSF
directs the differentiation of CD34+ bone marrow cells
and HL-60 cells into granulocytes without affecting STAT3α
activity, but only increasing STAT3β activity. These results
demonstrate that the balance of the two isoforms of STAT3 in
myeloid cells may influence gene activation and the ability of
these cells to differentiate in response to G-CSF (47). In addition, other researchers have
revealed that the expression and activation of STAT3s is altered in
a maturation stage-specific manner. During the process of
granulocyte differentiation, the ratio of isoforms shifts from
predominantly STAT3α to STAT3β. Concomitant with STAT3α protein
downregulation in different stages of granulocyte differentiation,
the expression of STAT3γ is upregulated inversely. STAT3δ protein
is expressed at low levels and is reduced with differentiation, but
is primarily phosphorylated during an intermediate stage of
maturation (25).
The role of STAT3 in the process of leukemia cell
differentiation into granulocytes is increasingly valued. Xu et
al (58) observed that the
fusion protein TAT-CT3 induced the committed differentiation of the
human acute promyelocytic leukemia (APL) cell line, HL-60, into
granulocytes, and TAT-CT3 transiently promoted an elevation in
STAT3 phosphorylation, which resulted in the STAT3 dimer entering
the nucleus and promoting the expression of the Bic gene.
STAT3 also plays a role in controlling the H3K27me3 modification of
the Bic gene promoter, through which it reduces Bic
transcription, and suppresses miR-155 transcription and
finally results in an elevation in SOCS-1 levels, which further
inhibits STAT3 phosphorylation in a feedback loop for a long-term
period to promote HL-60 cell differentiation into granulocytes
(58). Moreover, in another study,
in the process of the all-trans retinoic acid (ATRA)-induced
differentiation of the APL cell line, HT93A, into granulocytes, the
expression levels of total and phosphorylated STAT3 were
significantly reduced, which further suggested that the inhibition
of STAT3 expression and activation would promote the
granulocyte-oriented differentiation of leukemia cells (59). Thus, drugs or strategies that
target STAT3 may hold promising prospects in modulating the
granulocyte differentiation of leukemia cells (Fig. 4).
b) Involvement of STAT3 in
monocyte/macrophage differentiation and its role in leukemia
Monocytes originate from granulocyte-monocyte
progenitor cells in bone marrow and are transported through the
blood; they migrate into the tissues, where they are transformed
into macrophages, and macrophages can be polarized into two
subgroups: M1 and M2. M1 macrophages mainly secrete tumor necrosis
factor (TNF)-α, interleukin (IL)-6 and other cytokines, exerting
pro-inflammatory and antitumor effects; M2 macrophages, which are
characterized by the overexpression of arginase 1 and IL-4 receptor
α (IL-4Rα), mainly secrete IL-4, IL-10 and other cytokines, and
play important roles in diverse biological processes, including
anti-inflammation, tissue repair and tumor promotion (60–63).
It has been reported that IL-6, in combination with macrophage
colony stimulating factor (M-CSF), can induce CD14+
monocyte differentiation into M2-like phenotype macrophages in
vitro, and during the process, the phosphorylated levels of
STAT3 were found to be significantly increased, which suggests that
STAT3 can induce the committed differentiation of mononuclear cells
into M2 macrophages and exert pro-tumor functions (64). Moreover, Komohara et al
(65) reported that upon
co-culture with primary central nervous system lymphoma cells,
macrophages exhibited a greater STAT3 phosphorylation and acquired
the M2 phenotype, which indicated that STAT3 activation may also be
involved in the promotion of macrophage M2 polarization, and tumor
occurrence and development.
The roles of STAT3 in the process of leukemia cell
differentiation into monocytes/macrophages have been increasingly
investigated by researchers. It has been found that IL-6 activates
STAT3 to bind to and initiate the transcription of the target
genes, c-myc and c-myb, through the GAS sequence in
the promoter, and thus promotes M1-type mouse myeloid leukemia cell
differentiation into mononuclear cells. Moreover, leukemia
inhibitory factor (LIF) promotes M1 mouse myeloid leukemia cell
differentiation into macrophages by activating STAT3 and inducing
IL-3Rα and β subunit gene expression (23,66,67).
Furthermore, some studies have indicated that protein tyrosine
phosphatase (PTP) epsilon C inhibits the activation of
IL-6-mediated JAKs and STAT3 and the expression of their downstream
target gene, c-myc, thereby inhibiting the differentiation
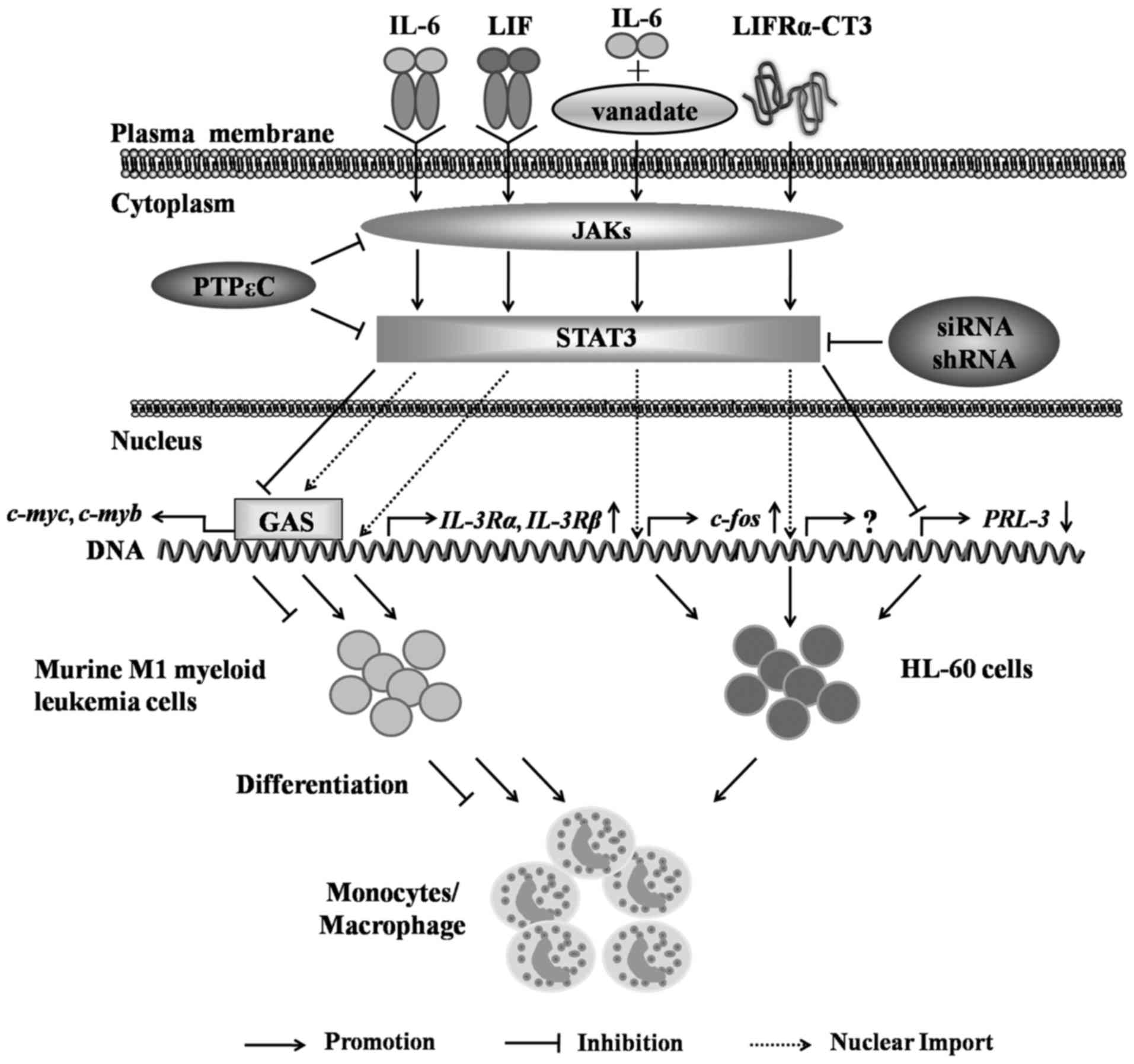
of M1 mouse myeloid leukemia cells into mononuclear cells (68) (Fig.
5).
In human HL-60 cells, the synergistic action of IL-6
with the PTP inhibitor, vanadate, can induce the expression of the
differentiation gene c-fos by stimulating the IL-6/JAK/STAT3
signaling pathway to promote the differentiation of HL-60 cells
into monocytes (69,70). Leukemia inhibitory factor
receptor-chain LIFRα-CT3 activates STAT3 through the JAK2/STAT3
pathway, which leads to the expression of CD11b and CD14 on the
surface of HL-60 cells and suggests that the activation of STAT3
can promote the differentiation of HL-60 cells into monocytes
(71). However, studies have also
found that the siRNA- or shRNA-mediated knockdown of STAT3 in HL-60
cells results in a decreased PRL-3 gene expression and an
increased expression of the cell surface markers, CD11b and CD14,
which indicates the dual roles of STAT3 in the regulation of
leukemia cell differentiation; thus, the specific mechanisms
warrant further investigation (72) (Fig.
5). However, STAT3 has yet not been validated to play a role in
the process of leukemia cell differentiation into M1/M2 subgroup
macrophages.
c) STAT3 in DC differentiation and its
role in leukemia
Dendritic cells (DCs) originate from multifunctional
hematopoietic stem cells, and are the most important
antigen-presenting cells (APCs) in vivo, which play crucial
roles in tumor surveillance, pathogenic microbial infection
resistance and internal environmental stability maintenance
(73,74). Patients with leukemia with a
defective DC number and function cannot effectively stimulate the
adaptive immune response or eliminate leukemia cells (75). Thus, if the committed
differentiation of leukemia cells into DCs could be induced, which
would probably express leukemia-specific antigens on the membrane,
this would directly present the leukemia antigens to the T-cells,
and then kill leukemia cells (76). DC development is highly dependent
upon fms-like tyrosine kinase 3 ligand/fms-like tyrosine kinase 3
(Flt3L/Flt3) signaling, and the interaction of Flt3L/Flt3 activates
STAT3 via JAK, which subsequently promotes the transcription of
regulatory factor E-box protein, and promotes DC differentiation
(77–80). Brady et al (81) observed that STAT3 phosphorylation
levels were significantly increased upon human
granulocyte-macrophage colony stimulating factor (GM-CSF) and IL-4
treatment during the process of differentiation into DCs for the
AML cell lines, HEL, KG-1 and MUTZ-3, which suggested that the
constitutive STAT3 activation may promote the differentiation of
leukemia cells into DCs and the intervention of STAT3 activity may
have great prospects in modulating the process of the committed
differentiation of leukemia cells into DCs for leukemia
treatment.
STAT3 and leukemia cell apoptosis
The occurrence of leukemia is closely related to the
apoptotic disorder of leukemia cells. STAT3 plays a key role in
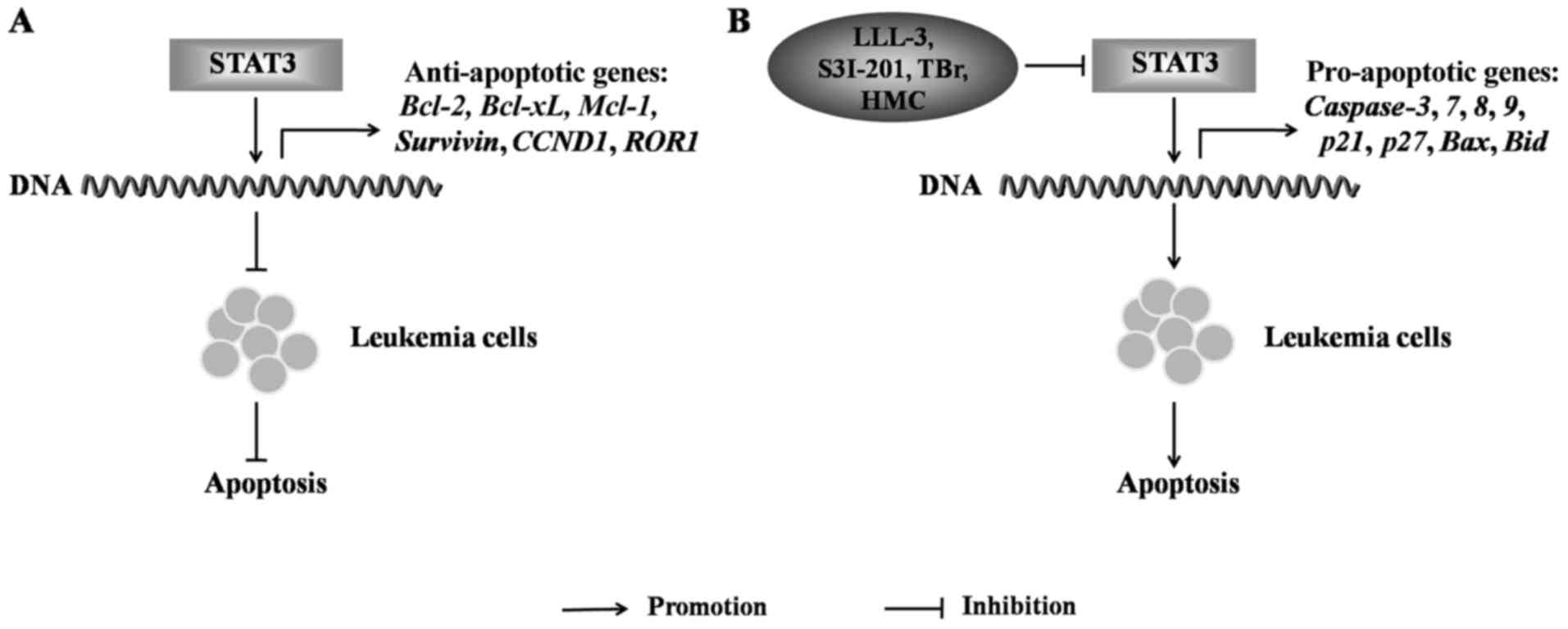
leukemia cell apoptosis. In general, STAT3 activation can inhibit
the apoptosis of leukemia cells by inducing anti-apoptotic gene
expression. For example, internal tandem duplication (ITD) in the
juxtamembrane in the kinase domain of Flt3 is a common genetic
lesion in AML, and the stimulation of the Flt3-ITD AML cells,
MV4-11, results in the elevation of p-STAT3 levels, which
upregulates the expression of the anti-apoptotic gene,
survivin, to protect AML cells from apoptosis (82). In the CML cell line, K562, the
expression levels of the anti-apoptotic genes, Bcl-2,
Bcl-xL, Mcl-1 and survivin and those of the
inhibitor of apoptosis protein, c-IAP2, have been shown to be
upregulated by STAT3 activation to protect K562 cells from
apoptosis (83). In CLL cells, the
expression levels of the anti-apoptotic genes, Bcl-2,
Bcl-xL, cyclin D1 (CCND1) and ROR1, were
upregulated following the activation of STAT3, and protected CLL
cells from apoptosis (84)
(Fig. 6A). The inhibition of STAT3
expression can upregulate the expression of pro-apoptotic genes and
induces leukemia cell apoptosis. For example, in K562 cells, the
STAT3-specific inhibitor, LLL-3, has been shown to induce leukemia
cell apoptosis by activating the caspase-3-and -7-mediated
pro-apoptotic pathway (85). In
TEL-AML1 fused ALL, the STAT3 inhibitor, S3I-201, has been
shown to induce leukemia cell apoptosis by upregulating the
expression of the pro-apoptotic genes, caspase-3, p27
and p21 (86). The
cytotoxic agent, bromo analogue (TBr), has been shown to enhance
the expression of the pro-apoptotic genes caspase-3,
-8 and -9, and to decrease the ratio of the
anti-apoptotic genes, Bcl-2/Bax, by downregulating
p-STAT3 and upregulating p-ERK to induce the apoptosis of HL-60
cells (87). Moreover,
(R)-5-hydroxy-2-methylchroman-4-one (HMC), isolated from a novel
endophytic fungus, can significantly reduce the level of p-STAT3,
which activates the pro-apoptotic genes, Bax, Bid,
caspase-3, -8 and -9 to induce leukemia cell
apoptosis (88) (Fig. 6B). However, a previous study
reported that the overexpression of STAT3 in CLL upregulates
caspase-3 expression, and induces CLL cell apoptosis
(89), suggesting that the
different activation status of STAT3 may play differential roles in
the regulation of leukemia cell apoptosis.
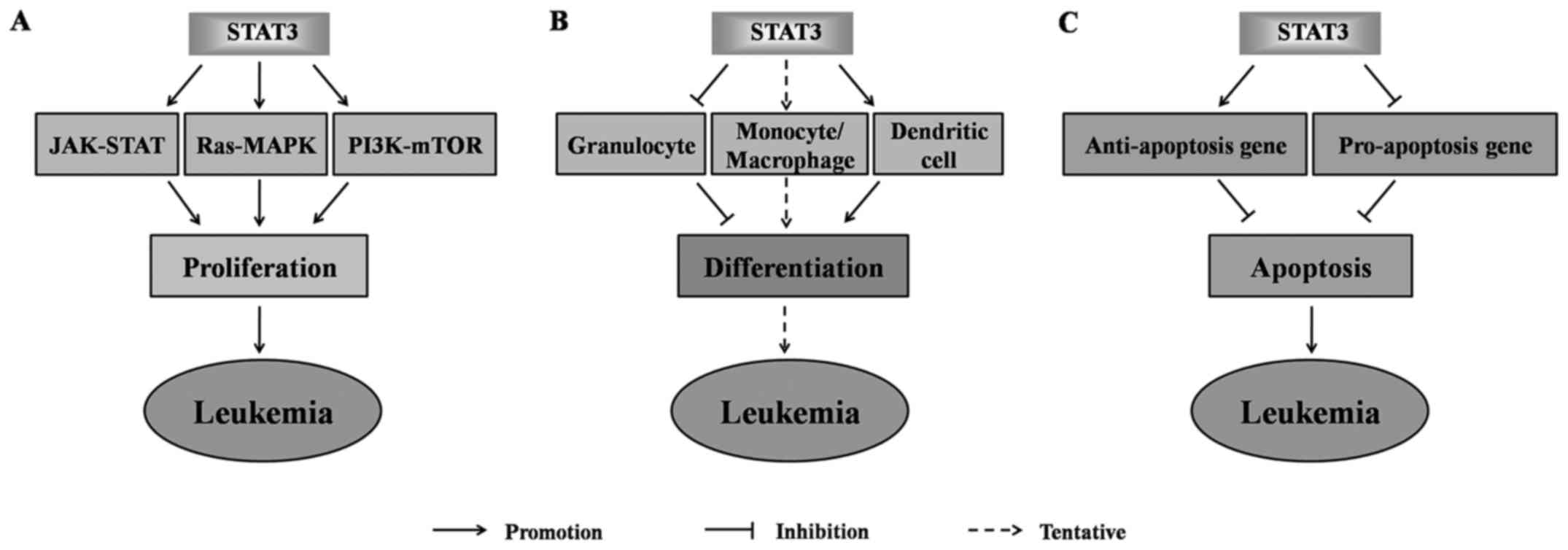
In summary, STAT3 is closely related to the
development of leukemia. It plays essential roles in the
development of leukemia by promoting the proliferation of leukemia
cells (Fig. 7A), regulating the
differentiation of granulocytes, monocytes/macrophages and
dendritic cells and blocking the apoptosis of leukemia cells
(Fig. 7B and C). The STAT3
expression and activation status may thus be an important target
for leukemia treatment and prognosis assessment.
4. The roles of STAT3 in the diagnosis,
treatment and prognosis of leukemia
The sustained activation of the STAT3 gene
plays an important role in the development of leukemia; it can be
used as an important target in the early diagnosis, treatment and
prognosis of leukemia.
The role of STAT3 in the diagnosis of
leukemia
STAT3 plays an important role in the diagnosis of
leukemia. For example, Xia et al (90) found that in patients newly
diagnosed with AML, 13 out of 17 patients were found to exhibit the
constitutive phosphorylation of STAT3 on Y705. Stevens et al
(91) demonstrated that the STAT3
pathway was more sensitive to ligand stimulation in patients with
relapsing AML. Benekli et al reported that constitutively
active STAT3 was detected in ten of 36 patients newly diagnosed
with AML (92), and in another
study, almost 78% of the patients (21 of 27) expressed STAT3β
protein (25).
Thus, collectively, it is suggested that STAT3 plays
an important diagnostic role in both newly diagnosed and relapsing
AML. T-cell large granular lymphocytic leukemia (T-LGLL) sustains
the phosphorylation of STAT3, and mutations of the STAT3
gene have been identified as a recurrent genetic abnormality in
T-LGLL (93–99).
STAT3 single nucleotide polymorphisms (SNPs) are
also prospective candidates for use in predicting susceptibility to
leukemia, which is associated with the individual sensitivity of
leukemia and is important for leukemia diagnosis. For example,
Zhong et al (100)
investigated association between STAT3 SNPs and leukemia, and found
that rs17886724 located on STAT3 intron 4 was closely related to
the pathogenesis of leukemia; Lautner-Csorba et al (101) found that two SNPs of rs3816769
and rs12949918 in the STAT3 gene, were associated with a
decreased risk of hyperdiploid ALL occurrence (≥50 chromosomes).
All these studies indicate the significance of STAT3 in the
diagnosis of leukemia.
The role of STAT3 in the treatment of
leukemia
STAT3 plays an important role in leukemia, and STAT3
intervention is currently a 'hot' topic of research, which may
reveal novel targets and strategies for leukemia treatment
(102,103). At present, different types of
STAT3 inhibitors have been implemented for leukemia treatment,
including STAT3 SH2 domain inhibitors, DNA-binding domain
inhibitors, STAT3 gene expression oligonucleotide inhibitors and
N-terminal domain inhibitors (104). For example, small molecule
C188-9, a STAT3 inhibitor, has been shown to induce the apoptosis
of multiple AML cell lines and primary cells (8). The STAT3 inhibitor, OPB-31121, has
also been shown to strongly inhibit STAT3 and STAT5
phosphorylation, and to exert a significant anti-proliferative
effect on human leukemia cells (105). In addition, a number of
chemotherapy drugs can also target STAT3 to exert a therapeutic
effect. For instance, decitabine has been shown to markedly inhibit
the proliferation of HL-60 cells and to enhance the cytotoxicity of
natural killer cells to HL-60 cells, which may be related to the
STAT3 signaling pathway (106).
Zoledronic acid and bortezomib can both inhibit the proliferation
of K562 cells and induce apoptosis via the STAT3 signaling pathway
(107,108). Chidamide has been shown to affect
AML cell viability by inhibiting the JAK2/STAT3 signaling pathway
(109). Although some inhibitors
have been shown to be effective in the treatment of leukemia in
vitro, few inhibitors of STAT3 have been approved for leukemia
therapy in clinical practice (Table
II).
| Table IISTAT3 inhibitors. |
Table II
STAT3 inhibitors.
| STAT3
inhibitor | Targeted
domain | Effect on leukemia
treatment (pre-clinical) |
|---|
| ST3-H2A2 | The N-terminal | Unknown |
| BP-1-102 | The SH2 | Unknown |
| BP-5-087 | The SH2 | Yes |
| Catechol | The SH2 | Unknown |
| C188-9 | The SH2 | Yes |
| ISS 610 | The SH2 | Unknown |
| LLL-3 | The SH2 | Yes |
| LLL12 | The SH2 | Unknown |
| MM-206 | The SH2 | Yes |
| OPB-31121 | The SH2 | Yes |
| OPB-51602 | The SH2 | Yes |
| STA-21 | The SH2 | Yes |
| Stattic | The SH2 | Yes |
| S3I-201 | The SH2 | Unknown |
| S3I-201.1066 | The SH2 | Unknown |
| S3I-M2001 | The SH2 | Unknown |
| STX-0119 | The SH2 | Yes |
| S3I-1757 | The SH2 | Unknown |
| 5,15-DPP | The SH2 | Unknown |
| CPA-1 | DNA binding | Unknown |
| CPA-7 | DNA binding | Unknown |
| DBD-1 | DNA binding | Unknown |
| InS3-54 | DNA binding | Unknown |
| IS3-295 | DNA binding | Unknown |
| Platinum (IV)
tetrachloride | DNA binding | Unknown |
| Atovaquone | Unknown | Yes |
Moreover, many natural extracts of traditional
Chinese medicine have also been shownt o inhibit the activation of
STAT3 to exert antitumor effects. For instance, the traditional
Chinese medicine, cucurbitacin B, has been shown to inhibit STAT3
activation along with the Raf/MEK/ERK pathway and to promote the
apoptosis of K562 cells (110).
It has also been demonstrated that dehydrocostus lactone
significantly suppresses the proliferation of K562 cells by
inhibiting STAT3 phosphorylation and the expression of downstream
target genes (111). Tanshinone
IIA and cryptotanshinone have been shown to induce K562 cell
apoptosis by modulating JAK/STAT3/5 and SHP1/2 signaling
distinctively (112). Matrine
suppresses the growth of human CLL cells via its inhibition of the
IL-6/JAK/STAT3 signaling (113).
On the whole, traditional Chinese medicine has great potential in
the treatment of leukemia. It has also been reported that certain
biological therapies can also be used to target STAT3 signaling
pathways for leukemia treatment. Lentivirus-mediated STAT3
silencing may inhibit the expression of its downstream genes,
c-myc, Bcl-xL and cyclin D1, to suppress the
malignant biological behaviors, and STAT3 silencing also inhibits
the leukemogenic potency of K562 cells in mice (114). Moreover, the suppression of the
survivin gene induced by the BCR/ABL/JAK2/STAT3 pathway
sensitizes imatinib-resistant CML cells to different cytotoxic
drugs (115). Thus, the role of
biological therapy in the treatment of leukemia cannot be
ignored.
The role of STAT3 in the prognosis of
leukemia
The roles of STAT3 in the prognosis of leukemia have
drawn increasing attentions, but are still controversial. Recent
studies have found that Mcl-1 gene expression and
indoleamine 2,3-dioxygenase 1 (IDO1) activity can both be regulated
by STAT3 which are associated with the poorer prognosis of leukemia
(116,117). Bruserud et al (103) examined the association between
STAT3 expression in bone marrow and the prognosis of patients with
AML, and found that STAT3 expression and Y705 activation had an
adverse prognostic impact on human AML. Moreover, Adamaki et
al (118) discovered that 8
out of 14 childhood patients with ALL, in which STAT1 and STAT3
were expressed at lower levels on day 33, were relapse-free with a
high survival rate, indicating that STAT3 gene upregulation
is a poor predictor of the clinical prognosis of childhood ALL.
Furthermore, Zhong et al (119) investigated the association
between genomic polymorphisms of STAT3 and AML patient response to
chemotherapy in a Chinese population, and found that there were
strong associations between unfavorable cytogenetics, partial
remission (and even no remission) and GG genotype frequency in
rs9909659, which suggested that the STAT3 GG genotype in rs9909659
may confer an enhanced resistance to standard chemotherapy, and
predict an unfavorable prognosis. Besides, it has also been
reported that constitutive STAT3 activation is associated with the
shortest disease-free survival in AML, and the subgroup of AML
patients, which expressed STAT3β and showed constitutive activity
of STAT3, experienced a particularly poor outcome (92).
By contrast, some studies have suggested that
p-STAT3 also has a beneficial effect on the prognosis of leukemia.
Redell et al (8,120) discovered that when the
phosphorylated level of STAT3 on Y705 and S727 increased in AML
upon cytokine stimulation, the disease-free and survival rates of
patients were improved. Levidou et al (121) discovered that the
immunoreactivity of tyrosine phosphorylated STAT3 was marginally
associated with a prolonged overall survival. In addition, the
upregulation of tyrosine phosphorylated STAT3 was associated with a
longer time to first treatment, and the absence of p-STAT3 was
associated with a shorter time to progression, which indicated that
p-STAT3 also has a favorable prognostic effect on leukemia.
Nevertheless, the upregulation of STAT3 is also associated with the
favorable prognosis of AL (122).
Collectively, the impact of STAT3 on the prognosis of leukemia
remains controversial. Due to the defects of current clinical
studies, including small sample sizes and so on, extensive clinical
studies are warranted in order to further confirm the role of STAT3
in the evaluation of the prognosis of leukemia.
5. Conclusion
STAT3 is an important signal transducer and
activator of transcription, which is widely involved in cell
physiological processes, such as cell proliferation,
differentiation and apoptosis through a variety of signal
transduction pathways, and plays key roles in the occurrence of
cancer and other diseases. In this review, the roles of STAT3 in
the pathogenesis, diagnosis, treatment and prognosis of leukemia
were reviewed and discussed in the aspects of cell proliferation,
committed cell differentiation and apoptosis. There is increasing
evidence to indicate that STAT3 expression and activity
abnormalities will lead to excessive proliferation, differentiation
disorders and the abnormal apoptosis of leukemia cells, which may
provide a novel new biomarker for the clinical diagnosis, treatment
and prognosis evaluation of leukemia. At present, the roles of
upstream factors modulating STAT3 expression and activity, and the
signaling pathways mediated by downstream target genes are not yet
clear during the process of leukemia development. The in-depth
exploration of this area, particularly in the perspective of
epigenetic regulation, may further reveal the roles of STAT3 in the
pathogenesis of leukemia and the essential regulatory
mechanisms.
Acknowledgments
Not applicable.
Funding
This study was supported by the Natural Science
Foundation of China (grant nos. 81373670, 81673981, 81573467,
81601442 and 81704116), the Primary Research and Development Plan
of Shandong Province (grant nos. 2016GSF202016, 2017GSF19118 and
2017GSF218013), the Project of Transformation in High-tech
Achievements (grant no. 2013ZHZX2A0405), the Natural Science
Foundation of Shandong Province (grant no. ZR2017PH008 and
ZR2018PH042), the Science and Technology Development Grant of the
State Administration of traditional Chinese medicine of Shandong
Province (grant nos. 2013-2016 and 2017-174), the Family Planning
Committee of Shandong Province [grant no. (2014)14], the Project
for Shandong Medical and Health Science and Technology Plan (grant
no. 2015WS0191 and 2016WS0524), the Project of Science and
Technology of Shandong Academy of Medical Sciences (grant nos.
2016-34, 2016-35 and 2017-15) and the Innovation Project of
Shandong Academy of Medical Sciences.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YS, ZZ, XQ, XZ and LZ searched the literature for
STAT3 roles in leukemia; RW, XY and YZ screened the literature for
STAT3 inhibitors; YS, ZZ, QG and LS drew the graphics; XL conceived
the project, and YS, ZZ and XL wrote the manuscript. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chen WQ, Shan BE, Zheng SR, Lin GZ, Chen
JZ, Chen JG and He YT: Analysis of incidence and mortality of
leukemia in registration areas of China from 2003 to 2007. Tumor.
32:251–255. 2012.In Chinese.
|
|
2
|
Amitay EL and Keinan-Boker L:
Breastfeeding and childhood leukemia incidence: Ameta-analysis and
systematic review. JAMA Pediatr. 169:e1510252015. View Article : Google Scholar
|
|
3
|
Redaelli A, Stephens JM, Laskin BL, Pashos
CL and Botteman MF: The burden and outcomes associated with four
leukemias: AML, ALL, CLL and CML. Expert Rev Anticancer Ther.
3:311–329. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Namanja AT, Wang J, Buettner R, Colson L
and Chen Y: Allosteric communication across STAT3 domains
associated with STAT3 function and disease-causing mutation. J Mol
Biol. 428:579–589. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yu H, Kortylewski M and Pardoll D:
Crosstalk between cancer and immune cells: Role of STAT3 in the
tumour microenvironment. Nat Rev Immunol. 7:41–51. 2007. View Article : Google Scholar
|
|
6
|
Yu H and Jove R: The STATs of cancer - new
molecular targets come of age. Nat Rev Cancer. 4:97–105. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Villarino AV, Kanno Y, Ferdinand JR and
O'Shea JJ: Mechanisms of Jak/STAT signaling in immunity and
disease. J Immunol. 194:21–27. 2015. View Article : Google Scholar :
|
|
8
|
Redell MS, Ruiz MJ, Alonzo TA, Gerbing RB
and Tweardy DJ: Stat3 signaling in acute myeloid leukemia:
Ligand-dependent and -independent activation and induction of
apoptosis by a novel small-molecule Stat3 inhibitor. Blood.
117:5701–5709. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Benekli M, Xia Z, Donohue KA, Ford LA,
Pixley LA, Baer MR, Baumann H and Wetzler M: Constitutive activity
of signal transducer and activator of transcription 3 protein in
acute myeloid leukemia blasts is associated with short disease-free
survival. Blood. 99:252–257. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rezvani K and Barrett J: STAT3: The
'Achilles' heel for AML? Blood. 123:1–2. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Redell MS: A STAT3 decoy lures AML out of
hiding. Blood. 127:1628–1629. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yue P and Turkson J: Targeting STAT3 in
cancer: How successful are we? Expert Opin Investig Drugs.
18:45–56. 2009. View Article : Google Scholar :
|
|
13
|
Zhang T, Kee WH, Seow KT, Fung W and Cao
X: The coiled-coil domain of Stat3 is essential for its SH2
domain-mediated receptor binding and subsequent activation induced
by epidermal growth factor and interleukin-6. Mol Cell Biol.
20:7132–7139. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Seidel HM, Milocco LH, Lamb P, Darnell JE
Jr, Stein RB and Rosen J: Spacing of palindromic half sites as a
determinant of selective STAT (signal transducers and activators of
transcription) DNA binding and transcriptional activity. Proc Natl
Acad Sci USA. 92:3041–3045. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Song L and Shen BF: New development on
Jak/STAT sigal transduction pathway. Immunol J. 16:68–71. 2000.In
Chinese.
|
|
16
|
Dewilde S, Vercelli A, Chiarle R and Poli
V: Of alphas and betas: Distinct and overlapping functions of STAT3
isoforms. Front Biosci. 13:6501–6514. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim BH, Yi EH and Ye SK: Signal transducer
and activator of transcription 3 as a therapeutic target for cancer
and the tumor microenvironment. Arch Pharm Res. 39:1085–1099. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ren Z, Mao X, Mertens C, Krishnaraj R, Qin
J, Mandal PK, Romanowski MJ, McMurray JS and Chen X: Crystal
structure of unphosphorylated STAT3 core fragment. Biochem Biophys
Res Commun. 374:1–5. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chakraborty A and Tweardy DJ: Stat3 and
G-CSF-induced myeloid differentiation. Leuk Lymphoma. 30:433–442.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ilaria RL Jr: STAT isoforms: Mediators of
STAT specificity or leukemogenesis? Leuk Res. 25:483–484. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Becker S, Groner B and Müller CW:
Three-dimensional structure of the Stat3beta homodimer bound to
DNA. Nature. 394:145–151. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Benekli M, Baer MR, Baumann H and Wetzler
M: Signal transducer and activator of transcription proteins in
leukemias. Blood. 101:2940–2954. 2003. View Article : Google Scholar
|
|
23
|
Coffer PJ, Koenderman L and de Groot RP:
The role of STATs in myeloid differentiation and leukemia.
Oncogene. 19:2511–2522. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chakraborty A and Tweardy DJ: Granulocyte
colony-stimulating factor activates a 72-kDa isoform of STAT3 in
human neutrophils. J Leukoc Biol. 64:675–680. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hevehan DL, Miller WM and Papoutsakis ET:
Differential expression and phosphorylation of distinct STAT3
proteins during granulocytic differentiation. Blood. 99:1627–1637.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pilati C and Zucman-Rossi J: Mutations
leading to constitutive active gp130/JAK1/STAT3 pathway. Cytokine
Growth Factor Rev. 26:499–506. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bowman T, Garcia R, Turkson J and Jove R:
STATs in oncogenesis. Oncogene. 19:2474–2488. 2000. View Article : Google Scholar
|
|
28
|
Groner B, Lucks P and Borghouts C: The
function of Stat3 in tumor cells and their microenvironment. Semin
Cell Dev Biol. 19:341–350. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Takeda K, Kaisho T, Yoshida N, Takeda J,
Kishimoto T and Akira S: Correction: Stat3 activation is
responsible for IL-6-dependent T cell proliferation through
preventing apoptosis: generation and characterization of T
cell-specific Stat3-deficient mice. J Immunol. 194:35262015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Levy DE and Lee CK: What does Stat3 do? J
Clin Invest. 109:1143–1148. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bournazou E and Bromberg J: Targeting the
tumor microenvironment: JAK-STAT3 signaling. JAK-STAT.
2:e238282013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Guo H, Zhou H, Lu J, Qu Y, Yu D and Tong
Y: Vascular endothelial growth factor: An attractive target in the
treatment of hypoxic/ischemic brain injury. Neural Regen Res.
11:174–179. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Leonard WJ and O'Shea JJ: Jaks and STATs:
Biological implications. Annu Rev Immunol. 16:293–322. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schindler C and Strehlow I: Cytokines and
STAT signaling. Adv Pharmacol. 47:113–174. 2000. View Article : Google Scholar
|
|
35
|
Kisseleva T, Bhattacharya S, Braunstein J
and Schindler CW: Signaling through the JAK/STAT pathway, recent
advances and future challenges. Gene. 285:1–24. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kang SK, Kang KS, Jee MK and Kim BS:
Vascular endothelial growth factor/kinase insult domain receptor
(KDR)/fetal liver kinase 1 (FLK1)-mediated skin-epithelial
progenitor cells reprogramming. Tissue Eng Part A. 16:2687–2697.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pastuschek J, Poetzsch J, Morales-Prieto
DM, Schleussner E, Markert UR and Georgiev G: Stimulation of the
JAK/STAT pathway by LIF and OSM in the human granulosa cell line
COV434. J Reprod Immunol. 108:48–55. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Marino VJ and Roguin LP: The granulocyte
colony stimulating factor (G-CSF) activates Jak/STAT and MAPK
pathways in a trophoblastic cell line. J Cell Biochem.
103:1512–1523. 2008. View Article : Google Scholar
|
|
39
|
Platanias LC: Mechanisms of type-I- and
type-II-interferon-mediated signalling. Nat Rev Immunol. 5:375–386.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kijima T, Niwa H, Steinman RA, Drenning
SD, Gooding WE, Wentzel AL, Xi S and Grandis JR: STAT3 activation
abrogates growth factor dependence and contributes to head and neck
squamous cell carcinoma tumor growth in vivo. Cell Growth Differ.
13:355–362. 2002.PubMed/NCBI
|
|
41
|
Ram PT and Iyengar R: G protein coupled
receptor signaling through the Src and Stat3 pathway: Role in
proliferation and transformation. Oncogene. 20:1601–1606. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ahmed ST and Ivashkiv LB: Inhibition of
IL-6 and IL-10 signaling and Stat activation by inflammatory and
stress pathways. J Immunol. 165:5227–5237. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Trengove MC and Ward AC: SOCS proteins in
development and disease. Am J Clin Exp Immunol. 2:1–29.
2013.PubMed/NCBI
|
|
44
|
Piessevaux J, Lavens D, Peelman F and
Tavernier J: The many faces of the SOCS box. Cytokine Growth Factor
Rev. 19:371–381. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Shuai K: Regulation of cytokine signaling
pathways by PIAS proteins. Cell Res. 16:196–202. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ma LD, Zhou M, Wen SH, Ni C, Jiang LJ, Fan
J and Xia L: Effects of STAT3 silencing on fate of chronic
myelogenous leukemia K562 cells. Leuk Lymphoma. 51:1326–1336. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chakraborty A, White SM, Schaefer TS, Ball
ED, Dyer KF and Tweardy DJ: Granulocyte colony-stimulating factor
activation of Stat3 alpha and Stat3 beta in immature normal and
leukemic human myeloid cells. Blood. 88:2442–2449. 1996.PubMed/NCBI
|
|
48
|
Quotti Tubi L, Canovas Nunes S, Brancalion
A, Doriguzzi Breatta E, Manni S, Mandato E, Zaffino F, Macaccaro P,
Carrino M, Gianesin K, et al: Protein kinase CK2 regulates AKT,
NF-κB and STAT3 activation, stem cell viability and proliferation
in acute myeloid leukemia. Leukemia. 31:292–300. 2017. View Article : Google Scholar
|
|
49
|
Mencalha AL, Binato R, Ferreira GM, Du
Rocher B and Abdelhay E: Forkhead box M1 (FoxM1) gene is a new
STAT3 transcriptional factor target and is essential for
proliferation, survival and DNA repair of K562 cell line. PLoS One.
7:e481602012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yuzugullu H, Von T, Thorpe LM, Walker SR,
Roberts TM, Frank DA and Zhao JJ: NTRK2 activation cooperates with
PTEN deficiency in T-ALL through activation of both the PI3K-AKT
and JAK-STAT3 pathways. Cell Discov. 2:160302016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cook AM, Li L, Ho Y, Lin A, Li L, Stein A,
Forman S, Perrotti D, Jove R and Bhatia R: Role of altered growth
factor receptor-mediated JAK2 signaling in growth and maintenance
of human acute myeloid leukemia stem cells. Blood. 123:2826–2837.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sasaki R, Ito S, Asahi M and Ishida Y:
YM155 suppresses cell proliferation and induces cell death in human
adult T-cell leukemia/lymphoma cells. Leuk Res. 39:1473–1479. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Avalos BR: Molecular analysis of the
granulocyte colony-stimulating factor receptor. Blood. 88:761–777.
1996.PubMed/NCBI
|
|
54
|
Hillmer EJ, Zhang H, Li HS and Watowich
SS: STAT3 signaling in immunity. Cytokine Growth Factor Rev.
31:1–15. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Panopoulos AD, Zhang L, Snow JW, Jones DM,
Smith AM, El Kasmi KC, Liu F, Goldsmith MA, Link DC, Murray PJ, et
al: STAT3 governs distinct pathways in emergency granulopoiesis and
mature neutrophils. Blood. 108:3682–3690. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Krönke M, Schlüter C and Pfizenmaier K:
Tumor necrosis factor inhibits MYC expression in HL-60 cells at the
level of mRNA transcription. Proc Natl Acad Sci USA. 84:469–473.
1987. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Johansen LM, Iwama A, Lodie TA, Sasaki K,
Felsher DW, Golub TR and Tenen DG: c-Myc is a critical target for
c/EBPalpha in granulopoiesis. Mol Cell Biol. 21:3789–3806. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Xu S, Xu Z, Liu B, Sun Q, Yang L, Wang J,
Wang Y and Liu H: LIFRα-CT3 induces differentiation of a human
acute myelogenous leukemia cell line HL-60 by suppressing miR-155
expression through the JAK/STAT pathway. Leuk Res. 38:1237–1244.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Uchino Y, Iriyama N, Hatta Y and Takei M:
Granulocyte colony-stimulating factor potentiates all-trans
retinoic acid-induced granulocytic differentiation in acute
promyelocytic leukemia cell line HT93A. Cancer Cell Int. 15:302015.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Xu N, Tang XH, Pan W, Xie ZM, Zhang GF, Ji
MH, Yang JJ, Zhou MT and Zhou ZQ: Spared nerve injury increases the
expression of microglia M1 markers in the prefrontal cortex of rats
and provokes depression-like behaviors. Front Neurosci. 11:2092017.
View Article : Google Scholar :
|
|
61
|
Allman WR, Dey R, Liu L, Siddiqui S,
Coleman AS, Bhattacharya P, Yano M, Uslu K, Takeda K, Nakhasi HL,
et al: TACI deficiency leads to alternatively activated macrophage
phenotype and susceptibility to Leishmania infection. Proc Natl
Acad Sci USA. 112:E4094–E4103. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zhou L, Zhuo H, Ouyang H, Liu Y, Yuan F,
Sun L, Liu F and Liu H: Glycoprotein non-metastatic melanoma
protein b (Gpnmb) is highly expressed in macrophages of acute
injured kidney and promotes M2 macrophages polarization. Cell
Immunol. 316:53–60. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Binnemars-Postma K, Storm G and Prakash J:
Nanomedicine strategies to target tumor-associated macrophages. Int
J Mol Sci. 18:9792017. View Article : Google Scholar :
|
|
64
|
Fu XL, Duan W, Su CY, Shen XQ, Zhuang Y,
Yu PW and Zhao YL: IL-6 cooperated with M-CSF induce
CD14+ monocytes differentiation into M2-like phenotype
macrophages in vitro. Immunol J. 32:1013–1018. 2016.In Chinese.
|
|
65
|
Komohara Y, Horlad H, Ohnishi K, Ohta K,
Makino K, Hondo H, Yamanaka R, Kajiwara K, Saito T, Kuratsu J, et
al: M2 macrophage/microglial cells induce activation of Stat3 in
primary central nervous system lymphoma. J Clin Exp Hematop.
51:93–99. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Mangan JK, Rane SG, Kang AD, Amanullah A,
Wong BC and Reddy EP: Mechanisms associated with IL-6-induced
up-regulation of Jak3 and its role in monocytic differentiation.
Blood. 103:4093–4101. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Iwamoto T, Senga T, Adachi K and Hamaguchi
M: Stat3-dependent induction of interleukin-3 receptor expression
in leukemia inhibitory factor-stimulated M1 mouse leukemia cells.
Cytokine. 25:136–139. 2004. View Article : Google Scholar
|
|
68
|
Tanuma N, Nakamura K, Shima H and Kikuchi
K: Protein-tyrosine phosphatase PTPepsilon C inhibits Jak-STAT
signaling and differentiation induced by interleukin-6 and leukemia
inhibitory factor in M1 leukemia cells. J Biol Chem.
275:28216–28221. 2000.PubMed/NCBI
|
|
69
|
Yao Y, Zhou Q and Ericson SG: Vanadate
stimulates monocytic differentiation activity of IL-6 by enhancing
actin filament polymerization in HL-60 cells. J Biomed Sci.
11:940–949. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Woetmann A, Nielsen M, Christensen ST,
Brockdorff J, Kaltoft K, Engel AM, Skov S, Brender C, Geisler C,
Svejgaard A, et al: Inhibition of protein phosphatase 2A induces
serine/threonine phosphorylation, subcellular redistribution, and
functional inhibition of STAT3. Proc Natl Acad Sci USA.
96:10620–10625. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Sun Q, Wang J, Xiong J, Yang L and Liu H:
Free LIF receptor α-chain distal cytoplasmic motifs enhance
Jak2-independent STAT3 phosphorylation and induce differentiation
in HL-60 cells. Oncol Rep. 26:399–404. 2011.PubMed/NCBI
|
|
72
|
Zhou J, Chong PS, Lu X, Cheong LL, Bi C,
Liu SC, Zhou Y, Tan TZ, Yang H, Chung TH, et al: Phosphatase of
regenerating liver-3 is regulated by signal transducer and
activator of transcription 3 in acute myeloid leukemia. Exp
Hematol. 42:1041–1052. e1–2. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Banchereau J, Briere F, Caux C, Davoust J,
Lebecque S, Liu YJ, Pulendran B and Palucka K: Immunobiology of
dendritic cells. Annu Rev Immunol. 18:767–811. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Liu YJ, Kanzler H, Soumelis V and Gilliet
M: Dendritic cell lineage, plasticity and cross-regulation. Nat
Immunol. 2:585–589. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
75
|
Mohty M, Jarrossay D, Lafage-Pochitaloff
M, Zandotti C, Brière F, de Lamballeri XN, Isnardon D, Sainty D,
Olive D and Gaugler B: Circulating blood dendritic cells from
myeloid leukemia patients display quantitative and cytogenetic
abnormalities as well as functional impairment. Blood.
98:3750–3756. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Claxton DF, McMannis J, Champlin R and
Choudhury A: Therapeutic potential of leukemia-derived dendritic
cells: Preclinical and clinical progress. Crit Rev Immunol.
21:147–155. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Wu L and Liu YJ: Development of
dendritic-cell lineages. Immunity. 26:741–750. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
McKenna HJ, Stocking KL, Miller RE, Brasel
K, De Smedt T, Maraskovsky E, Maliszewski CR, Lynch DH, Smith J,
Pulendran B, et al: Mice lacking flt3 ligand have deficient
hematopoiesis affecting hematopoietic progenitor cells, dendritic
cells, and natural killer cells. Blood. 95:3489–3497.
2000.PubMed/NCBI
|
|
79
|
Waskow C, Liu K, Darrasse-Jèze G,
Guermonprez P, Ginhoux F, Merad M, Shengelia T, Yao K and
Nussenzweig M: The receptor tyrosine kinase Flt3 is required for
dendritic cell development in peripheral lymphoid tissues. Nat
Immunol. 9:676–683. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Li HS, Yang CY, Nallaparaju KC, Zhang H,
Liu YJ, Goldrath AW and Watowich SS: The signal transducers STAT5
and STAT3 control expression of Id2 and E2-2 during dendritic cell
development. Blood. 120:4363–4373. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Brady MT, Miller A, Sait SN, Ford LA,
Minderman H, Wang ES, Lee KP, Baumann H and Wetzler M:
Down-regulation of signal transducer and activator of transcription
3 improves human acute myeloid leukemia-derived dendritic cell
function. Leuk Res. 37:822–828. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Zhou J, Bi C, Janakakumara JV, Liu SC,
Chng WJ, Tay KG, Poon LF, Xie Z, Palaniyandi S, Yu H, et al:
Enhanced activation of STAT pathways and overexpression of survivin
confer resistance to FLT3 inhibitors and could be therapeutic
targets in AML. Blood. 113:4052–4062. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Nair RR, Tolentino JH and Hazlehurst LA:
Role of STAT3 in transformation and drug resistance in CML. Front
Oncol. 2:302012. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Li P, Harris D, Liu Z, Rozovski U,
Ferrajoli A, Wang Y, Bueso-Ramos C, Hazan-Halevy I, Grgurevic S,
Wierda W, et al: STAT3-activated GM-CSFRα translocates to the
nucleus and protects CLL cells from apoptosis. Mol Cancer Res.
12:1267–1282. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Mencalha AL, Du Rocher B, Salles D, Binato
R and Abdelhay E: LLL-3, a STAT3 inhibitor, represses
BCR-ABL-positive cell proliferation, activates apoptosis and
improves the effects of Imatinib mesylate. Cancer Chemother
Pharmacol. 65:1039–1046. 2010. View Article : Google Scholar
|
|
86
|
Mangolini M, de Boer J,
Walf-Vorderwülbecke V, Pieters R, den Boer ML and Williams O: STAT3
mediates oncogenic addiction to TEL-AML1 in t(12;21) acute
lymphoblastic leukemia. Blood. 122:542–549. 2013. View Article : Google Scholar
|
|
87
|
Pathania AS, Kumar S, Guru SK, Bhushan S,
Sharma PR, Aithagani SK, Singh PP, Vishwakarma RA, Kumar A and
Malik F: The synthetic tryptanthrin analogue suppresses STAT3
signaling and induces caspase dependent apoptosis via ERK up
regulation in human leukemia HL-60 cells. PLoS One. 9:e1104112014.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Pathania AS, Guru SK, Ul Ashraf N,
Riyaz-Ul-Hassan S, Ali A, Abdullah Tasduq S, Malik F and Bhushan S:
A novel stereo bioactive metabolite isolated from an endophytic
fungus induces caspase dependent apoptosis and STAT-3 inhibition in
human leukemia cells. Eur J Pharmacol. 765:75–85. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Rozovski U, Harris DM, Li P, Liu Z, Wu JY,
Grgurevic S, Faderl S, Ferrajoli A, Wierda WG, Martinez M, et al:
At high levels, constitutively activated STAT3 induces apoptosis of
chronic lymphocytic leukemia cells. J Immunol. 196:4400–4409. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Xia Z, Sait SN, Baer MR, Barcos M, Donohue
KA, Lawrence D, Ford LA, Block AM, Baumann H and Wetzler M:
Truncated STAT proteins are prevalent at relapse of acute myeloid
leukemia. Leuk Res. 25:473–482. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Stevens AM, Ruiz MJ, Gerbing RB, Alonzo
TA, Gamis AS and Redell MS: Ligand-induced STAT3 signaling
increases at relapse and is associated with outcome in pediatric
acute myeloid leukemia: A report from the Children's Oncology
Group. Haematologica. 100:e496–e500. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Benekli M1, Xia Z, Donohue KA, Ford LA,
Pixley LA, Baer MR, Baumann H and Wetzler M: Constitutive activity
of signal transducer and activator of transcription 3 protein in
acute myeloid leukemia blasts is associated with short disease-free
survival. Blood. 2002:252–257. 2002. View Article : Google Scholar
|
|
93
|
Kristensen T, Larsen M, Rewes A,
Frederiksen H, Thomassen M and Møller MB: Clinical relevance of
sensitive and quantitative STAT3 mutation analysis using
next-generation sequencing in T-cell large granular lymphocytic
leukemia. J Mol Diagn. 16:382–392. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Jerez A, Clemente MJ, Makishima H, Koskela
H, Leblanc F, Peng Ng K, Olson T, Przychodzen B, Afable M,
Gomez-Segui I, et al: STAT3 mutations unify the pathogenesis of
chronic lymphoproliferative disorders of NK cells and T-cell large
granular lymphocyte leukemia. Blood. 120:3048–3057. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Rajala HL, Olson T, Clemente MJ, Lagström
S, Ellonen P, Lundan T, Hamm DE, Zaman SA, Lopez Marti JM,
Andersson EI, et al: The analysis of clonal diversity and therapy
responses using STAT3 mutations as a molecular marker in large
granular lymphocytic leukemia. Haematologica. 100:91–99. 2015.
View Article : Google Scholar :
|
|
96
|
Lamy T and Loughran TP Jr: How I treat LGL
leukemia. Blood. 117:2764–2774. 2011. View Article : Google Scholar :
|
|
97
|
Lamy T, Moignet A and Loughran TP Jr: LGL
leukemia: From pathogenesis to treatment. Blood. 129:1082–1094.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Matutes E: Large granular lymphocytic
leukemia. Current diagnostic and therapeutic approaches and novel
treatment options. Expert Rev Hematol. 10:251–258. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Ohgami RS, Ma L, Merker JD, Martinez B,
Zehnder JL and Arber DA: STAT3 mutations are frequent in
CD30+ T-cell lymphomas and T-cell large granular
lymphocytic leukemia. Leukemia. 27:2244–2247. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Zhong Y, Wu J, Chen B, Ma R, Cao H, Wang
Z, Cheng L, Ding J and Feng J: Investigation and analysis of single
nucleotide polymorphisms in Janus kinase/signal transducer and
activator of transcription genes with leukemia. Leuk Lymphoma.
53:1216–1221. 2012. View Article : Google Scholar
|
|
101
|
Lautner-Csorba O, Gézsi A, Semsei AF,
Antal P, Erdélyi DJ, Schermann G, Kutszegi N, Csordás K, Hegyi M,
Kovács G, et al: Candidate gene association study in pediatric
acute lymphoblastic leukemia evaluated by Bayesian network based
Bayesian multilevel analysis of relevance. BMC Med Genomics.
5:422012. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Sakamoto KM, Grant S, Saleiro D, Crispino
JD, Hijiya N, Giles F, Platanias L and Eklund EA: Targeting novel
signaling pathways for resistant acute myeloid leukemia. Mol Genet
Metab. 114:397–402. 2015. View Article : Google Scholar :
|
|
103
|
Bruserud Ø, Nepstad I, Hauge M, Hatfield
KJ and Reikvam H: STAT3 as a possible therapeutic target in human
malignancies: Lessons from acute myeloid leukemia. Expert Rev
Hematol. 8:29–41. 2015. View Article : Google Scholar
|
|
104
|
Munoz J, Dhillon N, Janku F, Watowich SS
and Hong DS: STAT3 inhibitors: Finding a home in lymphoma and
leukemia. Oncologist. 19:536–544. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Hayakawa F, Sugimoto K, Harada Y,
Hashimoto N, Ohi N, Kurahashi S and Naoe T: A novel STAT inhibitor,
OPB-31121, has a significant antitumor effect on leukemia with
STAT-addictive oncokinases. Blood Cancer J. 3:e1662013. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Zhu Z, Lu X, Jiang L, Sun X, Zhou H, Jia
Z, Zhang X and Ma L: STAT3 signaling pathway is involved in
decitabine induced biological phenotype regulation of acute myeloid
leukemia cells. Am J Transl Res. 7:1896–1907. 2015.PubMed/NCBI
|
|
107
|
Selvi N, Kaymaz BT, Gündüz C, Aktan C,
Kiper HD, Sahin F, Cömert M, Selvi AF, Kosova B and Saydam G:
Bortezomib induces apoptosis by interacting with JAK/STAT pathway
in K562 leukemic cells. Tumour Biol. 35:7861–7870. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Kiper HD, Tezcanli Kaymaz B, Gokbulut AA,
Selvi N, Avci CB, Kosova B, Iskender G, Yandim MK, Gunduz C, Sahin
F, et al: STAT pathway in the regulation of zoledronic acid-induced
apoptosis in chronic myeloid leukemia cells. Biomed Pharmacother.
67:527–532. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Zhao S, Guo J, Zhao Y, Fei C, Zheng Q, Li
X and Chang C: Chidamide, a novel histone deacetylase inhibitor,
inhibits the viability of MDS and AML cells by suppressing
JAK2/STAT3 signaling. Am J Transl Res. 8:3169–3178. 2016.PubMed/NCBI
|
|
110
|
Chan KT, Li K, Liu SL, Chu KH, Toh M and
Xie WD: Cucurbitacin B inhibits STAT3 and the Raf/MEK/ERK pathway
in leukemia cell line K562. Cancer Lett. 289:46–52. 2010.
View Article : Google Scholar
|
|
111
|
Cai H, Qin X and Yang C: Dehydrocostus
lactone suppresses proliferation of human chronic myeloid leukemia
cells through Bcr/Abl-JAK/STAT signaling pathways. J Cell Biochem.
118:3381–3390. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Jung JH, Kwon TR, Jeong SJ, Kim EO, Sohn
EJ, Yun M and Kim SH: Apoptosis induced by tanshinone IIA and
cryptotanshinone is mediated by distinct JAK/STAT3/5 and SHP1/2
signaling in chronic myeloid leukemia K562 cells. Evid Based
Complement Alternat Med. 2013:8056392013. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Ma L, Zhu Z, Jiang L, Sun X, Lu X, Zhou M,
Qian S and Jianyong L: Matrine suppresses cell growth of human
chronic myeloid leukemia cells via its inhibition of the
interleukin-6/Janus activated kinase/signal transducer and
activator of transcription 3 signaling cohort. Leuk Lymphoma.
56:2923–2930. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Jia X, Yang W, Han J and Xiong H: Effects
of lentivirus mediated STAT3 silencing on human chronic myeloid
leukemia cells and leukemia mice. Int J Clin Exp Med. 7:4031–4037.
2014.
|
|
115
|
Stella S, Tirrò E, Conte E, Stagno F, Di
Raimondo F, Manzella L and Vigneri P: Suppression of survivin
induced by a BCR-ABL/JAK2/STAT3 pathway sensitizes
imatinib-resistant CML cells to different cytotoxic drugs. Mol
Cancer Ther. 12:1085–1098. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Allen JC, Talab F, Zuzel M, Lin K and
Slupsky JR: c-Abl regulates Mcl-1 gene expression in chronic
lymphocytic leukemia cells. Blood. 117:2414–2422. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Folgiero V, Goffredo BM, Filippini P,
Masetti R, Bonanno G, Caruso R, Bertaina V, Mastronuzzi A, Gaspari
S, Zecca M, et al: Indoleamine 2,3-dioxygenase 1 (IDO1) activity in
leukemia blasts correlates with poor outcome in childhood acute
myeloid leukemia. Oncotarget. 5:2052–2064. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Adamaki M, Tsotra M, Vlahopoulos S,
Zampogiannis A, Papavassiliou AG and Moschovi M: STAT transcript
levels in childhood acute lymphoblastic leukemia: STAT1 and STAT3
transcript correlations. Leuk Res. 39:1285–1291. 2015. View Article : Google Scholar
|
|
119
|
Zhong Y, Feng J, Chen B, Cheng L, Li Y,
Qian J, Ding J, Gao F and Xia G: Signal transducer and activator of
transcription 3 (STAT3) gene polymorphisms are associated with
treatment outcomes in acute myeloid leukemia. Int J Lab Hematol.
34:383–389. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Redell MS, Ruiz MJ, Gerbing RB, Alonzo TA,
Lange BJ, Tweardy DJ and Meshinchi S; Children's Oncology Group:
FACS analysis of Stat3/5 signaling reveals sensitivity to G-CSF and
IL-6 as a significant prognostic factor in pediatric AML: A
Children's Oncology Group report. Blood. 121:1083–1093. 2013.
View Article : Google Scholar :
|
|
121
|
Levidou G, Sachanas S, Pangalis GA,
Kalpadakis C, Yiakoumis X, Moschogiannis M, Sepsa A, Lakiotaki E,
Milionis V, Kyrtsonis MC, et al: Immunohistochemical analysis of
IL-6, IL-8/CXCR2 axis, Tyr p-STAT-3, and SOCS-3 in lymph nodes from
patients with chronic lymphocytic leukemia: Correlation between
microvascular characteristics and prognostic significance. BioMed
Res Int. 2014:2514792014. View Article : Google Scholar
|
|
122
|
Danis E, Yamauchi T, Echanique K, Zhang X,
Haladyna JN, Riedel SS, Zhu N, Xie H, Orkin SH, Armstrong SA, et
al: Ezh2 controls an early hematopoietic program and growth and
survival signaling in early T cell precursor acute lymphoblastic
leukemia. Cell Rep. 14:1953–1965. 2016. View Article : Google Scholar : PubMed/NCBI
|