Introduction
Nasopharyngeal carcinoma (NPC) is a type of head and
neck cancer that exhibits an endemic distribution with a high
prevalence in Southern China and Southeast Asia (1,2).
Clinically, approximately 70% of patients with NPC at the time of
diagnosis present with locally advanced disease (3). To date, although NPC is sensitive to
radiotherapy, the prognosis of NPC remains dismal, with 5-year
survival rates of 34–52% (4). A
major cause for this lethality is attributed to significant rates
of relapse and distant metastasis following therapy (5). In this context, resistance to
radiotherapy and chemotherapy represents a major challenge in the
treatment of NPC (6,7). Therefore, understanding the
mechanisms behind therapeutic resistance in NPC is crucial.
Therapy-induced senescence occurs in response to
aberrant oncogenic signaling, oxidative stress and DNA damage
independent of telomere dysfunction (8–11).
From either the morphological or biochemical point of view,
therapy-induced senescence is highly analogous to premature
senescence that has been studied in other circumstances. Recently,
it has been reported that that cancer cells can undergo senescence
following treatment with anticancer agents, such as doxorubicin
(Dox), cisplatin (CDDP) and paclitaxel (TAX), suggesting that
senescence may represent one of the major cellular responses to
anticancer therapeutics (12,13).
Thus, therapy-induced senescence has been considered an indicator
of the efficacy of anticancer drugs, provoking considerable
interest in cancer treatment. In addition, there have been few
reports regarding the role of senescence in NPC. For example,
evidence indicates that CDDP induces senescence in NPC cells,
resulting in cell growth arrest (14), and a high expression of SHP-1 can
inhibit cellular senescence to induce radiotherapeutic resistance
in NPC (15). However, the
molecular mechanisms through which senescence participates in
chemoresistance in NPC are poorly understood.
Cyclooxygenase (COX) is a rate-limiting enzyme in
prostaglandin biosynthesis. There are two isoforms of COX, COX-1
and COX-2. COX-1 is constitutively expressed in a number of tissues
and mainly plays a role in tissue homeostasis. By contrast, COX-2
is an inducible enzyme responsible for the production of
prostaglandins at sites of inflammation and wound healing (16). Of note, COX-2 is highly expressed
in numerous types of human cancer, including lung, breast, ovarian,
colorectal cancer and NPC (17–20).
Functionally, COX-2 is thought to play a role in initiation and
progression of a number of human diseases (21,22)
Although COX-2 has also been demonstrated to contribute to the
resistance of tumor cells to conventional chemotherapy and
radiotherapy, i.e., by preventing DNA damage, increasing cytokine
secretion and stimulating the growth of cancer stem cells (23,24),
its role and mechanisms of action as regards chemotherapy-induced
senescence remain largely unknown, particularly in NPC.
Therefore, understanding the mechanisms behind
chemotherapeutic resistance in NPC and identifying novel
therapeutic targets are required in order to improve the treatment
of patients with NPC. In this study, we found that COX-2 expression
was positively associated with the recurrence and a poor prognosis
of patients with NPC, and we then observed the effect of COX-2
overexpression and knockdown on the proliferation and
chemoresistance of NPC cells. Finally, we explored the role of
COX-2 in the chemoresistance of NPC cells by inhibiting cellular
senescence through the p53 pathway.
Materials and methods
Patients and primary samples
In total, 11 chronic nasopharyngitis (CN) and 43
primary NPC samples were obtained by routine diagnostic biopsy
before treatment with informed consent at the Hunan Cancer Hospital
(Changsha, China) between February, 2014 and January, 2015. A total
of 9 paired paraffin-embedded specimens, obtained at diagnosis and
recurrence respectively, were provided by the Department of
Pathology of Hunan Cancer Hospital. All the specimens were examined
by two independent pathologists. The Research Ethics Committee of
Central South University approved our study, and all patients
provided written informed consent.
Cells, antibodies and reagents
The NPC human cell lines, CNE1, CNE2, HK1 and
C666-1, were cultured in RPMI-1640 medium (Gibco/Thermo Fisher
Scientific, Waltham, MA, USA) with 10% fetal bovine serum (FBS) at
37°C in an atmosphere containing 5% CO2. NP69, a normal
nasopharyngeal epithelial cell line, was cultured in K-SFM medium
(Gibco/Thermo Fisher Scientific) at 37°C in an atmosphere
containing 5% CO2. The mouse Lewis lung cancer (LLC)
cell line was kindly provided by Professor Juanjuan Xiang at the
Cancer Research Institute of Central South University, Changsha,
China and cultured in Dulbecco's modified Eagle's medium (DMEM;
Gibco/Thermo Fisher Scientific) with 10% FBS at 37°C in an
atmosphere containing 5% CO2. All NPC cell lines were
obtained from the Cancer Research Institute of Central South
University.
Anti-COX-2 (1:1,000, #12282) antibody was obtained
from Cell Signaling Technology (Danvers, MA, USA), anti-p53 (1:200,
sc-6243), anti-p21 (1:200, sc-817), anti-GAPDH (1:5,000, sc-166545)
and mouse or rabbit secondary antibodies were from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). The p53 inhibitor, Pifithrin-α
(PFT-α) powder, was purchased from Selleckchem (Houston, TX, USA)
and dissolved in dimethylsulfoxide (DMSO) at 100 mM as a stock
solution. CDDP and TAX solution were purchased from Harbin
Pharmaceutical Group Bio-engineering Co. (Harbin, China). The COX-2
inhibitor, NS-398 powder, was purchased from Cayman Chemical Co.
(Ann Arbor, MI, USA) and dissolved in DMSO at 50 mg/ml as a stock
solution.
Immunohistochemistry (IHC) and
immunofluorescence (IF) staining
IHC was performed using a standard
streptavidin-biotin-peroxidase complex method as previously
described (25). The tissue
sections were incubated with anti-COX-2 antibody (1:100, #12282;
Cell Signaling Technology) overnight at 4°C. The assessment of
COX-2 staining was carried out by determining both the intensity
(0, 1, 2 or 3) and extent of staining (0, 0%; 1, <10%; 2,
10–50%; 3, >50%). Scores for the intensity and extent of
staining were added to provide weighted levels of COX-2
expression.
Mouse fibroblasts on slides were fixed in 4%
paraformaldehyde for 30 min. After blocking with normal goat serum,
slides were incubated overnight with primary anti-p53 antibody
(1:100, sc-6243), followed by fluorescence-conjugated secondary
antibodies (1:200, sc-3739) (both from Santa Cruz Biotechnology).
Frozen sections of tumor tissue from LLC tumor-bearing mice were
treated with anti-COX-2 antibody (1:100, #12282; Cell Signaling
Technology) as described above. The fluorescent signals were
observed under an Olympus fluorescence microscope (BX43; Olympus
China Co. Ltd., Beijing, China).
Stable transfection and RNA
interference
The expression plasmid, pEnter-COX2, containing the
open reading frame (ORF) of the COX-2 gene and the control
plasmid, pEnter, were purchased from Vigene Bioscience (Shandong,
China). GV248 lentiviral vectors with a GFP label containing
short-hairpin RNA (shRNA) targeting COX-2 (AACTGCTCAACACCGGAATTT)
and a scramble sequence (TTCTCCGAACGTGTCACGT) as a control were
purchased from Genechem (Shanghai, China). The CNE1 and CNE2 cells
was transfected with these constructs using
Lipofectamine® 3000 transfection reagent (Invitrogen,
Carlsbad, CA, USA) as per the manufacturer's instructions. Cells
were selected using 1 µg/ml puromycin for 2 weeks, and
stable cell lines were obtained.
Colony formation assay
A total of 1×103 CNE1 cells transfected
with COX-2 overexpression vector or empty vector were seeded in
6-well plates and treated with 0.5 or 1 µg/ml TAX and 1 or 2
µg/ml CDDP, respectively at 24 h after plating. Following
incubation for 10 days at 37°C in a 5% CO2 incubator,
the cells were fixed with methanol and stained with crystal violet
(Sangon Biotech, Shanghai, China) at 25°C for 10 min. Colonies
containing at least 50 cells were counted under an Olympus
microscope (CKX53; Olympus China Co., Ltd.) with ImageJ software
and colony formation ratio was calculated. Colony-forming ability
was determined as the colony number/plated cell number ×100%. Each
experiment was repeated at least 3 times and 2 wells were used for
each cell line.
Analysis of cell viability
Cell viability was assessed using the Cell Counting
kit-8 (CCK-8; Bimake, Houston, TX, USA). Briefly, 1×103
cells were seeded in one well of 96-well microplates. Subsequently,
100 µl of 10% CCK-8 solution were added to each well, and
the cells were then incubated for 2 h at 37°C. The absorbance at
450 nm was measured with an enzyme-labeled instrument (DTX880;
Beckman Coulter, Brea, CA, USA) to determine cell viability.
IC50 values were calculated using the following formula:
Percentage inhibition = (optical density value of without
treatment−optical density value of test)/(optical density value of
without treatment−optical density value of total inhibition).
According to the fitting curve of percentage inhibition analyzed by
regression analysis using SPSS 16.0 software, the IC50
values were calculated.
Cell cycle analysis
To analyze cell cycle distribution, 1×106
cells were fixed in 70% ethanol and incubated with the Cell Cycle
kit [Multisciences (Lianke) Biotech Co., Ltd., Zhejiang, China] for
30 min. Cell cycle analysis was then carried out using flow
cytometry (FACSCanto II; BD Biosciences, San Jose, CA, USA). The
cell cycle phase distribution was calculated from the resultant DNA
histogram using Flowjo 7 software.
Western blot and immunoprecipitation
analyses
The cells were lysed using RIPA buffer (Beyotime,
Shanghai, China), containing 1X Protease Inhibitor Cocktail
(Selleckchem, China). Subsequently, after 30 min on ice and
centrifugation at 4°C, 10,000 × g for 15 min, the supernatant was
collected. Protein in the supernatant was measured using a
Bicinchoninic Acid Protein Assay kit (Beyotime), and the protein
was denatured at 100°C for 10 min with 2X Protein Loading Buffer
(Sangon Biotech). The denatured protein was then separated via
8–10% SDS polyacrylamide gel electrophoresis and transferred onto
PVDF membranes (GE Healthcare Life Sciences, Little Chalfont, UK).
The membranes were blocked with TBST containing 8% non-fat milk for
2 h at room temperature, incubated with specific primary antibodies
mentioned in the reagents section at 4°C overnight, and then
incubated with secondary antibodies labeled with horseradish
peroxidase (HRP) (sc-2004 and sc-2005; Santa Cruz Biotechnology)
for 1 h. Finally, an electrochemiluminescence (ECL) system (Tanon,
China) was used for detection.
Immunoprecipitation was carried out with anti-p53
(sc-6243; Santa Cruz Biotechnology) specific antibody using the
Co-immunoprecipitation kit (Thermo Scientific, San Jose, CA, USA)
as per the manufacturer's instructions. Rabbit IgG (CR1; Sino
Biological, Beijing, China) was used as a negative control.
Immunoprecipitated proteins were then detected by western blot
analysis using anti-p53 and anti-COX-2 antibodies.
Analysis of senescence-associated
β-galactosidase (SA-β-gal) activity
The cells were grown in 12-well plates and treated
with 2 µg/ml CDDP for 48 h. Frozen tissue sections of lung
and tumor tissue from LLC tumor-bearing mice were prepared for
detection. Cellular senescence was assessed using the Senescence
β-Galactosidase Staining kit (Cell Signaling Technology) as per the
manufacturer's instructions. In summary, the cells were washed and
fixed before staining with β-galactosidase staining solution at
37°C overnight. Images of the labeled cells were acquired under a
BioRevo9000 microscope (Keyene, Shanghai, China) and cells with
blue staining were counted in 3 different fields. The percentage of
staining cells in the counted cells was used to determine the
degree of cellular senescence.
Preparation of mouse fibroblasts from
COX-2 knockout mice
A total of 10 C57BL/6 mice, 12 weeks old, weighing
approximately 20 g with a COX-2 knockout allele were kindly
provided by Dr Ying Yu (Institute for Nutritional Sciences,
Shanghai, China). Fibroblasts were isolated from mouse skin and
lung tissues as previously described (26). Briefly, cells were obtained through
enzymatic disaggregation with 1 mg/ml collagenase for 1 h at 37°C,
followed by mechanical dissociation. Dissociated cells were then
suspended in DMEM supplemented with 10% FBS and 100 U/ml of
penicillin-streptomycin. Cells were characterized by detecting the
fibroblast marker, α-smooth muscle actin (SMA; 1:1,000, ab124964;
Abcam, Cambridge, MA, USA).
Animal experiments
All animal experiments were performed in accordance
with the guidelines of the Institutional Animal Care and local
Veterinary Office and Ethics Committee of the Central South
University (CSU), China [Animal experimental license no. LLSCC(LA)
2015-035 for Fig. 2D and LLSCC(LA)
2016-034 for Fig. 4E] under an
approved protocol and the mice were maintained on a 12 day/night
light cycle at 25°C with relative humidity of 50% and had
continuous access to food and water. A total of 6 female specific
pathogen-free (SPF) BALB/c nude mice obtained from Hunan SJA
Laboratory Animal Co., Ltd., weighing approximately 20 g, 6–8 weeks
old were randomly allocated to 2 groups. The stably transfected
cells (CNE2-Scramble and CNE2-COX-2 sh; (1×106 cells per
mouse) were suspended in 10 µl of RPMI serum-free medium and
injected via the tail vein. Following 8 weeks of observation, two
mice from the CNE2-Scramble group and one from the CNE2-COX-2 sh
group exhibited a >4 g loss in body weight and then all animals
were sacrificed. As it is difficult to establish primary NPC tumor
models in mice in the pharynx (27), we thus injected the NPC tumor cells
via the tail vein and validated the effects of COX2 in the lungs.
The lungs of the mice were excised for IHC analysis, using
anti-COX-2 (1:100, #12282) and anti-Ki-67 antibodies (1:100, #9027)
(both from Cell Signaling Technology) to detect protein
expression
Secondly, 8 female SPF wild-type (WT) C57BL/6 mice
obtained from Animal Center of the Third Hospital of Central South
University, weighing approximately 18 g at 4 weeks of age were
inoculated subcutaneously with 1×106 mouse LLC cells and
then randomly allocated to 2 groups. One group of mice was treated
with 100 µg/20 g body weight NS-398/PBS solution (containing
1% DMSO) via intraperitoneal injection, once every 3 days for 4
weeks, while the other group of mice was treated with PBS solution
(containing 1% DMSO) as a vehicle control. Lung tissues were
excised to examine senescence.
Data mining
To identify functional COX-2 in NPC, we downloaded a
set of gene expression profiling data for NPC (GSE12452) from the
GEO database.
Statistical analysis
Values represent the means ± SD from at least 3
independent experiments. The significance of differences between
experimental variables was determined using the Student's t-test
and a Bonferroni post hoc test. Overall survival (OS) was analyzed
with the log-rank method and a Kaplan Meier survival curve was
drawn using GraphPad Prism 5 software. A two-sided value of
P<0.05 was considered to indicate a statistically significant
difference. We downloaded a publically available gene expression
profiling (GEP) database (GSE12452) and analyzed COX-2 expression
in NPC.
Results
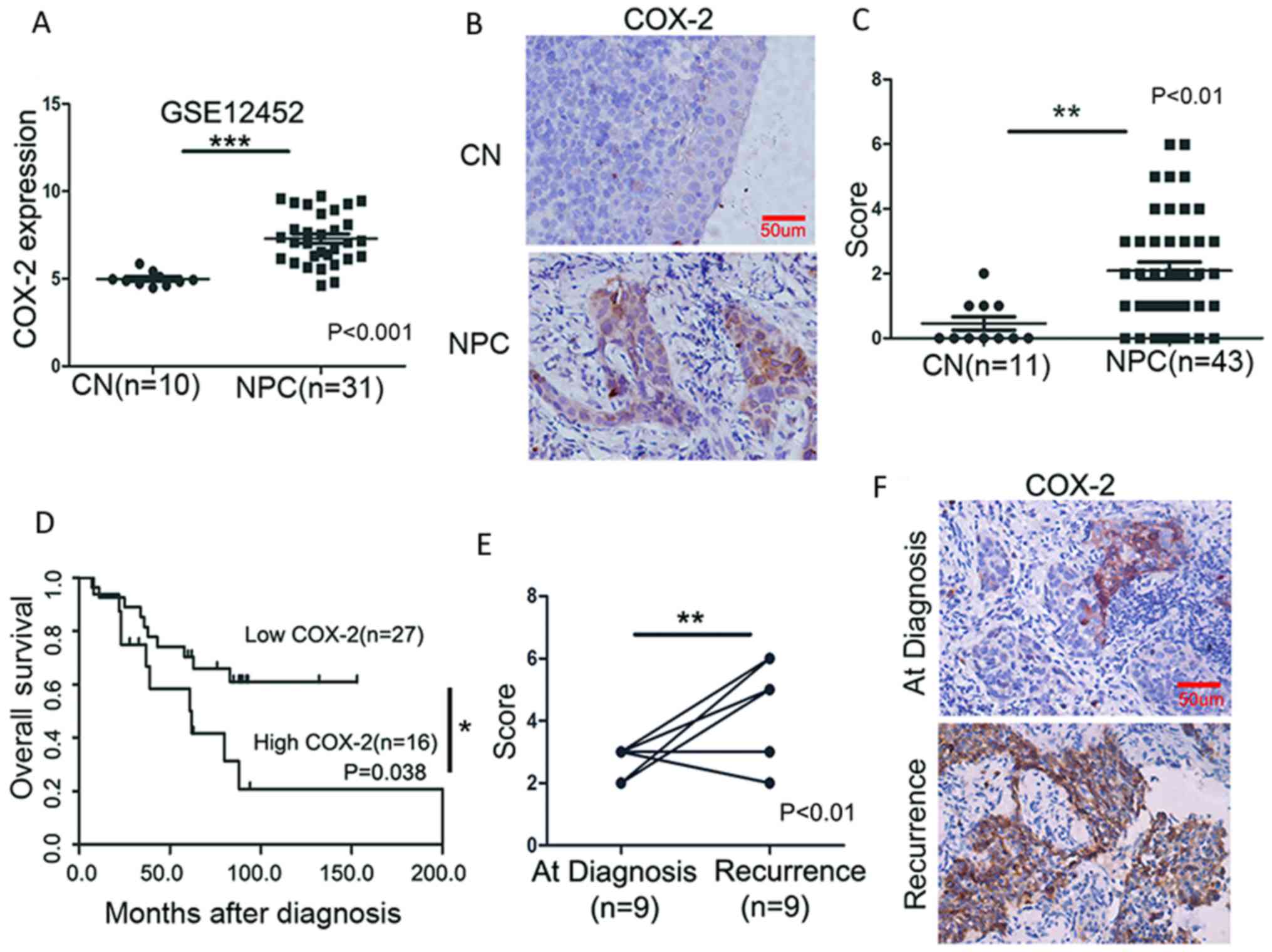
High COX-2 expression is associated with
a poor survival of patients with NPC
To evaluate the significance of COX-2 in NPC
pathogenesis, we analyzed the expression of COX-2 in NPC using a
publically available GEP database (GSE12452). As shown in Fig. 1A, COX-2 was highly expressed at the
mRNA level in the NPC tumor tissue when compared with the CN
tissues (P<0.001). To validate this result, we examined the
protein expression of COX-2 in paraffin-embedded sections by IHC in
a cohort of 43 NPC and 11 CN tissues. The positive immunostaining
of COX-2, primarily localized in the cytoplasm, was observed in 79%
of the NPC (34/43) and 36.3% of the CN (4/11) tissues. The average
staining scores in the NPC tissues were significantly higher than
those in the CN tissues (P<0.01) (Fig. 1B and C). Moreover, COX-2 expression
was significantly associated with lymph node (LN) metastasis
(P=0.042), tumor recurrence (P=0.024), and mortality (P=0.044)
(Table I). Of note, the patients
with NPC with a high COX-2 expression had a markedly shorter OS
(median OS, 60.5 months) than those with a low COX-2 expression
(median OS, >160 months; P=0.038) (Fig. 1D). A comparison was then made for
each of the 9 paired samples obtained at diagnosis and recurrence;
COX-2 expression was markedly increased after recurrence
(P<0.01) (Fig. 1E and F). Taken
together, these results suggest that COX-2 plays an important role
in the pathogenesis of NPC, as well as in disease progression
(e.g., metastasis) and recurrence.
 | Table IAssociation between COX-2 expression
and clinical characteristics of patients with NPC. |
Table I
Association between COX-2 expression
and clinical characteristics of patients with NPC.
|
Characteristics | COX-2
| P-value |
|---|
| High (n, %) | Low (n, %) |
|---|
| Sex | | | 0.782 |
| Male (28) | 10 (35.7) | 18 (64.3) | |
| Female (15) | 6 (40) | 9 (60) | |
| Age, years | | | 0.834 |
| <50 (26) | 10 (38.4) | 16 (61.6) | |
| ≥50 (17) | 6 (35.2) | 11 (64.8) | |
| Clinical stage | | | 0.744 |
| I–II (12) | 4 (33.3) | 8 (66.7) | |
| III–IV (31) | 12 (38.7) | 19 (61.3) | |
| LMN status | | | 0.042a |
| No LMN (6) | 0 (0) | 6 (100) | |
| LMN (37) | 16 (43.2) | 21 (56.8) | |
| Recurrence | | | 0.024a |
| No (28) | 7 (25) | 21 (75) | |
| Yes (15) | 9 (60) | 6 (40) | |
| Survival
status | | | 0.044a |
| Alive (22) | 5 (22.7) | 17 (77.3) | |
| Deceased (21) | 11 (52.3) | 10 (47.7) | |
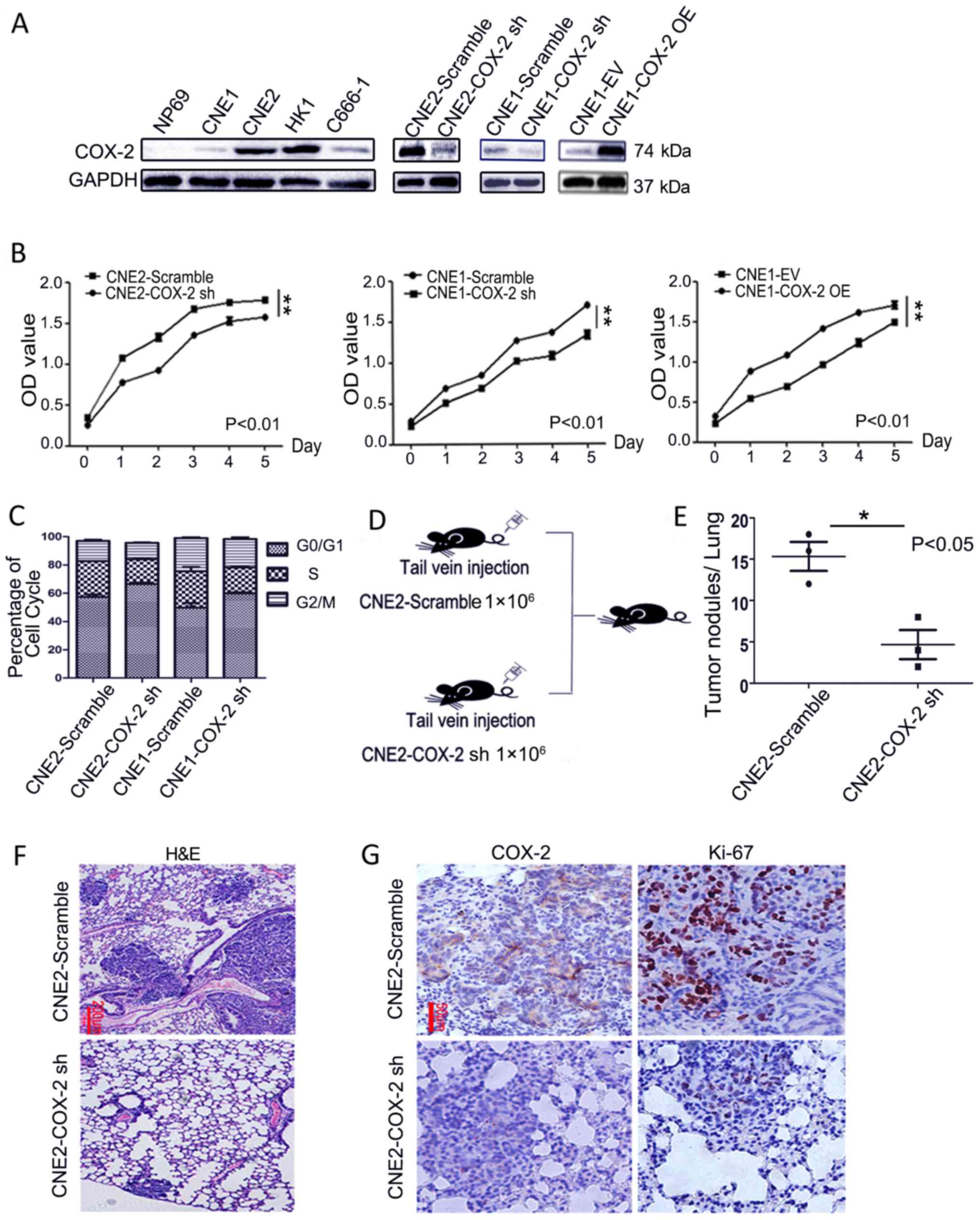
COX-2 knockdown attenuates the
proliferation of NPC cells in vitro and in vivo
To explore the functional role of COX-2 in NPC, we
detected the protein expression of COX-2 in NPC cell lines and the
NP69 normal cell line. The results of western blot analysis
revealed that the protein level of COX-2 was clearly higher in the
NPC cell lines than in the normal NP69 cells. Based on this
observation, the CNE1 cells with a lower COX-2 expression and the
CNE2 cells with a higher COX-2 expression were employed for use in
the following experiments to elucidate the function of COX-2 in
NPC. COX-2 was overexpressed in the CNE1 cells that displayed a low
basal level of COX-2, while it was knocked down by shRNA in both
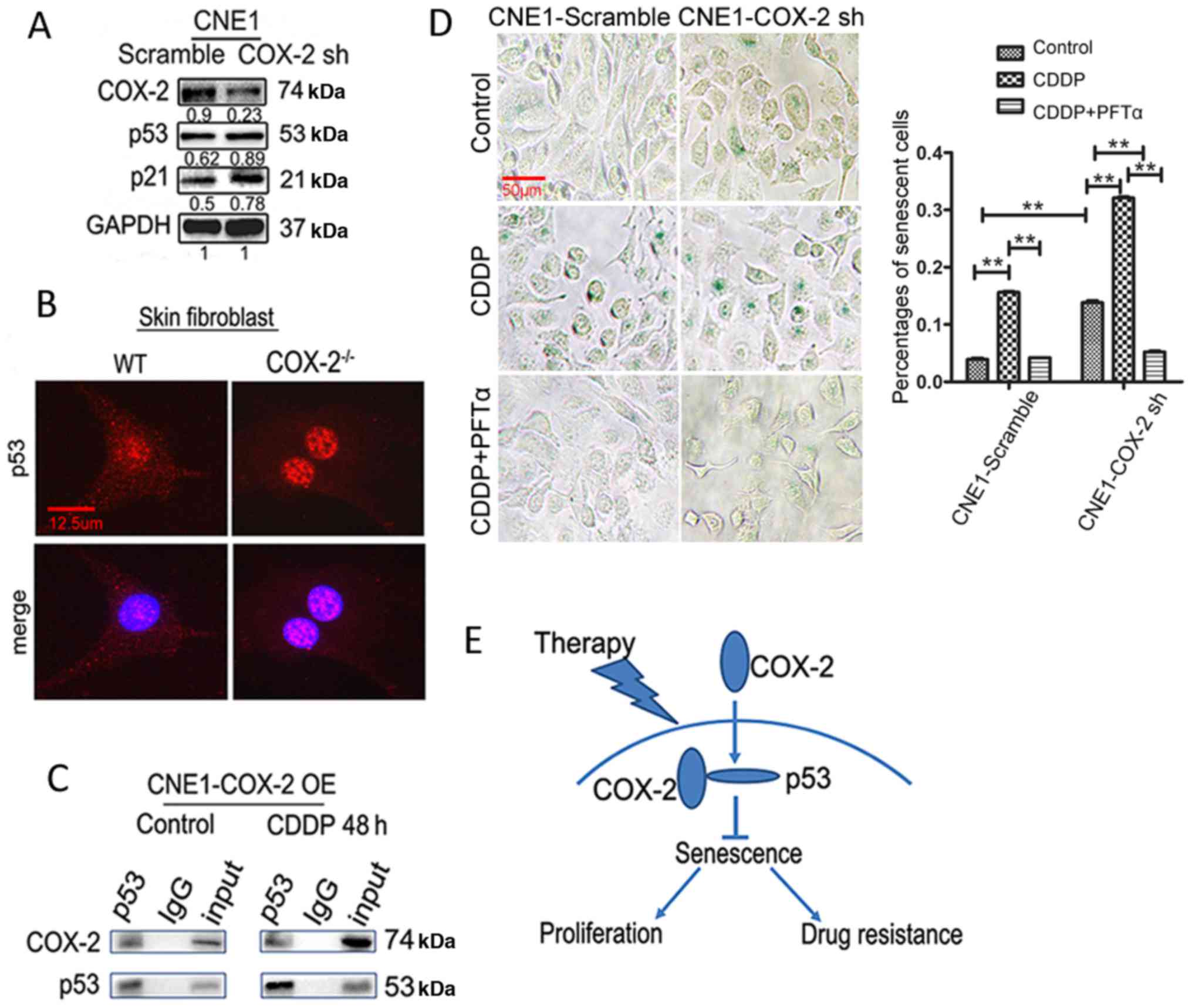
the CNE1 and CNE2 cells (Fig. 2A).
The results of CCK-8 assay revealed that the downregulation of
COX-2 using shRNA in either the CNE2 (CNE2-COX-2 sh) or CNE1
(CNE1-COX-2 sh) cells markedly inhibited cell proliferation,
compared to the scramble control (left and middle panels; P<0.01
for each case, Fig. 2B). By
contrast, COX-2 overexpression in the CNE1 cells (CNE1-COX-2 OE)
markedly increased the cell proliferation rate, compared to the
empty vector (CNE1-EV) control (right panel; P<0.01, Fig. 2B). In this context, the knockdown
of COX-2 in the CNE2 or CNE1 cells also resulted in cell cycle
arrest at the G0/G1 phase (P<0.05, Fig. 2C). To validate the effects of COX-2
knockdown on NPC tumor growth and metastasis to lung in
vivo, CNE2-Scramble and CNE2-COX-2 sh cells were injected via
the tail vein into nude mice (n=3 for each group, Fig. 2D). Furthermore, the number of tumor
nodules in the lung tissues from the mice in the CNE2-Scramble
group was greater than that from the mice in the CNE2-COX-2 sh
group at 8 weeks after inoculation (P<0.05, 15.3±1.7 vs.
4.7±1.7, Fig. 2E and F). As shown
in Fig. 2G, IHC staining revealed
a marked decrease in the number of Ki-67-positive cells in the
tumor nodules derived from the CNE2-COX-2 sh group cells, when
compared to those from the CNE2-Scramble group cells. Taken
together, these results indicate that COX-2 knockdown attenuates
the proliferation of NPC cells in vitro and in
vivo.
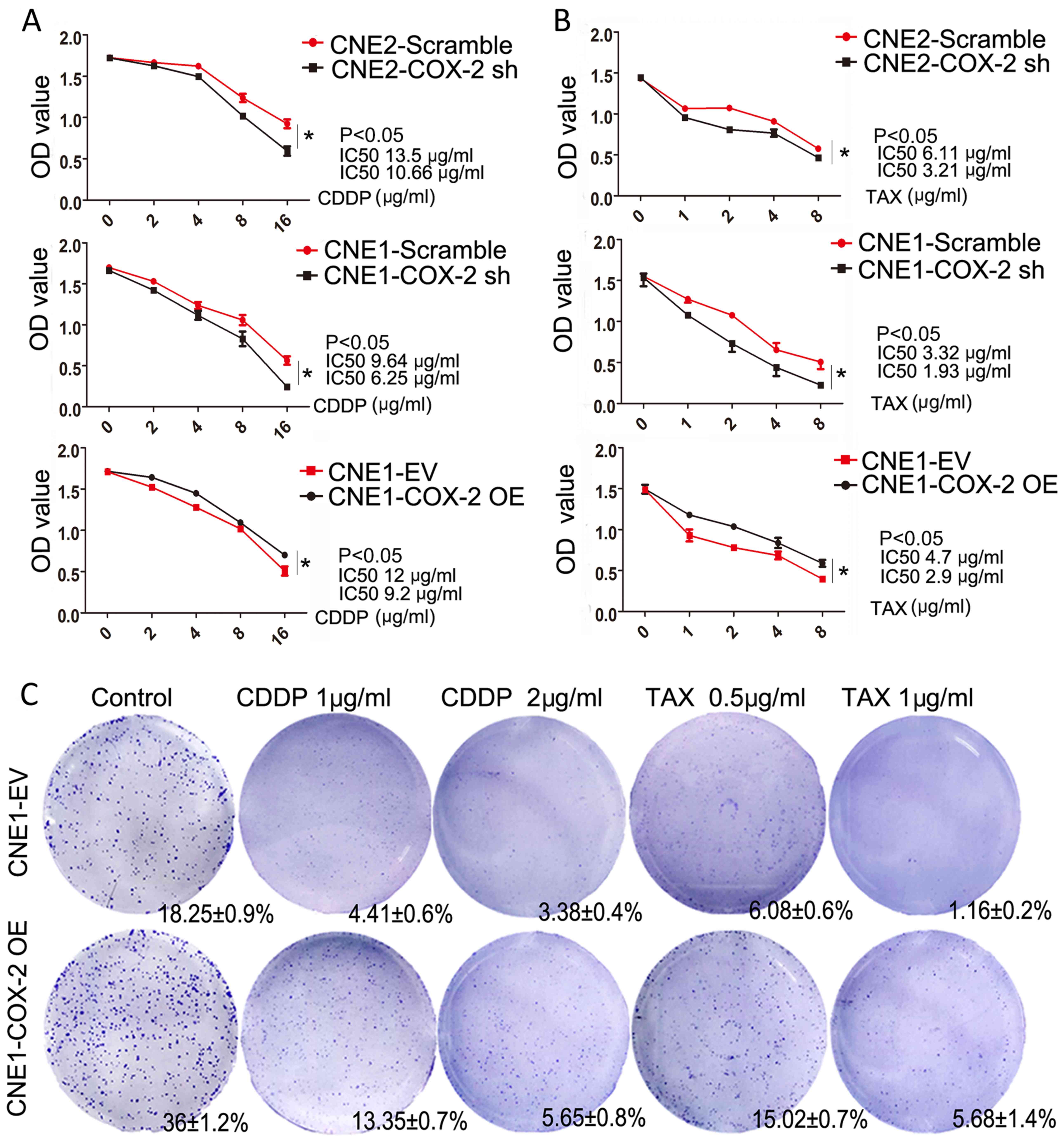
High COX-2 expression increases
chemoresistance in NPC cells
To explore the association between the expression of
COX-2 and chemotherapeutic resistance, cell viability and colony
formation ability were analyzed by CCK-8 and colony formation
assays, respectively in the CNE1 cells overexpressing COX-2
followed by CDDP or TAX treatment. Compared to the control cells,
the numbers of viable CNE1 and CNE2 cells in which COX-2 was
knocked down were markedly reduced following CDDP or TAX treatment,
while the numbers of viable CNE1 cells overexpressing COX-2 were
significantly higher than those of the control cells (P<0.05 for
each case, compared to the scramble or EV controls, Fig. 3A and B). Moreover, the results of
colony formation assay revealed a significant increase in the
colony-forming ability of the CNE1 cells overexpressing COX-2,
compared to the EV control (36±1.2 vs. 18.25±0.9%, P<0.05,
Fig. 3C). Following treatment with
CDDP, COX-2 overexpression reduced the lethality of the agent at
the two tested doses (3.0-fold, 13.35±0.7 vs. 4.41±0.6% and
1.6-fold, 5.65±0.8 vs. 3.38±0.4% for the EV control treated with 1
and 2 µg/ml CDDP, respectively, Fig. 3C). Following treatment with TAX,
COX-2 overexpression reduced the lethality of the agent at two
tested doses (2.5-fold, 15.02±0.7 vs. 6.08±0.6% and 4.8-fold,
5.68±1.4 vs. 1.16±0.2% for the EV control treated with 0.5 and 1
µg/ml TAX; P<0.05 for each case, respectively, Fig. 3C). Above all, these results
demonstrate that COX-2 expression leads to NPC cell
chemoresistance.
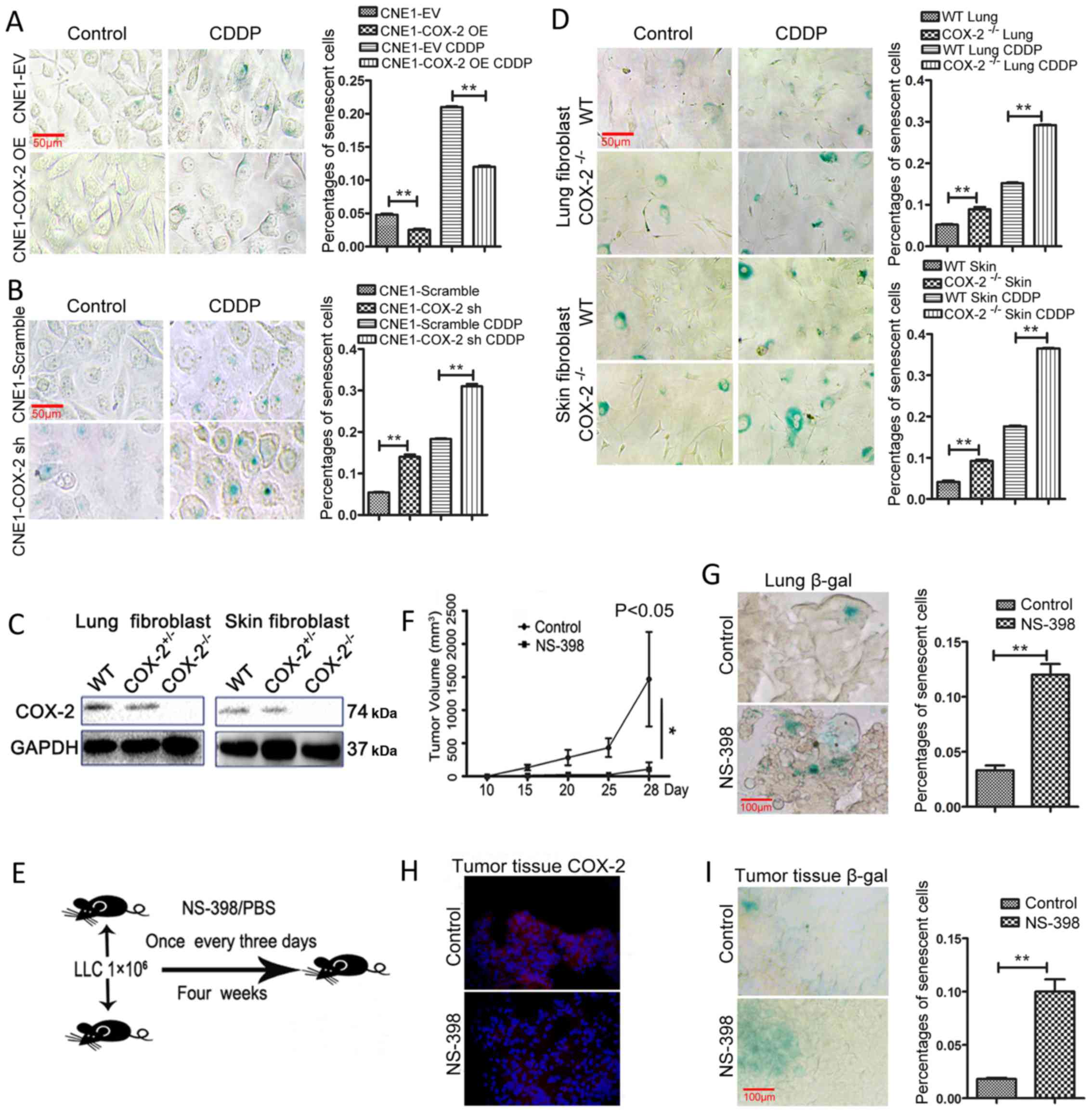
Inhibition of COX-2 increases
chemotherapy-induced senescence in vitro and in vivo
In order to elucidate the mechanisms underlying
COX-2-mediated resistance to chemotherapeutic agents, the role of
COX-2 in cellular senescence induced by chemotherapy was evaluated
following treatment with various concentrations of CDDP. The
results of SA-β-gal (28) assay
revealed that COX-2 overexpression reduced the percentage of
β-galactosidase-positive (blue) senescent CNE1 cells (2.5±0.3 vs.
4.8±0.3%; P<0.01, Fig. 4A),
while the knockdown of COX-2 with shRNA increased the percentage of
senescent cells (14±1 vs. 5.4±0.2%; P<0.01, Fig. 4B). Notably, whereas exposure to
CDDP induced a marked increase in the number of senescent cells,
this event was diminished by COX-2 overexpression (12±0.3 vs.
21±0.2%; P<0.01, Fig. 4A). By
contrast, exposure to CDDP induced a further increase in the number
of senescent cells in the CNE1 cells in which COX-2 was knocked
down (31±1 vs. 18.3±0.3%; P<0.01, Fig. 4B).
Previous studies have suggested that fibroblasts are
a valuable tool for studying cellular senescence (29). Thus, in this study, to further
validate the functional role of COX-2 in cellular senescence,
fibroblasts without COX-2 expression were obtained from COX-2
knockout (COX-2−/−) mice (30) (Fig.
4C). As shown in Fig. 4D, the
number of SA-β-gal-positive cells increased in both the lung (8.9±1
vs. 5.2±0.3% for WT) and skin fibroblasts (9.2±0.6 vs. 4.1±0.6% for
WT) from the COX-2−/− mice (P<0.01 for both cases).
Additionally, COX-2 gene knockout promoted the senescence induced
by CDDP, as reflected by the increased number of SA-β-gal-positive
cells, in the lung (29.2±0.2 vs. 15.2±0.3% for WT) and skin
fibroblasts (36.5±0.2 vs. 17.6±0.3% for WT; P<0.01 for each
case, Fig. 4D). As shown in
Fig. 4D that the numbers of
SA-β-gal-positive cells were increased in fibroblasts from
COX-2−/− mice (C57BL/6 background), which suggested that
senescence was induced in the COX-2−/− mouse model, this
prompted us to investigate the role of senescence in the mice with
the same genetic C57BL/6 background following treatment with
NS-398. As the LLC cell lines can lead to tumor bearing C57BL/6 WT
mice (31), while none of the NPC
cell lines could, we selected the LLC line for use in this in
vivo experiment. The COX-2 inhibitor, NS-398, was employed in
the mouse model established by subcutaneous inoculation with
1×106 LLC cells into C57BL/6 mouse (Fig. 4E). The results suggested that the
administration of NS-398 inhibited tumor growth, compared to the
vehicle control (P<0.05, n=4 for each group, Fig. 4F). Moreover, the number of
senescent cells increased in the lungs from LLC tumor-bearing mice
treated with NS-398 (P<0.01, 12±1.7 vs. 3.3±0.8%, compared to
the vehicle control, Fig. 4G). In
addition, COX-2 expression was weaker in the tumor tissue from the
LLC tumor-bearing mice treated with NS-398 (Fig. 4H). SA-β-gal staining revealed that
the number of senescent cells increased in the tumor tissue from
the mice treated with NS-398 (P<0.01, 10±2 vs. 1.8±0.2%,
compared to the vehicle control, Fig.
4I). Thus, these results support the notion that COX-2
negatively regulates cellular senescence and that COX-2 may serve
as a potential target for the further clinical treatment of
NPC.
COX-2 interacts with p53 to inhibit
therapy-induced senescence
It is well established that p53 plays a critical
role in cellular senescence (8).
Thus, in this study, to determine whether p53 is also involved in
the COX-2-mediated inhibition of senescence, the association
between COX-2 and p53 expression in NPC cells was investigated.
Although the expression of p53 exhibited no distinct change in the
CNE1-Scramble and CNE1-COX-2 sh cells, downstream p21 expression
was upregulated (Fig. 5A). In
addition, we found that p53 expression was markedly upregulated in
the nuclei of COX-2−/− skin fibroblasts, compared to
those from WT mice (Fig. 5B),
suggesting that p53 may play its role mainly through entering the
nucleus when COX-2 is downregulated. Furthermore,
co-immunoprecipitation assay revealed that COX-2 was precipitated
with p53 in he CNE1 cells overexpressing COX-2 with or without
exposure to 2 µg/ml CDDP (Fig.
5C), consistent with a previous finding that COX-2 interacts
with p53 in the nucleus (32).
Then, we examined whether inhibiting p53 would affect the function
of COX-2 in senescence. As shown in Fig. 5D, the knockdown of the expression
of COX-2 promoted senescence in the CNE1 cells (13.8±0.6 vs.
3.9±0.3% for the scramble control, P<0.01); CDDP induced a
further increase in the number of senescent cells (32.1±0.3 vs.
13.8±0.6% for the untreated control, P<0.01). Notably, increased
senescence in the CNE1-COX-2 sh cells was significantly blocked by
the p53 inhibitor, PFT-α (5.2±0.3 vs. 13.8±0.6% for untreated
control or vs. 32.1±0.3% for CDDP alone, P<0.01 for both cases,
Fig. 5D). Taken together, these
findings support a notion that COX-2 inhibits chemotherapy-induced
senescence in NPC cells, likely via binding p53 and inactivating
p53 function.
Discussion
The standard care for patients with locally advanced
NPC currently includes neoadjuvant chemotherapy combined with
radiotherapy. However, therapeutic resistance remains the most
important challenge in clinical practice. Inflammatory factors,
such as interleukin (IL)-6 and tumor necrosis factor (TNF)α not
only precede the development of NPC, but are also associated with a
poor survival of patients with NPC (33,34).
In this study, we found that the pro-inflammatory mediator, COX-2,
was expressed in NPC patient tissues at diagnosis and also highly
expressed in paired patients with recurrent NPC, which suggested
that COX-2 promotes NPC tumorigenesis and recurrence. In addition,
we identified the functional role of COX-2 in the pathogenesis of
NPC and unveiled the underlying mechanisms of COX-2-mediated
chemoresistance via the inhibition of chemotherapy-induced
senescence in NPC. Moreover, we also found that cellular senescence
significantly increased in fibroblasts from COX-2 knockout mice,
particularly when treated with CDDP. This study describes for the
first time, to the best of our knowledge, that COX-2 gene knockout
promotes senescence in a mouse model.
Therapeutic resistance includes intrinsic and
acquired resistance. Acquired resistance develops during treatment
often through the activation of alternative compensatory signaling
pathways that lead to the evasion of cell death. COX-2 may
contribute to the repopulation of tumors from cancer stem cells and
the activation of cancer-related genes, which contributes to
acquired resistance (35). Of
note, cellular senescence acts as a double-edged sword in the
pathogenesis and treatment of cancer (8). On the one hand, some senescent tumor
cells can bypass the cell cycle restriction and thereby survive
from therapy. On the other hand, therapy-induced senescence can
suppress tumor growth by irreversibly arresting cell proliferation.
For instance, Braumüller et al reported that IFN-γ and TNF
induced senescence in numerous murine and human cancers, and that
this may be a general mechanism for arresting cancer progression
(36). Goel et al also
demonstrated that CDK4/6 inhibitors overcame therapeutic resistance
in HER2-positive breast cancer by increasing G1 arrest and cellular
senescence through the suppression of Rb phosphorylation (37). In this study, we found that
chemotherapy induced cellular senescence in NPC cells, while the
senescence induced by the inhibition of COX-2 may confer acquired
therapeutic resistance.
To date, there are few studies available reporting
the function of COX-2 in the regulation of cellular senescence.
Furthermore, the role of COX-2 in therapy-induced senescence
remains almost unknown. Under physiological conditions, the
inhibition of prostaglandin E2 (PGE2)
production has been shown to be therapeutically beneficial in the
treatment of age-associated collagen deficits in human skin
(38). Kim et al found that
p16 and p53 expression levels were upregulated in the tissues of
COX-2 transgenic mice, suggesting that an increased COX-2
expression has an impact on the aging process (39). However, Han et al observed
that mouse lung fibroblasts (MLFs) derived from lung tissues
expressed less p21 in adult WT mice than in COX-2 knockout mice.
They also found that p53-induced COX-2 expression counteracted the
apoptosis mediated by p53 (40).
In the present study, we observed that COX-2 could bind to p53
protein, as determined by co-immunoprecipitation; however, it did
not influence the protein expression of p53, while the p53 protein
level was increased in the nuclei of fibroblasts from COX-2
knockout mice. This suggested that COX-2 may play a negative role
in the regulation of senescence in NPC cells, particularly induced
by chemotherapy, mostly likely via the inactivation of p53.
It should be noted that this study was partly based
on the use of CNE-1 and CNE-2 cells to investigate senescence in
NPC. These have been demonstrated to be contaminated with HeLa cell
line, and this may be a potential limitation of this study
(41).
In conclusion, the results of the present study
indicate that a high expression of COX-2 is associated with the
recurrence and a poor prognosis of patients with NPC, and that
COX-2 expression confers chemotherapeutic resistance in NPC.
Mechanistically, COX-2 functions to promote chemotherapeutic
resistance, mainly through the inhibition of senescence by
interacting with and inactivating p53 in NPC cells. Based on these
findings, we propose a working model for the mechanisms of action
of COX-2 in NPC, as shown in Fig.
5E. It is suggested that COX-2 may serve as a potential
predictor of the recurrence and therapeutic resistance, and may
serve as a therapeutic target for the development of targeted
therapy to eradicate resistant tumor cells in the treatment of
NPC.
Acknowledgments
The authors appreciate the discussions provided by
all the members in the laboratory of WZ.
References
|
1
|
Ma BB, Hui EP and Chan AT: Investigational
drugs for nasopharyngeal carcinoma. Expert Opin Investig Drugs.
26:677–685. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chua ML, Wee JT, Hui EP and Chan AT:
Nasopharyngeal carcinoma. Lancet. 387:1012–1024. 2016. View Article : Google Scholar
|
|
4
|
Shao JY, Wang HY, Huang XM, Feng QS, Huang
P, Feng BJ, Huang LX, Yu XJ, Li JT, Hu LF, et al: Genome-wide
allelotype analysis of sporadic primary nasopharyngeal carcinoma
from southern China. Int J Oncol. 17:1267–1275. 2000.PubMed/NCBI
|
|
5
|
Chan AT: Current treatment of
nasopharyngeal carcinoma. Eur J Cancer. 47(Suppl 3): S302–S303.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chan AT, Grégoire V, Lefebvre JL, Licitra
L, Hui EP, Leung SF and Felip E; EHNS-ESMO-ESTRO Guidelines Working
Group: Nasopharyngeal cancer: EHNS-ESMO-ESTRO Clinical Practice
Guidelines for diagnosis, treatment and follow-up. Ann Oncol.
23(Suppl 7): vii83–vii85. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen C, Chen T, Huang C, Wang J and Fei Z:
Experience of weekly cisplatin concurrent with intensity-modulated
radiotherapy for locally advanced nasopharyngeal carcinoma patients
with resistance to neoadjuvant chemotherapy. Medicine (Baltimore).
96. pp. e84342017, View Article : Google Scholar
|
|
8
|
Ewald JA, Desotelle JA, Wilding G and
Jarrard DF: Therapy-induced senescence in cancer. J Natl Cancer
Inst. 102:1536–1546. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Soto-Gamez A and Demaria M: Therapeutic
interventions for aging: The case of cellular senescence. Drug
Discov Today. 22:786–795. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Loaiza N and Demaria M: Cellular
senescence and tumor promotion: Is aging the key. Biochim Biophys
Acta. 1865.155–167. 2016.
|
|
11
|
Lee M and Lee JS: Exploiting tumor cell
senescence in anticancer therapy. BMB Rep. 47:51–59. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gordon RR and Nelson PS: Cellular
senescence and cancer chemotherapy resistance. Drug Resist Updat.
15:123–131. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Petrova NV, Velichko AK, Razin SV and
Kantidze OL: Small molecule compounds that induce cellular
senescence. Aging Cell. 15:999–1017. 2016. View Article : Google Scholar
|
|
14
|
Wang X, Wong SC, Pan J, Tsao SW, Fung KH,
Kwong DL, Sham JS and Nicholls JM: Evidence of cisplatin-induced
senescent-like growth arrest in nasopharyngeal carcinoma cells.
Cancer Res. 58:5019–5022. 1998.PubMed/NCBI
|
|
15
|
Sun Z, Pan X, Zou Z, Ding Q, Wu G and Peng
G: Increased SHP-1 expression results in radioresistance,
inhibition of cellular senescence, and cell cycle redistribution in
nasopharyngeal carcinoma cells. Radiat Oncol. 10:1522015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang J, Zou F, Tang J, Zhang Q, Gong Y,
Wang Q, Shen Y, Xiong L, Breyer RM, Lazarus M, et al:
Cyclooxygenase-2-derived prostaglandin E2 promotes
injury-induced vascular neointimal hyperplasia through the
E-prostanoid 3 receptor. Circ Res. 113:104–114. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schumacher Y, Aparicio T, Ourabah S,
Baraille F, Martin A, Wind P, Dentin R, Postic C and Guilmeau S:
Dysregulated CRTC1 activity is a novel component of PGE2
signaling that contributes to colon cancer growth. Oncogene.
35:2602–2614. 2016. View Article : Google Scholar
|
|
18
|
Wu K, Fukuda K, Xing F, Zhang Y, Sharma S,
Liu Y, Chan MD, Zhou X, Qasem SA, Pochampally R, et al: Roles of
the cyclooxygenase 2 matrix metalloproteinase 1 pathway in brain
metastasis of breast cancer. J Biol Chem. 290:9842–9854. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Qiu X, Cheng JC, Chang HM and Leung PC:
COX2 and PGE2 mediate EGF-induced E-cadherin-independent
human ovarian cancer cell invasion. Endocr Relat Cancer.
21:533–543. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pan J, Tang T, Xu L, Lu JJ, Lin S, Qiu S,
Chen G and K Tham IW: Prognostic significance of expression of
cyclooxygenase-2, vascular endothelial growth factor, and epidermal
growth factor receptor in nasopharyngeal carcinoma. Head Neck.
35:1238–1247. 2013. View Article : Google Scholar
|
|
21
|
Majumder M, Dunn L, Liu L, Hasan A,
Vincent K, Brackstone M, Hess D and Lala PK: COX-2 induces
oncogenic micro RNA miR655 in human breast cancer. Sci Rep.
8:3272018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Le CP, Nowell CJ, Kim-Fuchs C, Botteri E,
Hiller JG, Ismail H, Pimentel MA, Chai MG, Karnezis T, Rotmensz N,
et al: Chronic stress in mice remodels lymph vasculature to promote
tumour cell dissemination. Nat Commun. 7:106342016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhao CX, Luo CL and Wu XH: Hypoxia
promotes 786-O cells invasiveness and resistance to sorafenib via
HIF-2α/COX-2. Med Oncol. 32:4192015. View Article : Google Scholar
|
|
24
|
Kurtova AV, Xiao J, Mo Q, Pazhanisamy S,
Krasnow R, Lerner SP, Chen F, Roh TT, Lay E, Ho PL, et al: Blocking
PGE2-induced tumour repopulation abrogates bladder
cancer chemoresistance. Nature. 517:209–213. 2015. View Article : Google Scholar
|
|
25
|
Zhou W, Feng X, Ren C, Jiang X, Liu W,
Huang W, Liu Z, Li Z, Zeng L, Wang L, et al: Over-expression of
BCAT1, a c-Myc target gene, induces cell proliferation, migration
and invasion in nasopharyngeal carcinoma. Mol Cancer. 12:532013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Khamaisi M, Katagiri S, Keenan H, Park K,
Maeda Y, Li Q, Qi W, Thomou T, Eschuk D, Tellechea A, et al: PKCδ
inhibition normalizes the wound-healing capacity of diabetic human
fibroblasts. J Clin Invest. 126:837–853. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu T, Ding Y, Xie W, Li Z, Bai X, Li X,
Fang W, Ren C, Wang S, Hoffman RM, et al: An imageable metastatic
treatment model of nasopharyngeal carcinoma. Clin Cancer Res.
13:3960–3967. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Campisi J: Aging, cellular senescence, and
cancer. Annu Rev Physiol. 75:685–705. 2013. View Article : Google Scholar
|
|
29
|
Dirac AM and Bernards R: Reversal of
senescence in mouse fibroblasts through lentiviral suppression of
p53. J Biol Chem. 278:11731–11734. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yu Y, Fan J, Chen XS, Wang D, Klein-Szanto
AJ, Campbell RL, FitzGerald GA and Funk CD: Genetic model of
selective COX2 inhibition reveals novel heterodimer signaling. Nat
Med. 12:699–704. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhu H, Kauffman ME, Trush MA, Jia Z and Li
YR: A simple bioluminescence imaging method for studying cancer
cell growth and metastasis after subcutaneous injection of Lewis
lung carcinoma cells in syngeneic C57BL/6 mice. React Oxyg Species
(Apex). 5. pp. 118–125. 2018
|
|
32
|
Choi EM, Kim SR, Lee EJ and Han JA:
Cyclooxygenase-2 functionally inactivates p53 through a physical
interaction with p53. Biochim Biophys Acta. 1793:1354–1365. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liao Q, Zeng Z, Guo X, Li X, Wei F, Zhang
W, Li X, Chen P, Liang F, Xiang B, et al: LPLUNC1 suppresses
IL-6-induced nasopharyngeal carcinoma cell proliferation via
inhibiting the Stat3 activation. Oncogene. 33:2098–2109. 2014.
View Article : Google Scholar
|
|
34
|
Bourouba M, Zergoun AA, Maffei JS, Chila
D, Djennaoui D, Asselah F, Amir-Tidadini ZC, Touil-Boukoffa C and
Zaman MH: TNFα antagonization alters NOS2 dependent nasopharyngeal
carcinoma tumor growth. Cytokine. 74:157–163. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhou TJ, Zhang SL, He CY, Zhuang QY, Han
PY, Jiang SW, Yao H, Huang YJ, Ling WH, Lin YC, et al:
Downregulation of mitochondrial cyclooxygenase-2 inhibits the
stemness of nasopharyngeal carcinoma by decreasing the activity of
dynamin-related protein 1. Theranostics. 7:1389–1406. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Braumüller H, Wieder T, Brenner E, Aßmann
S, Hahn M, Alkhaled M, Schilbach K, Essmann F, Kneilling M,
Griessinger C, et al: T-helper-1-cell cytokines drive cancer into
senescence. Nature. 494:361–365. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Goel S, Wang Q, Watt AC, Tolaney SM,
Dillon DA, Li W, Ramm S, Palmer AC, Yuzugullu H, Varadan V, et al:
Overcoming therapeutic resistance in HER2-positive breast cancers
with CDK4/6 inhibitors. Cancer Cell. 29:255–269. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li Y, Lei D, Swindell WR, Xia W, Weng S,
Fu J, Worthen CA, Okubo T, Johnston A, Gudjonsson JE, et al:
Age-associated increase in skin fibroblast-derived prostaglandin E2
contributes to reduced collagen levels in elderly human skin. J
Invest Dermatol. 135:2181–2188. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim J, Vaish V, Feng M, Field K,
Chatzistamou I and Shim M: Transgenic expression of
cyclooxygenase-2 (COX2) causes premature aging phenotypes in mice.
Aging (Albany NY). 8:2392–2406. 2016. View Article : Google Scholar
|
|
40
|
Han JA, Kim JI, Ongusaha PP, Hwang DH,
Ballou LR, Mahale A, Aaronson SA and Lee SW: P53-mediated induction
of Cox-2 counteracts p53- or genotoxic stress-induced apoptosis.
EMBO J. 21:5635–5644. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chan SY, Choy KW, Tsao SW, Tao Q, Tang T,
Chung GT and Lo KW: Authentication of nasopharyngeal carcinoma
tumor lines. Int J Cancer. 122:2169–2171. 2008. View Article : Google Scholar : PubMed/NCBI
|