Introduction
Checkpoint kinases (Chks) are serine/threonine
kinases that serve a critical role in cell cycle control, DNA
damage responses and cell survival (1). Chks have two subtypes, Chk1 and Chk2,
and they are highly conserved from yeast to humans (2). The upregulation of Chks induces cell
cycle arrest, while their inhibition in the presence of DNA damage
induces abnormal DNA replication, followed by cell death (3). For cancer treatment, inhibitors of
Chks are used as chemotherapeutic agents, in many cases with DNA
damaging agents (4–6). However, it has been reported that Chk
inhibition alone may lead to apoptosis in tumor cells through
mutation and/or regulation of cellular tumor antigen p53 (p53) and
cyclin-dependent kinase inhibitor 1 (p21) tumor suppressor genes
(7). While Chk inhibitors may
require delivery to tumor cells, they are able to potentially
induce varying responses in other organs. The present study
evaluated the effect of AZD7762, a Chk1 and Chk2 inhibitor, on
three types of bone cells: Bone-resorbing osteoclasts, bone-forming
osteoblasts, and osteocytes.
Breast cancer accounts for 25% of all cancer cases
in women, and advanced breast cancer metastasizes to distant
organs, most commonly bone (8).
Thus, to treat breast cancer and protect bones from metastasis, the
desired function of chemotherapeutic agents is the inhibition of
tumor proliferation and osteoclast development. It is also
beneficial if the stimulation of osteoblast development is induced.
Employing an inhibitor of Chk1 and Chk2, AZD7762 (362 Da), in
addition to using PD407824 (328 Da) as a control Chk1 inhibitor,
the present study examined their effects on mammary tumor cells,
osteoclasts and osteoblasts/osteocytes. AZD7762 is a potent
inhibitor of Chk1 and Chk2 that potentiates antitumor activity in
xenograft models in a dose-dependent manner when simultaneously
administered with DNA-damaging agents (9). PD407824 is a selective inhibitor of
Chk1 (10), and its inhibitory
effect on tumor growth and bone resorption was recently reported in
connection to the regulation of stress in the endoplasmic reticulum
(ER) (11). Chk1 is a primary
gatekeeper of cell division at multiple cell cycle checkpoints,
including the S, G2/M and M phases. Chk2 prevents uncontrolled
rapid cell division, and inherited mutations in Chk2 are reported
to be associated with breast cancer (12,13).
The present study investigated the common effects of AZD7762 and
PD407824 as Chk1 inhibitors, in addition to the differential
effects of Chk2 inhibition.
It was previously reported that the elevated
phosphorylation of eukaryotic translation initiation factor-α
(eIF2α) reduces the proliferation of tumor cells and suppresses the
differentiation of osteoclasts (14,15).
Phosphorylation of eIF2α is regulated by phosphatases, including
protein phosphatase 1, in addition to four known kinases, including
protein kinase RNA-like endoplasmic reticulum kinase (PERK). PERK
is activated by stress in the ER (16). Using thapsigargin, an inducer of ER
stress, the present study evaluated the role of PERK and eIF2α in
response to AZD7762.
In the present study, the possible multiple roles of
AZD7762 in the suppression of tumor growth, inhibition of
osteoclast development and stimulation of osteoblast development
were investigated. Three bone cell lines were employed [RAW264.7
pre-osteoclast cells (17), MC3T3
osteoblast-like cells (18), and
MLO-A5 osteocyte-like cells (19)], together with 4T1.2 mammary tumor
cells. The 4T1.2 cell line was isolated from metastasized bone, and
represents a breast cancer model of efficient metastasis to bone
(20). The responses to AZD7762
and PD407824 were examined in monolayer cell cultures,
three-dimensional (3D) spheroids (21), and 3D bio-printed bone constructs
(22). While PD407824 is able to
inhibit tumor growth and osteoclastogenesis, its ability to promote
osteoblast development is limited (11). It has been reported that inhibition
of Chk2, and not Chk1, downregulates p53 expression (23). Since p53 is involved in osteoblast
development (24), it was
hypothesized that besides inhibitory effects on tumor cells and
osteoclasts, AZD7762 may have more potent effects on
osteoblastogenesis than PD407824 by a p53-dependent pathway.
Materials and methods
Cell culture
4T1.2 mouse mammary tumor cells (obtained from Dr R.
Anderson at Peter MacCallum Cancer Institute, Melbourne,
Australia), MDA-MB-231 human breast cancer cells [American Type
Culture Collection (ATCC), Manassas, VA, USA (25)], and MCF-7 human breast cancer cells
(26) were cultured in Dulbecco's
modified Eagle's medium (Corning Incorporated, Corning, NY, USA).
RAW264.7 pre-osteoclast cells (ATCC), MC3T3 osteoblast-like cells
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), and MLO-A5
osteocyte-like cells (obtained from Dr L. Bonewald at Indiana
University, Indianapolis, IN, USA) were grown in α minimum
essential medium (MEM; Gibco; Thermo Fisher Scientific. Inc.,
Waltham, MA, USA). Bone marrow-derived cells, obtained by flushing
the tibia and femur from one female 10-week old BALB/c mouse and
separating with low-density gradient centrifugation (27), were cultured in αMEM with 10 ng/ml
macrophage colony-stimulating factor (PeproTech, Inc., Rocky Hill,
NJ, USA) for 2 days, and the surface-attached cells were used as
osteoclast precursors. The experimental procedures using animals
were approved by the Indiana University Animal Care and Use
Committee and were in compliance with the Guiding Principles in the
Care and Use of Animals endorsed by the American Physiological
Society (28). The culture media
contained 10% fetal bovine serum (FBS; Atlanta Biologicals, Flowery
Branch, GA, USA) and antibiotics (50 U/ml penicillin and 50
µg/ml streptomycin; Life Technologies; Thermo Fisher
Scientific, Inc.). MLO-A5 cells were grown with 5% FBS and 5%
bovine calf serum. Cells were maintained at 37°C and 5%
CO2 in a humidified incubator, and treated with AZD7762
(0.1–10 µM), PD407824 (0.1–20 µM), or thapsigargin (1
µM; Tocris Bioscience, Bristol, UK).
Cellular proliferation was evaluated using an MTT
assay (Sigma-Aldrich; Merck KGaA), according to the manufacturer's
protocol. The relative cell proliferation was determined as a ratio
of the absorbance at 570 nm of each sample and control, as measured
using a plate reader. To quantify 3D spheroid cell viability, the
CellTiter-Glo 3D Cell Viability Assay (Promega Corporation,
Madison, WI, USA) was used, according to the manufacturer's
protocol, measuring luminescence with a plate reader.
3D spheroid assay and 3D
bio-printing
To induce spheroid formation, ~5,000 cells were
cultured in a U-bottom low-adhesion 96-well plate (S-Bio, Hudson,
NH, USA). MLO-A5 osteocyte spheroids were bio-printed onto a needle
array using a Regenova 3D Bioprinter (Cyfuse Biomedical K.K.,
Tokyo, Japan) (22) to generate
bone constructs mineralized in osteogenic medium (70 µM
ascorbic acid and 5 mM β-glycerophosphate) (27). Each construct contained 18
osteocyte spheroids (3×3×2 configuration).
Motility assay
To evaluate motility in a monolayer culture, a wound
healing scratch motility assay was performed as described
previously (29). In brief, cells
were plated in 12-well plates and, the following day, scratching
was performed using a plastic tip. The areas newly occupied with
cells in the scratched zone were determined 24 h post-scratching
using images obtained using a microscope (magnification, ×40; Nikon
Eclipse TS100, Nikon Corporation, Tokyo, Japan), which were scanned
using Adobe Photoshop (CS2; Adobe Systems, Inc., San Jose, CA, USA)
and quantified with Image J v1.51p (National Institutes of Health,
Bethesda, MD, USA).
Osteoclast differentiation assay and pit
formation assay
Using RAW264.7 pre-osteoclast cells, an osteoclast
differentiation assay was conducted in 24-well plates with 30 ng/ml
receptor activator of nuclear factor-κB ligand (RANKL) in the
presence and absence of 0.1–2 µM AZD7762 and PD407824. For
bone marrow-derived cells, cells were cultured in 10 ng/ml
macrophage colony stimulating factor (M-CSF; Peprotech, Inc., Rocky
Hill, NJ, USA) for 2 days prior to the addition of RANKL (27). During the 5-day experiments, the
culture medium was replaced once on day 3. Adherent cells were
fixed with a citrate, acetone and formaldehyde solution for 30 sec
at room temperature and stained for 20 min at 37°C with a tartrate
resistant acid phosphate (TRAP)-staining kit, according to the
manufacturer's protocol (Sigma-Aldrich; Merck KGaA). TRAP-positive
multinucleated cells (>3 nuclei) were identified as mature
osteoclasts (30).
A pit formation assay was conducted similarly using
24-well culture plates coated with hydroxyapatite (31). After 6 days, cells were removed
with 5% sodium hypochlorite for 5 min at room temperature, and
images of the wells were captured with a phase-contrast inverted
microscope at ×40 magnification. Using ImageJ, the areas of the
wells no longer coated with hydroxyapatite were measured, and the
pit area fraction was calculated by normalizing to the total image
area.
Alizarin red staining
MC3T3 cells were seeded on 24-well tissue-culture
plates at 6.5×104 cells/well. Upon becoming fully
confluent, osteogenic medium (70 µM ascorbic acid and 5 mM
β-glycerophosphate) was added, with 0–0.5 µM AZD7762, and
the cells were cultured for 5 weeks, changing the media every other
day. The effect of AZD7762 on mineralization was evaluated by
staining at room temperature for 3 min with 1% Alizarin Red S
solution (Sigma-Aldrich; Merck KGaA) and imaged at ×40
magnification with an inverted microscope. Alizarin red staining
was quantified by measuring the image grey-scale values using
ImageJ.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) and RNA interference
Total RNA was extracted using an RNeasy Plus mini
kit (Qiagen, Inc., Valencia, CA, USA) and RT was conducted (37°C
for 2 h; 85°C for 5 min) with a high-capacity cDNA reverse
transcription kit (Applied Biosystems; Thermo Fisher Scientific,
Inc.). RT-qPCR was performed using Power SYBR green PCR master mix
kits (Applied Biosystems; Thermo Fisher Scientific, Inc.) with the
PCR primers listed in Table I. RNA
interference was conducted using small interfering (si)RNA specific
to PERK (cat. no. 100334), Chk1 (cat. no. 160184) and Chk2 (cat.
no. 78549; Life Technologies; Thermo Fisher Scientific, Inc.). A
negative siRNA (Silencer Select #1; Life Technologies; Thermo
Fisher Scientific, Inc.) was used as a nonspecific negative
control. Cells were transiently transfected with siRNA (130 pmol
per 6-cm dish) using Lipofectamine® RNAiMAX (Life
Technologies; Thermo Fisher Scientific, Inc.) in Opti-MEM I medium.
The medium was replaced with regular culture medium after 24 h, and
the silencing efficiency was measured by immunoblotting 48 h
post-transfection.
 | Table IReverse transcription-quantitative
polymerase chain reaction primers used in the present study. |
Table I
Reverse transcription-quantitative
polymerase chain reaction primers used in the present study.
| Target | Forward primer | Reverse primer |
|---|
| ALP |
5′-CTGACTGACCCTTCGCTCTC-3′ |
5′-GGTCAATCCTGCCTCCTTCC-3′ |
| OCN |
5′-CCGGGAGCAGTGTGAGCTTA-3′ |
5′-AGGCGGTCTTCAAGCCATACT-3′ |
| Osterix |
5′-CCCTTCTCAAGCACCAATGG-3′ |
5′-AGGGTGGGTAGTCATTTGCATAG-3′ |
| GAPDH |
5′-TGCACCACCAACTGCTTAG-3′ |
5′-GGATGCAGGGATGATGTTC-3′ |
Western blot analysis
Cells were lysed in a radioimmuno-precipitation
assay buffer with protease inhibitors (Santa Cruz Biotechnology,
Inc., Dallas, TX, USA) and phosphatase inhibitors (Calbiochem;
Merck KGaA). Following measuring of the protein concentration using
a Bicinchoninic Acid Protein Assay kit (Thermo Fisher Scientific,
Inc.), ~10 µg isolated proteins/lane were fractionated using
10–15% SDS gels and electrotransferred to polyvinylidene
difluo-ride membranes (EMD Millipore, Billerica, MA, USA).
Membranes were blocked in 2% blotting-grade blocker (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) for 1 h at room temperature
or overnight at 4°C. Antibodies against alkaline phosphate (cat.
no. sc-28904; Santa Cruz Biotechnology, Inc.), cathepsin K (cat.
no. sc-48353; Santa Cruz Biotechnology, Inc.), PERK (cat. no.
sc-13073; Santa Cruz Biotechnology, Inc.), nuclear factor of
activated T cells cytoplasmic 1 (NFATc1; cat. no. sc-7294; Santa
Cruz Biotechnology, Inc.), Chk1 (cat. no. 2360; Cell Signaling
Technology, Inc., Danvers, MA, USA), phosphorylated (p)-Chk1 (cat.
no. 90178; Cell Signaling Technology, Inc.), Chk2 (cat. no. 2662;
Cell Signaling Technology, Inc.), eIF2α (cat. no. 9722; Cell
Signaling Technology, Inc.), p-eIF2α (cat. no. 9721; Cell Signaling
Technology, Inc.), microtubule-associated proteins 1A/1B light
chain (LC)3A/B II (cat. no. 4108; Cell Signaling Technology, Inc.),
caspase 3 (cat. no. 9662; Cell Signaling Technology, Inc.), cleaved
caspase 3 (cat. no. 9661; Cell Signaling Technology, Inc.), p38
(cat. no. 9212; Cell Signaling Technology, Inc.), p-p38 (cat. no.
9211; Cell Signaling Technology, Inc.), p-PERK (cat. no. 3179; Cell
Signaling Technology, Inc.), p53 (cat. no. MA5-12557; Invitrogen;
Thermo Fisher Scientific, Inc.), p-Chk2 (cat. no. SAB4504366;
Invitrogen; Thermo Fisher Scientific, Inc.), and β-actin (cat. no.
A5441; Sigma-Aldrich; Merck KGaA) were used. Primary antibodies
were incubated for 1 h at room temperature or overnight at 4°C at a
1:1,000 dilution, except Chk2, p-Chk2, p-PERK (all 1:500), p53
(1:200) and β-actin (1:10,000), while anti-rabbit (cat. no. 7074;
Cell Signaling Technology, Inc.) and anti-mouse (cat. no. 7076;
Cell Signaling Technology, Inc.) secondary antibodies were
incubated for 45 min at room temperature at a 1:2,000 dilution.
Protein expression levels were assayed using a SuperSignal west
femto maximum sensitivity substrate (Thermo Fisher Scientific,
Inc.).
Fluorescence resonance energy transfer
(FRET) imaging
To evaluate the potential role of GTPases in
cellular motility, the activity of RhoA GTPase was determined by
FRET imaging. In response to 2 µM AZD7762 and PD407824,
energy transfer was measured in a RhoA-specific cyan fluorescent
protein (CFP)-yellow fluorescent protein (YFP) biosensor (32). The filter sets (Semrock; IDEX
Health & Science, LLC, Rochester, NY, USA) were chosen for CFP
excitation at 438±24 nm (center wavelength ± bandwidth), CFP
emission at 483±32 nm and YFP emission at 542±27 nm. Using a
fluorescence microscope (Nikon Corporation, Tokyo, Japan)
time-lapse images were acquired at an interval of 5 min. The
emission ratio of YFP/CFP for individual cells was computed to
determining the activity levels using NIS-Elements software v3.2
(Nikon Corporation).
Micro-computed tomography (CT)
imaging
Micro-CT was performed using Skyscan 1172
(Bruker-MicroCT, Kontich, Belgium) (33). The printed bone constructs were
wrapped in Parafilm to maintain hydration and placed in a plastic
tube. Scans were performed at pixel size 6 µm. Using
manufacturer-provided software, the images were reconstructed
(nRecon v1.6.9.18), rotated (DataViewer v1.5.0), and the bone
mineral density of the mineralized bone construct was determined by
calculating the mean attenuation coefficient and calibrated to two
hydroxyapatite phantoms of known density (CTAn v1.11.4.2).
Statistical analysis
The data are expressed as the mean ± standard
deviation of three to four replicates. One-way analysis of variance
was employed to examine the statistical significance among groups,
and Fisher's protected least significant difference was conducted
as a post hoc test to evaluate the pairwise comparisons using R
3.4.3 (R Foundation for Statistical Computing, Vienna, Austria).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Inhibitory effects of AZD7762 on 4T1.2
mammary tumor cells
The present study first examined the effects of
AZD7762, an inhibitor of Chk1 and Chk2, on the proliferation and
migration of 4T1.2 mammary tumor cells. As the representative
images of 4T1.2 cells illustrate, in response to 1 µM
AZD7762, the cells underwent apoptosis (blebbing and cell
shrinkage) and autophagy (increase in vacuole-like structures in
the cytoplasm and membrane expansion), and the relative
proliferation of the tumor cells was significantly reduced in a
dose-dependent manner (Fig. 1A and
B). Western blot analysis revealed that p-eIF2α was elevated in
addition to cleaved caspase 3 (apoptosis marker) and LC3A/B II
(autophagy marker) (Fig. 1C and
D), while the expression levels of p-Chk1 and p-Chk2 were
reduced. The scratch assay for examining cellular motility
demonstrated that the healing of the wound area was decreased by
0.5 and 1 µM AZD7762 (Fig. 1E
and F), indicating that AZD7762 reduces the motility of tumor
cells. Notably, the wound area was also enlarged as a result of
AZD7762-driven cell death.
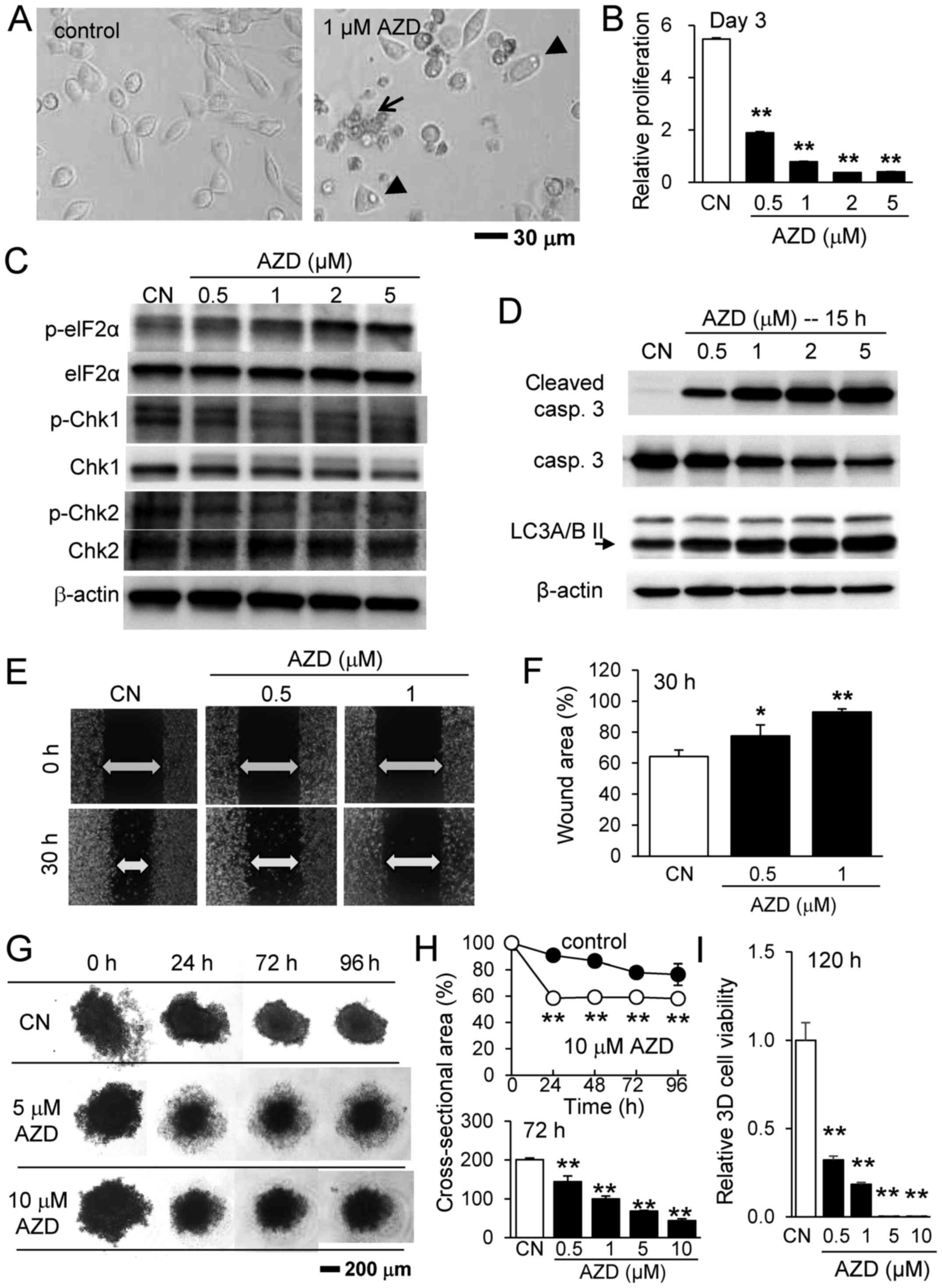 | Figure 1Inhibitory effects of AZD7762 on
4T1.2 mammary tumor cells. (A) Representative images of 4T1.2 cells
in response to 1 µM AZD7762 for 24 h. The arrow and arrow
heads indicate apoptotic and autophagic cells, respectively,
assessed according to cellular morphology. (B) Relative
proliferation of 4T1.2 cells in response to 0.5, 1, 2 and 5
µM AZD7762. (C) Upregulation of p-eIF2α, and downregulation
of p-Chk1 and p-Chk2. (D) Upregulation of cleaved caspase 3
(apoptosis marker) and LC3A/B II (autophagy marker). (E) Scratch
assay and (F) wound area in response to 0.5 and 1 µM
AZD7762. (G) 3D tumor spheroids in response to 5 and 10 µM
AZD7762. (H) Cross-sectional area and (I) cell viability of 3D
tumor spheroids in response to AZD7762. *P<0.05;
**P<0.01 vs. CN. CN, control; AZD, AZD7762; 3D,
three-dimensional; eIF2α, eukaryotic translation initiation
factor-α; p, phosphorylated; Chk, checkpoint kinase inhibitor,
casp, caspase; LC3A/B II, microtubule-associated proteins 1A/1B
light chain 3A/B II. |
Since tumor growth may differ according to the
culture conditions, the present study subsequently examined the
effects of AZD7762 on 3D spheroids solely composed of 4T1.2 cells
(Fig. 1G). Initially, the
spheroids were irregularly shaped with a rough surface. They
gradually compacted, forming a necrotic core at the center and a
quiescent shell around the core with a proliferating surface. In
response to 0.5 to 10 µM AZD7762, the spheroids
significantly altered in appearance. While the spheroids exhibited
extended quiescent zones at the lower concentrations (0.5 and 1
µM AZD7762), it increased the necrotic core at the higher
concentrations (5 and 10 µM AZD7762). AZD7762 reduced the
cross-sectional area of the tumor spheroids and decreased 3D cell
viability (Fig. 1H and I).
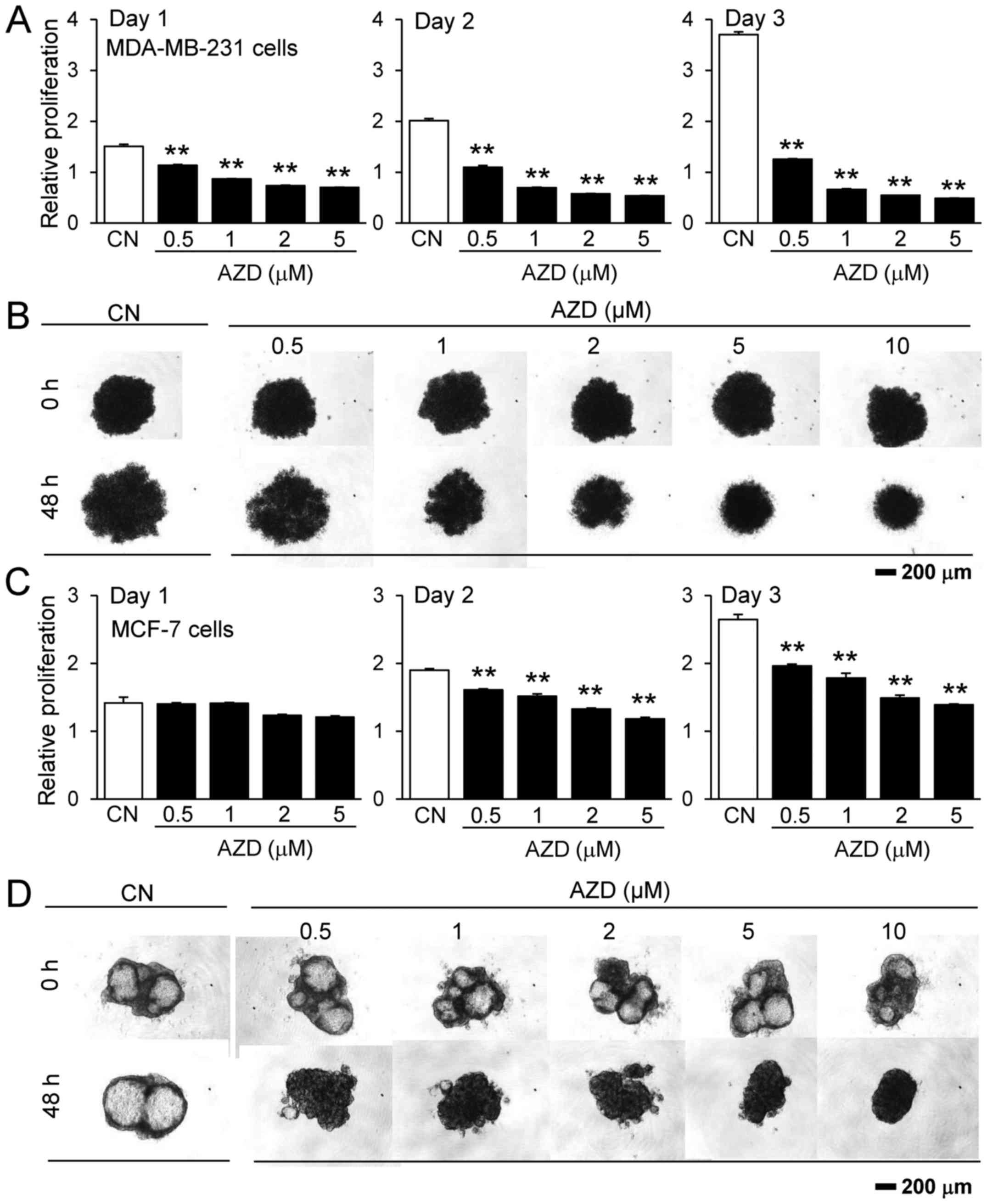
To evaluate its effect on other types of breast
cancer, the present study also examined the responses of two human
breast cancer cell lines, MDA-MB-231 and MCF-7, to AZD7762.
MDA-MB-231 cells are estrogen receptor-negative, while MCF-7 cells
are estrogen receptor-positive. The results revealed that AZD7762
inhibited the proliferation of the two cell lines, as measured by
MTT assay, in a monolayer cell culture, and decreased the sizes of
the 3D tumor spheroids, although 4T1.2 and MDA-MB-231 cells were
more sensitive to AZD7762 compared with MCF-7 cells (Fig. 2).
Inhibitory effects of AZD7762 on RAW264.7
pre-osteoclast cells
Osteoclasts serve a pivotal role in bone
homeostasis. Using RAW264.7 pre-osteoclast cells and osteoclast
precursors from bone marrow-derived cells, the present study
evaluated the effect of AZD7762 on the proliferation and
differentiation of osteoclasts. Treatment with AZD7762
significantly reduced the proliferation of RAW264.7 cells and
primary osteoclast precursors (Fig. 3A
and B). The half-maximal inhibitory concentration for AZD7762
in bone marrow-derived osteoclasts was estimated as 0.54 µM
by fitting an exponential curve to the MTT results (data not
shown). In RAW cells, AZD7762 also downregulated NFATc1, a master
transcription factor for osteoclastogenesis, in addition to
cathepsin K, a marker of bone resorption (Fig. 3C). It was additionally observed
that the expression level of p-eIF2α was elevated, together with
cleaved caspase 3, in RAW cells (Fig.
3C and D). In primary osteoclast precursors, AZD7762 suppressed
the RANKL-driven upregulation of NFATc1 and elevated cleaved
caspase 3 expression (Fig. 3E). As
demonstrated by TRAP staining for identifying multi-nucleated
osteoclasts, AZD7762 significantly reduced the number of
multi-nucleated TRAP-positive osteoclast cells in a dose-dependent
manner (Fig. 3F and G).
Furthermore, AZD7762 decreased the 3D cell viability of RAW264.7
spheroids in a dose-dependent manner (Fig. 3H). Consistent with the inhibitory
effect of AZD7762 on RAW264.7 cultured cells, AZD7762 at 0.5, 1, 5
and 10 µM decreased the cross-sectional area of RAW264.7
spheroids at 24–72 h (Fig. 3I).
Finally, osteoclast activity measured by a pit formation assay was
reduced by 0.5 and 1 µM AZD7762 (Fig. 3J).
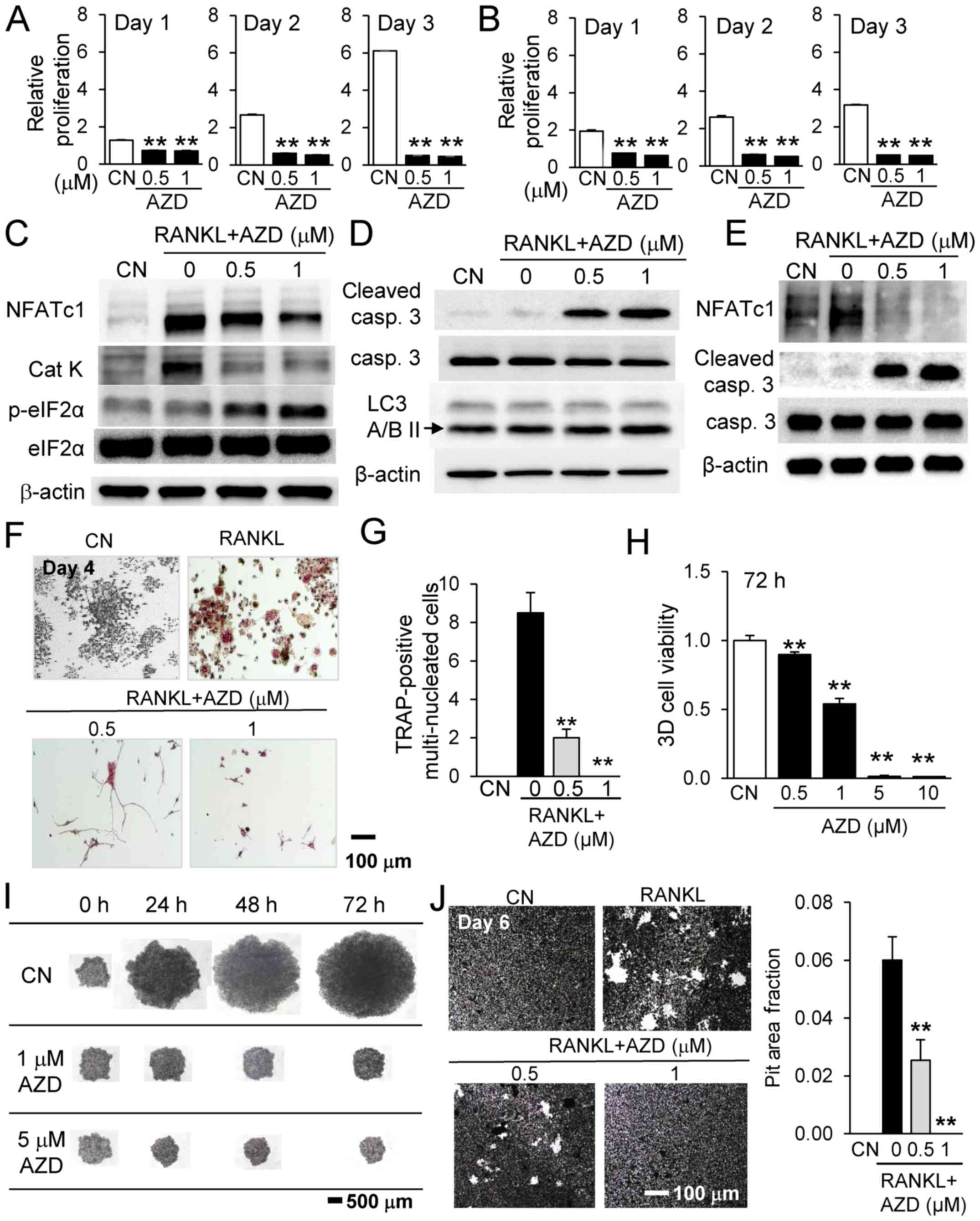 | Figure 3Inhibitory effects of AZD7762 on
RAW264.7 pre-osteoclast cells and osteoclast precursors from bone
marrow-derived cells. Relative proliferation of (A) RAW264.7 cells
and (B) osteoclast precursors from bone marrow-derived cells in
response to 0.5 and 1 µM AZD7762 on days 1, 2 and 3,
respectively. (C) Downregulation of NFATc1 and Cat K, and
upregulation of p-eIF2α in RAW cells. (D) Upregulation of cleaved
caspase 3 in RAW cells. (E) Downregulation of NFATc1 and
upregulation of cleaved caspase 3 in osteoclast precursors from
bone marrow-derived cells. (F) Visualization and (G) quantification
of the reduction in the number of multi-nucleated TRAP-positive
osteoclast cells in response to 0.5 and 1 µM AZD7762. (H)
Dose-dependent reduction in 3D cell viability in RAW264.7
spheroids. (I) RAW264.7 spheroids in response to 0.5, 1, 5 and 10
µM AZD7762 for 24, 48 and 72 h. (J) Reduction of osteoclast
activity by 0.5 and 1 µM AZD7762. **P<0.01 vs.
CN. CN, control; AZD, AZD7762; Cat K, cathepsin K; casp, caspase;
eIF2α, eukaryotic translation initiation factor-α; RANKL, receptor
activator of nuclear factor-κB ligand; 3D, three-dimensional;
NFATc1, nuclear factor of activated T cells cytoplasmic 1; LC3A/B
II, microtubule-associated proteins 1A/1B light chain 3A/B II. |
Development of MC3T3 osteoblast-like
cells in response to AZD7762
The present study additionally examined the effect
of AZD7762 on osteoblast activity. AZD7762 reduced the relative
proliferation of MC3T3 osteoblast-like cells in response to 0.1,
0.5, 1 and 2 µM, although the degree of reduction was
smaller compared with RAW264.7 pre-osteoclast cells (Fig. 4A). AZD7762 increased the mRNA
levels of osteogenesis-associated genes, including osterix,
osteocalcin (OCN), and alkaline phosphatase (ALP) at 24 and 48 h
(Fig. 4B and C); it also elevated
the protein expression levels of p-eIF2α, p-p38 and ALP (Fig. 4D and E).
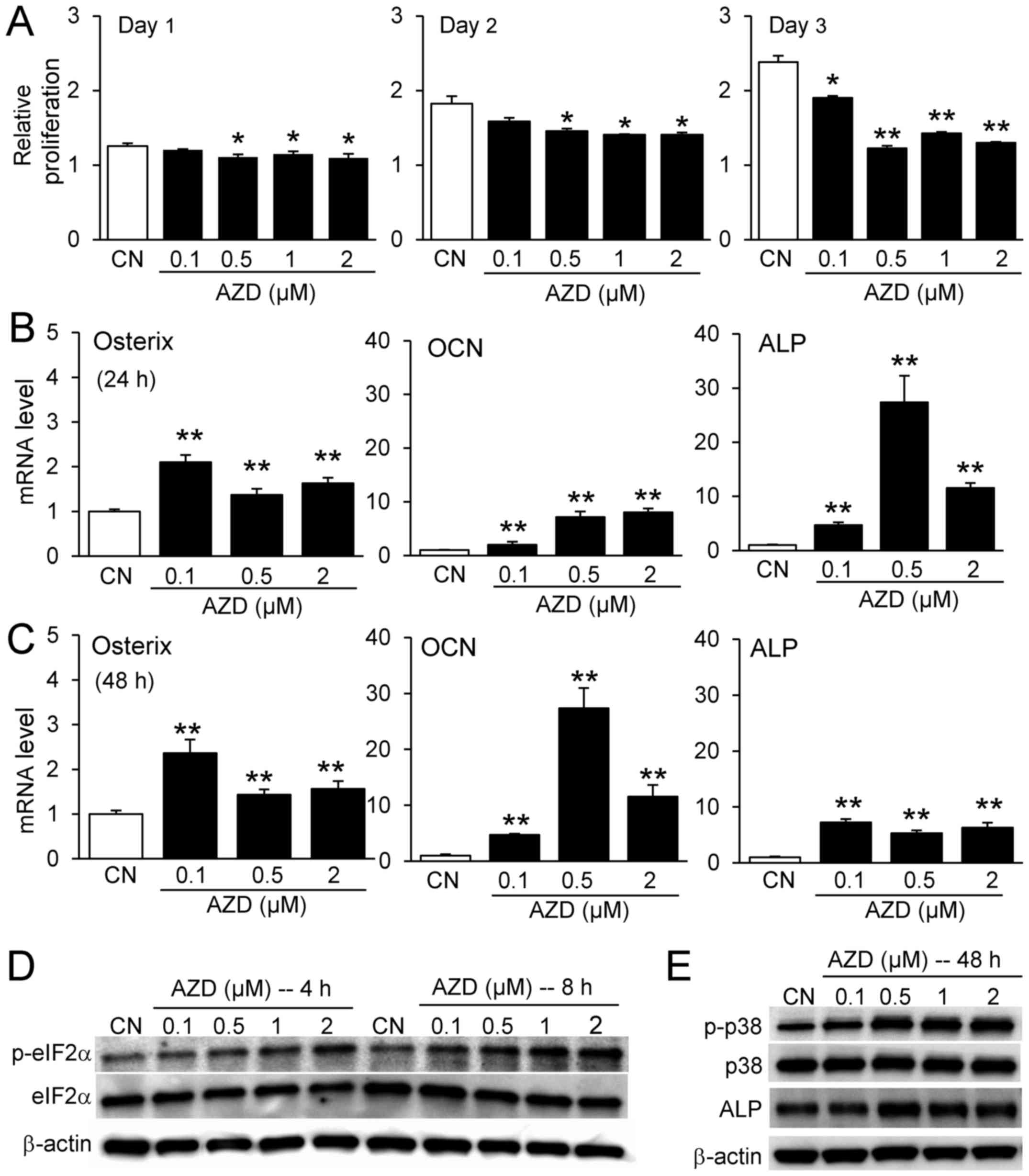 | Figure 4Responses of MC3T3 osteoblast-like
cells to AZD7762. (A) Relative proliferation of MC3T3 cells in
response to 0.1, 0.5, 1, and 2 µM AZD7762 on days 1, 2, and
3. Increase in the mRNA expression levels of osterix, OCN, and ALP
in response to 0.1, 0.5, and 2 µM AZD7762 at (B) 24 h and
(C) 48 h. (D) Elevation of p-eIF2α, (E) p-p38 and ALP in response
to 0.1, 0.5, 1, and 2 µM AZD7762. *P<0.05;
**P<0.01 vs. CN. CN, control; AZD, AZD7762; OCN,
osteocalcin; ALP, alkaline phosphatase; p, phosphorylated; eIF2α,
eukaryotic translation initiation factor-α. |
Comparison of the effect of AZD7762 and
PD407824 on tumor cells
In order to evaluate any differential effects of
Chk1 and Chk2 inhibition, PD407824, a selective inhibitor of Chk1,
was used. The responses to PD407824 were largely similar to those
to AZD7762. PD407824 at 0.1, 0.5 and 2 µM reduced the
relative proliferation of 4T1.2 cells (Fig. 5A), and the expression level of
p-eIF2α was elevated in a dose-dependent manner (Fig. 5B). Furthermore, the scratch assay
with 4T1.2 mammary tumor cells revealed that PD407824 suppressed
cell motility by inhibiting the healing of the wounded area
(Fig. 5C). To evaluate the
potential mechanism of p-eIF2α upregulation, the expression of
p-PERK, a kinase induced by stress to the (ER), was evaluated.
First, thapsigargin, an ER stress inducer, elevated p-eIF2α and
p-PERK expression after 3 h of incubation (Fig. 5D). Second, partial silencing of
PERK by RNA interference downregulated p-eIF2α (Fig. 5E), and PD407824 and AZD7762
elevated the expression level of p-PERK (Fig. 5F). Collectively, these data
indicated that the elevation of p-eIF2α may be induced by p-PERK,
an eIF2α selective kinase.
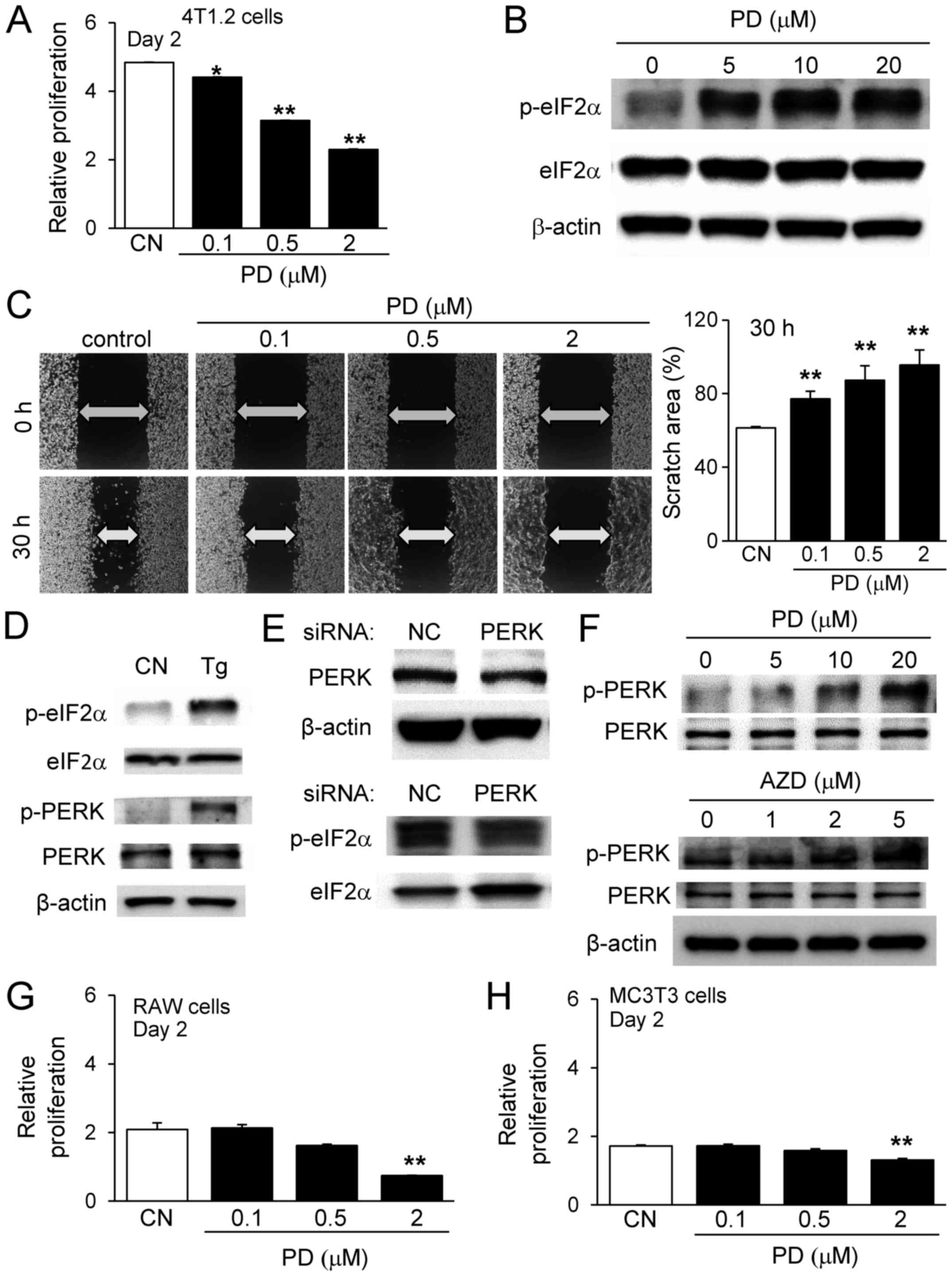 | Figure 5Effects of PD407824 and AZD7762 on
4T1.2 tumor cells, RAW264.7 pre-osteoclast cells and MC3T3
osteoblast-like cells. (A) Relative proliferation of 4T1.2 cells in
response to 0.1, 0.5, and 2 µM PD407824 on day 2. (B)
Upregulation of p-eIF2α by PD407824. (C) Scratch assay of 4T1.2
cells in response to 0.1, 0.5, and 2 µM PD407824. (D)
Elevation of p-eIF2α and p-PERK by Tg. (E) Downregulation of
p-eIF2α by PERK siRNA. (F) Upregulation of p-PERK by PD407824 and
AZD7762. Relative proliferation of (G) RAW264.7 cells and (H) MC3T3
cells, respectively, in response to 0.1, 0.5, and 2 µM
PD407824 on day 2. **P<0.01 vs. CN. CN, control; AZD,
AZD7762; p, phosphorylated; eIF2α, eukaryotic translation
initiation factor-α; PERK, protein kinase RNA-like endoplasmic
reticulum kinase; Tg, thapsigargin; siRNA, small interfering RNA;
PD, PD407824. |
Effects of PD407824 on osteoclasts and
osteoblasts
In addition to its effect on tumor cells, PD407824
affects RAW264.7 pre-osteoclasts similarly to AZD7762. PD407824 at
0.1, 0.5 and 2 µM reduced the relative proliferation of
RAW264.7 cells (Fig. 5G). In MC3T3
cells, PD407824 at 0.1 and 0.5 µM did not significantly
alter the relative cell proliferation, although a higher dosage of
2 µM decreased the number of MC3T3 cells (Fig. 5H). The RANKL-driven differentiation
of RAW264.7 cells was also suppressed by 0.1, 0.5 and 2 µM
PD407824 in a dose-dependent manner (Fig. 6A and B).
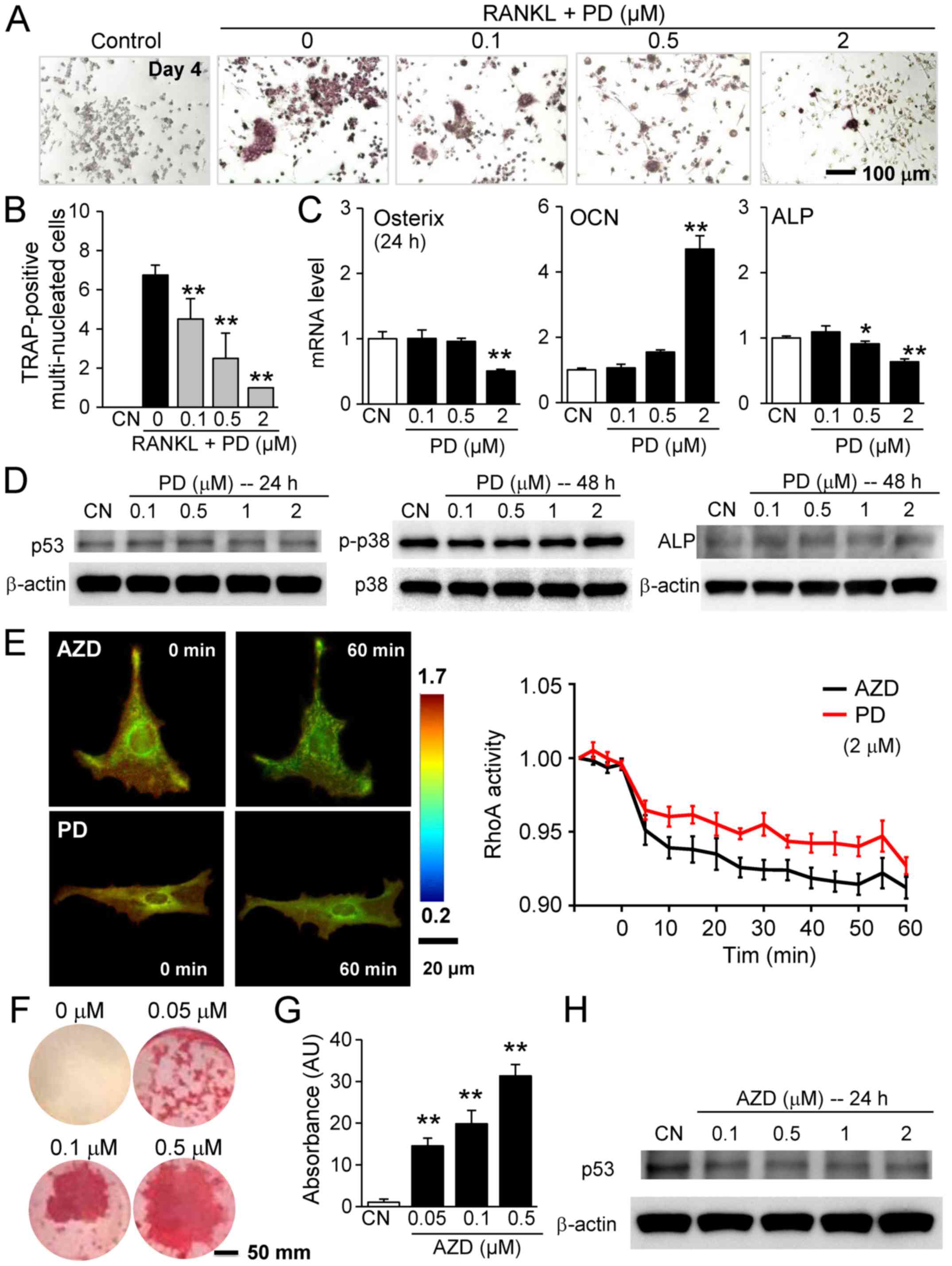 | Figure 6Effects of PD407824 and AZD7762 on
RAW264.7 pre-osteoclast cells and MC3T3 osteoblast-like cells. (A)
Suppression of RANKL-driven differentiation of RAW264.7 cells by
0.1, 0.5, and 2 µM PD407824, as demonstrated by (B) the
numbers of TRAP-positive multinucleated cells. (C) Alterations in
the mRNA expression levels of osterix, OCN and ALP by 0.1, 0.5, and
2 µM PD407824. (D) No significant alterations were observed
in the protein expression levels of p53, p-p38 and ALP in response
to 0.1 to 2 µM PD407824. (E) Reduction in RhoA activity in
MC3T3 cells in response to 2 µM AZD7762 and PD407824. (F)
Alizarin red staining of mineralized MC3T3 cells in response to
0.05, 0.1, and 0.5 µM AZD7762 for 5 weeks. (G)
Dose-dependent intensity of alizarin red staining by AZD7762. (H)
Downregulation of p53 by AZD7762. *P<0.05;
**P<0.01 vs. CN. CN, control; AZD, AZD7762; PD,
PD407824; OCN, osteocalcin; ALP, alkaline phosphatase; TRAP,
tartrate resistant acid phosphate; p, phosphorylated; p53, cellular
tumor antigen p53. |
Mineralization of MC3T3 and MLO-A5 cells
by AZD7762
A major difference between AZD7762 and PD407824 was
their effects on genes involved in bone formation. While AZD7762
elevated the mRNA expression levels of osterix and ALP, PD407824 at
2 µM reduced their expression levels (Fig. 6C). Furthermore, the effect of
PD407824 on the protein expression levels of p53, p-p38, and ALP
was not notable (Fig. 6D). To
evaluate the potential involvement of RhoA GTPase in the regulation
of cellular motility with Chk1 inhibitors, its activity level was
evaluated using a RhoA GTPase FRET biosensor. In response to 2
µM AZD7762 and PD407824, significant reductions in RhoA
activity were observed after 60 min (Fig. 6E).
The present study demonstrated that AZD7762 serves
an inhibitory role in bone-resorbing osteoclasts. To examine its
role in bone-forming osteoblasts, Alizarin red staining was
performed using MC3T3 cells. The results demonstrated that in
response to 0.05, 0.1 and 0.5 µM AZD7762 for 5 weeks,
staining intensity was elevated in a dose-dependent manner
(Fig. 6F and G). It was also
observed that AZD7762 decreased the expression of p53 (Fig. 6H). Furthermore, 3D bone constructs
with MLO-A5 osteocyte-like cell spheroids were bio-printed, and
X-ray imaging was conducted after a 2-week incubation in osteogenic
medium. The results revealed that bone mineral density was elevated
in response to 0.05 and 0.1 µM AZD7762 (Fig. 7A–C). To evaluate the mediators of
the differential effects of AZD7762 and PD407824 on p53 expression,
partial silencing of Chk1 and Chk2 was conducted via siRNA in MC3T3
cells. Following silencing of Chk2, the AZD7762-mediated
upregulation of ALP and downregulation of p53 were muted. However,
the same alterations were not observed in Chk1-silenced cells
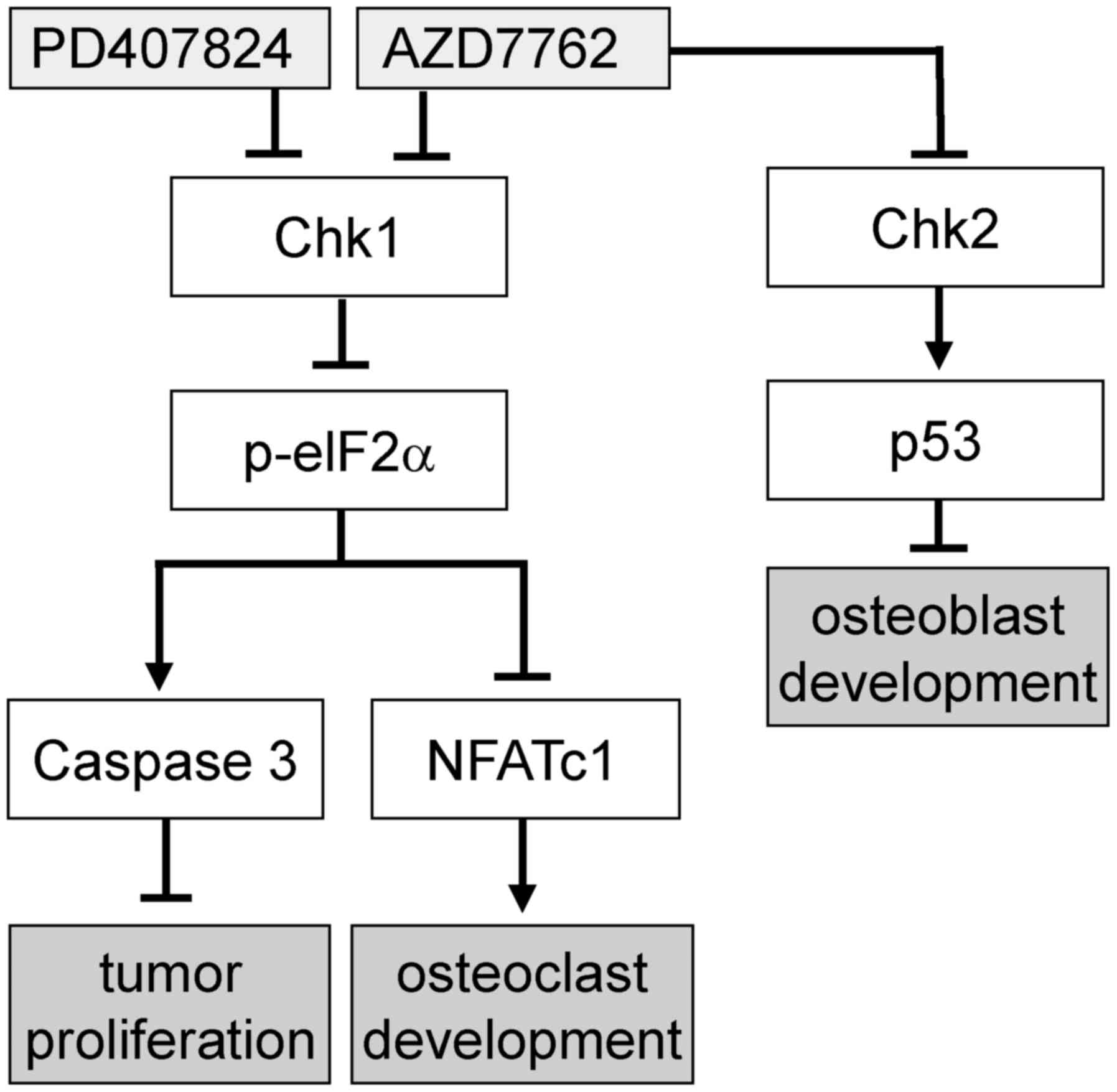
(Fig. 7D). A proposed regulatory
mechanism for AZD7762 is illustrated in Fig. 8, in which Chk1 and Chk2 are
inhibited by AZD7762, as opposed to only Chk1 inhibition by
PD407824.
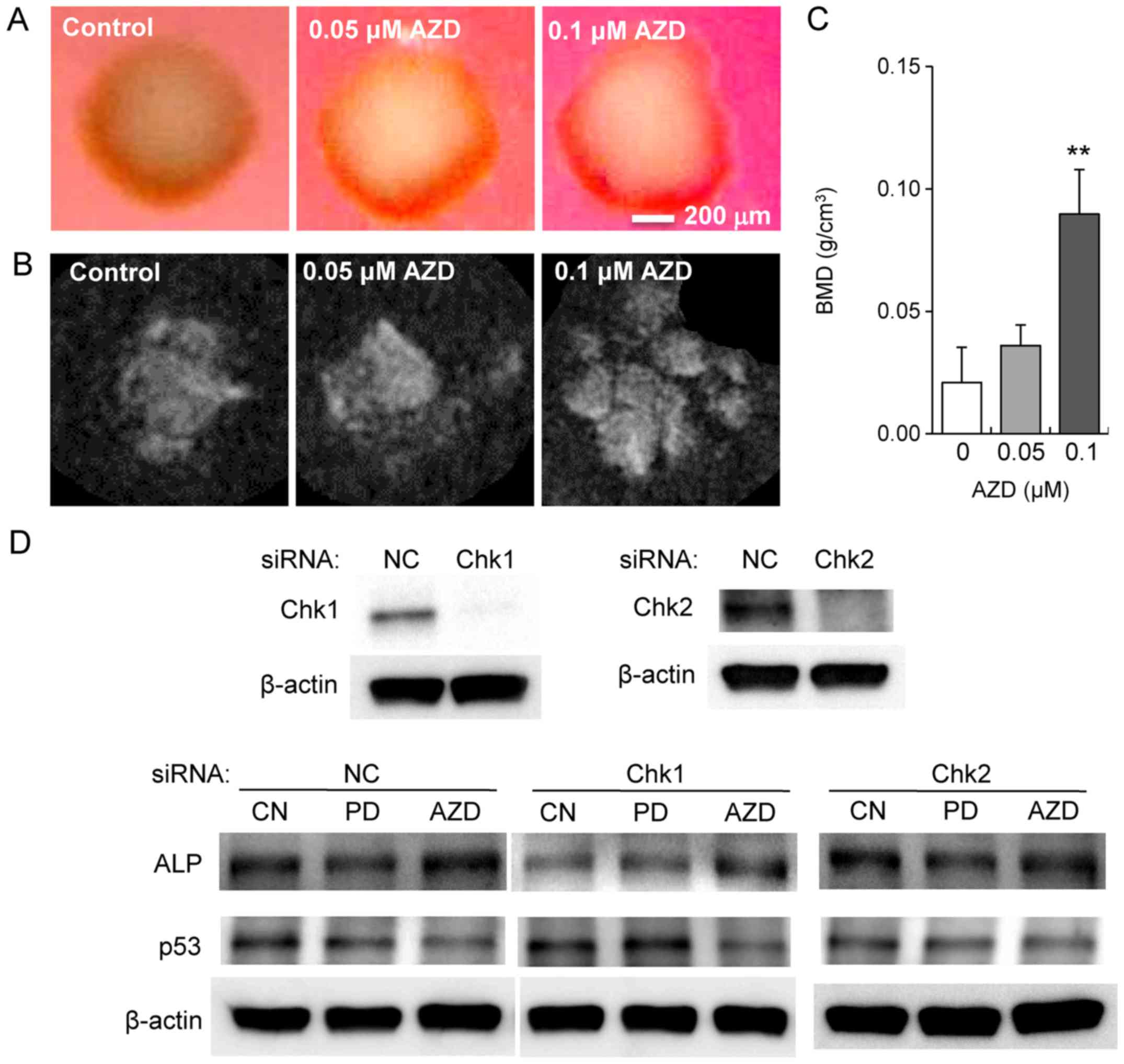 | Figure 7Mineralization of MC3T3
osteoblast-like cells and MLO-A5 osteocyte-like cells by AZD7762.
(A) Optical and (B) microCT-derived X-ray images of mineralized
spheroids from MLO-A5 osteocyte-like cells in response to 0.05 and
0.1 µM AZD7762 for 2 weeks. (C) BMD of MLO-A5 spheroid
constructs derived using microCT volumetric reconstruction. (D)
Modulation of the response to PD407824 and AZD7762 by Chk1 and Chk2
siRNA in the protein level of ALP and p53. **P<0.01
vs. 0 µM AZD. CN, control; AZD, AZD7762; PD, PD407824; BMD,
bone mineral density; CT, computed tomography; Chk, checkpoint
kinase; siRNA, small interfering RNA; ALP, alkaline phosphatase;
p53, cellular tumor antigen; NC, negative control. |
Discussion
The present study demonstrated that the Chk1 and
Chk2 inhibitor AZD7762 suppressed tumor cell proliferation,
inhibited osteoclast activity and stimulated osteoblast
mineralization. Treatment with AZD7762 decreased the proliferation
and spheroid formation of three different breast cancer cell lines
(4T1.2, MDA-MB-231 and MCF-7). The 4T1.2 and MDA-MB-231 cell lines
are metastatic and estrogen receptor-negative, while MCF-7 cells
are non-metastatic and estrogen receptor-positive. Treatment with
AZD7762 decreased the size and 3D cell viability of 4T1.2 spheroids
in a dose-dependent manner. The inhibitory effect of AZD7762 was
associated with the elevated protein expression of an apoptotic
marker (caspase 3) and an autophagic marker (LC3A/BII). Of note,
the role of autophagy is complex, and it is currently considered to
function in tumor suppression and as a protective cell survival
mechanism (34).
AZD7762 and PD407824 suppressed the 2D motility of
4T1.2 cells in the wound healing assay, although the apparent
reduction of motility was also due to cell death. The reduction in
RhoA GTPase activity by AZD7762 and PD407824 was consistent with
their inactivation of cellular motility. The inhibitory effects of
these two agents were evident without simultaneous application of
DNA damaging agents. It has been reported that the effect of Chk
inhibition on tumor growth may be dependent on the expression of
and mutations in p53 and/or p21 (7). It was previously identified that
inhibitors of eIF2α de-phosphorylation (salubrinal and guanabenz)
may suppress tumor proliferation and osteoclast differentiation
(14,15). In the present study, p-eIF2α was
upregulated by AZD7762 and PD407824. Due to the observed
upregulation of p-PERK, one possibility is that Chk1 and Chk2
inhibitors induce stress to the ER and elevate p-PERK and p-eIF2α
expression. It is also possible that other kinases or phosphatases
may be involved in the regulation of p-eIF2α.
Similar to its effects on tumor cells, AZD7762 also
inhibited the proliferation of RAW264.7 pre-osteoclast cells in
monolayer cultures and in 3D spheroids. Staining of TRAP-positive
multi-nucleated osteoclasts and measuring the resorbed mineral area
demonstrated that RANKL-induced osteoclast differentiation and
activity was inhibited by treatment with AZD7762 in a
dose-dependent manner. Treatment with AZD7762 also decreased the
protein expression of NFATc1, a master transcription factor of
osteoclastogenesis, and cathepsin K, a protease involved in bone
resorption, while increasing the phosphorylation of eIF2α and the
expression of cleaved caspase 3. PD407824 also decreased osteoclast
differentiation. Metastatic tumor cells that invade the bone may
accelerate osteoclast-driven bone resorption, triggering a
dangerous cycle (35). These
results demonstrated that targeting Chk signaling may provide a way
to slow tumor growth and metastasis-associated bone
degradation.
While the effects of AZD7762 and PD407824 on tumor
cells and osteoclast cells were similar, their effects on
osteoblasts diverged. AZD7762 upregulated the mRNA expression
levels of bone the formation markers osterix and ALP, while
PD407824 downregulated their mRNA expression levels. Furthermore,
AZD7762 elevated the protein expression level of p-p38 and reduced
that of p53, and PD407824 did not significantly alter the levels of
p-p38 or p53. RNA interference of Chk1 and Chk2 in osteoblasts
suggested that this difference may have been caused by the
inhibitory effect of AZD7762 on Chk2, thereby downregulating p53
(23), a negative regulator of
osterix (24). Therefore,
targeting Chk2 may be a way to attenuate bone degradation caused by
metastasis. However, p53 is also a tumor suppressor (36), and the p53-dependent effects of the
inhibitory effect ofAZD7762 on tumor growth and bone remodeling via
Chk2 require further study. A previous study indicated the
complexity of the action of p53 as a crucial tumor suppressor, in
addition to as a modulator of stem cell maintenance, invasion and
metastasis (37).
While the present results revealed a novel role for
AZD7762 in bone remodeling and bone metastasis, the present study
has certain limitations. The experiments herein employed mouse and
human cell lines (11), and gene
expression was primarily examined using monolayer cell cultures.
Although clinical trials of AZD7762 are not going forward due to
unpredictable cardiac toxicity, inhibition of Chk remains an
important therapeutic target (38). Future work may aim to identify
other Chk inhibitors that exhibit similar effects to AZD7762, and
test their efficacy in a mouse model of bone metastasis. In
conclusion, the present study demonstrated that Chk1/2 inhibitors
may be used to suppress tumor proliferation and to prevent bone
loss via inhibition of osteoclastogenesis and stimulation of
osteoblastogenesis.
Acknowledgments
The authors would like to thank Ms. T. Truong Vo for
technical support.
References
|
1
|
Smits VA and Gillespie DA: DNA damage
control: Regulation and functions of checkpoint kinase 1. FEBS J.
282:3681–3692. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sanchez Y, Wong C, Thoma RS, Richman R, Wu
Z, Piwnica-Worms H and Elledge SJ: Conservation of the Chk1
checkpoint pathway in mammals: Linkage of DNA damage to Cdk
regulation through Cdc25. Science. 277:1497–1501. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Narayanaswamy PB, Tkachuk S, Haller H,
Dumler I and Kiyan Y: CHK1 and RAD51 activation after DNA damage is
regulated via urokinase receptor/TLR4 signaling. Cell Death Dis.
7:e23832016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
McNeely S, Beckmann R and Bence Lin AK:
CHEK again: Revisiting the development of CHK1 inhibitors for
cancer therapy. Pharmacol Ther. 142:1–10. 2014. View Article : Google Scholar
|
|
5
|
Kuo C-Y, Zupkó I, Chang F-R, Hunyadi A, Wu
C-C, Weng T-S and Wang H-C: Dietary flavonoid derivatives enhance
chemotherapeutic effect by inhibiting the DNA damage response
pathway. Toxicol Appl Pharmacol. 311:99–105. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Massey AJ, Stokes S, Browne H, Foloppe N,
Fiumana A, Scrace S, Fallowfield M, Bedford S, Webb P, Baker L, et
al: Identification of novel, in vivo active Chk1 inhibitors
utilizing structure guided drug design. Oncotarget. 6:35797–35812.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Origanti S, Cai SR, Munir AZ, White LS and
Piwnica-Worms H: Synthetic lethality of Chk1 inhibition combined
with p53 and/or p21 loss during a DNA damage response in normal and
tumor cells. Oncogene. 32:577–588. 2013. View Article : Google Scholar
|
|
8
|
Gokulnath M, Swetha R, Thejaswini G,
Shilpa P and Selvamurugan N: Transforming growth factor-β1
regulation of ATF-3, c-Jun and JunB proteins for activation of
matrix metal-loproteinase-13 gene in human breast cancer cells. Int
J Biol Macromol. 94:370–377. 2017. View Article : Google Scholar
|
|
9
|
Zabludoff SD, Deng C, Grondine MR, Sheehy
AM, Ashwell S, Caleb BL, Green S, Haye HR, Horn CL, Janetka JW, et
al: AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint
abrogation and potentiates DNA-targeted therapies. Mol Cancer Ther.
7:2955–2966. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Azorsa DO, Gonzales IM, Basu GD, Choudhary
A, Arora S, Bisanz KM, Kiefer JA, Henderson MC, Trent JM, Von Hoff
DD, et al: Synthetic lethal RNAi screening identifies sensitizing
targets for gemcitabine therapy in pancreatic cancer. J Transl Med.
7:432009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu S, Liu Y, Minami K, Chen A, Wan Q, Yin
Y, Gan L, Xu A, Matsuura N, Koizumi M, et al: Inhibiting checkpoint
kinase 1 protects bone from bone resorption by mammary tumor in a
mouse model. Oncotarget. 9:9364–9378. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zannini L, Delia D and Buscemi G: CHK2
kinase in the DNA damage response and beyond. J Mol Cell Biol.
6:442–457. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu X, Webster SR and Chen J:
Characterization of tumor-associated Chk2 mutations. J Biol Chem.
276:2971–2974. 2001. View Article : Google Scholar
|
|
14
|
Hamamura K, Minami K, Tanjung N, Wan Q,
Koizumi M, Matsuura N, Na S and Yokota H: Attenuation of malignant
phenotypes of breast cancer cells through eIF2α-mediated
downregulation of Rac1 signaling. Int J Oncol. 44:1980–1988. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hamamura K, Tanjung N and Yokota H:
Suppression of osteoclastogenesis through phosphorylation of
eukaryotic translation initiation factor 2 alpha. J Bone Miner
Metab. 31:618–628. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu Z, Lv Y, Zhao N, Guan G and Wang J:
Protein kinase R-like ER kinase and its role in endoplasmic
reticulum stress-decided cell fate. Cell Death Dis. 6:e18222015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Raschke WC, Baird S, Ralph P and Nakoinz
I: Functional macrophage cell lines transformed by Abelson leukemia
virus. Cell. 15:261–267. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang D, Christensen K, Chawla K, Xiao G,
Krebsbach PH and Franceschi RT: Isolation and characterization of
MC3T3-E1 preosteoblast subclones with distinct in vitro and in vivo
differentiation/mineralization potential. J Bone Miner Res.
14:893–903. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kato Y, Boskey A, Spevak L, Dallas M, Hori
M and Bonewald LF: Establishment of an osteoid preosteocyte-like
cell MLO-A5 that spontaneously mineralizes in culture. J Bone Miner
Res. 16:1622–1633. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lelekakis M, Moseley JM, Martin TJ, Hards
D, Williams E, Ho P, Lowen D, Javni J, Miller FR, Slavin J, et al:
A novel orthotopic model of breast cancer metastasis to bone. Clin
Exp Metastasis. 17:163–170. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen A, Wang L, Li B-Y, Sherman J, Ryu JE,
Hamamura K, Liu Y, Nakshatri H and Yokota H: Reduction in migratory
phenotype in a metastasized breast cancer cell line via
down-regulation of S100A4 and GRM3. Sci Rep. 7:34592017. View Article : Google Scholar
|
|
22
|
Moldovan NI, Hibino N and Nakayama K:
Principles of the Kenzan method for robotic cell spheroid-based
three-dimensional bioprinting. Tissue Eng Part B Rev. 23:237–244.
2017. View Article : Google Scholar
|
|
23
|
Hirao A, Kong YY, Matsuoka S, Wakeham A,
Ruland J, Yoshida H, Liu D, Elledge SJ and Mak TW: DNA
damage-induced activation of p53 by the checkpoint kinase Chk2.
Science. 287:1824–1827. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang X, Kua H-Y, Hu Y, Guo K, Zeng Q, Wu
Q, Ng HH, Karsenty G, de Crombrugghe B, Yeh J, et al: p53 functions
as a negative regulator of osteoblastogenesis, osteoblast-dependent
osteoclastogenesis, and bone remodeling. J Cell Biol. 172:115–125.
2006. View Article : Google Scholar
|
|
25
|
Cailleau R, Young R, Olivé M and Reeves WJ
Jr: Breast tumor cell lines from pleural effusions. J Natl Cancer
Inst. 53:661–674. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Soule HD, Vazguez J, Long A, Albert S and
Brennan M: A human cell line from a pleural effusion derived from a
breast carcinoma. J Natl Cancer Inst. 51:1409–1416. 1973.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yokota H, Hamamura K, Chen A, Dodge TR,
Tanjung N, Abedinpoor A and Zhang P: Effects of salubrinal on
development of osteoclasts and osteoblasts from bone marrow-derived
cells. BMC Musculoskelet Disord. 14:1972013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
National Research Council (US) Committee:
Guide for the Care and Use of Laboratory Animals. 8th edition.
National Academies Press; Washington, DC: 2011
|
|
29
|
Ascione F, Vasaturo A, Caserta S,
D'Esposito V, Formisano P and Guido S: Comparison between
fibroblast wound healing and cell random migration assays in vitro.
Exp Cell Res. 347:123–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang J, Zhu L and Peng B: Effect of
BioAggregate on osteoclast differentiation and inflammatory bone
resorption in vivo. Int Endod J. 48:1077–1085. 2015. View Article : Google Scholar
|
|
31
|
Miyazaki T, Miyauchi S, Anada T, Imaizumi
H and Suzuki O: Evaluation of osteoclastic resorption activity
using calcium phosphate coating combined with labeled polyanion.
Anal Biochem. 410:7–12. 2011. View Article : Google Scholar
|
|
32
|
Hamamura K, Swarnkar G, Tanjung N, Cho E,
Li J, Na S and Yokota H: RhoA-mediated signaling in
mechanotransduction of osteoblasts. Connect Tissue Res. 53:398–406.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wei W, Clockaerts S,
Bastiaansen-Jenniskens YM, Gierman LM, Botter SM, Bierma-Zeinstra
SM, Weinans H, Verhaar JA, Kloppenburg M, Zuurmond AM, et al:
Statins and fibrates do not affect development of spontaneous
cartilage damage in STR/Ort mice. Osteoarthritis Cartilage.
22:293–301. 2014. View Article : Google Scholar
|
|
34
|
Dalby KN, Tekedereli I, Lopez-Berestein G
and Ozpolat B: Targeting the prodeath and prosurvival functions of
autophagy as novel therapeutic strategies in cancer. Autophagy.
6:322–329. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Clezardin P and Teti A: Bone metastasis:
Pathogenesis and therapeutic implications. Clin Exp Metastasis.
24:599–608. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Vogelstein B, Lane D and Levine AJ:
Surfing the p53 network. Nature. 408:307–310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bieging KT, Mello SS and Attardi LD:
Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev
Cancer. 14:359–370. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sausville E, Lorusso P, Carducci M, Carter
J, Quinn MF, Malburg L, Azad N, Cosgrove D, Knight R, Barker P, et
al: Phase I dose-escalation study of AZD7762, a checkpoint kinase
inhibitor, in combination with gemcitabine in US patients with
advanced solid tumors. Cancer Chemother Pharmacol. 73:539–549.
2014. View Article : Google Scholar : PubMed/NCBI
|