1. Introduction
Cancer therapy resistance is mediated by several
mechanisms, including intrinsic and extrinsic factors, and those
originating from the tumor microenvironment (TME) (1). Most of the widely used
chemotherapeutic agents and γ-radiation utilize apoptosis or
autophagy as common death pathways. Thus, a better understanding of
the molecular mechanisms behind tumor biology and cancer therapy
resistance is a mandatory step to propose novel approaches aiming
to bypass chemotherapy and radiotherapy resistance and associated
gene products. Among several markers associated with response to
therapy, osteopontin (OPN) has been identified as a key molecule
(2,3).
OPN is a multi-functional chemokine-like
matricellular phosphoglycoprotein. Depending on its intracellular
or extracellular localization, OPN is involved in a series of
physiological roles, including inflammation, cell adhesion and
migration, differentiation, cell survival and apoptosis, as well as
regulation of bone matrix mineralization. These diverse biological
roles are partly due to its capacity to interact with several
molecules, including cell surface receptors, such as integrin and
cluster of differentiation (CD44), intracellular signaling
molecules, calcium and heparin. OPN is produced by distinct cell
types, such as epithelial, stromal, immune system, bone and
endothelial cells (4). High OPN
expression has also been detected in adipose tissues and body
fluids (4).
In multiple cancer types, OPN expression is
upregulated (3). In tumors, OPN
regulates tumorigenesis, tumor progression and metastasis formation
(5), by activating cell migration,
inhibiting apoptosis (6,7), stimulating angiogenesis (8) and metabolism (9), and modulating the tumor
microenvironment (10) and the
immune system (11). Notably, OPN
can also promote cell survival by negatively regulating apoptosis
in response to stress conditions, including exposure to anticancer
agents effect (12). OPN performs
these roles by binding to cell surface receptors, including
integrin and CD44 cell receptors (13).
Several aberrantly activated signaling pathways can
activate OPN expression in cancer cells, such as activator
protein-1, mycA, phosphatidylinositol 3 kinase (PI3K),
serine/threonine kinase (AKT) and nuclear factor‑қB (NF‑қB)
(14, 15). Notably, OPN has been described as a
key modulator of cancer hallmarks, by regulating the main aspects
of tumor progression in several tumor models (16,17).
OPN is able to promote tumor cell migration by interacting with
integrin receptors, especially αvβ3 integrin, stimulating cell
adhesion and tumor cell migration properties (3). It has been demonstrated previously
that OPN downregulation in breast cancer cell lines inhibits OPN
interaction with αvβ3 integrin, thereby impairing cell migration,
invasion and apoptosis (18). It
was demonstrated that these effects have been mediated by
PI3K/AKT/mechanistic target of rapamycin (mTOR) signaling,
promoting the upregulation of light chain 3 and beclin-3, then
favoring apoptotic cell death, while inhibiting aggressive
phenotype of these breast cancer cells. It has also been
demonstrated that alteration of OPN expression levels may influence
tumor growth, migration and cell cycle in human nasopharyngeal
CNE-2 carcinoma cell lines (18).
When downregulating OPN expression, diminished levels of matrix
metalloproteinase MMP-2 and MMP-9 have been observed, evidencing
that OPN may also induce MMP expression levels through the
activation of NF‑қB signaling in this tumor model (19). OPN expression levels can also
modulate mitochondrial mediated apoptotic cell death, involving
cytochrome c, apoptotic protease activating factor 1,
cleaved caspase-3 and B cell lymphoma (Bcl)-2/Bcl-2-associated X
protein, resulting in lower expression levels of proteins
associated with cell invasion, such as MMP-2 and urokinase-type
plasminogen activator (uPA) (20).
Then, downregulation of OPN expression levels can promote apoptotic
cell death and cell invasion properties in a
mitochondrial-dependent pathway (20). OPN can also promote tumorigenesis
and tumor progression by evading apoptotic cell death, mainly by
interacting with CD44 cell receptors (21). Furthermore, OPN interactions with
immune and inflammatory cells from the TME perform essential roles
on tumor development and progression. Besides being produced by
tumor cells, OPN can also be secreted by stromal and infiltrating
inflammatory cells that can affect the TME and its corresponding
cell roles. Stromal fibroblasts can also be influenced by OPN
(22,23). Given its roles in angiogenesis,
extracellular matrix remodeling and metastasis, their presence and
influence by OPN can also favor tumor growth (22,23).
It has been reported that when interacting with α9β1, OPN activates
p38 and extracellular signal-regulated kinase (ERK) signaling,
which then can promote the expression of cyclooxygenase-2 and
prostaglandin E (PGE), favoring melanoma tumor cell migration
(24). In addition, macrophages
from the TME, when activated by OPN can promote angiogenesis via
PGE2 and stimulate the expression of MMP-9, consequently promoting
tumor progression in this tumor model (24). When recruiting macrophages to the
tumor inflammatory environment, OPN can also stimulate
tumorigenesis (25).
OPN also induces the expression and activity of
MMPs, which can contribute to tumor metastasis by degrading the
extracellular matrix (ECM) and promoting cell invasion. Conversely,
OPN biological activity can also be modulated by MMP-induced
cleavage (26). It is well-known
that OPN stimulates tumor cell invasion and migration possibly by
inhibiting apoptotic cell death and by regulating the activity of
MMP-2 and MMP-9 that degrade the ECM (27). OPN can stimulate the activity of
MMP-9, modulating multiple signaling pathways, such as focal
adhesion kinase (FAK), ERK and NF‑қB, which then can regulate
cytoskeleton architecture, cell growth, motility and extracellular
matrix (ECM) (28,29). By activating PI3K/AKT signaling,
OPN can similarly regulate hypoxia-inducible factor (HIF)-1α
expression via αvβ3 integrin interaction and promoting ECM
degradation through uPA and MMP-9, further mediating metastasis
formation in ovarian cancer cells (30).
Also in the context of OPN roles on modulating the
TME and the immune system, OPN has been described as a
multifactorial cytokine activated by T lymphocytes, monocytes,
macrophages, epithelial cells, fibroblasts and a promoter of
cell-mediated immune responses (15,28).
Similarly, OPN is a typical angiogenesis stimulating
factor, sustaining tumor progression and metastatic growth. The
role of OPN in angiogenesis is mainly associated with OPN
interaction with αvβ3 integrin, a central angiogenesis marker
(31), but is also associated with
several other factors, including vascular endothelial growth factor
(VEGF) (32,33). OPN interactions with VEGF are also
mediated by aberrant signaling pathways, such as PI3K/AKT and
ERK1/ERK2 (32,33). It has also been demonstrated in
acute leukemia that the expression of OPN and VEGF are strongly
correlated with the occurrence and development of this non-solid
tumor. In this model, OPN can regulate VEGF expression and promote
angiogenesis besides favoring disease progression (31). It was also found that OPN
interaction with αvβ3 activates signaling pathways, such as breast
tumor kinase/NF‑қB/activating transcription factor (TF)-4,
promoting cell migration, tumor growth, endothelial cell migration
and angiogenesis (34). Our group
also demonstrated that OPNb and OPNc splicing isoforms favored
tumor growth, cell proliferation, invasion and migration by
modulating VEGF, MMP-2 and MMP-9 expression levels through PI3K
signaling (35).
It has also been demonstrated that OPN can modulate
metabolism by signaling through the activation of oxidoreductase
gene expression, associated with the mitochondrial respiratory
chain, the hexose monophosphate shunt or the regulation of the
hexose monophosphate shunt (9).
Furthermore, it has been reported that OPN can disrupt liver
cholesterol metabolism (36).
The majority of the data regarding the role of OPN
in cancer cell refers to total OPN, which includes the sum of all
OPN isoforms, including those generated by alternative splice,
post‑translational modifications and alternative translation
(17,37).
OPN primary transcript suffers alternative splicing,
generating at least five splice variants, named OPNa (which contain
all coding exons), OPNb (exon 5 is deleted), OPNc (exon 4 is
deleted), OPN4 (in which both exon 4 and exon 5 are deleted) and
OPN5 (which contains an additional exon originated from inclusion
of a region from intron 3; Fig.
1). OPN splicing isoforms (OPN-SI) are aberrantly expressed in
cancer cells (17). Particularly,
the expression and functional roles of OPNa, OPNb and OPNc have
been broadly studied in distinct tumor models, in which they were
demonstrated to present tissue and tumor specific roles (17,37).
Notably, the same OPN-SI might activate or inhibit tumor
progression, depending on the tumor type (17,37).
However, data regarding the expression and functional roles of OPN4
and OPN5 in tumor cells are scarce. One previous report described
that these recently described OPN-SI are frequently co-expressed in
esophageal carcinoma tissues (38), but functional studies evaluating
these splice variants are still lacking.
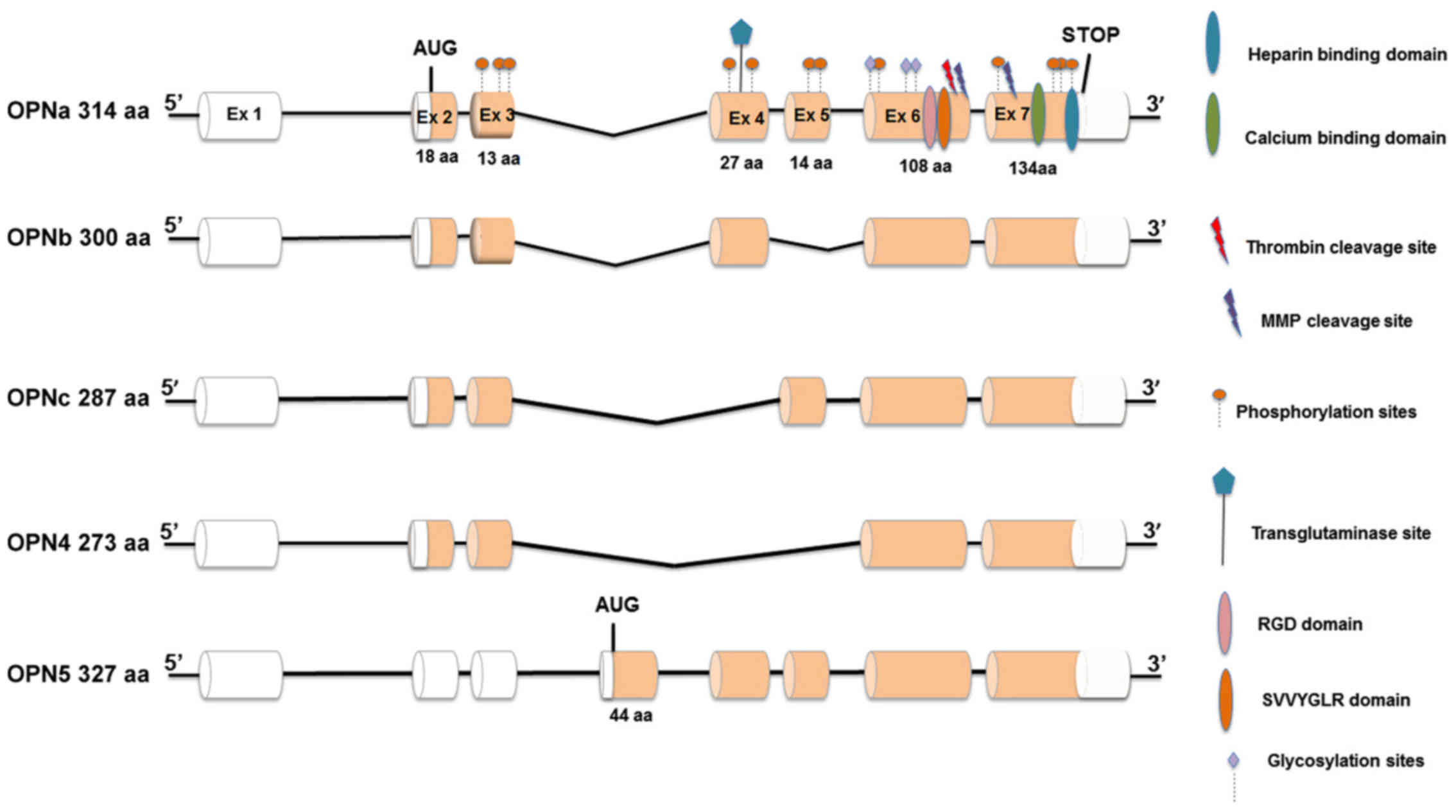 | Figure 1Structure of OPN isoforms. Exonic
regions represented by white cylinders correspond to 5’ and 3’
untranslated regions. Each translated Ex is represented by orange
cylinders. Ex total lengths are also presented. OPN functional
domains are also presented above each Ex, such as RGD, SVVYGLR and
calcium binding domain, as well as post-translational
modifications, such as phosphorylation and glycosylation sites.
Also presented are protein cleavage sites for thrombin and MMPs.
The initiation translation codon (AUG) and the stop codon are also
represented. OPNa, the full length isoform, contains Ex 2-7,
whereas OPNb and OPNc lack Ex 5 and 4, respectively. Ex 4 and 5 are
deleted in the OPN4 isoform, whereas OPN5 contains an extra Ex
resulting from an inclusion of an intronic region located between
Ex 3 and 4, corresponding with the longer isoform. Total length of
each isoform of each isoform is also presented. OPN, osteopontin;
aa, amino acids; MMP, metalloproteases; Ex, exon. |
In the present review, among the broadly known OPN
roles on activating tumor progression, additional features involved
in tumor resistance are highlighted. These typically include OPN
functions on modulating cell survival, apoptosis and autophagy, as
well as cell plasticity and sustaining stem-like properties of
cancer cells (39), which are key
factors associated with tumor resistance.
A molecular mechanism frequently associated with
failure in the treatment of malignant carcinomas is the biological
reprogramming of epithelial cells called epithelial mesenchymal
plasticity. More recently, it has been defined as a dynamic process
implicated in epithelial-mesenchymal transition (EMT) and its
reverse program, mesenchymal-epithelial transition (MET) or
intermediate phenotypes, known as partial or intermediate EMT
(40,41). EMT renders cancer cells the ability
to lose epithelial traits, while gaining mesenchymal features.
During EMT, cells also acquire stem cell-like properties and are
able to disseminate and colonize to distant organ sites, where they
may exhibit elevated resistance to cancer therapies (42,43).
Aberrant activation of oncogenic signaling pathways, including
Wnt/β-catenin, hedgehog, Notch, PI3K-AKT, tumor necrosis factor-α
and transforming growth factor (TGF)-β has a critical role in EMT
(44) and occurs during
acquisition of EMT phenotype and resistance to therapy.
Furthermore, induction of resistance is also mediated by several
genes regulated by the TF NF-κB, including Bcl-2, Bcl-xL, X-linked
inhibitor of apoptosis (XIAP), survivin and AKT, which have been
reported to mediate chemoresistance and radioresistance in numerous
types of tumor cells (45).
In the present report, current knowledge regarding
the OPN roles on mediating chemoresistance and radioresistance were
reviewed, which are the primary cancer treatment approaches that
are currently used. The distinct mechanisms by which OPN can
promote resistance to specific drugs and their associated signaling
pathways are also explored. Based on these data, putative treatment
approaches targeting OPN that have been proposed to overcome
resistance or inhibit tumor progression, and the particular
contribution of OPN splice variants on the resistant phenotype are
then discussed.
2. OPN and resistance to chemotherapy
OPN is able to mediate resistance to distinct
chemotherapeutic drugs and in several cancer types. Table I summarizes these data and the
corresponding signaling pathways and molecular mechanisms
involved.
 | Table IOPN roles in mediating
chemoresistance and radioresistance. |
Table I
OPN roles in mediating
chemoresistance and radioresistance.
| Author, year | Drug or
therapy | Tumor type | Biological
material | Main findings | Signaling pathways
involved |
Refs. |
|---|
| Zhang et al,
2014 | PTL | AML | AML cells resistant
to DNR | OPN promotes
resistance to PTL in CD34+/CD123+ leukemia
stem cell population resistant to DNR | PTL treatment
downregulates OPN, and inhibits AKT, mTOR, β-catenin and PTEN | (18) |
| Zhang et al,
2010 | Sorafenib | AML | TCGA data; MV4-11
human acute leukemia cell line (FLT3-ITD
mutation) | OPN binds to
integrin αvβ3 and decrease sorafenib sensitivity in FLT3-ITD
mutated AML cells | Sorafenib
sensitivity involves OPN/αvβ3 integrin/PI3K/ AKT/GSK3β/β-catenin
pathway | (20) |
| Gu et al,
2009 | CDDP | Human small cell
lung cancer | Stable OPN
overexpression in SBC-3 cells (SBC-3/OPN) | Intracellular OPN
mediates resistance to CDDP in SBC-3 cells | OPN induces
anti-apoptotic Bcl-2 while blocking caspase-3 and -9 | (5) |
| Polyak et
al, 2009 | CDDP | OSCC | SAS cells | OPN overexpression
promotes resistance to CDDP | Not
investigated | (23) |
| Luo et al,
2015 | 5-FU | OSCC | SAS cells
overexpressing OPN | OPN-αvβ3 integrin
axis mediates resistance to 5-FU | OPN-mediated
resistance involves αvβ3 axis | (50) |
| Ng et al,
2015 | Oxaliplatin | Colorectal
cancer | DLD1 cells
overexpressing OPN | OPN expression and
chemoresistance in patients treated with oxaliplatin | OPN enhances
stemness of cancer cells through CD44v6, HGF and SDF-1 | (51) |
| Lin et al,
2015 | Temozolomide and
CDDP | Glioma | U251 human glioma
cells | OPN mediates
resistance to temozolomide and CDDP | OPN-mediated
resistance involves NF-κB/Bcl-2 pathway | (52) |
| Shao et al,
2017 | CTX | Breast cancer | MDA-MB-231 | OPN knockdown
sensitizes cells to CTX | OPN-mediated
resistance involves p38 MAPK pathway | (26) |
| Guarneri et
al, 2017 | Vinorelbin,
etoposide and gemcitabine | Malignant pleural
mesothelioma | ACC-MESO-1/OPN
cells overexpressing OPN | OPN is involved in
multi drug resistance by enhancing the CD44 binding to HA | CD44 and HA binding
and activation of PI3K signaling | (27) |
| Wang et al,
2011 | Radiotherapy | Lung cancer | NSCLC cell lines
and xenograft tumors | OPN and EGFR
pathway, have been associated with radiation resistance in lung
cancer cells | Radiation
resistance in tumors harboring KRAS mutations involves MLCC and
CSC-like phenotypes | (31) |
| Song et al,
2008 | Radiotherapy | Lung cancer | A549 lung cancer
cells | Beclin1-induced
autophagy abrogates radioresistance by suppressing OPN | Radioresistance is
mediated by inhibition of Beclin1-induced autophagy | (30) |
| Castello et
al, 2017 | Radiotherapy | Cervical
cancer | Radiation resistant
and sensitive LACSCC paraffin tissues | OPN and E-cadherin
aberrant expression indicates radiation resistance | OPN-mediated EMT
mediates radioresistance | (32) |
In non-solid tumors, such as leukemia, OPN has been
reported to mediate resistance to parthenolide and sorafenib. The
natural compound parthenolide was demonstrated to induce apoptosis
in daunorubicin-resistant stem-like leukemic cells through OPN
downregulation and modulation of AKT, mTOR and β-catenin signaling
(46). Particularly in acute
myeloid leukemia, OPN was demonstrated to be upregulated in
leukemic blasts, being correlated with a poor clinical outcome
(47). Consistently, OPN promoted
resistance to sorafenib by binding to αvβ3 integrin and by inducing
β-catenin expression in an AKT and glycogen synthase kinase
(GSK)3β-dependent manner (48).
Furthermore, there are several examples in which OPN
mediates resistance in solid tumors, most of which focused on the
roles of OPN in hepatocellular carcinomas (HCC). In these tumors,
high OPN expression level is correlated with increased metastatic
potential and resistance to taxanes or cisplatin. In lung cancer
cells, intracellular OPN upregulation promotes cisplatin resistance
by inducing the expression of anti-apoptotic protein Bcl-2 and by
blocking caspase-3 and caspase-9 from activation (5). Also, OPN is able to stimulate HCC
cell survival and autophagy, which then can favor stem cell-like
properties and the resistant phenotype in response to epirubicin
and cisplatin via binding with its receptor integrin αvβ3 and
sustaining Forkhead box (Fox)O3a stability (42). It has been proposed that OPN may
promote a cancer stem cell (CSC)-like phenotype via the
αvβ3-NF-κB-HIF-1α pathway (49).
Other studies using human samples with oral squamous
cell carcinoma demonstrated that OPN expression levels are
correlated with therapy response and shorter overall survival. OPN
upregulation in these tumors promoted resistance to cisplatin and
to 5-fluorouracil and also involved the OPN-integrin αvβ3 axis
(50). In addition, several
reports described the association between upregulated OPN
expression and chemoresistance in patients treated with oxaliplatin
in colorectal cancer (51),
cisplatin in lung cancer (5),
temozolomide and cisplatin in glioma (52), cyclophosphamide in breast cancer
(53) and vinorelbin, etoposide
and gemcitabine in malignant pleural mesothelioma (54). Similarly to other tumor models,
resistance to these chemotherapeutic drugs mainly involves the
regulated expression of octamer-binding TF 4 and sex determining
region Y-box (SOX)2 (modulators of stemness), Bcl-2, caspase-3 and
caspase-9 (apoptosis regulators) (5,52),
as well as NF-κB, AKT and p38/mitogen-activated protein kinase
(MAPK) signaling (49,51). Furthermore, it has been
demonstrated in malignant pleural mesothelioma that OPN could
regulate chemosensitivity to vinorelbine, etoposide and gencitabin
through the alteration of CD44 binding to hyaluronate (HA)
(54). HA is a linear
glycosaminoglycan that interacts with cell surface receptors,
including CD44, facilitating cell adhesion, cell motility, cellular
proliferation and tumor progression (54). OPN is strongly involved in
multidrug resistance in this tumor by enhancing the CD44 binding to
HA and PI3K/AKT signaling, thereby promoting cell survival and
chemoresistance (54).
3. OPN and association to
radioresistance
In addition to OPN roles in mediating
chemoresistance, reports describing the functional association
between OPN and radiotherapy resistance are scarce (Table I). However, a number of reports
have described the roles of OPN as a marker of response to
radiotherapy (55,56). OPN expression, in conjunction with
the epidermal growth factor receptor pathway, have been associated
with radiation resistance and poor prognosis in lung cancer,
particularly in patients presenting tumors with KRAS mutations
(57). It has also been reported
in lung cancer that OPN is an indicator of resistance to
radiotherapy. In a previous study, overexpression of beclin-1
induced cell death by autophagy in human lung cancer cells,
reversing radioresistance (58).
Radioresistance in these tumors has been associated with a stem
cell-like phenotype and invasive potential (57). In cervical cancer, high OPN and low
E-cadherin expression levels correlate with a radiation-resistant
phenotype (59), further
implicating OPN as a key molecule in the interface of
radioresistance and cellular plasticity. Stem cell-like features
and radioresistance in glioma cells can be promoted by OPN possibly
via activation of CD44 signaling through its intracellular domain
by enhancing HIF-2α activity (60). Fig.
2 summarizes the mechanisms and signaling pathways activated by
OPN on promoting chemoresistance and radioresistance.
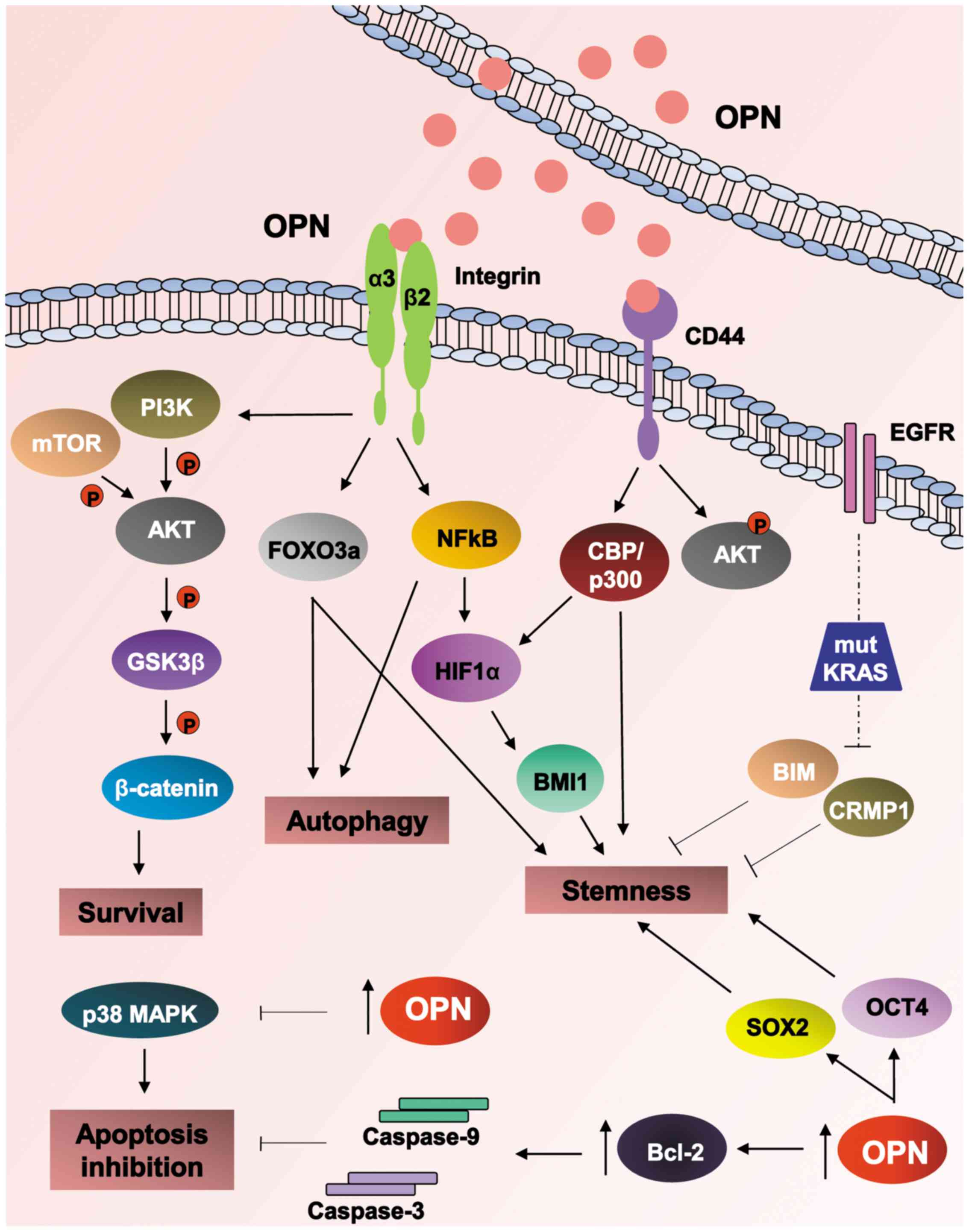 | Figure 2Cancer treatment resistance
mechanisms mediated by OPN. Numerous and intricate mechanisms
developed by cancer cells have been described at different levels
in response to intracellular or extracellular OPN. Upon OPN binding
to integrin receptors, especially by αV/β3 heterodimers, the
PI3K/AKT/mTOR/β-catenin signaling pathway is triggered, leading to
cell survival and chemotherapeutic resistance. OPN can also confer
stem cell-like features to cancer cells by sustaining FoxO3a
stability-induced autophagy as well as activating the
αv/β3/NF-κB/HIF-1α pathway. Accordingly, OPN can modulate cancer
stemness by altering CD44 receptor binding to hyaluronate and
inducing AKT phosphorylation via CD44, thereby promoting cell
survival and chemoresistance. In conjunction with the EGFR pathway,
OPN expression, has been associated with radiation resistance
particularly in tumors presenting KRAS mut, which was closely
associated with the modulation of BIM and CRMP1 expression and
thus, stem cell phenotype. Consistently, intracellular OPN was
demonstrated to regulate the expression of Oct4 and Sox2, well
known modulators of stemness. OPN expression levels have been
demonstrated to negatively regulate p38 MAPK and correlate with
caspase-3 and -9 blocking and expression of Bcl-2 anti-apoptotic
protein, further contributing towards an apoptosis inactivation
phenotype. Therefore, OPN has been demonstrated to modulate
signaling pathways associated with chemoresistance and
radioresistance, through promoting cell survival and inhibiting
apoptosis as well as modulating autophagy and stemness in tumor
cells. OPN, osteopontin; PI3K, phosphoinositide 3-kinase; AKT,
protein kinase B; mTOR, mechanistic target of rapamycin; Fox,
Forkhead box; NF-κB; nuclear factor-κB; HIF-1α, hypoxia-inducible
factor-1α; Oct4, octamer-binding transcription factor 4; CD,
cluster of differentiation; EGFR, epidermal growth factor receptor;
mut, mutations; Bcl-2, B cell lymphoma-2; BIM, Bcl-2-like protein
11; CRMP1, collapsin response mediator protein 1; Sox2, sex
determining region Y-box 2; MAPK, mitogen-activated protein kinase;
GSK3β, glycogen synthase kinase 3β; CBP, cAMP response element
binding protein binding protein. |
4. OPN in the interface of epithelial
plasticity and therapeutic resistance
EMT corresponds with the dynamic
transdifferentiation of epithelial into mesenchymal cells, in which
cells lose their epithelial features and become more motile
mesenchymal cells (44).
Increasing evidence has demonstrated that there is a close
association between EMT and resistance to therapy (59,60),
which may be caused by an enhancement of cancer cell survival, cell
fate transition, and/or upregulation of drug resistance-related
genes during the EMT transition.
The EMT process is mediated by several EMT-inducing
TFs (EMT-TF), including TWIST1/2, SNAIL1/2 and zinc finger E-box
binding homeobox (ZEB)1/2. In addition to these established
EMT-TFs, other TFs have been demonstrated to induce or regulate
EMT, including Fox TFs, GATA family, SOX, ovo-like transcriptional
repressor 1/2 and grainyhead-like 2 (61,62).
Mechanistically, EMT-TFs suppress the expression of key epithelial
markers, such as E-cadherin, while activating the expression of
mesenchymal genes, such as those coding for vimentin and
fibronectin. Collectively, these regulatory networks control the
integrity of and the balance between the epithelial and mesenchymal
phenotypes. In chemoresistant cells, EMT-TFs increase cell survival
in response to therapy-induced programmed cell-death by
upregulating anti-apoptotic genes, while downregulating gene
products performing pro-apoptotic roles (52). Certain EMT-TFs can also contribute
to hormone therapy resistance by modulating the expression of their
corresponding receptors (61).
These findings make the EMT process an attractive target for
reducing chemotherapy resistance (61).
It has also been demonstrated that EMT-TFs act
cooperatively with changes at the RNA level that regulate EMT
progression, such as alternative splicing and microRNA (miRNA or
miR) and long non-coding RNA mediated control of EMT (61-64).
EMT process is further controlled by multiple signaling pathways,
in which multiple morphogenetic and environmental signals, such as
TGF-β, WNT, epidermal and platelet-derived growth factors,
inflammatory cytokines and integrin receptor ligands, have been
demonstrated to promote EMT in response to extracellular signals
(61). Among these molecules,
NF-κB and matrix MMPs have also been identified as specific
inducers of EMT. NF-κB has been described as a contributor to EMT,
by combining with oncogenic gene products, such as RAS in order to
protect cells from apoptosis (65). MMPs induce EMT associated with
malignant transformation via a pathway dependent upon production of
reactive oxygen species (ROS) and degrading matrix proteins, then
favoring tumor cell migration and motility (65).
TGF-β, mostly TGF-β1 isoform, is the most
well-studied cytokine in the induction of EMT. As reviewed
elsewhere (61), TGF-β mediates
EMT through mothers against decapentaplegic homolog
(SMAD)-dependent or SMAD-independent pathways The SMAD-dependent
pathway is initiated by binding of TGF-β1 to TGF-β receptor (TβR)II
and TβRI, which then activate SMAD protein complex, which then
enter the nucleus to induce the expression of lymphoid enhancer
binding factor-1 TF. This TF binds to β-catenin, suppressing
transcription of epithelial markers, while promoting the expression
of mesenchymal markers. SMAD complexes not only activate the
expression, but also increase the activity of EMT-TFs. Other
changes in gene expression during EMT occur without directly
requiring EMT-TFs, but are rather controlled by TGF-β-activated
SMADs, which can directly activate the expression of certain
mesenchymal genes. TGF-β also induces signaling through cytoplasmic
expression of β-catenin, which is also modulated by the Wnt
pathway. In addition to TGF-β-activated SMAD signals, TGF-β also
induces EMT signaling through RHO-like GTPases, PI3K/AKT and MAPK
pathways. Activation of RHO, RAC and cell division control protein
42 homolog GTPases drives actin reorganization, and lamellipodia
and filopodia formation. PI3K signaling activation by TGF-β
promotes the activation of mammalian TOR complex (mTORC1 and
mTORC2). Notably, mTORC1 and mTORC2 modulate cell size, protein
synthesis, motility and invasion. AKT decreases the level of SNAIL1
expression, attenuating E-cadherin repression and the activation of
MMP-9 expression. AKT also phosphorylates GSK3β, resulting in
SNAIL-1 stabilization. TGF-β also activates ERK, p38 and JUN
N-terminal kinase/MAPK pathways, which then also increases
TGF-β-induced transcription, leading to increased E-cadherin
repression and activation of N-cadherin and MMP expression. EMT
signaling has been demonstrated to induce the expression of genes
coding for MMP-2 and MMP-9, which cleave Type IV collagen in the
basal lamina to promote post-EMT invasion of underlying tissues.
MMP-3 has also been demonstrated to directly induce EMT through
activation of RAC1 GTPase-reactive oxygen species signaling, which
promotes SNAIL1 expression. Additionally, SNAIL1/2 can also promote
breakdown of the ECM via upregulation of MMPs (61). Furthermore, MMPs are capable of
degrading E-cadherin in the cell membrane. All these processes
enable cells to acquire a mesenchymal phenotype (61).
In this context, OPN has previously been reported as
a master regulator of epithelial-mesenchymal plasticity, once it
has an important regulatory role in the expression of key EMT
regulators (66-71). OPN expression also shares
functional interplay with the previously mentioned traditional EMT
activators, such as TGF-β, TWIST 1/2, ZEB1/2 and SNAIL-family
members. Importantly, OPN is able to guide EMT through specific
cellular signaling pathways and by restructuring the TME to modify
EMT processes (66-71).
OPN overexpression induces TWIST phosphorylation
and/or activation through MAPK, AKT and/or RAC/AKT signaling
pathways (71,72). Besides, OPN upregulates HIF-1α to
induce EMT through TWIST activation and by maintaining the tumor
cell stemness (66). TWIST also
serves in OPN-mediated metastasis through activation of the
PI3K/AKT pathway (66). Signaling
mediated by OPN interacts directly or indirectly with ZEB TF family
members, as well as leading to EMT. Notably, it is known that OPN
is a potent activator of NF-κB (73), which induces the expression of both
ZEB1/2 and therefore can regulate NF-κB/ZEB dependent EMT.
Likewise, OPN can regulate ZEB-related EMT through non-NF-κB
pathways, such as by upregulating the expression of miR-200 family
members that inhibit ZEB1/2 initiated EMT (71). Otherwise, NF-κB has also been
indicated as a key player of OPN and MMP-9 activation (74). Furthermore, OPN regulates EMT by
overexpressing SNAIL EMT-TF. In addition, OPN co-regulates the
expression of glioma-associated oncogene, a TF that mediates Sonic
hedgehog signaling, which then induces the expression of SNAIL
(71). Vimentin upregulation has
also been induced by OPN (66).
Notably, OPN induces the expression of TGF-β (71).
It has been reported that OPN is able to modify the
tissue and TME to support EMT and hence can also indirectly modify
EMT (66). During tumor
progression, limited tumor oxygen availability and tumor metabolic
demands induce hypoxia. As reviewed previously (60,61),
the activities of HIF-1α and HIF-2α are enhanced in response to
prolonged TGF-β and certain tyrosine kinase receptors. HIF-1α and
HIF-2α can then activate EMT-TF expression and/or subcellular
localization in tumor cells. Immune and stromal cells from the TME
are also sources of EMT-inducing cytokine stimulation. It has been
demonstrated that chemotherapy and radiotherapy promote oxidative
and inflammatory stress in tumor tissues, contributing to increased
expression of inflammatory cytokines and subsequent induction of
the EMT program. In the TME, post-translational modifications of
EMT-TF can also regulate the half-life of EMT-TFs (61). Malignant signals from the TME with
long-lasting effects on associated cancer cells may also modulate
epithelial plasticity and then sustain the metastatic potential and
tumor chemoresistance. OPN modulates tumor-specific EMT by
generating cancer-associated fibroblasts (CAFs), which secrete a
multitude of factors in the TME that support tumor invasiveness and
metastases, including TGF-β and interleukin-6. Tumor-derived OPN
and exogenous OPN are able to induce transformation of CAFs from
mesenchymal stem cells by stimulating the production of TGF-β. OPN
also enhances the migration and invasion of malignant tumor cells
through both the inhibition of apoptosis and by regulating the
activities of MMP-2 and MMP-9, which degrade the ECM. OPN has been
described to upregulate MMP-9 activity, modulating multiple
signaling pathways via focal FAK, ERK and NF-κB that regulate
cytoskeletal organization, cell motility, cell growth, and also
cell migration, ECM invasion and tumor growth (61,62).
In this context, it has been indicated that NF-κB may be a key
player in OPN and MMP-9 activation (27). OPN is a substrate for several
extracellular proteases and is cleaved in vitro and in
vivo by MMPs. MMP-9, for instance, is known to cleave OPN
(75), and it is also known that
OPN-regulated signaling leads upregulation of MMP-2 in prostate
cancer cells (74). Similarly, OPN
can mediate the activation of MMP-9 during migration of prostate
cancer and melanoma cells (74).
It has also been reported that OPN has an important
role in regulating TGF-β-mediated processes and likely also
regulates TGF-β-mediated EMT (70). Considering the emerging roles of
OPN in modulating the EMT process, thus contributing to a drug
resistant phenotype, the analysis of OPN expression has been
considered as a potential target for future interventions to
overcome tumor resistance. Notably, it has been reported that,
unlike secreted OPN, which is able to trigger the EMT to initiate
cancer metastasis, nuclear OPN is able to induce MET, contributing
to metastasis establishment at secondary sites. In this model, OPN
interacted with HIF-2α, latterly impacting on the AKT/mir-29/ZEB
cascade (71). It has also
demonstrated that VEGF in the TME is able to induce OPN nuclear
translocation (71).
5. OPN as a target to overcome resistance to
cancer therapy
Considering the emerging roles of OPN in several
processes associated with chemoresistant and radioresistant
phenotype, and its pleiotropic roles in the tumor cascade, the
analysis of OPN expression and associated signaling has been
considered as a potential target for future interventions trying to
overcome tumor resistance, both to early and advanced tumors.
OPN-associated signaling pathways include PI3K/AKT,
mTOR, β-catenin or phosphatase and tensin homolog (PTEN) gene
expression (72), as well as
αvβ3-NF-κB-HIF-1α (49), caspases
and Bcl-2 (5,52), p38/MAPK (54) and GSK3β (48), among other targets. Advances in the
understanding of the biology of tumor resistance, particularly the
signaling pathways associated with this phenotype, may enable the
development of novel approaches to overcome resistance to
therapy.
Sp eci f ic a nd t a rgeted i n h ibit ion of t hese
resistance-associated proteins and signaling pathways may
potentially increase sensitivity of cancer cells to the cytotoxic
action of chemotherapeutic agents and to radiation exposure
(44,45). As an example of this approach to
further sensitize cells to chemotherapy by targeting OPN and
associated signaling, isolated primary
CD34+/CD38- bone marrow derived acute myeloid
leukemia (AML) cells have been treated with curcumin and
daunorubicin in combination. This strategy induced AML cell growth
inhibition and increased cytotoxicity by upregulating AKT, mTOR,
β-catenin or PTEN. Notably, these effects were stronger when OPN
expression was specifically knocked-down (72). It has also been reported that
OPN/NF-κB-mediated autophagy is required for the maintenance of the
stemness state of pancreatic cancer cells, which is associated with
survival and chemoresistance (76). These data demonstrated that the
blockade of autophagy by downregulating autophagy markers or by
treating these pancreatic cells with an autophagy inhibitor reduced
the pancreatic CSC populations and associated features, such as the
expression of CD44, CD24, CD133 and aldehyde dehydrogenase 1
(76). Once OPN is able to
stimulate autophagy and the expression of CSC markers (which
includes integrin and CD44 receptors), these cell populations could
be prevented by OPN downregulation approaches in order to sensitize
pancreatic cancer cells to current chemotherapeutic drugs, such as
gemcitabine (76).
Therapeutic application of small interfering RNA
molecules targeting OPN (77,78)
or neutralizing antibodies associated with OPN epitopes (79) have been tested in order to
downregulate OPN expression levels and the results have been
promising. An additional approach to sensitize cells to
chemotherapy or radiotherapy would be using miRNA molecules
targeting OPN or additional gene products associated with
chemoresistance, such as those modulating EMT (80,81),
drug transporters (78) and cell
survival (81). Oncogenic miRNAs
are the miRNAs with a defined role in cancer. Several miRNAs are
deregulated in cancer cells and correlated with tumor features.
Specifically, miRNAs have been reported to influence several
tumor-related processes, such as EMT, tumor invasion, metastasis
and resistance to therapy (80,82).
Among currently tested miRNAs targeting OPN, a
number are able to modulate tumorigenicity, tumor growth and
metastasis, such as miR-127-5p (83), hsa-miR-299-5p (80) and miR-181a (84). However, to the best of our
knowledge, there is no report describing the specific effects of
miRNAs on sensitizing tumor cells to chemotherapy. Conversely, a
number of miRNAs that, via targeting OPN, may be promising tools to
regulate chemoresistance and radioresistance mediated by OPN. It
has been demonstrated that miR-127-5p and hsa-miR-299-5p are able
to regulate OPN expression and can respectively modulate human
chondrocyte cell proliferation and tumorigenicity, and also display
vasculogenic mimicry of spheroid-forming breast cancer cells
(80). Similarly, miR-181a
regulation of OPN expression provides a novel mechanism of
suppressing metastasis in cancer cell lines (84). Furthermore, three lentiviral
vectors encoding miRNA against OPN have been reported to inhibit
tumor growth and metastasis of human hepatocellular carcinoma, by
decreasing MMP-2 and uPA expression, thus leading to inhibition of
lung metastasis (85).
Furthermore, RNA aptamers have also been proposed to target OPN and
it has been demonstrated to decrease EMT and tumor growth (86,87).
Future studies aiming to improve the effects of
chemotherapy and radiotherapy could propose similar strategies
targeting OPN signaling and additional pathways associated with
cancer cell survival and resistance.
6. OPN splice variants and their potential
role in tumor resistance
Although previous studies have reported the
expression and roles of OPN-SI regarding distinct aspects of tumor
progression, data reporting the association between their
expression and resistance to therapy are limited. A recent report
proposed that OPNb and OPNc splicing isoforms are aberrantly
expressed in leukemia cells in response to distinct
chemotherapeutic drugs, such as daunorubicin, idarubicin and
cytarabine, further mediating resistance to these drugs (88). However, it was not clearly
demonstrated that specific knockdown of these splice variants
reverted chemoresistance. Our group pioneered the studies of OPN-SI
and their relation to chemoresistance, demonstrating that prostate
cancer cells that ectopically overexpress OPNb or OPNc are more
resistant to docetaxel (DXT) and display higher survival rates
(89). The DXT-resistant phenotype
was also associated with EMT features in which cells overexpressing
OPNb or OPNc exhibited upregulated mesenchymal markers, as opposed
to epithelial markers (89). In
summary, these data demonstrated that OPN-SI differently modulate
chemoresistance. As nuclear OPNc has been demonstrated as a
prognostic marker in breast cancer (90) and also reported to be correlated
with relapse (90,91) and poor survival (92), it is possible that nuclear OPNc may
also be a potential marker of response to chemotherapy or
radiotherapy, as has been previously reported (90). Future studies should further
investigate the roles of OPN-SI in chemoresistance in distinct
tumor models and also explore their involvement in
radioresistance.
7. Conclusions
Growing evidence has pointed to the crucial role
that OPN has in many aspects of cancer progression, including the
acquisition of drug resistance. OPN not only induces integrin
receptor-mediated oncogenic signaling pathways but also modulates
the epithelial-mesenchymal phenotype and stemness, conferring
cancer cells the ability to survive, proliferate, evade from cell
death, migrate and colonize other tissues. Consequently,
OPN-overexpressing cells are refractory to current treatment
options as well as exhibit invasive and metastatic potential.
Together, these findings provide evidence that OPN-triggered
signaling pathways can be targeted to specifically induce cell
death in chemo- and radio-insensitive cancer cells in order to
overcome the therapeutic resistant phenotype. Future studies will
uncover the role of specific OPN isoforms in the acquisition of
treatment resistance and also address whether OPN and its isoforms
may be reliable markers for cancer progression and poor response to
standard therapy.
Funding
The present study has been funded by FAPERJ (grant
nos. E-26/ 210.394/2014, E-26/010.002007/2014 and
E-26/203.204/2015), CNPq (grant no. 310591/2014-7); Ministério da
Sáude and UFF/Proppi (L'ORÉAL-UNESCO-ABC for Women in Science).
Availability of data and materials
Not applicable.
Authors' contributions
ERPG designed the manuscript, provided financial
support, wrote the manuscript, prepared figures, edited and
performed major revisions. MCB contributed to figure preparation
and revision, as well as writing and editing the manuscript. GNM
provided financial support, prepared figures, and contributed to
writing and editing the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Gottesman MM, Lavi O, Hall MD and Gillet
J-P: Toward a better understanding of the complexity of cancer drug
resistance. Annu Rev Pharmacol Toxicol. 56:85–102. 2016. View Article : Google Scholar
|
|
2
|
Bergman PJ and Harris D: Radioresistance,
chemoresistance, and apoptosis resistance The past, present, and
future. Vet Clin North Am Small Anim Pract. 27:47–57. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shevde LA and Samant RS: Role of
osteopontin in the pathophysiology of cancer. Matrix Biol.
37:131–141. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Clemente N, Raineri D, Cappellano G,
Boggio E, Favero F, Soluri MF, Dianzani C, Comi C, Dianzani U and
Chiocchetti A: Osteopontin bridging innate and adaptive immunity in
autoimmune diseases. J Immunol Res. 2016:76754372016. View Article : Google Scholar
|
|
5
|
Gu T, Ohashi R, Cui R, Tajima K, Yoshioka
M, Iwakami S, Sasaki S, Shinohara A, Matsukawa T, Kobayashi J, et
al: Osteopontin is involved in the development of acquired
chemo-resistance of cisplatin in small cell lung cancer. Lung
Cancer. 66:176–183. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Huang RH, Quan YJ, Chen JH, Wang TF, Xu M,
Ye M, Yuan H, Zhang CJ, Liu XJ and Min ZJ: Osteopontin promotes
cell migration and invasion, and inhibits apoptosis and autophagy
in colorectal cancer by activating the p38 MAPK signaling pathway.
Cell Physiol Biochem. 41:1851–1864. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu Y, Jiang W, Wang Y, Wu J, Saiyin H,
Qiao X, Mei X, Guo B, Fang X, Zhang L, et al: Breast cancer
metastasis suppressor 1 regulates hepatocellular carcinoma cell
apoptosis via suppressing osteopontin expression. PLoS One.
7:e429762012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu XL, Lin KJ, Bai AP, Wang WX, Meng XK,
Su XL, Hou MX, Dong PD, Zhang JJ, Wang ZY, et al: Osteopontin
knockdown suppresses the growth and angiogenesis of colon cancer
cells. World J Gastroenterol. 20:10440–10448. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shi Z, Wang B, Chihanga T, Kennedy MA and
Weber GF: Energy metabolism during anchorage-independence Induction
by osteopontin-c. PLoS One. 9:e1056752014. View Article : Google Scholar
|
|
10
|
Malaponte G, Hafsi S, Polesel J,
Castellano G, Spessotto P, Guarneri C, Canevari S, Signorelli SS,
McCubrey JA and Libra M: Tumor microenvironment in diffuse large
B-cell lymphoma: Matrixmetalloproteinases activation is mediated by
osteopontin overexpression. Biochim Biophys Acta. 1863:483–489.
2016. View Article : Google Scholar
|
|
11
|
Caputo S and Bellone M: Osteopontin and
the immune system: Another brick in the wall. Cell Mol Immunol.
15:405–407. 2018. View Article : Google Scholar
|
|
12
|
Mohammadi S, Ghaffari SH, Shaiegan M,
Zarif MN, Nikbakht M, Akbari Birgani S, Alimoghadam K and
Ghavamzadeh A: Acquired expression of osteopontin selectively
promotes enrichment of leukemia stem cells through
AKT/mTOR/PTEN/β-catenin pathways in AML cells. Life Sci.
152:190–198. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bellahcène A, Castronovo V, Ogbureke KUE,
Fisher LW and Fedarko NS: Small integrin-binding ligand N-linked
glycoproteins (SIBLINGs): Multifunctional proteins in cancer. Nat
Rev Cancer. 8:212–226. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Phillips RJ, Helbig KJ, Van der Hoek KH,
Seth D and Beard MR: Osteopontin increases hepatocellular carcinoma
cell growth in a CD44 dependant manner. World J Gastroenterol.
18:3389–3399. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rabenstein M, Vay SU, Flitsch LJ, Fink GR,
Schroeter M and Rueger MA: Osteopontin directly modulates cytokine
expression of primary microglia and increases their survival. J
Neuroimmunol. 299:130–138. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cook AC, Tuck AB, McCarthy S, Turner JG,
Irby RB, Bloom GC, Yeatman TJ and Chambers AF: Osteopontin induces
multiple changes in gene expression that reflect the six ‘hallmarks
of cancer’ in a model of breast cancer progression. Mol Carcinog.
43:225–236. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gimba ER and Tilli TM: Human osteopontin
splicing isoforms: Known roles, potential clinical applications and
activated signaling pathways. Cancer Lett. 331:11–17. 2013.
View Article : Google Scholar
|
|
18
|
Zhang H, Guo M, Chen JH, Wang Z, Du XF,
Liu PX and Li WH: Osteopontin knockdown inhibits αv,β3
integrin-induced cell migration and invasion and promotes apoptosis
of breast cancer cells by inducing autophagy and inactivating the
PI3K/Akt/mTOR pathway. Cell Physiol Biochem. 33:991–1002. 2014.
View Article : Google Scholar
|
|
19
|
Yang L, Wei L, Zhao W, Wang X, Zheng G,
Zheng M, Song X and Zuo W: Down-regulation of osteopontin
expression by RNA interference affects cell proliferation and
chemotherapy sensitivity of breast cancer MDA-MB-231 cells. Mol Med
Rep. 5:373–376. 2012.
|
|
20
|
Zhang A, Liu Y, Shen Y, Xu Y and Li X:
Osteopontin silencing by small interfering RNA induces apoptosis
and suppresses invasion in human renal carcinoma Caki-1 cells. Med
Oncol. 27:1179–1184. 2010. View Article : Google Scholar
|
|
21
|
Naor D, Wallach-Dayan SB, Zahalka MA and
Sionov RV: Involvement of CD44, a molecule with a thousand faces,
in cancer dissemination. Semin Cancer Biol. 18:260–267. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
De Wever O, Demetter P, Mareel M and
Bracke M: Stromal myofibroblasts are drivers of invasive cancer
growth. Int J Cancer. 123:2229–2238. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Polyak K, Haviv I and Campbell IG:
Co-evolution of tumor cells and their microenvironment. Trends
Genet. 25:30–38. 2009. View Article : Google Scholar
|
|
24
|
Kale S, Raja R, Thorat D, Soundararajan G,
Patil TV and Kundu GC: Osteopontin signaling upregulates
cyclooxygenase-2 expression in tumor-associated macrophages leading
to enhanced angiogenesis and melanoma growth via α9β1 integrin.
Oncogene. 33:2295–2306. 2014. View Article : Google Scholar
|
|
25
|
Lin CN, Wang CJ, Chao YJ, Lai M-D and Shan
Y-S: The significance of the co-existence of osteopontin and
tumor-associated macrophages in gastric cancer progression. BMC
Cancer. 15:1282015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shao L, Zhang B, Wang L, Wu L, Kan Q and
Fan K: MMP-9-cleaved osteopontin isoform mediates tumor immune
escape by inducing expansion of myeloid-derived suppressor cells.
Biochem Biophys Res Commun. 493:1478–1484. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Guarneri C, Bevelacqua V, Polesel J,
Falzone L, Cannavò PS, Spandidos DA, Malaponte G and Libra M: NF-κB
inhibition is associated with OPN/MMP 9 downregulation in cutaneous
melanoma. Oncol Rep. 37:737–746. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu GX, Sun JT, Yang MX, Qi XM, Shao QQ,
Xie Q, Qu X, Wei FC and Sun SZ: OPN promotes survival of activated
T cells by up-regulating CD44 in patients with oral lichen planus.
Clin Immunol. 138:291–298. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Matušan-Ilijaš K, Damante G, Fabbro D,
Dorđević G, Hadžisejdić I, Grahovac M, Marić I, Spanjol J, Grahovac
B, Jonjić N, et al: Osteopontin expression correlates with nuclear
factor-κB activation and apoptosis downregulation in clear cell
renal cell carcinoma. Pathol Res Pract. 207:104–110. 2011.
View Article : Google Scholar
|
|
30
|
Song G, Cai QF, Mao YB, Ming YL, Bao SD
and Ouyang GL: Osteopontin promotes ovarian cancer progression and
cell survival and increases HIF-1alpha expression through the
PI3-K/Akt pathway. Cancer Sci. 99:1901–1907. 2008.PubMed/NCBI
|
|
31
|
Wang HL, Ruan LH and Zhao XQ: Expression
of osteopontin and VEGF in acute leukemia and their relationship
with angiogenesis. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 19:926–929.
2011.In Chinese. PubMed/NCBI
|
|
32
|
Castello LM, Raineri D, Salmi L, Clemente
N, Vaschetto R, Quaglia M, Garzaro M, Gentilli S, Navalesi P,
Cantaluppi V, et al: Osteopontin at the crossroads of inflammation
and tumor progression. Mediators Inflamm. 2017:40490982017.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dai J, Peng L, Fan K, Wang H, Wei R, Ji G,
Cai J, Lu B, Li B, Zhang D, et al: Osteopontin induces angiogenesis
through activation of PI3K/AKT and ERK1/2 in endothelial cells.
Oncogene. 28:3412–3422. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chakraborty G, Jain S and Kundu GC:
Osteopontin promotes vascular endothelial growth factor-dependent
breast tumor growth and angiogenesis via autocrine and paracrine
mechanisms. Cancer Res. 68:152–161. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tilli TM, Mello KD, Ferreira LB, Matos AR,
Accioly MT, Faria PA, Bellahcène A, Castronovo V and Gimba ER: Both
osteopontin-c and osteopontin-b splicing isoforms exert
pro-tumorigenic roles in prostate cancer cells. Prostate.
72:1688–1699. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nuñez-Garcia M, Gomez-Santos B, Buqué X,
García-Rodriguez JL, Romero MR, Marin JJG, Arteta B, García-Monzón
C, Castaño L, Syn WK, et al: Osteopontin regulates the cross-talk
between phosphatidylcholine and cholesterol metabolism in mouse
liver. J Lipid Res. 58:1903–1915. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Briones-Orta MA, Avendaño-Vázquez SE,
Aparicio-Bautista DI, Coombes JD, Weber GF and Syn W-K: Osteopontin
splice variants and polymorphisms in cancer progression and
prognosis. Biochim Biophys Acta Rev Cancer. 1868:93–108. 108.A2017.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lin J, Myers AL, Wang Z, Nancarrow DJ,
Ferrer-Torres D, Handlogten A, Leverenz K, Bao J, Thomas DG, Wang
TD, et al: Osteopontin (OPN/SPP1) isoforms collectively enhance
tumor cell invasion and dissemination in esophageal adenocarcinoma.
Oncotarget. 6:22239–22257. 2015.PubMed/NCBI
|
|
39
|
Choi SI, Kim SY, Lee JH, Kim JY, Cho EW
and Kim IG: Osteopontin production by TM4SF4 signaling drives a
positive feedback autocrine loop with the STAT3 pathway to maintain
cancer stem cell-like properties in lung cancer cells. Oncotarget.
8:101284–101297. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sui H, Zhu L, Deng W and Li Q:
Epithelial-mesenchymal transition and drug resistance: Role,
molecular mechanisms, and therapeutic strategies. Oncol Res Treat.
37:584–589. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ye X and Weinberg RA:
Epithelial-mesenchymal plasticity: A central regulator of cancer
progression. Trends Cell Biol. 25:675–686. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu X and Fan D: The
epithelial-mesenchymal transition and cancer stem cells: Functional
and mechanistic links. Curr Pharm Des. 21:1279–1291. 2015.
View Article : Google Scholar
|
|
43
|
Li L and Li W: Epithelial-mesenchymal
transition in human cancer: Comprehensive reprogramming of
metabolism, epigenetics, and differentiation. Pharmacol Ther.
150:33–46. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gonzalez DM and Medici D: Signaling
mechanisms of the epithelial-mesenchymal transition. Sci Signal.
7:re82014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li F and Sethi G: Targeting transcription
factor NF-kappaB to overcome chemoresistance and radioresistance in
cancer therapy. Biochim Biophys Acta. 1805:167–180. 2010.PubMed/NCBI
|
|
46
|
Mohammadi S, Zahedpanah M, Ghaffari SH,
Shaiegan M, Nikbakht M and Nikugoftar M: Osteopontin plays a unique
role in resistance of CD34+/CD123+ human
leukemia cell lines KG1a to parthenolide. Life Sci. 189:89–95.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Liersch R, Shin JW, Bayer M, Schwöppe C,
Schliemann C, Berdel WE, Mesters R and Detmar M: Analysis of a
novel highly metastatic melanoma cell line identifies osteopontin
as a new lymphangiogenic factor. Int J Oncol. 41:1455–1463.
2012.PubMed/NCBI
|
|
48
|
Yi H, Zeng D, Shen Z, Liao J, Wang X, Liu
Y, Zhang X and Kong P: Integrin alphavbeta3 enhances β-catenin
signaling in acute myeloid leukemia harboring Fms-like tyrosine
kinase-3 internal tandem duplication mutations: Implications for
micro-environment influence on sorafenib sensitivity. Oncotarget.
7:40387–40397. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cao L, Fan X, Jing W, Liang Y, Chen R, Liu
Y, Zhu M, Jia R, Wang H, Zhang X, et al: Osteopontin promotes a
cancer stem cell-like phenotype in hepatocellular carcinoma cells
via an integrin-NF-κB-HIF-1α pathway. Oncotarget. 6:6627–6640.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Luo SD, Chen YJ, Liu CT, Rau KM, Chen YC,
Tsai HT, Chen CH and Chiu TJ: Osteopontin involves cisplatin
resistance and poor prognosis in oral squamous cell carcinoma.
BioMed Res Int. 2015:5085872015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ng L, Wan T, Chow A, Iyer D, Man J, Chen
G, Yau TC, Lo O, Foo CC, Poon JT, et al: Osteopontin overexpression
induced tumor progression and chemoresistance to oxaliplatin
through induction of stem-like properties in human colorectal
cancer. Stem Cells Int. 2015:2478922015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Qian C, Li P, Yan W, Shi L, Zhang J, Wang
Y, Liu H and You Y: Downregulation of osteopontin enhances the
sensitivity of glioma U251 cells to temozolomide and cisplatin by
targeting the NF-κB/Bcl 2 pathway. Mol Med Rep. 11:1951–1955. 2015.
View Article : Google Scholar
|
|
53
|
Pang H, Cai L, Yang Y, Chen X, Sui G and
Zhao C: Knockdown of osteopontin chemosensitizes MDA-MB-231 cells
to cyclophosphamide by enhancing apoptosis through activating p38
MAPK pathway. Cancer Biother Radiopharm. 26:165–173. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Tajima K, Ohashi R, Sekido Y, Hida T, Nara
T, Hashimoto M, Iwakami S, Minakata K, Yae T, Takahashi F, et al:
Osteopontin-mediated enhanced hyaluronan binding induces multidrug
resistance in mesothelioma cells. Oncogene. 29:1941–1951. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wohlleben G, Scherzad A, Güttler A,
Vordermark D, Kuger S, Flentje M and Polat B: Influence of hypoxia
and irradiation on osteopontin expression in head and neck cancer
and glioblastoma cell lines. Radiat Oncol. 10:1672015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ostheimer C, Schweyer F, Reese T, Bache M
and Vordermark D: The relationship between tumor volume changes and
serial plasma osteopontin detection during radical radiotherapy of
non-small-cell lung cancer. Oncol Lett. 12:3449–3456. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wang M, Han J, Marcar L, Black J, Liu Q,
Li X, Nagulapalli K, Sequist LV, Mak RH, Benes CH, et al: Radiation
resistance in KRAS-mutated lung cancer is enabled by stem-like
properties mediated by an osteopontin-EGFR pathway. Cancer Res.
77:2018–2028. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Chang SH, Minai-Tehrani A, Shin JY, Park
S, Kim JE, Yu KN, Hong SH, Hong CM, Lee KH, Beck GR Jr, et al:
Beclin1-induced autophagy abrogates radioresistance of lung cancer
cells by suppressing osteopontin. J Radiat Res (Tokyo). 53:422–432.
2012. View Article : Google Scholar
|
|
59
|
Huang X, Qian Y, Wu H, Xie X, Zhou Q, Wang
Y, Kuang W, Shen L, Li K, Su J, et al: Aberrant expression of
osteopontin and E-cadherin indicates radiation resistance and poor
prognosis for patients with cervical carcinoma. J Histochem
Cytochem. 63:88–98. 2015. View Article : Google Scholar :
|
|
60
|
Pietras A, Katz AM, Ekström EJ, Wee B,
Halliday JJ, Pitter KL, Werbeck JL, Amankulor NM, Huse JT and
Holland EC: Osteopontin-CD44 signaling in the glioma perivascular
niche enhances cancer stem cell phenotypes and promotes aggressive
tumor growth. Cell Stem Cell. 14:357–369. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
van Staalduinen J, Baker D, Ten Dijke P
and van Dam H: Epithelial-mesenchymal-transition-inducing
transcription factors: New targets for tackling chemoresistance in
cancer? Oncogene. Jul 12–2018.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Shibue T and Weinberg RA: EMT, CSCs, and
drug resistance: The mechanistic link and clinical implications.
Nat Rev Clin Oncol. 14:611–629. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Vega S, Morales AV, Ocaña OH, Valdés F,
Fabregat I and Nieto MA: Snail blocks the cell cycle and confers
resistance to cell death. Genes Dev. 18:1131–1143. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Cichon MA and Radisky DC: ROS-induced
epithelial-mesenchymal transition in mammary epithelial cells is
mediated by NF-κB-dependent activation of Snail. Oncotarget.
5:2827–2838. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kothari AN, Arffa ML, Chang V, Blackwell
RH, Syn WK, Zhang J, Mi Z and Kuo PC: Osteopontin-A Master
Regulator of Epithelial-Mesenchymal Transition. J Clin Med.
5:52016. View Article : Google Scholar
|
|
67
|
Yu X, Zheng Y, Zhu X, Gao X, Wang C, Sheng
Y, Cheng W, Qin L, Ren N, Jia H, et al: Osteopontin promotes
hepatocellular carcinoma progression via the PI3K/AKT/Twist
signaling pathway. Oncol Lett. 16:5299–5308. 2018.PubMed/NCBI
|
|
68
|
Li NY, Weber CE, Mi Z, Wai PY, Cuevas BD
and Kuo PC: Osteopontin up-regulates critical
epithelial-mesenchymal transition transcription factors to induce
an aggressive breast cancer phenotype. J Am Coll Surg. 217:17–26.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Dong Q, Zhu X, Dai C, Zhang X, Gao X, Wei
J, Sheng Y, Zheng Y, Yu J, Xie L, et al: Osteopontin promotes
epithelial-mesenchymal transition of hepatocellular carcinoma
through regulating vimentin. Oncotarget. 7:12997–13012. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Weber CE, Li NY, Wai PY and Kuo PC:
Epithelial-mesenchymal transition, TGF-β, and osteopontin in wound
healing and tissue remodeling after injury. J Burn Care Res.
33:311–318. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Jia R, Liang Y, Chen R, Liu G, Wang H,
Tang M, Zhou X, Wang H, Yang Y, Wei H, et al: Osteopontin
facilitates tumor metastasis by regulating epithelial-mesenchymal
plasticity. Cell Death Dis. 7:e25642016. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zahed Panah M, Nikbakht M, Sajjadi SM,
Rostami S, Norooznezhad AH, Kamranzadeh Fumani H, Ghavamzadeh A and
Mohammadi S: Anti-apoptotic effects of osteopontin via the
up-regulation of AKT/mTOR/β-catenin loop in acute myeloid leukemia
cells. Int J Hematol Oncol Stem Cell Res. 11:148–157.
2017.PubMed/NCBI
|
|
73
|
Li X, Jiang Z, Li X and Zhang X: SIRT1
overexpression protects non-small cell lung cancer cells against
osteopontin-induced epithelial-mesenchymal transition by
suppressing NF-κB signaling. OncoTargets Ther. 11:1157–1171. 2018.
View Article : Google Scholar
|
|
74
|
Castellano G, Malaponte G, Mazzarino MC,
Figini M, Marchese F, Gangemi P, Travali S, Stivala F, Canevari S
and Libra M: Activation of the osteopontin/matrix
metalloproteinase-9 pathway correlates with prostate cancer
progression. Clin Cancer Res. 14:7470–7480. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Tan TK, Zheng G, Hsu TT, Lee SR, Zhang J,
Zhao Y, Tian X, Wang Y, Wang YM, Cao Q, et al: Matrix
metalloproteinase-9 of tubular and macrophage origin contributes to
the pathogenesis of renal fibrosis via macrophage recruitment
through osteopontin cleavage. Lab Invest. 93:434–449. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Yang MC, Wang HC, Hou YC, Tung HL, Chiu TJ
and Shan YS: Blockade of autophagy reduces pancreatic cancer stem
cell activity and potentiates the tumoricidal effect of
gemcitabine. Mol Cancer. 14:1792015. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Huang G, Du M-Y, Zhu H, Zhang N, Lu ZW,
Qian LX, Zhang W, Tian X, He X and Yin L: MiRNA-34a reversed
TGF-β-induced epithelial-mesenchymal transition via suppression of
SMAD4 in NPC cells. Biomed Pharmacother. 106:217–224. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Cui H, Zhang AJ, Chen M and Liu JJ: ABC
transporter inhibitors in reversing multidrug resistance to
chemotherapy. Curr Drug Targets. 16:1356–1371. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Zhang F, Luo W, Li Y, Gao S and Lei G:
Role of osteopontin in rheumatoid arthritis. Rheumatol Int.
35:589–595. 2015. View Article : Google Scholar
|
|
80
|
Shevde LA, Metge BJ, Mitra A, Xi Y, Ju J,
King JA and Samant RS: Spheroid-forming subpopulation of breast
cancer cells demonstrates vasculogenic mimicry via hsa-miR-299-5p
regulated de novo expression of osteopontin. J Cell Mol Med.
14:1693–1706. 2010. View Article : Google Scholar
|
|
81
|
Bhattacharya SD, Mi Z, Kim VM, Guo H,
Talbot LJ and Kuo PC: Osteopontin regulates epithelial mesenchymal
transition-associated growth of hepatocellular cancer in a mouse
xenograft model. Ann Surg. 255:319–325. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Wang J, Yang M, Li Y and Han B: The role
of microRNAs in the chemoresistance of breast cancer. Drug Dev Res.
76:368–374. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Liang J, Xu L, Zhou F, Liu AM, Ge HX, Chen
YY and Tu M: MALAT1/miR-127-5p regulates osteopontin (OPN)-mediated
proliferation of human chondrocytes through PI3K/Akt pathway. J
Cell Biochem. 119:431–439. 2018. View Article : Google Scholar
|
|
84
|
Boguslawska J, Sokol E, Rybicka B, Czubaty
A, Rodzik K and Piekielko-Witkowska A: microRNAs target SRSF7
splicing factor to modulate the expression of osteopontin splice
variants in renal cancer cells. Gene. 595:142–149. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Sun BS, Dong QZ, Ye QH, Sun HJ, Jia HL,
Zhu XQ, Liu DY, Chen J, Xue Q, Zhou HJ, et al: Lentiviral-mediated
miRNA against osteopontin suppresses tumor growth and metastasis of
human hepatocellular carcinoma. Hepatology. 48:1834–1842. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Hunter C, Bond J, Kuo PC, Selim MA and
Levinson H: The role of osteopontin and osteopontin aptamer
(OPN-R3) in fibroblast activity. J Surg Res. 176:348–358. 2012.
View Article : Google Scholar :
|
|
87
|
Talbot LJ, Mi Z, Bhattacharya SD, Kim V,
Guo H and Kuo PC: Pharmacokinetic characterization of an RNA
aptamer against osteopontin and demonstration of in vivo efficacy
in reversing growth of human breast cancer cells. Surgery.
150:224–230. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Mirzaei A, Mohammadi S, Ghaffari SH,
Nikbakht M, Bashash D, Alimoghaddam K and Ghavamzadeh A:
Osteopontin b and c isoforms: Molecular Candidates Associated with
Leukemic Stem Cell Chemoresistance in Acute Myeloid Leukemia. Asian
Pac J Cancer Prev. 18:1707–1715. 2017.PubMed/NCBI
|
|
89
|
Nakamura KDM, Tilli TM, Wanderley JL,
Palumbo A Jr, Mattos RM, Ferreira AC, Klumb CE, Nasciutti LE and
Gimba ER: Osteopontin splice variants expression is involved on
docetaxel resistance in PC3 prostate cancer cells. Tumour Biol.
37:2655–2663. 2016. View Article : Google Scholar
|
|
90
|
Zduniak K, Ziolkowski P, Ahlin C, Agrawal
A, Agrawal S, Blomqvist C, Fjällskog ML and Weber GF: Nuclear
osteopontin-c is a prognostic breast cancer marker. Br J Cancer.
112:729–738. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Ortiz-Martínez F, Perez-Balaguer A,
Ciprián D, Andrés L, Ponce J, Adrover E, Sánchez-Payá J, Aranda FI,
Lerma E and Peiró G: Association of increased osteopontin and
splice variant-c mRNA expression with HER2 and
triple-negative/basal-like breast carcinomas subtypes and
recurrence. Hum Pathol. 45:504–512. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Patani N, Jiang W and Mokbel K:
Osteopontin C mRNA expression is associated with a poor clinical
outcome in human breast cancer. Int J Cancer. 122:26462008.
View Article : Google Scholar : PubMed/NCBI
|














