1. Introduction
Breast cancer (BC) is the most common epidermal
neoplasia worldwide, representing approximately 12% of all new
cancer cases and 25% of all cancer cases in women. Belgium has the
highest rate of BC, followed by Denmark and France, while the
lowest incidence has been observed in Asia and Africa (1). The Breast Cancer Organization
estimates that approximately 12% of women from the United States
will develop invasive BC during the course of their lifetime. Over
the past decade, tremendous progress has been made in identifying
BC etiological factors, understanding tumor biology, developing
tools for better detection and determining the most effective
treatments. Nevertheless, this disease still represents an
important cause of morbi-mortality due to cancer (1,2). It
is widely known that the oncogenesis phenomenon is a complex
biological mechanism not yet well understood. Cancer development is
a multistep process, involving genome instability, which leads to
sustained proliferative signaling, the evasion of growth
suppressors, resistance to cell death, tumor angiogenesis, invasion
and metastasis (3). Other
hallmarks of the cancer cell are the capacity to evade the immune
system and to generate reprogramming of the energy metabolism
system (4,5). As regards the latter hallmark, the
most well-known tumor cell metabolic abnormality is the Warburg
effect (4).
Otto Warburg observed that cancer cells prefer
aerobic glycolysis to oxidative phosphorylation; however, these
cells do not uptake oxygen like normal tissue cells, even under a
normal oxygen environment (4,6).
Subsequently, there is ample evidence to support the hypothesis
that tumor-related metabolic abnormalities involve defects in
mitochondrial function, since many of the metabolic genes whose
mutations can cause cancers are mitochondrial genes or nuclear
encoded genes that are relevant to mitochondrial biogenesis and
function. As an example, the metabolic enzyme, succinate
dehydrogenase (SDH), is frequently mutated in cancer (7). In this review, we provide an overview
of mitochondrial mutations and polymorphisms in cancer, emphasizing
those in BC.
2. Mitochondria
Mitochondria are involved in fundamental cellular
processes, which include the generation of adenosine triphosphate
(ATP) for cellular energy, calcium storage for cell signaling, and
mediating cell growth and death processes. Mitochondria,
descendants of endosymbiotic α-protobacteria that become
established in a host cell, are double-membraned and
semi-autonomous organelles in eukaryotic cells that can
auto-replicate and are maternally inherited. This organelle has its
own genome and is organized into DNA-protein complexes termed
mitochondrial nucleoids, which are relatively stable genetic
elements. Human cells have approximately 2,000 mitochondria per
cell and an average of between 1.4 and 7.5 mitochondrial DNA
(mtDNA) molecules per nucleoid (8). mtDNA is a negatively supercoiled
double-stranded circular molecule of 16,569 bp in size. Based on
sedimentation properties, one strand is termed light (L) and the
other one heavy (H). With only <7% of the sequence considered
non-coding, mtDNA contains 37 genes, 28 of which are encoded in the
H strand and 9 in the L strand. Thirteen of these genes code for
essential proteins for mitochondrial oxidative phosphorylation
(OXPHOS). These include 7 of nearly 45 polypeptides of the electron
transport chain complex I (ND1-3, ND4L and ND4-6); 1 of the 11
polypeptides of complex III (Cytb), 3 of the 13 polypeptides of
complex IV (COI-III), and 2 of the 15 polypeptides of complex V
(ATP6 and ATP8). mtDNA also encodes 22 transfer RNAs (tRNAs), as
well as the 18S and 16S ribosomal RNAs (rRNA). tRNAs and rRNAs are
necessary for the translation of the respiratory subunit mRNAs
within the mitochondrial matrix (Table
I) (8-10). The hypervariable region (HVR1:
16024-16383 nt and HVR2: 57-333 nt) or D-loop region is a
non-coding sequence; however, it contains the origin of replication
of the mtDNA (8-11). The proximity of mtDNA to the site
of production of reactive oxygen species (ROS) and the absence of
protective histones increase mtDNA susceptibility for acquiring
mutations and oxidative damage, causing a mutation rate of
100-1,000-fold higher for mtDNA than nuclear DNA (nDNA)
(1.1-1.7×10−8 per nucleotide site per generation).
Moreover, mtDNA damage induced by ROS is more extensive and
persists longer than ROS-nDNA injury in human cells (12,13).
Mitochondrial DNA encodes only a small number of proteins needed
for its molecular architecture and biological functions; hence,
additional nuclear-encoded mitochondrial (NEMt) proteins (NEMtPs)
are required for mitochondrial biogenesis, assembly of the
respiratory chain and the maintenance of mtDNA (Fig. 1) (14). The biogenesis of these organelles,
which implies variations in number, size and mass, is highly
influenced by factors, such as cell division, renewal,
differentiation, oxidative stress, exercise, caloric restriction,
low temperature, and mutations in both nDNA and mtDNA (14,15).
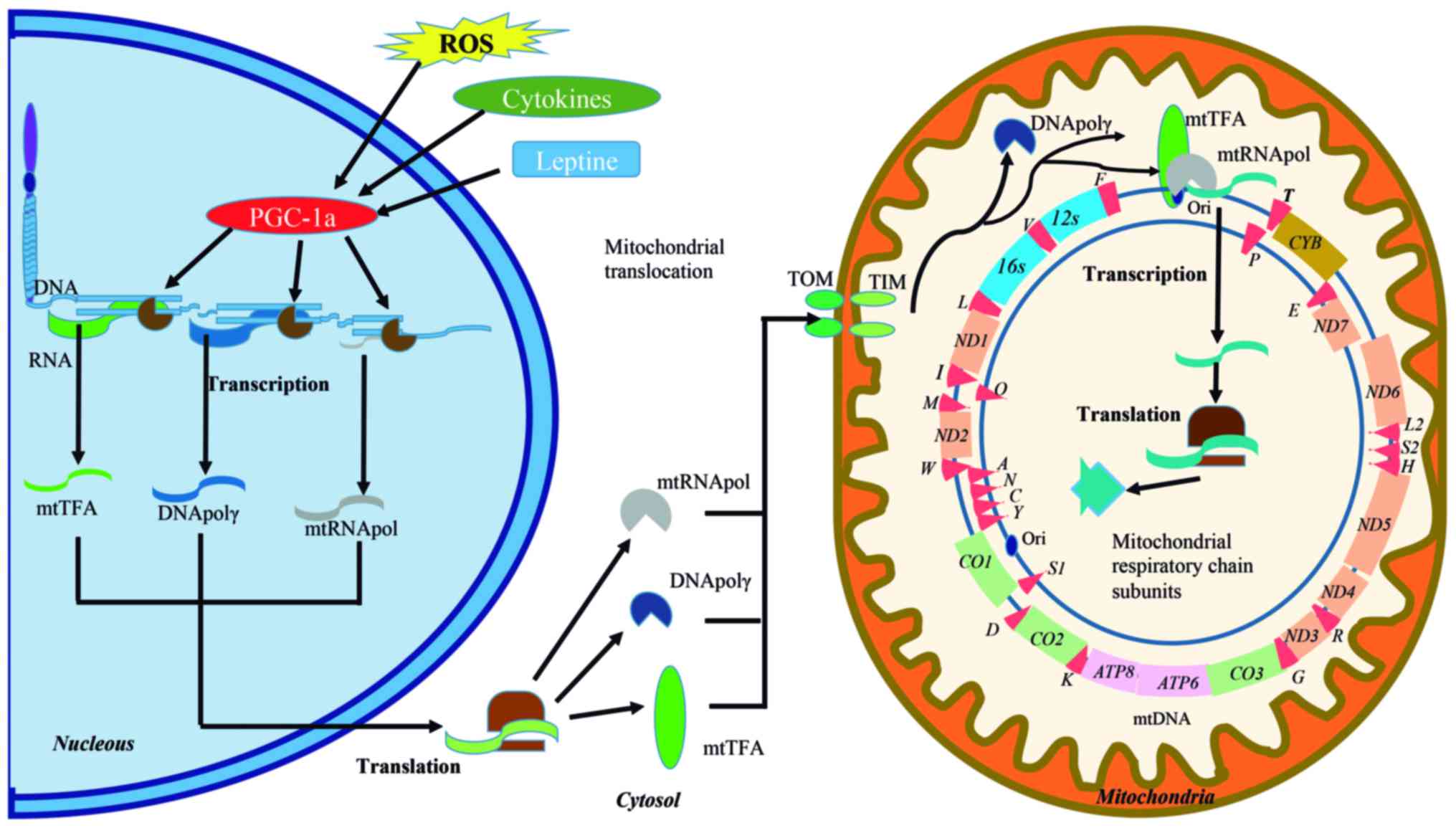 | Figure 1Signaling pathways involved in
mitochondrial function and the human mitochondrial DNA (mtDNA) map.
Activators, such as reactive oxygen species (ROS), cytokines and
leptin activate peroxisome proliferator-activated receptor gamma
coactivator-1 (PGC-1a) to promote the transcription of genes that
are involved in the mitochondrial transcriptional machinery
(DNApolγ, DNA polymerase γ; mtTFA, mitochondria transcription
factor A; mtRNApol, mitochondrial RNA polymerase, etc.). The mRNAs
are directed to the cytoplasm for translation and the resulting
proteins are immediately imported into the mitochondria by the
translocase of the outer and inner mitochondrial membrane (TOM and
TIM, respectively) system. DNApolγ is responsible for replication,
whereas mtTFA and mtRNApol are actively involved in the initiation
of transcription. The formed mRNAs give rise to mitoribosomal
subunits, tRNAs, or are translated into the mitoribosomes to
generate protein subunits of the mitochondrial respiratory chain
complex. mtDNA is a double-stranded (heavy chain: H and a light
chain: L) circular DNA, containing 37 genes, of which, 28 are
located in the H strand and nine in the L strand. Thirteen of these
genes code for essential components of the electron transport
complexes (ETC), 22 are transfer RNAs (tRNAs) and two ribosomal
RNAs (rRNA). ND1, NADH-ubiquinone oxidoreductase chain 1;
ND2, NADH dehydrogenase 2; COI, cytochrome c
oxidase I; COII, cytochrome c oxidase 2; ATP8,
ATP synthase 8; ATP6, ATP synthase 6; COIII,
cytochrome c oxidase 3; ND3, NADH dehydrogenase 3;
ND4L, NADH-ubiquinone oxidoreductase chain 4L; ND4, NADH
dehydrogenase 4; ND5, NADH dehydrogenase 5; NAD6, NADH
dehydrogenase 6; CYB, cytochrome b. ND genes are
indicated by orange boxes, CO genes by green boxes, the
CYB gene by a gold box and ATP genes by pink boxes.
rRNA is indicated by blue boxes; and tRNA by red triangles. |
 | Table INuclear DNA and mitochondrial DNA
features. |
Table I
Nuclear DNA and mitochondrial DNA
features.
| Features | Nuclear DNA | Mitochondrial
DNA |
|---|
| Base pairs
(bp) | 3.2 billion | 16,569 |
| Strand | Double, lineal,
forward (5′-3′) and reverse (3′-5′) | Double, circular,
light and heavy |
| Organization | Chromosomes (23
pairs) | Nucleoids |
| Coding region | 2-3% | 90% |
| Genes | | |
| Number | 22,000 approx. | 37 |
| | L (forward): 8 tRNA
and 1 polypeptide |
| | H (reverse): 12
polypetides, 14 tRNAs, 2 RNAs |
| Structure | Exon, intron,
intergenic regions | Polycistronic |
| Variants | SNPs, indel, CNVs,
STRs, VNTRs, translocations, inversions | SNPs, Indels,
CN |
| Ploidy | Diploid | Polyploid |
| Genetic code | | |
| Transduction
codons | 32 | 24 |
| Initiation
codon | AUG | AUG, AUA, AUU |
| Stop codon | UAA, UAG, UGA | AGA, AGG |
| Transcription | Multiple | ITL,
ITH1, 1TH2 |
| Genotype | Homozygote and
heterozygote | Homoplasmic,
heteroplasmic |
| Inheritance | Maternal and
paternal | Maternal |
| Recombination | Homologous
chromosomes | No evidence |
Nuclear-encoded genes that participate in
mitochondrial biogenesis and function
In addition to the 13 mitochondrial encoded genes,
to function properly the mitochondria require >1,500 NEMtPs.
These proteins are required by the mitochondria to perform DNA
replication, transcription and translation processes, as well as to
carry out biochemical functions and to maintain its molecular
architecture (Fig. 1). Almost 1%
of the products contained in the mitochondria are synthesized in
the matrix and the remainder, in the cytosol. Mitochondrial
proteins synthesized in the cytosol are encoded by nuclear genes
derived from prokaryotic genes that were transferred to the nucleus
or that originated during the evolution of eukaryotic cells. Since
NEMt genes (NEMtGs or mitonuclear genes) include proteins and
enzymes involved in mtDNA replication, transcription and
translation, catabolytic pathways, and are components of the
mitochondrial import and folding machineries, most of them are
strictly essential for life (16,17).
One of the most extensively studied NEMtG is the mitochondrial
transcription factor A (TFAM) that regulates mtDNA
replication/transcription and is implicated in the dynamics of
mtDNA nucleoids (18-21). TFAM expression is promoted
by the nuclear respiratory factor transcriptional regulators
(NRF)-1 and NRF-2, which are activators of nuclear genes necessary
for multiple mitochondrial functions. For example, the biogenesis
of the mitochondria is regulated by the induction of peroxisome
proliferator-activated receptor γ coactivator-1 (PGC-1) (18,19,22).
Additionally, it has been reported that the absence of TFAM
in knockout mice is responsible for a severe OXPHOS defect, marked
reduction in mtDNA content and embryonic lethality (20). Furthermore, the loss of
PolgA, encoding a subunit of mitochondrial DNA polymerase,
also results in lethality in mice (23). This observation suggests that
mutations in NEMtGs can affect functions, such as electron
transport and oxidative phosphorylation, protein translocation and
mitochondrial biogenesis. The translocase of the outer membrane
(TOM) complex is a clear example of the relevant role of
mitonuclear genes in the cell, since nearly all mitochondrial
pre-proteins are imported via a TOM entry gate (24). From the cytosol to the TOM complex,
pre-proteins are guided by molecular chaperones (heat shock protein
90 or heat shock cognate 70) (25); there, TOM20, TOM22 and TOM70
recognize the mitochondrial targeting signals of cytosolic
preproteins (Fig. 1). Hence, it is
clear that errors in the import and assembly of mitochondrial
proteins can result in a disease state in humans (26). Additionally, nDNA genes have also
been found to be involved in >30 different modified mt-tRNAs,
and interestingly, Bandiera et al (27) found a large number of
nuclear-encoded miRNAs, designated 'mitomiRs', with differential
expression in the mitochondria and cytosol, demonstrating that the
majority of mitochondrial miRNAs have both nuclear and
mitochondrial-encoded targets.
3. Mitochondrial DNA variants in human
diseases
Mitochondria are involved in the fundamental
cellular process of generating ATP for cellular energy. However,
energy metabolism is not the only essential role of mitochondria,
since they are also crucial for stress response, cell survival and
death, immune response and cell signaling. In consequence,
mitochondrial impairment contributes to a wide spectrum of
heterogeneous human conditions (mental disorders, cardiomyopathies,
metabolic diseases, immune mediated diseases, aging and cancer);
preferentially affecting those tissues with high-energy demands,
such as brain, muscle and liver (12,28,29).
The strongest evidence linking a mtDNA mutation to a human
phenotype is the increased risk of developing blindness in subjects
harboring mutations in the mitochondrial encoded complex I genes
(ND1-6 and ND4L). These mutations are known to cause Leber
hereditary optic neuropathy (LHON), synergistically interacting
with a primary LHON mutation to cause a defect in OXPHOS complex I
activity (30).
Mitochondrial disorders also cause an overlapping
spectrum of diseases. In a case-control study, Hudson et al
(29) analyzed mtDNA single
nucleotide polymorphisms (SNPs) from 51,106 subjects from the
Wellcome Trust Case Consortium, which included patients with
ankylosing spondylitis, ischemic stroke, multiple sclerosis,
Parkinson's disease, primary biliary cirrhosis, psoriasis,
schizophrenia, ulcerative colitis, coronary artery disease,
hypertension and type 2 diabetes. The authors observed that mtDNA
polymorphisms modify the risk of developing one or more of these
diseases. Of note, high-risk alleles were more frequent than
protective alleles, indicating that mtDNA is not at equilibrium in
the human population, and that recent mutations interact with
nuclear loci to modify the risk of developing diseases (29). Notably, polymorphisms that increase
the risk of developing two or more diseases were limited to
mitochondrial cytochrome b (CYB: H16R,
T158A) and subunit 3 of cytochrome c oxidase
(COIII: V91L, N154S) genes. The CYB
variants H16R and T158A were previously associated with several
diseases (31,32). The only non-synonymous polymorphism
associated with a reduced risk of various entities was the G10398A
(T114A) of ND3, which occurs twice on the human mtDNA
phylogeny and has previously been associated with Parkinson's
disease and cardiomyopathies (33-36).
This polymorphism has been shown to reduce complex I activity,
cytosolic calcium levels, and mitochondrial membrane potential, and
thus may reduce the levels of ROS (33).
As regards NEMtGs, mutations in >100 genes have
been implicated in mitochondrial human diseases (37-40).
As an example, mutations in elaC ribonuclease Z2 (ELAC2)
have been found in individuals suffering from infantile-onset
hypertrophic cardiomyopathy and complex I deficiency (39,41).
Similarly, defects in any step of protein transport due to TOM
complex mutations lead to oxidative stress, neurodegenerative
diseases, and metabolic disorders (26). Irregularities in mitochondrial
targeting signals have also been identified as a cause of pyruvate
dehydrogenase (PDH) deficiency. These mutations were described in
the amino-terminal targeting signal (N-MTS) of the PDH, which is a
subunit of the mitochondrial matrix protein complex. X-linked
mutations in the PDH E1α subunit are the most common cause of PDH
deficiency and have been associated to microcephaly and cerebral
atrophy (42). Moreover, mutations
in nuclear-encoded components of the mitochondrial translation
machinery have been shown to be involved in other human diseases
(31). To mention a few, tRNA
modifying enzymes (PUS1, TRMU, MTO1),
aminoacyl-tRNA synthetases (RARS2, DARS2,
YARS2, SARS2, HARS2, AARS2,
MARS2, EARS2, FARS2), ribosomal proteins
(MRPS16, MRPS22, MRPL3, MRPL12),
elongation factors (GFM1, EFTs, EFTu),
translational activators (LRPPRC, TACO1),
C12orf62, etc. (43).
4. Mitochondria variants and cancer
In pathological conditions, such as cancer,
malignant transformation induces reprogramming of cell metabolism
and permanent fission and fusion of mitochondria to maintain
daughter cells upon cell division. During oncogenic processes,
tumor cells not only acquire persistent changes in metabolism to
support cell growth, but also other types of mtDNA damage (12). mtDNA mutations have been identified
in all types of human tumors, both in the non-coding and coding
regions of the mtDNA (Fig. 2).
Larman et al, studying different cancer types, found that
the frequency of somatic mtDNA mutations ranged from 13% in
glioblastoma to 63% in rectal adenocarcinomas (44). Notably, the majority of the mtDNA
mutations appeared to be homoplasmic (same sequence in all mtDNA
molecules) in nature; nevertheless, heteroplasmy (the presence of
>1 mtDNA sequence in a cell) could contribute to heterogeneous
mitochondrial morphologies and correlate with metabolic flexibility
and cancer metastasis (28,45).
According to their percentage in the cell, mutations may be
homoplasmic (>95 to 100%), high heteroplasmic (>20 to
<95%), low heteroplasmic (>0.5 to 20%) or rare (0.5% or less)
(46). Surprisingly, 72% of the
reported tumor-specific mtDNA mutations are also found in non-tumor
cells (germline variants) of healthy subjects (44). As mtDNA is highly susceptible to
acquiring mutations from ROS, in addition to inherited mtDNA
mutations, multiple subpopulations of mtDNA could arise during the
lifespan, enriching the heteroplasmic condition (mainly in cells
with high division rates). Of note, although there is co-occurrence
of mtDNA germline variants and somatic mutations, a high proportion
of these become homoplasmic in cancer cells (47,48).
However, the heteroplasmic threshold effect and the mechanisms
underlying their selection towards a homoplasmic state during tumor
development are not yet well understood (48).
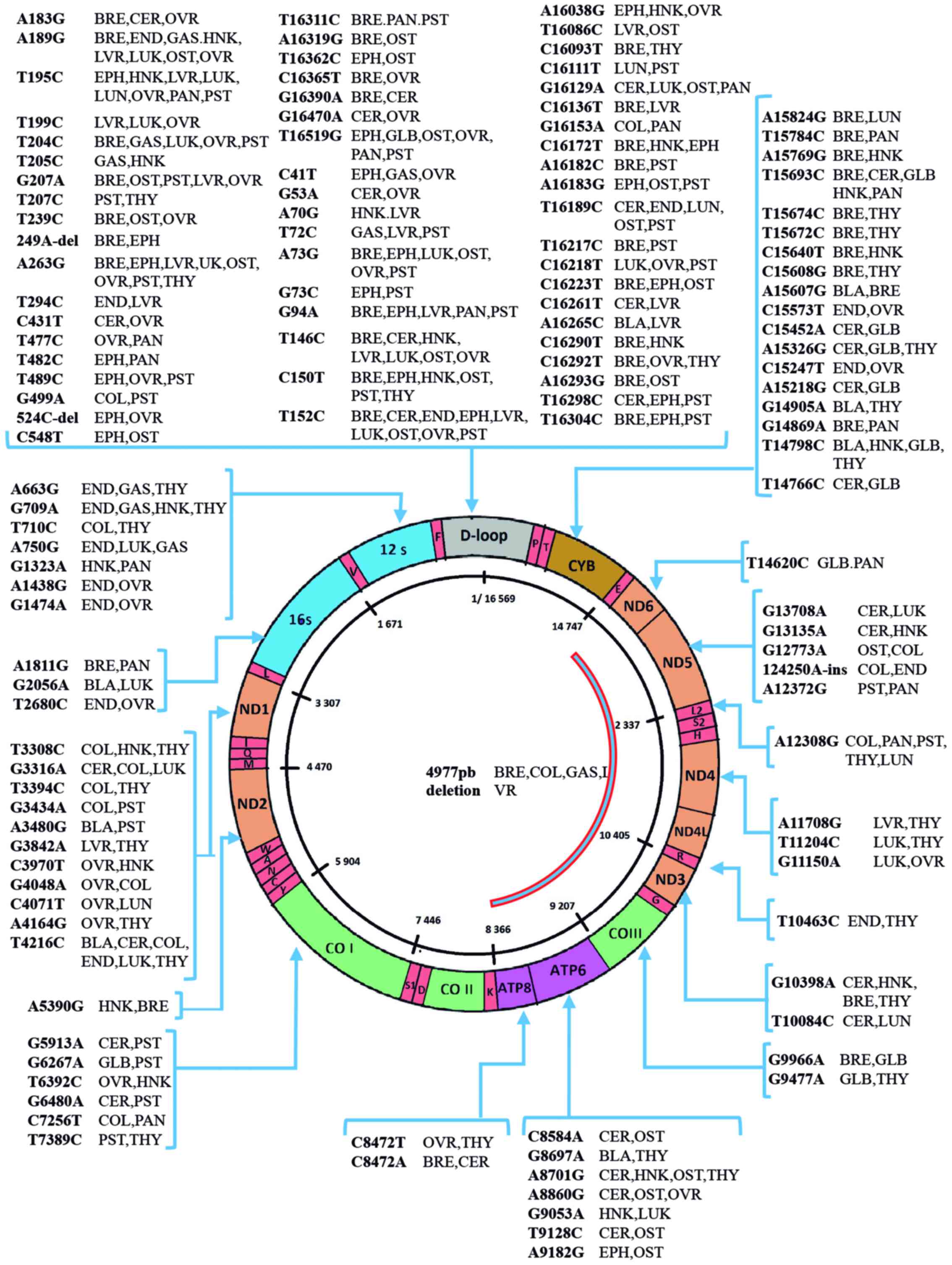 | Figure 2Mitochondrial DNA mutations reported
in different types of cancer. The figure shows the mitochondrial
(mtDNA) genome diagram and mutations reported in human cancer.
mtDNA is a double-stranded circular DNA comprised by 16,569 bases
and contain 37 genes. Of these, 13 encode essential components of
the electron transport complexes (ETC), 22 are tRNAs and 2 rRNAs.
Mutations are named by the locations of the mutated base (C:
cytosine, G: guanine, T: thymine, A: adenine). The non-coding
D-loop region (gray color) exhibits the higher number of mutations.
ND1, NADH-ubiquinone oxidoreductase chain 1; ND2,
NADH dehydrogenase 2; COI, cytochrome c oxidase I;
COII, cytochrome c oxidase 2; ATP8, ATP
synthase 8; ATP6, ATP synthase 6; COIII, cytochrome
c oxidase 3; ND3, NADH dehydrogenase 3; ND4L,
NADH-ubiquinone oxidoreductase chain 4L; ND4, NADH
dehydrogenase 4; ND5, NADH dehydrogenase 5; NAD6,
NADH dehydrogenase 6; CYB, cytochrome b; BLA, bladder
cancer; BRE, breast cancer; CER, cervical cancer; COL, colon
cancer; END, endometrial cancer; EPH, esophageal cancer; GAS,
gastric cancer; GLB, glioblastoma; HNK, head and neck cancer; LUK,
leukemia; LVR, liver cancer; LUN, lung cancer; OTS, osteosarcoma;
OVR, ovarian cancer; PNC, pancreatic cancer; PST, prostate cancer;
THY, thyroid cancer. |
Warburg's observation was the cornerstone for the
study of mitochondrial function in tumor cells. Later, it was
observed that the mRNA levels for defined mtDNA encoded genes were
upregulated in many human cancer types (44,49-51).
In this regard, the first significant finding was the observation
that hepatoma cells express hexokinase II, while normal hepatic
cells express hexokinase IV (glucokinase) (49). Metabolic deregulation and its
associated consequences induce a strong selective pressure on tumor
cells. Hence, acquiring somatic mtDNA mutations that impact
oxidative phosphorylation seems to be an alternate mechanism for
enhancing tumor growth (44).
Another important observation is that mutations in OXPHOS genes and
accumulation of ROS can lead to the oxidation of proteins and their
missfolding. To balance this, metastatic cells show high expression
of the unfolded protein response (UPRmt), which allows
them to survive in this highly stressed environment (45,52).
The mtDNA of cancer cells was first studied by
Polyak et al in 1998 (47).
They sequenced the mtDNA of 10 human colorectal cancer (CC) cell
lines. The authors identified mutations in 70% of all studied cell
lines; many of these occurred in rRNA genes or were missense
mutations, with only one frame-shift and one chain-termination
mutation (47). Another
interesting finding was that tumor cells frequently contained a
relatively small number of mitochondria with few mtDNA molecules
per mitochondrion. In addition, most of the mtDNA mutations were
homoplasmic, derived from a mtDNA somatic mutation initially
represented in a heteroplasmic state (44). These data suggested that at least
one of the observed tumoral mtDNA mutations could confer a
selective advantage to the cancer cell, further supporting the
hypothesis that mtDNA somatic mutations play an important role in
promoting the proliferation of several types of tumor cells
(47). Moreover, Habano et
al (53) reported that changes
in the length of the polycytosine tract of the HVR2 in the
mitochondrial D-loop are the most frequent (44%) mutations in
colorectal tumors. Currently, it is well known that this region is
highly mutated in a variety of human cancers (ovarian, thyroid,
kidney, liver, lung, colon, gastric, brain, bladder, head and neck,
prostate, breast and leukemia), although most of these mutations do
not have pathogenic implications, as they are not generating
stop-codons, frame-shifts or altering tRNA structures (44). Furthermore, Larman et al
(44), analyzing whole-genome data
generated by The Cancer Genome Atlas Research Network (CGARN),
reported that the majority of coding mutations occurred in the
subunits of the electron transport chain complex (NADH
dehydrogenase, cytochrome b and cytochrome c
oxidase). The atlas includes paired tumor and normal tissue samples
from 226 individuals with 5 types of cancer: Colon adenocarcinoma
(COAD), rectal adenocarcinoma (READ), acute myeloid leukemia (AML),
glioblastoma (GLB) and ovarian serous cystadenocarcinoma (OVR).
Highly variable proportions of deleterious tumor-specific mutations
across tumor types were found, ranging from 13% in GLB to 63% in
READ. Another interesting finding was the differential distribution
of nonsynonymous (missense and truncating) and synonymous mutations
between inherited variants and somatic mutations in all types of
cancer (COAD, READ, AML, GLB, OVR). The nonsynonymous mutations
were in higher proportion in the tumor mtDNA (86%) in comparison
with the germline mtDNA (31%). The majority of tumors (94%) that
harbored somatic mtDNA mutations carried at least one nonsynonymous
mutation and only 37% bearer somatic nonsynonymous mutations.
Tumors that carried synonymous mutations, but no nonsynonymous
mutations accounted for only 3%. OVR (93%) exhibited the highest
proportion of nonsynonymous mtDNA somatic mutations compared to the
rest of the tumor types (COAD, 89%; READ, 80%; AML, 78%; and GLB,
80%).
The first direct causal connection between a mtDNA
mutation and cancer was reported by Petros et al (54) who identified the T8993G (L156R)
germline mutation in the ATP6 gene as heteroplasmic in a
patient with prostate cancer and demonstrated its tumorigenic
capacity in a nude mouse prostate cancer model. The T8993G mutation
in ATP6 is known to induce enhanced ROS production, which
may be the cause of enhanced tumorigenicity, since ROS are known to
affect transcription factors, signal transduction kinases,
regulatory phosphatases and angiogenic factors (44). Recently developed techniques, such
as next generation sequencing (NGS) have improved our knowledge of
the nature of mtDNA mutations thanks to the comparison of tumor and
adjacent normal tissue or blood in a wide variety of cancer types.
mtDNA variants commonly observed either as germline polymorphisms
(or germline mutations) or somatic mutations (cancer cells) include
point mutations, mono- or dinucleotide repeats, insertions,
deletions and mtDNA copy number variations (50,51,54).
The most common mtDNA polymorphisms which have been associated with
an increased risk of cancer are T16189C (D-Loop), G10398A (A114T at
ND3 gene) and the deletion ΔmtDNA4977 (55-59).
ΔmtDNA4977 is the most common deletion in the mitochondria, which
occurs between nucleotides 8,470 and 13,477 of the human mtDNA.
This deletion includes 5 tRNA genes, 4 genes encoding subunits of
NADH dehydrogenases, COIII and ATPase genes, creating a
smaller mtDNA molecule that leads to a decrease in ATP production
and to abnormal ROS generation (Fig.
2) (60).
In addition to the structural abnormalities in
mtDNA, fluctuations in mtDNA copy number are frequently described
in cancer. For instance, an elevated mtDNA content has been found
in primary head and neck squamous cell carcinoma, papillary thyroid
carcinoma and endometrial cancer, in contrast to gastric cancer,
which exhibits mtDNA depletion (61-63).
The mtDNA copy number in cancer may depend upon the specific site
of mutation associated with that cancer. Thus, an increase in mtDNA
copy number can occur as a compensatory response to mitochondrial
dysfunction or to mutations in nuclear genes indirectly involved in
controlling mtDNA copy number. Conversely, mutations in the D-loop
region, which control mtDNA replication, would be expected to
result in a decrease in copy number. In many cases, fewer copies of
mtDNA are accompanied by a decrease in the expression of
mitochondrial genes, suggesting a suppressed mitochondrial activity
in these tumor types. mtDNA impairment may result, not only in
mtDNA copy number alterations, but also in the acquisition of new
point mutations and deletions in mtDNA, damaged mitochondrial
function and changes in cell and tissue viability. Defects in the
components of the mtDNA repair machinery, which mediate mtDNA
replication and repair, may increase the accumulation of mtDNA
mutations, even though mitochondria have nDNA repair pathways, such
as base excision repair, miss-match repair and recombinational
repair (12,13). Indeed, Reznik et al
(64) found that the mtDNA copy
number is related to the incidence of key driver mutations that
cause cells to become cancerous (13,14).
Of note, although the mtDNA copy number can influence the level of
transcription of mtDNA genes, not all cancer types exhibit a
correlation between the expression of respiratory genes and mtDNA
copy number (64). Exogenous and
environmental factors, such as industrial by-products, ultraviolet
and ionizing radiation, tobacco smoke, chemicals, environmental
toxins and therapeutic drugs may also affect mtDNA expression
(13,14).
Haplogroups and cancer risk
Ancient mtDNA SNPs are grouped into haplogroups,
which are groups of haplotypes that share several polymorphisms
acquired during the expansion and migration of the human
population. Haplogroups reflect specific ancestral populations and
are the result of sequential accumulation of mitochondrial
mutations through maternal lineages; hence, they present
continent-specific distributions. The molecular mechanisms
underlying the association between mtDNA haplogroups and cancer
development remains unclear and controversial results between
populations have been reported (65-73).
However, specific mtDNA haplogroups have been associated with the
risk of prostate cancer, BC, CC, nasopharyngeal cancer, gastric
cancer, myelodysplastic syndromes (MS), acute lymphoblastic
leukemia, etc. (65-72,74).
For example, an over-representation of haplogroup I in Polish
cancer patients has been reported (75), haplogroup U has been shown to be
associated with an increased risk of developing renal and prostate
cancers (65), and haplogroup JT
has been shown to be associated with MS susceptibility in North
American caucasion subjects (70).
Additionally, haplogroups H, N and L have been shown to be
associated with the risk of developing pancreatic cancer, whereas
haplogroup K has been identified as protective for this type of
cancer in participants from the San Francisco Bay Area in
California (76). In China,
haplogroups D4a and D5a have been found to be associated with an
increased the risk of esophageal cancer, while the N and M
haplogroups have been reported as biomarkers for the good and poor
survival of patients with gastric cancer, respectively (67,69).
Another study, including 1,503 autopsy cases from Japan (76), reported an association between the
haplogroup M7b2, previously associated with leukemia (77) and an increased risk of developing
hematologic cancers (76).
Nevertheless, some of the most robust association data have been
reported for haplogroup T (M184) and the risk of CC in
European-American descendants, following the analysis of 2,453 CC
cases and 11,930 multi-ethnic controls (68).
Nuclear-encoded genes, mtDNA biogenesis
and function in cancer
While a number of studies on cancer have focused on
nDNA mutations, less research has been directed at determining the
influence of germline variants and somatic mutations in NEMtGs and
mitochondrial genes on organelle dysfunction and cancer
development. It is well known that mutations in nuclear-encoded
components of the mitochondrial translation machinery accumulate
non-repaired nDNA and mtDNA that may promote cellular
transformation and tumor development. Mutations in nuclear genes
responsible for mtDNA biosynthesis and integrity maintenance, such
as POLG, mtTFA, PGC-1, NRF-1,
NRF-2, isocitrate dehydrogenase (IDH), SDH
family members (SDHA, SDHB, SDHC and
SDHD), fumarate hydratase (FH), SIRT1-3,
POLG, TWINKLE and OPA1 are frequently
implicated in carcinogenic processes (74,78,79).
In fact, heterozygous germline mutations in SDHB,
SDHC, SDHD and SDHAF2 cause hereditary
paragangliomas and pheochromocytomas (80). POLG is known as the only DNA
polymerase that functions in human mitochondria. Due to its ability
to bypass lesions by translation synthesis, POLG is vital
for maintaining the genetic integrity of mtDNA. Mutations in this
gene can promote mtDNA mutations, the accumulation of unrepaired
lesions or can even block DNA replication in cancer (4,6-13).
Furthermore, IDH1 and IDH2, mitochondrial and TCA
cycle-related proteins, are frequently mutated in tumors, such as
glioblastoma. In fact, studies on GLB and AML have identified
recurrent missense mutations that impact specific residues of the
nuclear genes IDH1 and IDH2 (78,79).
Notably, it has been observed that tumors carrying IDH
mutations have few or no somatic mtDNA mutations (79).
The abnormal expression of several NEMtGs is a
common finding in all steps of cancer development and progression.
For example, TNF receptor-associated protein 1 (TRAP1) is a protein
that plays a role in the regulation of proteostasis, mitochondrial
apoptosis and suppression of ROS production in the mitochondria of
tumor cells, through regulation of folding and stability of
selective proteins. In fact, it has been reported that TRAP1
overexpression favors resistance to standard chemotherapeutic
agents (81). The overexpression
of genes involved in the activation of the UPRmt has
also been observed in metastatic cells. The UPRmt system
protects from protein misfolding and oxidation; however, it might
also maintain deleterious mtDNA mutations conferring advantages
that facilitate cancer cell metastasis (52,82).
5. Mitochondrial DNA variants in breast
cancer
The recognition of the importance of mtDNA mutations
as a relevant feature in BC development, metastasis and resistance
to treatment, has occurred over the past decade (48,83,84).
Both mtDNA germline variants and tumor mutations have been involved
in BC (Table II). As with other
types of cancer, the D-loop region (16024-576 nt), mainly the HVR1
and HVR2 regions, is an important mitochondrial mutation hotspot
(48,83,85).
This region has been reported to be ~60-fold more susceptible to
mutations than other regions of the mitochondrial genome (55,86-88).
The T16189C, C16207T, T16519C, T239C and A263G polymorphisms,
located in the D-loop region, are frequently associated with the
risk of BC (85-87). By contrast, the T16183C, C16223T,
T16362C, A73G and C150T polymorphisms, have been found to be
enriched in controls compared to BC patients, suggesting that they
could protect for this disease (86). Of these, T16189C is the most common
polymorphism that is associated with the risk of BC (75,89,90).
The T to C change in the wild-type (C5TC4) generates a long run of
C residues considered to be a source of enhanced instability
(55,85,87,88,91).
This abnormality often leads to heteroplasmic length variation of
the poly-C tract (>10 cytosines) and a correlation has been
observed between the poly-C variants and the mtDNA copy numbers
(59), as subjects who have an
interrupted poly-C carry higher mtDNA copy number, in contrast to
those with an uninterrupted poly-C, suggesting that the T16189C
variant may affect mtDNA replication (92).
| Table IICommon germline variants associated
with breast cancer. |
Table II
Common germline variants associated
with breast cancer.
| Population | Mutation | Gene | OR (CI 95%) | P-value | Refs. |
|---|
|
African-American | G10398A | ND3 | 1.6
(1.10-2.31) | 0.013 | (57) |
| Canadian | 310C-ins | D-loop | - | 0.0001 | (106) |
| Chinese | 2463A-del | RNR2 | 8.05
(1.023-63.34) | 0.022 | (66,87,96) |
| C6296T | COI | 4.464
(0.55-35.90) | 0.038 | |
| 6298T-del | COI | 4.464
(0.55-35.90) | 0.038 | |
| 8460-13477del | ATP8,
ATP6, CO3, TG, ND3, TR, | – | – | |
| ND4L,
ND4, TH, TS2, TL2, ND5 | | | |
| A8860 | ATP6 | 5.254
(0.65-41.92) | 0.021 | |
| A10397G | ND3 | 3.11
(1.07-9.06) | 0.03 | |
| G10398A | ND3 | 1.77
(1.00-3.14) | 0.05 | |
| 13237A-del | ND5 | 4.85
(0.60-38.86) | 0.037 | |
| T16189C | D-loop | 2.36
(1.14-4.88) | 0.019 | |
|
European-American | T3197C | RNR2 | 0.31
(0.13-0.75) | 0.004 | (85,98) |
| G9055A | ATP6 | 3.03
(1.63-5.63) | 0.0004 | |
| A10397G | ND3 | – | 0.03 | |
| G10398A | ND3 | 1.79
(1.14-2.81) | 0.01 | |
| G13708A | ND5 | 0.47
(0.24-0.92) | 0.002 | |
| T16519C | D-loop | 1.98
(1.25-3.12) | 0.003 | |
| T16519C | D-loop | 1.98
(1.25-3.12) | 0.003 | |
| Indian | G10398A | ND3 | 1.73
(1.13-2.66) | 0.01 | (99) |
| Italian | A153G | D-loop | 19 (1.8-201.9) | 0.009 | (107,113) |
| T195C | D-loop | 6 (1.12-31.99) | 0.04 | |
| G225A | D-loop | 12.7
(1.18-136.28) | 0.03 | |
| T226C | D-loop | 12.7
(1.18-136.28) | 0.03 | |
| G3918A | ND1 | – | – | |
| G11719A | ND4 | 13.2
(2.13-82.13) | 0.005 | |
| G11900A | ND4 | – | – | |
| T12344A | ND5 | – | – | |
| G13708A | ND5 | – | – | |
| G14869A | CYB | – | – | |
| C16093T | D-loop | – | – | |
| T16183C | D-loop | 12.7
(1.18-136.28) | 0.03 | |
| C16278T | D-loop | 7.3
(1.14-4.88) | 0.03 | |
| T16519C | D-loop | 21
(2.15-204.6) | 0.003 | |
| Malay | G10398A | ND3 | 2.29
(1.25-4.20) | 0.007 | (100) |
| Polish | A73G | D-loop | – | 0.001 | (75,94) |
| C150T | D-loop | – | 0.001 | |
| T152C | D-loop | – | 0.059 | |
| T239C | D-loop | – | 0.001 | |
| A263G | D-loop | – | 0.001 | |
| A4727G | ND2 | – | – | |
| G9947A | COIII | – | – | |
| A10044G | TG | – | – | |
| A10283G | ND3 | – | – | |
| T11233C | ND4 | – | – | |
| C11503T | ND4 | – | – | |
| T16183C | D-loop | – | 0.036 | |
| T16189C | D-loop | – | 0.004 | |
| C16192T | D-loop | – | 0.023 | |
| T16207C | D-loop | – | 0.023 | |
| C16223T | D-loop | – | 0.001 | |
| T16362C | D-loop | – | 0.001 | |
| T16519C | D-loop | – | 0.003 | |
| Spanish | G3010A | RNR2 | 0.73
(0.44-1.00) | 0.047 | (116) |
| T3197C | RNR2 | 2.72
(1.14-7.18) | 0.015 | |
| A12308G | TL2 | 0.66
(0.46-0.94) | 0.019 | |
| T16519C | D-loop | 0.74
(0.55-0.99) | 0.039 | |
| Taiwanese | mtDNA
depletion | All | – | 0.0008 | (125) |
Coding mitochondrial variants have also been
implicated in the risk of BC. On one hand, the A4727G, G9055A,
C6296A, G9947A, A8860G, A10044G, A10283G, T11233C and C11503T
variants increase the risk, and on the other hand, the T3197C and
G13708A SNPs decrease the risk of developing this neoplasia
(85,86,88,93-95).
In terms of deletions, the 2463delA, 6298delT and the ΔmtDNA4977
polymorphisms have been identified as susceptibility factors for
BC; however, ΔmtDNA4977 is the most commonly implicated in the
breast carcinogenesis process (88,96).
The mechanisms that promote tolerance to these deletions or
contribute to their propagation in the tumor cells are unknown
(82). Additionally, differential
contribution and interaction between germline variants have been
reported to increase BC susceptibility. For example, G10398A has
been shown to be associated with an increased risk of BC in
European-American, Polish and Malay populations, but not in Hindu
and African-American women (57,75,84,97-100). Furthermore, T4216C confers a risk
of BC only in combination with the G10398A germline variant
(97).
It is relevant to state that in addition to the
G6267A somatic mutation, the germline variants T14110C, G14207A and
D310 (from nucleotide positions 303 to 315 and interrupted by a T
in position 310) are very common in BC cases (96,101,102). The poly-cytidine stretch D310 is
the most common, reaching 92% of the tumor samples; thus, it has
been suggested as a potential starting point for the clonal
expansion of malignant cells and may be a clinical marker for
breast tumorigenesis (85,88,101,103-108). Alhomidi et al, in 2013,
supported this hypothesis, reporting that 32.5% of patients from
India with BC were D310 carriers (108).
Apart from somatic mtDNA mutations, studies have
identified mtDNA mutation (varying degrees, different sites, and
different types) in nearly 60% of BCs tumors (87,88,95,101); however, the question of whether
the mutations help drive the tumor or are bystander events remains
unanswered (76,83). McMahon and LaFramboise (83), in a study that included 99
normal-tumor paired BC samples, identified 142 somatic mutations;
however, many of these have been reported as germ-line variants.
G567A, G766A, G1333A, A2085G, G2621A, G5043 and G11346A were
identified in only one patient, as examples of somatic mutations
(described exclusively in breast tumor tissue) (83,109). Other matched blood-tumor mtDNA
sample studies have also shown that most somatic mutations are
singletons arising in a single patient and interestingly, are
present in a heteroplasmic state in the tumor, but not in mtDNA
from normal tissues. It has been suggested that many putative
somatic mutations are in undetected germline heteroplasmies that
have undergone clonal expansion in the tumor (46,83,110).
With regard to the impact of mtDNA content in BC, a
lower mtDNA content has been observed in BC tumors when compared to
their surrounding normal epithelium; however, these data are
inconsistent (111).
Haplogroups and breast cancer
The important role of haplogroups in different
human conditions such as aging, neurodegenerative diseases,
metabolic diseases, as well as BC (Table III) have also been explored.
Currently, it is considered that metabolism and sensitivity to
oxidative stress differs among mitochondrial haplogroups (112).
| Table IIIFrequency of haplogroups associated
with breast cancer. |
Table III
Frequency of haplogroups associated
with breast cancer.
| Haplo-group | SNPs | Population | N
Controls/cases | Frequency (%) | Effect | OR (CI 95%) | P-value | Refs. |
|---|
| D5 | C150T, T1107,
A5301G, A10397G | Chinese | 104/114 | 12.9 | Increased risk | 3.11
(1.07-9.06) | 0.030 | (66) |
| K | A10550G, T11299C,
T14798C, T16224C, T16311C | European-
American | 156/260 | 18.6 | Increased risk of
hereditary cancer | 3.03
(1.63-5.63) | 0.0004 | (85) |
| H | G2706A, T7028C | Polish | 44/100 | 36 | Protection | 0.636
(0.51-0.74) | 0.019 | (75) |
| | Italian | 10/10 | 40 | Increased risk of
BRCA2 mutation carriers | – | 0.05 | (113) |
| I | T10034C,
G16129A | Polish | 44/100 | 14 | Increased risk | – | 0.017 | (75) |
| M | T489C, C10400T,
T14783C, G15043A | Chinese | 104/114 | 66.3 | Increased risk.
Protection against metastasis | 1.77
(1.03-3.07) | 0.040 | (62) |
| N | G8701A, C9540T,
G10398A, C10873T, A15301G | Chinese | 34/70 | 47 | Risk of
metastasis | 0.39
(0.17-0.94) | 0.036 | (66) |
| U | A11467G, A12308G,
G12372A | European-
American | 156/260 | 7.7 | Decreased risk of
hereditary cancer | 0.37
(0.19-0.73) | 0.0023 | (85) |
| X | T6221C, C6371T,
A13966, T14470C, T16189C, C16278T | Italian | 10/10 | 60 | Increased risk of
BRCA1 mutation carriers | – | 0.04 | (113) |
| T1a1 | T9899C, A16163G,
C16186T, T152C, T195C | Multi-
national | 3989/3698 | 9.3 | Decreased risk of
BRCA2 mutations carriers | 0.62
(0.40-0.95) | 0.03 | (114) |
Studies performed on the Polish population have
demonstrated the overrepresentation and under-representation of
haplogroups I and H, respectively, in patients with BC, in
comparison with healthy groups. These data suggest that the
presence of different polymorphisms given in the haplogroup may be
fundamental for the carcinogenic process (75). Furthermore, an association between
haplogroups and BRCA1 and BRCA2 mutations has been
reported. Tommasi et al (113) found that haplogroups X and H were
significantly more frequent in subjects bearing mutations in the
BRCA1 and BRCA2 genes, respectively. Additionally, it
has recently been observed that haplogroup T1a1 may modify the
individual risk of acquiring the disease in BRCA2 mutation
carriers (114). Likewise, Rao
et al (115), studying 92
triple-negative BC (TNBC) patients of African-American (n=31),
caucasion (n=31) and Hispanic (n=30) origin, found that Nigerian
(29%), Cameroon (26%), Sierra Leone (16%) and Guinea-Bissau (13%)
haplogroups were highly prevalent in African-American patients;
while the H (29%), U (23%), K (13%), V (7%), and J (7%) haplogroups
accounted for 79% of all haplogroups identified in
caucasian-derived populations. As has been previously reported,
haplogroups A (34%), B (17%) and C (13%) are the most frequent in
Hispanic patients. Notably, the authors found that non-caucasian
haplogroups represent almost 64% of TNBC patients, which is in
accordance with data showing that TNBC is more common in African-
and Hispanic-derived populations (115). Although some of these studies
have provided controversial findings and statistically underpowered
results, the European haplogroup N, defined by the G10398A germline
variant, has been reported as a risk factor for BC, particularly in
women of African-American and Indian origin (96,116-118). Additionally, Bai et al
(85), studying a caucasian
population, found that haplogroup K (G10398A and T16189C) increased
the risk of BC, whereas haplogroup U conferred protection against
this disease. Moreover, Darvishi et al (99) re-analyzed approximately 1,000
mitochondrial DNA sequences and found an association between the
distribution of haplogroup N and the incidence of sporadic BC. This
was corroborated through a case control study, where they also
observed association of the A10398C germline variant with sporadic
BC.
Mutations in nuclear-encoded genes that
participate in mtDNA biogenesis and BC
It is well known that mutations in nuclear encoded
genes, such as BRCA1, BRCA2, TP53,
PTEN, CHEK2 and CHEK1, are commonly associated
with a high risk of developing BC. Moreover, as the majority of
mitochondrial proteins are nuclear encoded and post-translationally
imported in the mitochondria, the nuclear/mitochondrial connection
in BC is also altered. However, the most notable association
between the mitochondrial function and nuclear encoded genes
relates to the OXPHOS system since 79 of its components are encoded
by nDNA. In fact, none of the subunits of complex II are encoded by
mtDNA (74,78,119). Hossein and Houshmand (120) used algorithms to determine that
the mitochondrial deletion ΔmtDNA4977 in association with
alterations in nuclear genes (such as BRCA, ER and
TP53 genes) led to a phenotype of premature aging and BC
(95). Moreover, Jandova et
al (121) reported that the
copy number of mtDNA can produce differential expression levels of
matrix metalloproteinase 9 (MMP9) and collagen, type I, a1
(Colla), which are capable of triggering the malignancy.
Notably, MMP9 has been shown to be involved in several types
of cancer and metastasis. Additionally, expression analyses have
also revealed an association between the expression of NEMtGs and
BC subtypes. Furthermore, mitochondrial membrane respiratory chain
complex I and IV and ATP synthesis were reduced, in both epithelial
and stromal cancer cells compared to normal breast cells.
Additionally, MRPL12, POLG and RNASEH1 were
upregulated in cancerous stromal cells (122).
mtDNA variants and BC subtypes and
survival
Few studies have been conducted to clarify whether
there is a correlation between mtDNA germline variants or somatic
mutations and BC subtypes. Thus far, there has been no direct
association of mutations with the intrinsic subtypes of this
neoplasia; however, Lin et al (123) observed that the D310 germline
variant was associated with an advanced stage of the tumor and with
negative Her2/neu expression. Tommasi et al (113) found that the T16126C germline
variant was more frequent, but not exclusive, in estrogen- and
progesterone receptor-negative tumors. Nevertheless, the authors
detected that C150T was exclusive to patients with hereditary BC,
as was T16519C, which was found in 90% of individuals carrying
mutations in BRCA1. Blein et al (124) sequenced blood mtDNA from 436
women, with a positive familial BC history, but negative for
BRCA1/2 pathogenic mutations, diagnosed with BC, and found
an enrichment of mutations in MT-ATP6 (86.5 variants/Mb) and
MT-CYB (84.1 variants/Mb), but no association between the
rate or type of mutations. Moreover, McMahon and LaFramboise
(83) observed that alterations in
heteroplasmy levels were more significant in HER2-positive
tumors than in HER2-negative tumors.
The impact of the mtDNA mutation rate and mtDNA
content and their correlation with prognosis and survival have also
been investigated. A higher mutational burden and a low copy number
of mtDNA have been associated with a worse overall survival, in
contrast to patients carrying a low mutation rate or high mtDNA
content; however, these results remain inconclusive (111,125).
6. Mitochondria DNA mutations and response
to therapy in breast cancer
Widespread attention has been paid to mitochondria
mutations. This is a not a recent phenomenon, and it fits into the
broader historical interest in studying the size, evolution and
function of genomes. The first complete human mtDNA sequenced was
in 1981; currently, >3,000 complete mitochondrial genomes have
been sequenced (109). The unique
structure and function of mitochondria make them an excellent model
system to identify and answer biological and medical issues where
other approaches exhibit technical difficulties. As the
mitochondria control both energy metabolism and susceptibility
towards apoptotic cell death, mtDNA mutations have been suggested
to play important roles in breast carcinogenesis and
drug-resistance. Thus, the landscape of driving tumor mtDNA
mutations could be used for the design of clinical trials and
development of new anti-neoplasic therapies. In this regard, the
role of mitochondrial dysfunction and cancer therapy has been
widely investigated. Recently, Farnie et al (126) demonstrated that tumors with high
mitochondrial mass (mito-high) were specifically enriched in the
number of cancer stem-like cells (CSCs), which are involved in
tumor recurrence and metastasis. Furthermore, these mito-high
breast CSCs exhibited resistance to DNA damage following
antineoplastic treatment. Based on the 'Endo-symbiotic Theory of
Mitochondrial Evolution', it is suggested that antibiotics against
mitochondria could be used to modify mitochondrial mass within
CSCs. In fact, the FDA has already approved drugs/antibiotics that
target mitochondria to eradicate CSC activity (metformin and
pyrvinium pamoate, salinomycin) as well as inhibitors of
mitochondrial biogenesis and mitochondrial translation
(erythromycins, tetracyclines and glycylcyclines) (126,127). Notably, doxycycline and
azithromycin have already exhibited significant efficacy in
treatment-resistant cancer patients with lymphoma and non-small
cell lung tumors, respectively (128,129).
The steep rise in interest in the study of
mitochondrial mutations is also due to the fact that mtDNA
mutations play a role in many other human diseases, opening a
paradigm shift that suggests that mitochondrial mutations will
contribute to our understanding of tumor biology in the mammary
gland. In fact, Teixeira et al (130) found that the mitochondria were
also involved in stem cell differentiation, showing that blocking
any of the 13 key proteins linked to ATP synthase disrupted or
stalled egg development in fruit flies. Certainly, the recognition
of the widespread applicability of studying the role of
mitochondria and the biological implications of mtDNA may will
strongly improve the development of new means with which to treat
human common diseases, including BC.
7. Conclusions
As mtDNA mutations and mtDNA copy number have been
found to play important roles in breast carcinogenesis, the
landscape of driving tumor mtDNA mutations may be used for the
design of clinical trials and the development of novel
anti-neoplastic therapies. Moreover, the suggestion that mtDNA copy
number is associated with the severity of the disease could
identify cases with better or worse survival. It is clear that NGS
technologies have resulted in an unprecedented ability to
understand mtDNA structure; however, the challenge is to determine
whether and which mutations or germline variants carry biological
significance. In addition, meriting further investigation are other
aspects of mitochondrial genomes which undoubtedly are involved in
human diseases, these aspects include chromosome structure,
transcriptional and translational architecture, and modes of repair
and replication.
Abbreviations:
|
mtDNA
|
mitochondria DNA
|
|
BC
|
breast cancer
|
|
OXPHOS
|
oxidative phosphorylation
|
|
NEMtG
|
nuclear-encoded mitochondrial
genes
|
Acknowledgments
Not applicable.
References
|
1
|
World Cancer Research Fund International:
BC Statistics: http://www.wcrf.org/int/cancer-facts-figures/data-specific-cancers/breast-cancer-statistics
Accessed January 19, 2018.
|
|
2
|
Breastcancer.org: U.S. BC statistics:
http://www.breastcancer.org/symptoms/understand_bc/statistics.
Accessed January 19, 2018.
|
|
3
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Havas KM, Milchevskaya V, Radic K, Alladin
A, Kafkia E, Garcia M, Stolte J, Klaus B, Rotmensz N, Gibson TJ, et
al: Metabolic shifts in residual breast cancer drive tumor
recurrence. J Clin Invest. 127:2091–2105. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu W and Zhao S: Metabolic changes in
cancer: Beyond the Warburg effect. Acta Biochim Biophys Sin
(Shanghai). 45:18–26. 2013. View Article : Google Scholar
|
|
7
|
Bardella C, Pollard PJ and Tomlinson I:
SDH mutations in cancer. Biochim Biophys Acta. 1807:1432–1443.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bogenhagen DF: Mitochondrial DNA nucleoid
structure. Biochim Biophys Acta. 1819:914–920. 2012. View Article : Google Scholar
|
|
9
|
Andrews RM, Kubacka I, Chinnery PF,
Lightowlers RN, Turnbull DM and Howell N: Reanaly-sis and revision
of the Cambridge reference sequence for human mitochondrial DNA.
Nat Genet. 23:1471999. View
Article : Google Scholar
|
|
10
|
Wallace DC and Chalkia D: Mitochondrial
DNA genetics and the heteroplasmy conundrum in evolution and
disease. Cold Spring Harb Perspect Biol. 5:a0212202013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stewart JB and Chinnery PF: The dynamics
of mitochondrial DNA heteroplasmy: Implications for human health
and disease. Nat Rev Genet. 16:530–542. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Akhmedov AT and Marín-García J:
Mitochondrial DNA maintenance: An appraisal. Mol Cell Biochem.
409:283–305. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yakes FM and Van Houten B: Mitochondrial
DNA damage is more extensive and persists longer than nuclear DNA
damage in human cells following oxidative stress. Proc Natl Acad
Sci USA. 94:514–519. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Egea PF, Stroud RM and Walter P: Targeting
proteins to membranes: Structure of the signal recognition
particle. Curr Opin Struct Biol. 15:213–220. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Baker MJ, Frazier AE, Gulbis JM and Ryan
MT: Mitochondrial protein-import machinery: Correlating structure
with function. Trends Cell Biol. 17:456–464. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Neupert W and Herrmann JM: Translocation
of proteins into mitochondria. Annu Rev Biochem. 76:723–749. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lill R and Mühlenhoff U: Maturation of
iron-sulfur proteins in eukaryotes: Mechanisms, connected
processes, and diseases. Annu Rev Biochem. 77:669–700. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Virbasius JV and Scarpulla RC: Activation
of the human mitochondrial transcription factor A gene by nuclear
respiratory factors: A potential regulatory link between nuclear
and mitochondrial gene expression in organelle biogenesis. Proc
Natl Acad Sci USA. 91:1309–1313. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hendrickson SL, Lautenberger JA, Chinn LW,
Malasky M, Sezgin E, Kingsley LA, Goedert JJ, Kirk GD, Gomperts ED,
Buchbinder SP, et al: Genetic variants in nuclear-encoded
mitochondrial genes influence AIDS progression. PLoS One.
5:e128622010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Scarpulla RC: Nucleus-encoded regulators
of mitochondrial function: Integration of respiratory chain
expression, nutrient sensing and metabolic stress. Biochim Biophys
Acta. 1819:1088–1097. 2012. View Article : Google Scholar :
|
|
21
|
Kasashima K and Endo H: Interaction of
human mitochondrial transcription factor A in mitochondria: Its
involvement in the dynamics of mitochondrial DNA nucleoids. Genes
Cells. 20:1017–1027. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee HC and Wei YH: Mitochondrial
biogenesis and mitochondrial DNA maintenance of mammalian cells
under oxidative stress. Int J Biochem Cell Biol. 37:822–834. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hance N, Ekstrand MI and Trifunovic A:
Mitochondrial DNA polymerase gamma is essential for mammalian
embryogenesis. Hum Mol Genet. 14:1775–1783. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mokranjac D and Neupert W: Cell biology:
Architecture of a protein entry gate. Nature. 528:201–202. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fan AC, Bhangoo MK and Young JC: Hsp90
functions in the targeting and outer membrane translocation steps
of Tom70-mediated mitochondrial import. J Biol Chem.
281:33313–33324. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
MacKenzie JA and Payne RM: Mitochondrial
protein import and human health and disease. Bi-ochim Biophys Acta.
1772:509–523. 2007.
|
|
27
|
Bandiera S, Rüberg S, Girard M, Cagnard N,
Hanein S, Chrétien D, Munnich A, Lyonnet S and Henrion-Caude A:
Nuclear outsourcing of RNA interference components to human
mitochondria. PLoS One. 6:e207462011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Stefano GB and Kream RM: Mitochondrial DNA
heteroplasmy in human health and disease. Biomed Rep. 4:259–262.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hudson G, Gomez-Duran A, Wilson IJ and
Chinnery PF: Recent mitochondrial DNA mutations increase the risk
of developing common late-onset human diseases. PLoS Genet.
10:e10043692014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hudson G, Carelli V, Spruijt L, Gerards M,
Mowbray C, Achilli A, Pyle A, Elson J, Howell N, La Morgia C, et
al: Clinical expression of Leber hereditary optic neuropathy is
affected by the mitochondrial DNA-haplogroup background. Am J Hum
Genet. 81:228–233. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Soini HK, Moilanen JS, Vilmi-Kerälä T,
Finnilä S and Majamaa K: Mitochondrial DNA variant m.15218A >G
in Finnish epilepsy patients who have maternal relatives with
epilepsy, sensorineural hearing impairment or diabetes mellitus.
BMC Med Genet. 14:732013. View Article : Google Scholar
|
|
32
|
Fragaki K, Procaccio V, Bannwarth S, Serre
V, O'Hearn S, Potluri P, Augé G, Casagrande F, Caruba C, Lambert
JC, et al: A neonatal polyvisceral failure linked to a de novo
homoplasmic mutation in the mitochondrially encoded cytochrome b
gene. Mitochondrion. 9:346–352. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ghezzi D, Marelli C, Achilli A, Goldwurm
S, Pezzoli G, Barone P, Pellecchia MT, Stanzione P, Brusa L,
Bentivoglio AR, et al: Mitochondrial DNA haplogroup K is associated
with a lower risk of Parkinson's disease in Italians. Eur J Hum
Genet. 13:748–752. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huerta C, Castro MG, Coto E, Blázquez M,
Ribacoba R, Guisasola LM, Salvador C, Martínez C, Lahoz CH and
Alvarez V: Mitochondrial DNA polymorphisms and risk of Parkinson's
disease in Spanish population. J Neurol Sci. 236:49–54. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Galmiche L, Serre V, Beinat M, Assouline
Z, Lebre AS, Chretien D, Nietschke P, Benes V, Boddaert N, Sidi D,
et al: Exome sequencing identifies MRPL3 mutation in mitochondrial
cardiomyopathy. Hum Mutat. 32:1225–1231. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Götz A, Tyynismaa H, Euro L, Ellonen P,
Hyötyläinen T, Ojala T, Hämäläinen RH, Tommiska J, Raivio T, Oresic
M, et al: Exome sequencing identifies mitochondrial alanyl-tRNA
synthetase mutations in infantile mitochondrial cardiomyopathy. Am
J Hum Genet. 88:635–642. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bayat V, Thiffault I, Jaiswal M, Tétreault
M, Donti T, Sasarman F, Bernard G, Demers-Lamarche J, Dicaire MJ,
Mathieu J, et al: Mutations in the mitochondrial methionyl-tRNA
synthetase cause a neurodegenerative phenotype in flies and a
recessive ataxia (ARSAL) in humans. PLoS Biol. 10:e10012882012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Elo JM, Yadavalli SS, Euro L, Isohanni P,
Götz A, Carroll CJ, Valanne L, Alkuraya FS, Uusimaa J, Paetau A, et
al: Mitochondrial phenylalanyl-tRNA synthetase mutations underlie
fatal infantile Alpers encephalopathy. Hum Mol Genet. 21:4521–4529.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Haack TB, Kopajtich R, Freisinger P,
Wieland T, Rorbach J, Nicholls TJ, Baruffini E, Walther A,
Danhauser K, Zimmermann FA, et al: ELAC2 mutations cause a
mitochondrial RNA processing defect associated with hypertrophic
cardiomyopathy. Am J Hum Genet. 93:211–223. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Soiferman D, Ayalon O, Weissman S and
Saada A: The effect of small molecules on nuclear-encoded
translation diseases. Biochimie. 100:184–191. 2014. View Article : Google Scholar
|
|
41
|
Powell CA, Nicholls TJ and Minczuk M:
Nuclear-encoded factors involved in post-transcriptional processing
and modification of mitochondrial tRNAs in human disease. Front
Genet. 6:792015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Takakubo F, Cartwright P, Hoogenraad N,
Thorburn DR, Collins F, Lithgow T and Dahl HH: An amino acid
substitution in the pyruvate dehydrogenase E1 alpha gene, affecting
mitochondrial import of the precursor protein. Am J Hum Genet.
57:772–780. 1995.PubMed/NCBI
|
|
43
|
Weraarpachai W, Sasarman F, Nishimura T,
Antonicka H, Auré K, Rötig A, Lombès A and Shoubridge EA: Mutations
in C12orf62, a factor that couples COX I synthesis with cytochrome
c oxidase assembly, cause fatal neonatal lactic acidosis. Am J Hum
Genet. 90:142–151. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Larman TC, DePalma SR, Hadjipanayis AG,
Protopopov A, Zhang J, Gabriel SB, Chin L, Seidman CE, Kucherlapati
R and Seidman JG; Cancer Genome Atlas Research Network: Spectrum of
somatic mitochondrial mutations in five cancers. Proc Natl Acad Sci
USA. 109:14087–14091. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kenny TC, Hart P, Ragazzi M, Sersinghe M,
Chipuk J, Sagar MA, Eliceiri KW, LaFramboise T, Grandhi S, Santos
J, et al: Selected mitochondrial DNA landscapes activate the SIRT3
axis of the UPRmt to promote metastasis. Oncogene. 36:4393–4404.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ahn EH, Hirohata K, Kohrn BF, Fox EJ,
Chang CC and Loeb LA: Detection of ultrarare mitochondrial
mutations in breast stem cells by duplex sequencing. PLoS One.
10:e01362162015. View Article : Google Scholar
|
|
47
|
Polyak K, Li Y, Zhu H, Lengauer C, Willson
JK, Markowitz SD, Trush MA, Kinzler KW and Vogelstein B: Somatic
mutations of the mitochondrial genome in human colorectal tumours.
Nat Genet. 20:291–293. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yadav N and Chandra D: Mitochondrial DNA
mutations and breast tumorigenesis. Biochim Bi-ophys Acta.
1836:336–344. 2013.
|
|
49
|
Pedersen PL, Mathupala S, Rempel A,
Geschwind JF and Ko YH: Mitochondrial bound type II hexokinase: A
key player in the growth and survival of many cancers and an ideal
prospect for therapeutic intervention. Biochim Biophys Acta.
1555:14–20. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Brandon M, Baldi P and Wallace DC:
Mitochondrial mutations in cancer. Oncogene. 25:4647–4662. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kirches E: Mitochondrial and nuclear genes
of mitochondrial components in cancer. Curr Genomics. 10:281–293.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kenny TC and Germain D: mtDNA, metastasis,
and the mitochondrial unfolded protein response (UPRmt). Front Cell
Dev Biol. 5:372017. View Article : Google Scholar :
|
|
53
|
Habano W, Sugai T, Yoshida T and Nakamura
S: Mitochondrial gene mutation, but not large-scale deletion, is a
feature of colorectal carcinomas with mitochondrial microsatellite
instability. Int J Cancer. 83:625–629. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Petros JA, Baumann AK, Ruiz-Pesini E, Amin
MB, Sun CQ, Hall J, Lim S, Issa MM, Flanders WD, Hosseini SH, et
al: mtDNA mutations increase tumorigenicity in prostate cancer.
Proc Natl Acad Sci USA. 102:719–724. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yu M, Shi Y, Zhang F, Zhou Y, Yang Y, Wei
X, Zhang L and Niu R: Sequence variations of mitochondrial DNA
D-loop region are highly frequent events in familial breast cancer.
J Biomed Sci. 15:535–543. 2008. View Article : Google Scholar
|
|
56
|
Liu VW, Wang Y, Yang HJ, Tsang PCK, Ng TY,
Wong LC, Nagley P and Ngan HY: Mitochondrial DNA variant
16189T>C is associated with susceptibility to endometrial
cancer. Hum Mutat. 22:173–174. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Canter JA, Kallianpur AR, Parl FF and
Millikan RC: Mitochondrial DNA G10398A polymorphism and invasive
breast cancer in African-American women. Cancer Res. 65:8028–8033.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhang J, Asin-Cayuela J, Fish J, Michikawa
Y, Bonafe M, Olivieri F, Passarino G, De Benedictis G, Franceschi C
and Attardi G: Strikingly higher frequency in centenarians and
twins of mtDNA mutation causing remodeling of replication origin in
leukocytes. Proc Natl Acad Sci USA. 100:1116–1121. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Liou CW, Lin TK, Chen JB, Tiao MM, Weng
SW, Chen SD, Chuang YC, Chuang JH and Wang PW: Association between
a common mitochondrial DNA D-loop polycytosine variant and
alteration of mitochondrial copy number in human peripheral blood
cells. J Med Genet. 47:723–728. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Peng TI, Yu PR, Chen JY, Wang HL, Wu HY,
Wei YH and Jou MJ: Visualizing common deletion of mitochondrial
DNA-augmented mitochondrial reactive oxygen species generation and
apoptosis upon oxidative stress. Biochim Biophys Acta.
1762:241–255. 2006. View Article : Google Scholar
|
|
61
|
Su X, Wang W, Ruan G, Liang M, Zheng J,
Chen Y, Wu H, Fahey TJ III, Guan M and Teng L: A comprehensive
characterization of mitochondrial genome in papillary thyroid
cancer. Int J Mol Sci. 17:15942016. View Article : Google Scholar :
|
|
62
|
Wang Y, Liu VW, Xue WC, Tsang PC, Cheung
AN and Ngan HY: The increase of mitochondrial DNA content in
endometrial adenocarcinoma cells: A quantitative study using
laser-captured microdissected tissues. Gynecol Oncol. 98:104–110.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Singh B, Owens KM, Bajpai P, Desouki MM,
Srinivasasainagendra V, Tiwari HK and Singh KK: Mitochondrial DNA
polymerase POLG1 disease mutations and germline variants promote
tumorigenic properties. PLoS One. 10:e01398462015. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Reznik E, Miller ML, Şenbabaoğlu Y, Riaz
N, Sarungbam J, Tickoo SK, Al-Ahmadie HA, Lee W, Seshan VE, Hakimi
AA, et al: Mitochondrial DNA copy number variation across human
cancers. eLife. 5:e107692016. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Booker LM, Habermacher GM, Jessie BC, Sun
QC, Baumann AK, Amin M, Lim SD, Fernan-dez-Golarz C, Lyles RH,
Brown MD, et al: North American white mitochondrial haplogroups in
prostate and renal cancer. J Urol. 175:468–473. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Fang H, Shen L, Chen T, He J, Ding Z, Wei
J, Qu J, Chen G, Lu J and Bai Y: Cancer type-specific modulation of
mitochondrial haplogroups in breast, colorectal and thyroid cancer.
BMC Cancer. 10:4212010. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Hu SP, Du JP, Li DR and Yao YG:
Mitochondrial DNA haplogroup confers genetic susceptibility to
nasopharyngeal carcinoma in Chaoshanese from Guangdong, China. PLoS
One. 9:e877952014. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Li Y, Beckman KB, Caberto C, Kazma R,
Lum-Jones A, Haiman CA, Le Marchand L, Stram DO, Saxena R and Cheng
I: Association of genes, pathways, and haplogroups of the
mitochondrial genome with the risk of colorectal cancer: The
multiethnic Cohort. PLoS One. 10:e01367962015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Jiang J, Zhao JH, Wang XL, Di JI, Liu ZB,
Li GY, Wang MZ, Li Y, Chen R and Ge RL: Analysis of mitochondrial
DNA in Tibetan gastric cancer patients at high altitude. Mol Clin
Oncol. 3:875–879. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Poynter JN, Richardson M, Langer E, Hooten
AJ, Roesler M, Hirsch B, Nguyen PL, Cioc A, Warlick E and Ross JA:
Association between mitochondrial DNA haplogroup and
myelodysplastic syndromes. Genes Chromosomes Cancer. 55:688–693.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Singh KK and Kulawiec M: Mitochondrial DNA
polymorphism and risk of cancer. Methods Mol Biol. 471:291–303.
2009. View Article : Google Scholar
|
|
72
|
Yacoub HA, Mahmoud WM, El-Baz HA, Eid OM,
El-Fayoumi RI, Mahmoud MM, Harakeh S and Abuzinadah OH: New
haplotypes of the ATP synthase subunit 6 gene of mitochondrial DNA
are associated with acute lymphoblastic leukemia in Saudi Arabia.
Asian Pac J Cancer Prev. 15:10433–10438. 2014. View Article : Google Scholar
|
|
73
|
Cano D, Gomez CF, Ospina N, Cajigas JA,
Groot H, Andrade RE and Torres MM: Mitochondrial DNA haplogroups
and susceptibility to prostate cancer in a colombian population.
ISRN Oncol. 2014:5306752014.PubMed/NCBI
|
|
74
|
Yu FY, Xu Q, Wu DD, Lau AT and Xu YM: The
Prognostic and Clinicopathological roles of Sirtuin-3 in various
cancers. PLoS One. 11:e01598012016. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Czarnecka AM, Krawczyk T, Plak K, Klemba
A, Zdrozny M, Arnold RS, Kofler B, Golik P, Szybinska A, Lubinski
J, et al: Mitochondrial genotype and breast cancer predisposition.
Oncol Rep. 24:1521–1534. 2010.PubMed/NCBI
|
|
76
|
Verma M, Naviaux RK, Tanaka M, Kumar D,
Franceschi C and Singh KK: Meeting report: Mitochondrial DNA and
cancer epidemiology. Cancer Res. 67:437–439. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
van Gisbergen MW, Voets AM, Starmans MH,
de Coo IF, Yadak R, Hoffmann RF, Boutros PC, Smeets HJ, Dubois L
and Lambin P: How do changes in the mtDNA and mitochondrial
dysfunction influence cancer and cancer therapy? Challenges,
opportunities and models. Mutat Res Rev Mutat Res. 764:16–30. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Parker SJ and Metallo CM: Metabolic
consequences of oncogenic IDH mutations. Pharmacol Ther. 152:54–62.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Reitman ZJ, Jin G, Karoly ED, Spasojevic
I, Yang J, Kinzler KW, He Y, Bigner DD, Vogelstein B and Yan H:
Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations
on the cellular metabolome. Proc Natl Acad Sci USA. 108:3270–3275.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Burnichon N, Brière JJ, Libé R, Vescovo L,
Rivière J, Tissier F, Jouanno E, Jeunemaitre X, Bénit P, Tzagoloff
A, et al: SDHA is a tumor suppressor gene causing paraganglioma.
Hum Mol Genet. 19:3011–3020. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Zhang B, Wang J, Huang Z, Wei P, Liu Y,
Hao J, Zhao L, Zhang F, Tu Y and Wei T: Aberrantly upregulated
TRAP1 is required for tumorigenesis of breast cancer. Oncotarget.
6:44495–44508. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Lin YF, Schulz AM, Pellegrino MW, Lu Y,
Shaham S and Haynes CM: Maintenance and propagation of a
deleterious mitochondrial genome by the mitochondrial unfolded
protein response. Nature. 533:416–419. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
McMahon S and LaFramboise T: Mutational
patterns in the breast cancer mitochondrial genome, with clinical
correlates. Carcinogenesis. 35:1046–1054. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Yu Y, Lv F, Lin H, Qian G, Jiang YS, Pang
LX, Wang YP, Wang XF, Kang YM, Li CB, et al: Mitochondrial ND3
G10398A mutation: A biomarker for breast cancer. Genet Mol Res.
14:17426–17431. 2015. View Article : Google Scholar
|
|
85
|
Bai RK, Leal SM, Covarrubias D, Liu A and
Wong LJ: Mitochondrial genetic background modifies breast cancer
risk. Cancer Res. 67:4687–4694. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Zhu W, Qin W, Bradley P, Wessel A, Puckett
CL and Sauter ER: Mitochondrial DNA mutations in breast cancer
tissue and in matched nipple aspirate fluid. Carcinogenesis.
26:145–152. 2005. View Article : Google Scholar
|
|
87
|
Wang Y, Liu VW, Tsang PC, Chiu PM, Cheung
AN, Khoo US, Nagley P and Ngan HY: Microsatellite instability in
mitochondrial genome of common female cancers. Int J Gynecol
Cancer. 16(Suppl 1): 259–266. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Bianchi MS, Bianchi NO and Bailliet G:
Mitochondrial DNA mutations in normal and tumor tissues from breast
cancer patients. Cytogenet Cell Genet. 71:99–103. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Shakhssalim N, Houshmand M, Kamalidehghan
B, Faraji A, Sarhangnejad R, Dadgar S, Mobaraki M, Rosli R and
Sanati MH: The mitochondrial C16069T polymorphism, not
mitochondrial D310 (D-loop) mononucleotide sequence variations, is
associated with bladder cancer. Cancer Cell Int. 13:1202013.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Tipirisetti NR, Govatati S, Pullari P,
Malempati S, Thupurani MK, Perugu S, Guruvaiah P, Rao KL, Digumarti
RR, Nallanchakravarthula V, et al: Mitochondrial control region
alterations and breast cancer risk: A study in South Indian
population. PLoS One. 9:e853632014. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Fendt L, Niederstätter H, Huber G, Zelger
B, Dünser M, Seifarth C, Röck A, Schäfer G, Klock-er H and Parson
W: Accumulation of mutations over the entire mitochondrial genome
of breast cancer cells obtained by tissue microdis-section. Breast
Cancer Res Treat. 128:327–336. 2011. View Article : Google Scholar
|
|
92
|
Ebner S, Lang R, Mueller EE, Eder W,
Oeller M, Moser A, Koller J, Paulweber B, Mayr JA, Sperl W, et al:
Mitochondrial haplogroups, control region polymorphisms and
malignant melanoma: A study in middle European Caucasians. PLoS
One. 6:e271922011. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Wang CY, Wang HW, Yao YG, Kong QP and
Zhang YP: Somatic mutations of mitochondrial genome in early stage
breast cancer. Int J Cancer. 121:1253–1256. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Czarnecka AM, Klemba A, Krawczyk T,
Zdrozny M, Arnold RS, Bartnik E and Petros JA: Mitochondrial
NADH-dehydrogenase polymorphisms as sporadic breast cancer risk
factor. Oncol Rep. 23:531–535. 2010.PubMed/NCBI
|
|
95
|
Weigl S, Paradiso A and Tommasi S:
Mitochondria and familial predisposition to breast cancer. Curr
Genomics. 14:195–203. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Li L, Chen L, Li J, Zhang W, Liao Y, Chen
J and Sun Z: Correlational study on mitochondrial DNA mutations as
potential risk factors in breast cancer. Oncotarget. 7:31270–31283.
2016.PubMed/NCBI
|
|
97
|
Rohan TE, Wong LJ, Wang T, Haines J and
Kabat GC: Do alterations in mitochondrial DNA play a role in breast
carcinogenesis? J Oncol. 2010:6043042010. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Covarrubias D, Bai RK, Wong LJ and Leal
SM: Mitochondrial DNA variant interactions modify breast cancer
risk. J Hum Genet. 53:924–928. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Darvishi K, Sharma S, Bhat AK, Rai E and
Bamezai RN: Mitochondrial DNA G10398A polymorphism imparts maternal
Haplogroup N a risk for breast and esophageal cancer. Cancer Lett.
249:249–255. 2007. View Article : Google Scholar
|
|
100
|
Tengku Baharudin N, Jaafar H and Zainuddin
Z: Association of mitochondrial DNA 10398 polymorphism in invasive
breast cancer in malay population of peninsular malaysia. Malays J
Med Sci. 19:36–42. 2012.PubMed/NCBI
|
|
101
|
Tan DJ, Bai RK and Wong LJ: Comprehensive
scanning of somatic mitochondrial DNA mutations in breast cancer.
Cancer Res. 62:972–976. 2002.PubMed/NCBI
|
|
102
|
Gallardo ME, Moreno-Loshuertos R, López C,
Casqueiro M, Silva J, Bonilla F, Rodríguez de Córdoba S and
Enríquez JA: m.6267G>A: a recurrent mutation in the human
mitochondrial DNA that reduces cytochrome c oxidase activity and is
associated with tumors. Hum Mutat. 27:575–582. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Aral C, Kaya H, Ataizi-Celikel C, Akkiprik
M, Sonmez O, Gulluoglu BM and Ozer A: A novel approach for rapid
screening of mitochondrial D310 polymorphism. BMC Cancer. 6:212006.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Ma Y, Bai RK, Trieu R and Wong LJ:
Mitochondrial dysfunction in human breast cancer cells and their
transmitochondrial cybrids. Biochim Biophys Acta. 1797:29–37. 2010.
View Article : Google Scholar
|
|
105
|
Santos GC Jr, Góes AC, Vitto H, Moreira
CC, Avvad E, Rumjanek FD and Moura Gallo CV: Genomic instability at
the 13q31 locus and somatic mtDNA mutation in the D-loop site
correlate with tumor aggressiveness in sporadic Brazilian breast
cancer cases. Clinics (São Paulo). 67:1181–1190. 2012. View Article : Google Scholar
|
|
106
|
Xu C, Tran-Thanh D, Ma C, May K, Jung J,
Vecchiarelli J and Done SJ: Mitochondrial D310 mutations in the
early development of breast cancer. Br J Cancer. 106:1506–1511.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Parrella P, Xiao Y, Fliss M,
Sanchez-Cespedes M, Mazzarelli P, Rinaldi M, Nicol T, Gabriel-son
E, Cuomo C, Cohen D, et al: Detection of mitochondrial DNA
mutations in primary breast cancer and fine-needle aspirates.
Cancer Res. 61:7623–7626. 2001.PubMed/NCBI
|
|
108
|
Alhomidi MA, Vedicherla B, Movva S, Rao
PK, Ahuja YR and Hasan Q: Mitochondrial D310 instability in Asian
Indian breast cancer patients. Tumour Biol. 34:2427–2432. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Lott MT, Leipzig JN, Derbeneva O, Xie HM,
Chalkia D, Sarmady M, Procaccio V and Wallace DC: mtDNA variation
and analysis using mitomap and mitomaster. Curr Protoc
Bioinformatics. 44:1.23.1–1.23.26. 2013. View Article : Google Scholar
|
|
110
|
Payne BA, Wilson IJ, Yu-Wai-Man P, Coxhead
J, Deehan D, Horvath R, Taylor RW, Samuels DC, Santibanez-Koref M
and Chinnery PF: Universal heteroplasmy of human mitochondrial DNA.
Hum Mol Genet. 22:384–390. 2013. View Article : Google Scholar
|
|
111
|
Weerts MJ, Sieuwerts AM, Smid M, Look MP,
Foekens JA, Sleijfer S and Martens JW: Mitochondrial DNA content in
breast cancer: Impact on in vitro and in vivo phenotype and patient
prognosis. Oncotarget. 7:29166–29176. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Mueller EE, Brunner SM, Mayr JA, Stanger
O, Sperl W and Kofler B: Functional differences between
mitochondrial haplogroup T and haplogroup H in HEK293 cybrid cells.
PLoS One. 7:e523672012. View Article : Google Scholar
|
|
113
|
Tommasi S, Favia P, Weigl S, Bianco A,
Pilato B, Russo L, Paradiso A and Petruzzella V: Mitochondrial DNA
variants and risk of familial breast cancer: An exploratory study.
Int J Oncol. 44:1691–1698. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Blein S, Bardel C, Danjean V, McGuffog L,
Healey S, Barrowdale D, Lee A, Dennis J, Kuchenbaecker KB, Soucy P,
et al: Breast Cancer Family Registry; EMBRACE; GEMO Study
Col-laborators; HEBON: An original phylogenetic approach identified
mitochondrial haplogroup T1a1 as inversely associated with breast
cancer risk in BRCA2 mutation carriers. Breast Cancer Res.
17:612015. View Article : Google Scholar
|
|
115
|
Rao R, Rivers A, Rahimi A, Wooldridge R,
Rao M, Leitch M, Euhus D and Haley BB: Genetic ancestry using
mitochondrial DNA in patients with triple-negative breast cancer
(GAMiT study). Cancer. 123:107–113. 2017. View Article : Google Scholar
|
|
116
|
Mosquera-Miguel A, Alvarez-Iglesias V,
Carracedo A, Salas A, Vega A, Carracedo A, Milne R, de León AC,
Benitez J, Carracedo A, et al: Is mitochondrial DNA variation
associated with sporadic breast cancer risk. Cancer Res.
68:623–625; author reply 624. 2008. View Article : Google Scholar
|
|
117
|
van Oven M and Kayser M: Updated
comprehensive phylogenetic tree of global human mitochondrial DNA
variation. Hum Mutat. 30:E386–E394. 2009. View Article : Google Scholar
|
|
118
|
Salas A, García-Magariños M, Logan I and
Bandelt HJ: The saga of the many studies wrongly associating
mitochondrial DNA with breast cancer. BMC Cancer. 14:6592014.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Walsh T, Casadei S, Coats KH, Swisher E,
Stray SM, Higgins J, Roach KC, Mandell J, Lee MK, Ciernikova S, et
al: Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in
families at high risk of breast cancer. JAMA. 295:1379–1388. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Hossein R and Houshmand M: Diagnostic
algorithm for identification of individuals with hereditary
predisposition to breast cancer. Lik Sprava. 1–2:103–108. 2008.
|
|
121
|
Jandova J, Janda J and Sligh JE: Changes
in mitochondrial DNA alter expression of nuclear encoded genes
associated with tumorigenesis. Exp Cell Res. 318:2215–2225. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Zhang Q, Liang Z, Gao Y, Teng M and Niu L:
Differentially expressed mitochondrial genes in breast cancer
cells: Potential new targets for anti-cancer therapies. Gene.
596:45–52. 2017. View Article : Google Scholar
|
|
123
|
Lin CS, Chang SC, Ou LH, Chen CM, Hsieh
SS, Chung YP, King KL, Lin SL and Wei YH: Mitochondrial DNA
alterations correlate with the pathological status and the
immunological ER, PR, HER-2/neu, p53 and Ki-67 expression in breast
invasive ductal carcinoma. Oncol Rep. 33:2924–2934. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Blein S, Barjhoux L, Damiola F, Dondon MG,
Eon-Marchais S, Marcou M, Caron O, Lortholary A, Buecher B, Vennin
P, et al GENESIS investigators: Targeted sequencing of the
mitochondrial genome of women at high risk of breast cancer without
detectable mutations in BRCA1/2. PLoS One. 10:e01361922015.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Hsu CW, Yin PH, Lee HC, Chi CW and Tseng
LM: Mitochondrial DNA content as a potential marker to predict
response to anthra-cycline in breast cancer patients. Breast J.
16:264–270. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Farnie G, Sotgia F and Lisanti MP: High
mitochondrial mass identifies a sub-population of stem-like cancer
cells that are chemo-resistant. Oncotarget. 6:30472–30486. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Ferreri AJ, Ponzoni M, Guidoboni M, Resti
AG, Politi LS, Cortelazzo S, Demeter J, Zallio F, Palmas A, Muti G,
et al: Bacteria-eradicating therapy with doxycycline in ocular
adnexal MALT lymphoma: A multicenter prospective trial. J Natl
Cancer Inst. 98:1375–1382. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Lamb R, Harrison H, Hulit J, Smith DL,
Lisanti MP and Sotgia F: Mitochondria as new therapeutic targets
for eradicating cancer stem cells: Quantitative proteomics and
functional validation via MCT1/2 inhibition. Oncotarget.
5:11029–11037. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Ferreri AJ, Ponzoni M, Guidoboni M, De
Conciliis C, Resti AG, Mazzi B, Lettini AA, De-meter J, Dell'Oro S,
Doglioni C, et al: Regression of ocular adnexal lymphoma after
Chlamydia psittaci-eradicating antibiotic therapy. J Clin Oncol.
23:5067–5073. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Teixeira FK, Sanchez CG, Hurd TR, Seifert
JRK, Czech B, Preall JB, Hannon GJ and Lehmann R: ATP synthase
promotes germ cell differentiation independent of oxidative
phosphorylation. Nat Cell Biol. 17:689–696. 2015. View Article : Google Scholar : PubMed/NCBI
|














