Introduction
Hypoxia is defined as a low oxygen concentration of
<2% (1). Hypoxia occurs in
inflammation, stroke, tissue ischemia and tumor growth. In the
context of hypoxia, cells adapt to the energy production process by
minimizing oxygen consumption and converting to corresponding
anaerobic processes to stimulate cell growth, anti-apoptotic and
angiogenic signaling pathways (2,3).
Angiogenesis is a key step in tumor growth and metastasis (4). Several angiogenic molecules,
including vascular endothelial growth factor (VEGF), are secreted
from cancer cells in response to hypoxic stress; in particular,
secreted VEGF increases endothelial cell proliferation, migration
and differentiation, thereby affecting cancer progression (5).
Over the past decade, interest in tumor metabolism
has increased. Major hallmarks of cancer cells include abnormal
metabolism, energy production processes involving the mitochondrial
tricarboxylic acid (TCA) cycle and anaerobic digestion processes in
the cytoplasm, rather than oxidative phosphorylation, which can use
oxygen. This phenomenon was first described by Warburg (6,7) and
allows cancer cells to proliferate under hypoxic conditions (≤2%
O2) (8). Notably,
hypoxia involves a shift from adenosine triphosphate (ATP)
production through oxidative phosphorylation to that through
glycolysis under normal oxygen conditions. As a result, cancer
cells acquire large amounts of energy by converting the majority of
glucose to lactic acid. This phenomenon is known as the ‘Warburg
effect’. In addition, specific mutations in mitochondrial metabolic
enzymes have been shown to initiate carcinogenesis (9-11).
Isocitrate dehydrogenase (IDH)1 and IDH2 are
homodimeric enzymes that convert isocitrate to α-ketoglutarate
(α-KG), reduce NADP+ to NADPH, and release carbon
dioxide in glutamine, glucose and fatty acid metabolism (12). IDH1 is localized in the cytoplasm,
whereas IDH2 is localized in the mitochondria (13,14).
Somatic mutations in IDH1 were first observed in patients
with glioblastoma (15).
IDH2 mutations have also been identified in glioma, albeit
much less frequently than IDH1 mutations. Moreover, various
IDH1/2 point mutations have been identified in acute myeloid
leukemia (16), chondrosarcoma
(17), intrahepatic
cholangiocarcinoma (18),
angioimmunoblastic T-cell lymphoma (19), gastric cancer (20), colorectal cancer (21) and other types of cancer (22,23).
Almost all IDH1/2 mutations cause a single amino acid
substitution, Arg132 in IDH1 (to 1 of 6 amino acid residues:
His, Cys, Leu, Ile, Ser, Gly and Val), or corresponding Arg172 in
IDH2 (to 1 of 4 different residues: Lys, Met, Gly and Trp)
(24). Major mutations are R132H
in IDH1 and R172K in IDH2. A new reaction product,
2-hydroxyglutarate (2HG), has also been identified in the presence
of IDH1/2 mutations. 2HG has two enantiomers, d (or R)-2HG and l (or S)-2HG. Only d-2HG is generated by mutations
in IDH1/2. d-2HG, a
competitive inhibitor of α-KG, inhibits α-KG-dependent
dioxygenases, such as ten-eleven translocation methylcytosine
dioxygenase DNA demethylases, JmjC demethylases and proline
hydroxylases, resulting in epigenetic alterations (25-27).
d-2HG has been known
as an oncometabolite owing to its involvement in diverse biological
processes related to tumorigenesis (25-27).
This metabolite also directly induces epithelial-mesenchymal
transition and increases metastatic potential in colorectal cancer
(28). However, d-2HG inhibits hypoxia-inducible
factor (HIF) expression by promoting the activity of the HIF-1
prolyl hydroxylases Egl nine homolog (EGLN)1 and EGLN2, suggesting
that d-2HG has dual
functions as an oncometabolite and tumor suppressor (29). Hypoxia increases the formation of
l-2HG independent of
the IDH mutation, and elevated l-2HG stabilizes HIF-1α (30). Therefore, whether d-2HG increases angiogenesis
in vitro and in vivo remains unclear, and the
mechanisms mediating the angiogenic activities of d-2HG have not yet been
elucidated.
Accordingly, in this study, we compared metabolites
in the hypoxic state and in human breast cancer cells
overexpressing mutant IDH2. Following the identification of
d-2HG in this
analysis, we further examined the roles of this metabolite in
angiogenesis in vitro and ex vivo. The findings of
this study may provide important insight into the effects of
hypoxia and IDH2 mutations on metabolism and angiogenesis in
cancer.
Materials and methods
Cell culture
HepG2 human liver cancer (Korean Cell Line Bank,
Seoul, Korea), The MDA-MB-231 human breast cancer cells, A549 human
lung cancer cells, 293T cells (all from ATCC, Manassas, VA, USA)
and bovine aortic endothelial cells (BAECs; passages 5-14; Lonza
Biosciences, Basel, Switzerland) were grown on culture plates in
Dulbecco's modified Eagle's medium (DMEM; HyClone, Logan, UT, USA)
supplemented with 10% fetal bovine serum (FBS; HyClone), 1%
antibiotics (100 U/ml penicillin, 100 mg/ml streptomycin; Thermo
Fisher Scientific, Waltham, MA, USA). The cells were maintained at
37˚C in an incubator under a humidified atmosphere of 5%
CO2 and 95% air.
Lentiviral transduction of IDH2
mutants
The lentiviral expression vector for mutant IDH2
[pLenti6.2/V5-IDH2 (R172K)] was kindly provided by Professor Hai
Yan of Duke University. Lentiviral particles were packaged by
transfecting the 293T cells with pLenti6.2/V5 or pLenti6.2/V5-IDH2
(R172K) (kindly provided by Dr Hai Yan, Duke University Durham, NC,
USA) using Polyfect reagent (Qiagen, Valencia, CA, USA) according
to the manufacturer's instructions. Following 48 h of culture in
DMEM supplemented with 10% FBS, viral supernatants were collected
and filtered through a 0.45-µm syringe filter. The
MDA-MB-231 cells were infected with the viral supernatants in the
presence of polybrene (10 µg/ml) for 6 h.
Metabolite profiling of cancer cells
The targeted quantitative analysis of metabolites
was performed by Human Metabolome Technologies America (HMT,
Boston, MA, USA) based on methods previously described (31), using capillary electrophoresis mass
spectrometry (Agilent Technologies, Santa Clara, CA, USA).
Metabolites were extracted from the MDA-MB-231 cells treated under
normoxic or hypoxic conditions for 16 h and MDA-MB-231 cells that
were transduced with IDH2-R172K mutant using lentivirus, as
described above, under normoxic conditions. In our results, 116 of
the analyzed metabolites were involved in glycolysis; the pentose
phosphate pathway (PPP); the TCA (citric acid/Krebs) cycle; the
urea cycle; and polyamine, creatine, purine, glutathione,
nicotinamide, choline and amino acid metabolism.
Analysis of d-2HG levels in cells
The MDA-MB-231 cells were transduced with lentiviral
particles by transfection with pLenti6.2/V5-IDH2 (R172K) or exposed
to hypoxic conditions for 24 h. d-2HG contents in cell lysates or
medium were then determined using a d-2HG assay kit according to the
manufacturer's instructions (BioVision Inc., San Francisco, CA,
USA).
Cell counting kit-8 (CCK-8) assays
The BAECs were seeded at 5×103 cells/well
in 96-well plates and incubated for 24 h. After 24 h, the medium
was replaced with low-serum medium (0.5% FBS in DMEM/low glucose),
and the cells were starved for 16 h. Subsequently, the medium was
changed to medium containing VEGF (40 ng/ml) or various
concentrations of d-2HG (Sigma-Aldrich, St. Louis,
MO, USA). The BAECs were incubated for 24 h, and CCK-8 reagent (10
µl; Dojindo Laboratories, Seoul, Korea) was added to each
well. The cells were then incubated for 3 h at 37˚C, and the
optical density was measured with a microplate reader (Infinite
M200 Pro; Tecan, Mannedorf, Switzerland) at 450 nm.
Enzyme-linked immunosorbent assay
(ELISA)
The amount of VEGF protein secreted by the A549
human lung cancer cells into the medium was determined using a VEGF
ELISA kit (R&D Systems, Wiesbaden, Germany). The cells were
plated in 6-well plates, cultured until reaching 80-90% confluence,
starved with starvation medium (0.5% FBS in DMEM) for 16 h, and
then treated with d-2HG (250 µM or 5 mM) for
24 h. The medium was collected, and the amount of VEGF secreted
into the medium was quantified using a microplate reader (Infinite
M200 Pro; Tecan).
BrdU cell proliferation assays
The BAECs were seeded at 2×103 cells/well
in 96-well culture plates and then incubated for 24 h. The cells
were then starved with 0.5% FBS in DMEM/low glucose for 16 h. The
cells were subsequently incubated with VEGF (40 ng/ml) for 24 h in
the presence of various concentrations of d-2HG in DMEM/low glucose
containing 0.5% FBS. Cell proliferation was measured with a Cell
Proliferation ELISA BrdU kit (Roche, Mannheim, Germany).
Ki67 immunofluorescence staining
The BAECs were seeded at 1×104 cells/well
on coverslips in 4-well plates and allowed to attach for 24 h. The
cells were then starved with 0.5% FBS in DMEM/low glucose for 16 h.
The cells were incubated with VEGF (40 ng/ml) for 24 h in the
presence of various concentrations of d-2HG in DMEM/low glucose
containing 0.5% FBS. The cells were then fixed with 4%
paraformaldehyde solution and permeabilized with 0.1% Triton
X-100/phosphate-buffered saline (PBS). Fixed cells were blocked
with 1% bovine serum albumin (BSA) in PBS and incubated with
anti-Ki67 antibodies (1/200; sc-23900; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA) for 3 h at room temperature. The cells were
then incubated for 1 h at room temperature with Alexa Fluor
488-conjugated anti-rabbit IgG as secondary antibodies (1/200;
A-11008; Thermo Fisher Scientific) and Hoechst to label the nuclei
(green) and observed under a Leica TCS SP5 Ⅱ confocal microscope
(Leica Microsystems, Wetzlar, Germany).
Transwell migration assay
The chemotactic migration ability of the BAECs was
examined using a 24-Transwell system (6.5 mm diameter, 8 µm
pore size; Corning Costar, Lowell, MA, USA). The BAECs were starved
in 0.5% FBS in DMEM/low glucose for 16 h and seeded at
1×105 cells/well into the upper chambers of the plates
with 0.5% FBS in DMEM/low glucose. The lower chambers were filled
with 600 µl of 0.5% FBS in DMEM/low glucose with or without
VEGF (40 ng/ml) and d-2HG (250 µM) as
chemo-attractants. Following incubation at 37°C for 24 h, the
migrated cells were fixed with 100% methanol (Merck, Kenilworth,
NJ, USA) and stained with hematoxylin and eosin
(Sigma-Aldrich).
Wound healing assay
The BAECs were plated at 4×105 cells/well
on 12-well plate and allowed to reach 90% confluence. The BAECs
were then starved in 0.5% FBS in DMEM/low glucose for 16 h and
treated with mitomycin C (1 µg/ml) for 1 h. Thereafter, cell
layers were scratched with a 1-ml micropipette tip. Plates were
rinsed with PBS and incubated at 37°C for 24 h with starvation
medium with or without VEGF (40 ng/ml) or d-2HG (250 µM). Wound
closure was monitored and photographed at 0 and 24 h using an
inverted phase-contrast microscope (CKX41SF, Olympus, Tokyo,
Japan). The distance of cell migration was measured based on the
original location of the cells immediately after the cell layers
were scratched. These experiments were repeated at least twice with
similar results.
In vitro tube formation assay
Growth factor-reduced Matrigel (BD) was added to
96-well plates and polymerized for 30 min at 37°C. The BAECs were
starved in 0.5% FBS in DMEM/low-glucose for 16 h, seeded at
3×104 cells on the surface of the Matrigel, and
incubated for 8-16 h with or without VEGF (40 ng/ml) or
d-2HG (250 µM)
in DMEM/low-glucose containing 0.5% FBS. Morphological changes were
observed under a microscope (CKX41SF, Olympus) and photographed at
×100 magnification. The results are presented as the mean number of
branching points.
Chick embryo chorioallantoic membrane
(CAM) assay
Fertilized chicken eggs (CJ Corp., Seoul, Korea)
were maintained in a humidified incubator (Eunjo Incubator Company,
Pocheon, Korea) at 37°C for 3 days. CAM assay was performed as
previously described (3). Briefly,
on day 4.5, chemical-loaded Thermanox coverslips (Nunc, Rochester,
NY, USA) were applied to the CAM surface. Two days later, a 20% fat
emulsion (SMOFlipid, Fresenius Kabi Austria GmbH, Hafnerstra,
Austria) was injected beneath the CAM and observed under a
stereomicroscope. Phorbol 12-myristate 13-acetate (PMA) was used as
a positive control. Fifteen eggs per group were used in each
experiment, and experiments were performed in triplicate.
Gelatin zymography
Supernatants from cell cultures were analyzed for
gelatin degradation activity of matrix metalloproteinase (MMP) by
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) under non-reducing conditions. Gelatin (1 mg/ml) was
pre-polymerized on 10% polyacrylamide gels as a substrate.
Electrophoresis was carried out at room temperature. The gels were
washed with 2.5% Triton X-100 washing buffer twice, followed by
incubation with a zymography reaction buffer at 37°C overnight. The
samples were stained with Coomassie Blue (Thermo Fisher Scientific)
for 10 min and dried at room temperature.
Whole-cell extract preparation and
western blot analysis
Cell extract preparation and western blot analysis
were performed as previously described (32). Briefly, the cells were lysed with
PRO-PREP protein extraction solution (iNtRON Biotechnology, Seoul,
Korea), and the lysates protein concentrations were measured using
a Pierce BCA protein assay kit (Thermo Fisher Scientific). Proteins
(50 µg) were loaded and separated by 10% SDS-PAGE and
transferred onto nitrocellulose membranes (Whatman Inc., Maidstone,
UK). After blocking for 30 min with 5% skim milk at room
temperature, the nitrocellulose membranes were incubated with
primary antibodies for overnigh at 4°C and secondary antibodies for
1 h at room temperature. The immune complexes were detected using
enhanced chemiluminescence reagents (34577; Thermo Fisher
Scientific). Primary antibodies used for western blot analysis were
as follows: HIF-1α (1/500; Cat. no. 610958; BD Biosciences, San
Diego, CA, USA), IDH1 (1/1,000; Cat. no. 3997; Cell Signaling
Technology, Danvers, MA, USA), IDH2 (1/1,000; Cat. no. ab131263;
Abcam, Cambridge, UK), IDH2-172K (1/500; Cat. no. D-328-3; MBL Co.
Ltd., Nagoya, Japan), phospho-VEGFR2 (1/500; Cat. no. 2478; Cell
Signaling Technology), VEGFR2 (1/1,000; Cat. no. 2479; Cell
Signaling Technology), phospho-ERK (1/1,000; Cat. no. 9101; Cell
Signaling Technology), ERK (1/1,000; Cat. no. 9102; Cell Signaling
Technology), phospho-FAK (1/1,000; Cat. no. sc-81493; Santa Cruz
Biotechnology), FAK (1/1,000; Cat. no. ab40794; Abcam), phospho-AKT
(1/1,000; Cat. no. 9271; Cell Signaling Technology), AKT (1/1,000;
Cat. no. 4691; Cell Signaling Technology) and β-actin (1/1,000;
sc-47778; Santa Cruz Biotechnology). HRP-linked secondary
antibodies were obtained from Cell Signaling Technology (1/5,000;
Cat. nos. 7076 and 7074).
RNA isolation and reverse
transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from IDH2-R172K
mutant cells overexpressing HepG2 using TRIzol reagent
(Invitrogen/Thermo Fisher Scientific) according to the
manufacturer's instructions, and cDNA synthesis was carried out
with ReverTra Ace qPCR Master Mix (Toyobo, Osaka, Japan).
Semi-quantitative PCR was performed as previously described using
AccuPower PCR premix (Bioneer, Daejeon, Korea). The PCR conditions
were as follows: 30 cycles of denaturation (94°C/1 min), annealing
(60°C/1 min), and extension (72°C/1 min), and final extension
(72°C/10 min). The primers used for HIF-1α were as follows:
forward, 5'-CAGAAGATACAAGTAGCCTC-3' and reverse,
5'-GGCCCGGTGCAGCACCACCA-3'; and those for β-actin were as
follows: forward, 5'-GACTACCTCATGAAGATC-3' and reverse,
5'-GATCCACATCTGCTGGAA-3'. PCR products were separated on 1.5%
agarose gels in 1X TAE buffer, stained with ethidium bromide and
visualized under UV light.
Statistical analysis
All data are expressed as the means ± standard
deviations (SD) from at least 3 samples. Statistical comparisons
were determined by a Student's t-test using GraphPad Prism 5.04
(GraphPad Software, Inc., La Jolla, CA, USA). Comparisons among
multiple groups were analyzed using one-way analysis of variance
(ANOVA) with the Newman-Keuls post hoc test. Differences with
P<0.05 were considered statistically significant.
Results
2HG levels are increased by hypoxia and
IDH2 mutations in breast cancer cells
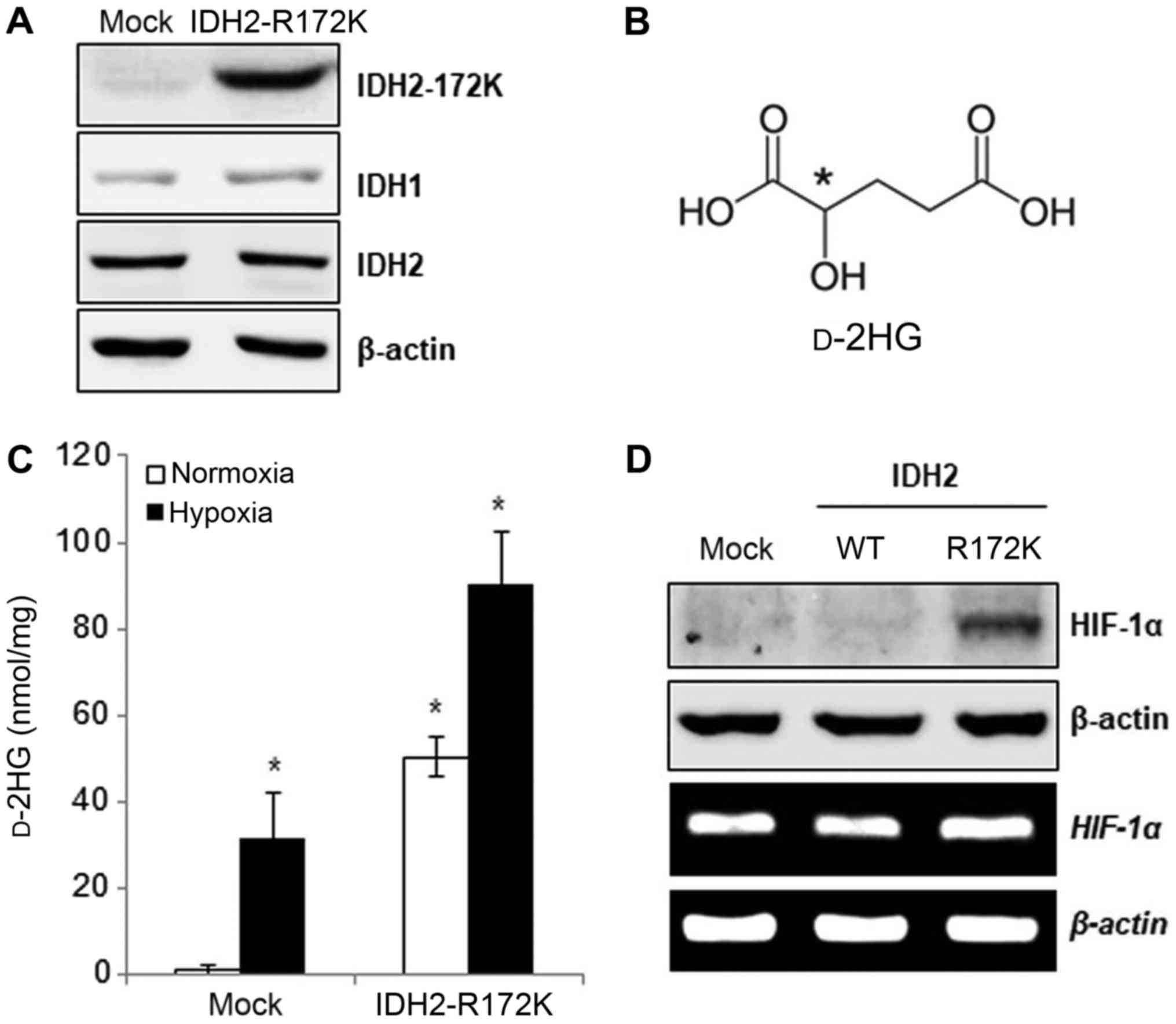
We infected the MDA-MB-231 human breast cancer cells
with lentiviral particles following transfection with IDH2-R172K
mutant-lentiviral vector in 293T cells, and the overexpression of
IDH2-R172K mutants was identified by western blot analysis using
anti-IHD2-R172K antibodies (Fig.
1A). We then analyzed altered metabolites in MDA-MB-231 cancer
cells due to hypoxia and IDH2 mutation using metabolite profiling.
The analysis identified a total of 116 up- or downregulated
compounds, which were involved in glycolysis; the PPP; the TCA
cycle; the urea cycle; and polyamine, creatinine, purine,
glutathione, nicotinamide, choline and amino acid metabolism, as
annotated on the HMT metabolite database. Among these compounds, 28
metabolites, including 2HG, a well-known metabolite in IDH2
mutants, were upregulated (Table
I). Hypoxia increased 2HG levels by 4.1-fold, whereas IDH2
mutation increased 2HG levels by 1.7-fold. HMT metabolite analysis
did not distinguish the d-form or l-form of 2HG. As hypoxia usually
increases l-2HG
levels, the increase in 2HG levels by hypoxia may primarily involve
l-2HG. Of note, many
metabolites were overlapped under hypoxic conditions and IDH
mutants, and the levels of many intermediate metabolites in
glycolytic pathways, such as glucose-6-phosphate, fructose
1,6-diphosphate and lactic acids, were increased in the mutants
(Table I). Some metabolites
involved in creatinine synthesis and purine metabolism were also
upregulated. Notably, hypoxia and IDH2 mutation resulted in the
downregulation of glutathione, β-hydroxy β-methylglutaryl-CoA and
putrescine. Among these downregulated metabolites, glutathione is
the main substrate affecting cellular antioxidant potential
(33) (Table II), suggesting that IDH2 mutation
and hypoxia suppress antioxidant potential.
 | Table IRepresentative increased metabolites
in hypoxic or IDH2R172K mutant MDA-MB-231 cells. |
Table I
Representative increased metabolites
in hypoxic or IDH2R172K mutant MDA-MB-231 cells.
| Rank | Metabolites | Nomoxia | Hypoxia | Fold | Rank | Metabolites | Nomoxia | IDH2-R172K | Fold |
|---|
| 2-Hydroxyglutaric
acid | 52 | 213 | 4.1 | | 2-Hydroxyglutaric
acid | 52 | 86 | 1.7 |
| 1 | Fructose
6-phosphate | 16 | 23 | 85.0 | 1 | Glucose
1-phosphate | N.D. | 27 | 27.0 |
| 2 | N-Carbamoylaspartic
acid | 24 | 439 | 18.4 | 2 | Succinic acid | 58 | 343 | 6.0 |
| 3 | Glucose
1-phosphate | N.D. | 9.9 | 9.9 | 3 | Uric acid | 5.7 | 33 | 5.8 |
| 4 | Inosine
monophosphate | 4.5 | 31 | 6.8 | 4 | Citrulline | 327 | 1,405 | 4.3 |
| 5 | Glycerol
3-phosphate | 59 | 143 | 2.4 | 5 | 2-Phosphoglyceric
acid | 8.7 | 36 | 4.2 |
| 6 | Guanosine
monophosphate | 5.6 | 13 | 2.3 | 6 | Lactic acid | 8,669 | 31,288 | 3.6 |
| 7 | Fructose
1,6-diphosphate | 440 | 811 | 1.8 | 7 | 3-Phosphoglyceric
acid | 139 | 454 | 3.3 |
| 8 | Ornithine | 103 | 187 | 1.8 | 8 | Fructose
6-phosphate | 16 | 52 | 3.2 |
| 9 | Lysine | 153 | 262 | 1.7 | 9 | Glucose
6-phosphate | 74 | 225 | 3.0 |
| 10 | Glutamine | 853 | 1,343 | 1.6 | 10 | Ornithine | 103 | 289 | 2.8 |
| 11 | Guanosine
5'-phosphate | 20 | 30 | 1.5 | 11 | Phosphoenolpyruvic
acid | 26 | 69 | 2.7 |
| 12 | Succinic acid | 58 | 86 | 1.5 | 12 | Alanine | 242 | 613 | 2.5 |
| 13 | Folic acid | 1.3 | 2.0 | 1.5 | 13 | Guanosine
monophosphate | 5.6 | 12 | 2.1 |
| 14 | Phenylalanine | 87 | 127 | 1.5 | 14 | Lysine | 153 | 301 | 2.0 |
| 15 | 3-Phosphoglyceric
acid | 139 | 191 | 1.4 | 15 | Guanosine
5'-phosphate | 20 | 37 | 1.9 |
| 16 | Creatinine | 16 | 22 | 1.4 | 16 | Phenylalanine | 87 | 157 | 1.8 |
| 17 | Histidine | 111 | 148 | 1.3 | 17 | Glycerol
3-phosphate | 59 | 96 | 1.6 |
| 18 | Lactic acid | 8,669 | 11,418 | 1.3 | 18 | Inosine
monophosphate | 4.5 | 7.2 | 1.6 |
| 19 | Phosphoenolpyruvic
acid | 26 | 34 | 1.3 | 19 | Histidine | 111 | 165 | 1.5 |
| 20 | GTP | 530 | 691 | 1.3 | 20 | Folic acid | 1.3 | 1.9 | 1.5 |
| 21 | 2-Phosphoglyceric
acid | 8.7 | 11 | 1.3 | 21 | Fructose
1,6-diphosphate | 440 | 638 | 1.5 |
| 22 | Citrulline | 327 | 381 | 1.2 | 22 | Acetyl CoA | 9.3 | 13 | 1.4 |
| 23 | Acetyl CoA | 9.3 | 11 | 1.1 | 23 | Creatinine | 16 | 21 | 1.3 |
| 24 | Uric acid | 5.7 | 6.5 | 1.1 | 24 | Guanosine
5'-triphosphate | 530 | 673 | 1.3 |
| 25 | Alanine | 242 | 272 | 1.1 | 25 |
N-Carbamoylaspartic acid | 24 | 28 | 1.2 |
| 26 | Glucose
6-phosphate | 74 | 80 | 1.1 | 26 | Glutamine | 853 | 1,003 | 1.2 |
| 27 | Ribulose
5-phosphate | 32 | 33 | 1.1 | 27 | Ribulose
5-phosphate | 32 | 36 | 1.1 |
| Table IIRepresentative decreased metabolites
in hypoxic or IDH2R172K mutants MDA-MB-231 cells. |
Table II
Representative decreased metabolites
in hypoxic or IDH2R172K mutants MDA-MB-231 cells.
| Rank | Metabolites | Nomoxia | Hypoxia | Fold | Rank | Metabolites | Nomoxia | IDH2-R172K | Fold |
|---|
| 2-Hydroxyglutaric
acid | 52 | 213 | 1 | | 2-Hydroxyglutaric
acid | 52 | 86 | 1 |
| 1 | Glutathione
(GSH) | 5,494 | 2,731 | 2.0 | 1 | Glutathione
(GSH) | 5,494 | 205 | 26.9 |
| 2 | Putrescine | 83 | 44 | 1.9 | 2 | Putrescine | 83 | 19 | 4.3 |
| 3 | HMG CoA | 2.4 | 2.1 | 1.1 | 3 | HMG CoA | 2.4 | 2.0 | 1.2 |
To determine whether the d-form of 2HG was increased, we
evaluated the amount of d-2HG in the MDA-MB-231 cells
exposed to hypoxia or harboring the IDH2-R172K mutation. Compared
with the normoxic controls, hypoxia significantly increased the
d-2HG levels. The
IDH2-R172K mutants also exhibited significant increases in
intracellular d-2HG
contents (Fig. 1C). Koivunen et
al (2012) reported that D-2HG increased EGLN activity, leading
to diminished HIF levels (29).
However, in this study, we observed that HIF-1α protein expression
was increased, while its mRNA level was not altered in the
IDH2-R172K mutant overexpressing HepG2 human liver cancer
cells (Fig. 1D), suggesting that
HIF-1α is stabilized by the D-2HG same as the L-2HG in cancer
cells.
d-2HG induces endothelial cell
proliferation
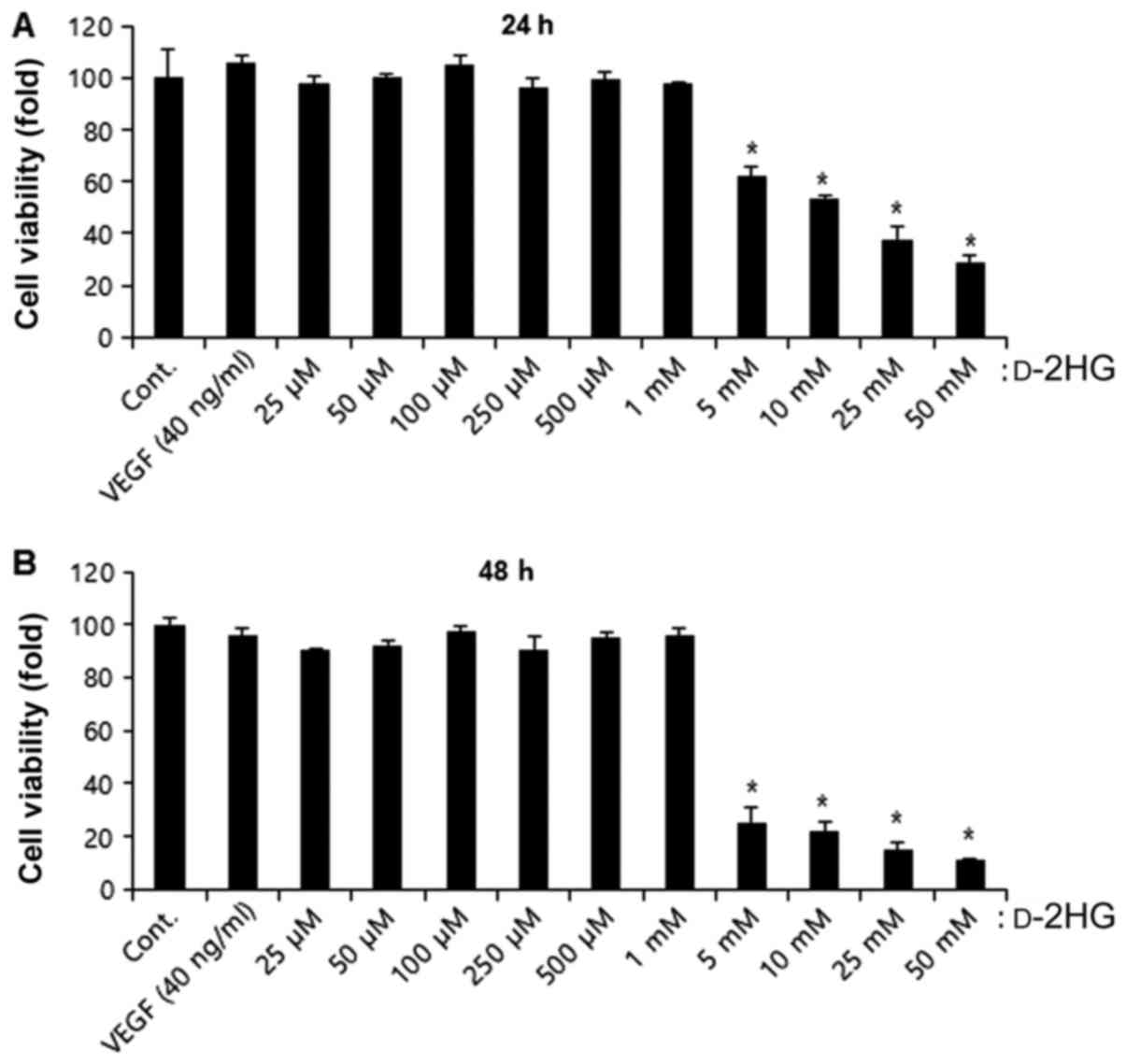
To determine the non-toxic concentrations of
d-2HG in endothelial
cells, we examined cell viability using CCK-8 assays in the BAECs.
From the concentration of 25 µM to 1 mM, d-2HG was not toxic to the
endothelial cells; however, at concentrations >5 mM,
cytotoxicity increased significantly at both 24 and 48 h (Fig. 2). Thus, we used concentrations
<1 mM to identify the angiogenic activity.
As d-2HG inhibits proline
hydroxylases (25), we
hypothesized that this metabolite may also increase VEGF protein
levels via the inhibition of HIF-1 degradation. To confirm this
hypothesis, we evaluated the secretion of VEGF in serum-starved
medium following treatment of the cancer cells with d-2HG. We found that d-2HG increased VEGF secretion
from the cells (Fig. 3A). To
determine whether d-2HG affecs endothelial cell
proliferation to modulate angiogenesis (5), we performed BrdU cell proliferation
assays and immunostaining for Ki67 in the endothelial cells.
d-2HG increased
endothelial cell proliferation in a concentration-dependent manner
from the concentrations of 25 to 250 µM, but did not exert
concentration-dependent effects at the concentrations of 500
µM and 1 mM, although increased proliferation was still
observed (Fig. 3B). Therefore, we
selected the concentrations of 250 µM d-2HG to reveal the maximum
effects on angiogenic activity for the subsequent experiments.
Immunostaining of the proliferating cells with anti-Ki67 antibodies
indicated that the increase in cell proliferation induced by
d-2HG (250 µM)
was similar to that induced by VEGF (40 ng/ml; Fig. 3C).
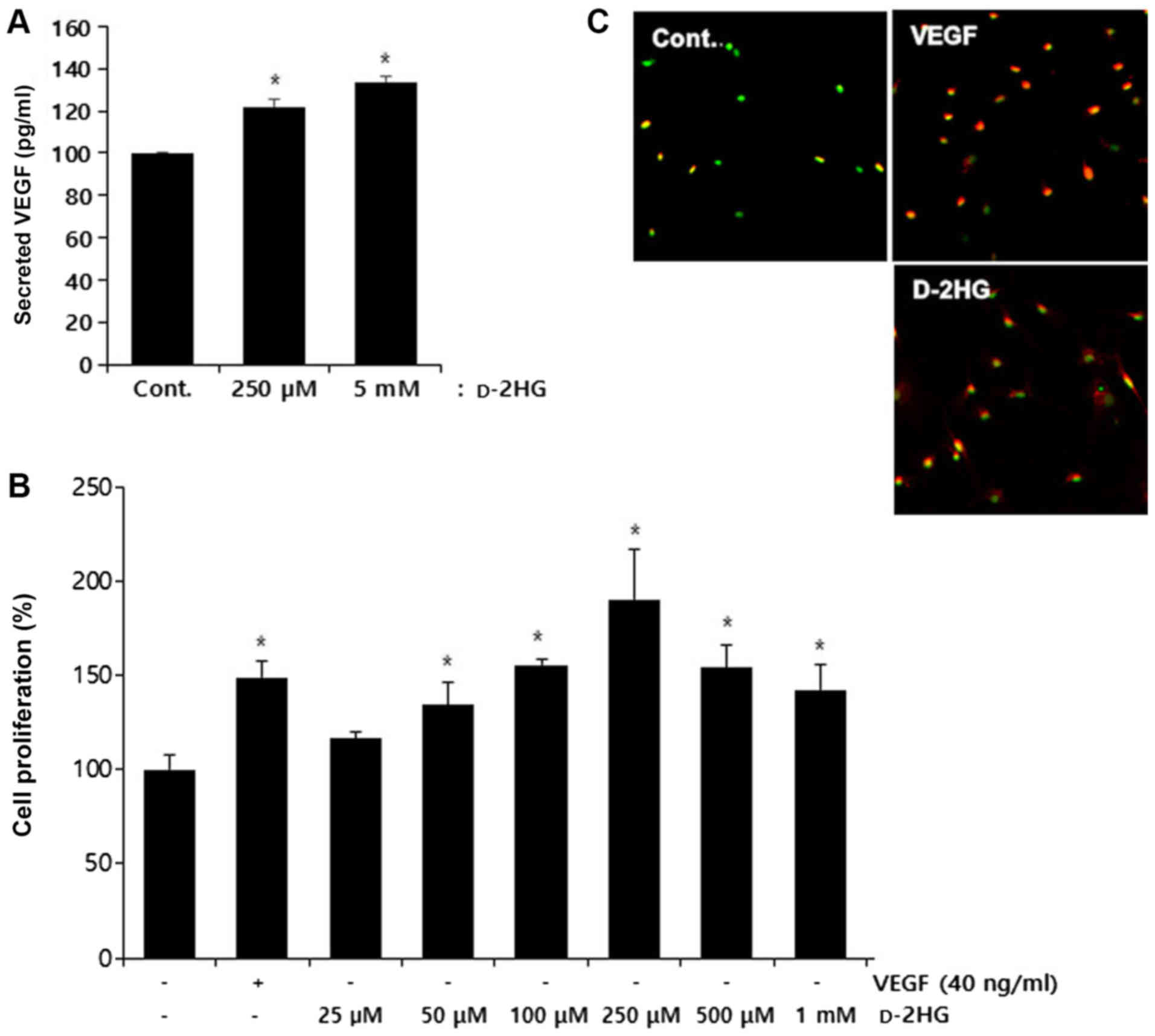 | Figure 3d-2HG increases VEGF secretion
and the proliferation of BAECs. (A) A549 cells were starved for 16
h in 0.5% FBS-containing medium, treated with d-2HG (250 µM and 5 mM),
collected and analyzed for VEGF protein levels using a VEGF ELISA
kit. *P<0.05 versus the control. (B) BAECs were
seeded at 2×103 cells/well in 96-well culture plates,
then incubated for 24 h, and starved in 0.5% FBS in DMEM/low
glucose for 16 h. The cells were treated with d-2HG at the indicated
concentrations or VEGF (40 ng/ml) in 0.5% FBS in DMEM/low glucose.
Cell proliferation was assayed using a BrdU kit, and the percentage
relative to the control was calculated. *P<0.05
versus the control. (C) BAECs were starved with 0.5% FBS in
DMEM/low glucose for 16 h, incubated with various concentrations of
d-2HG or VEGF (40
ng/ml) for 24 h in DMEM/low glucose containing 0.5% FBS, fixed with
4% paraformaldehyde solution, and permeabilized with 0.1% Triton
X-100/PBS. Fixed cells were blocked with 1% BSA in PBS and
incubated with anti-Ki67 antibodies. Green color indicates Hoechst
dye staining the nucleus and red color indicates Ki67-stained
cells. BAECs, bovine aortic endothelial cells; d-2HG, d-2-hydroxyglutarate; VEGF,
vascular endothelial growth factor. |
d-2HG induces endothelial cell
migration and tube formation capacity
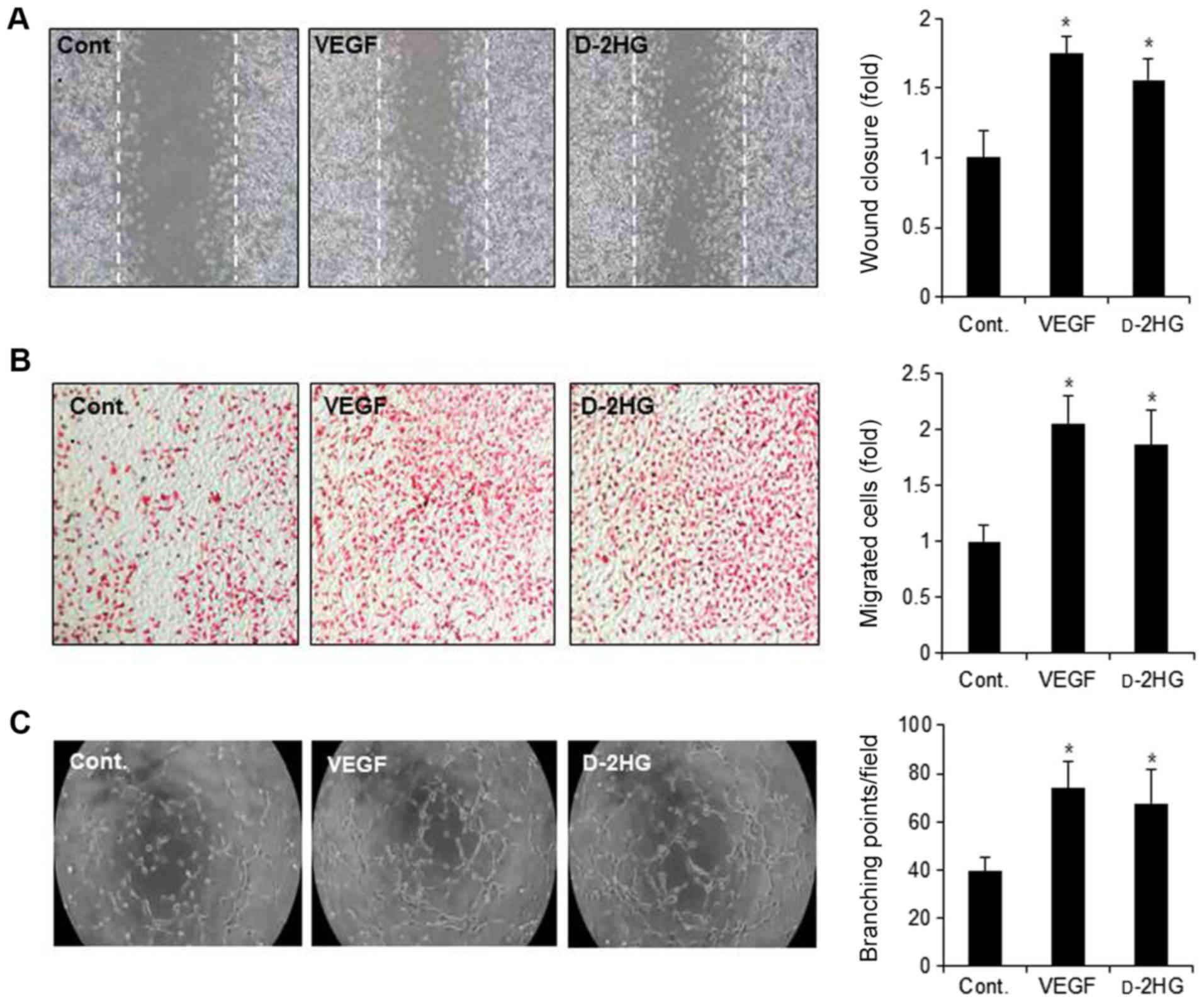
As endothelial cell migration is a key step in
angio-genesis (5), in this study,
we performed both wound healing motility and Transwell migration
assays. When wound closure was monitored for 24 h, d-2HG increased the cell
migratory capacity, similar to the effects observed with VEGF
(Fig. 4A). Wound closure was
increased to 1.5 fold by d-2HG treatment compared with the
control cells. The cell migratory capacity induced by d-2HG examined by Transwell assay
was also significantly increased by approximately 2-fold, similar
to that induced by VEGF (Fig.
4B).
The reorganization of angiogenesis is involved in
tube formation (5). In this study,
to determine the ability of d-2HG to induce the formation of
capillary-like structures, we performed tube formation assays using
the endothelial cells. d-2HG increased the formation of
capillary-like structures, as did VEGF (Fig. 4C). The number of branching points
was significantly increased by VEGF and d-2HG to 1.6- and 2-fold that of
the control, respectively (Fig.
4C).
d-2HG induces the formation of
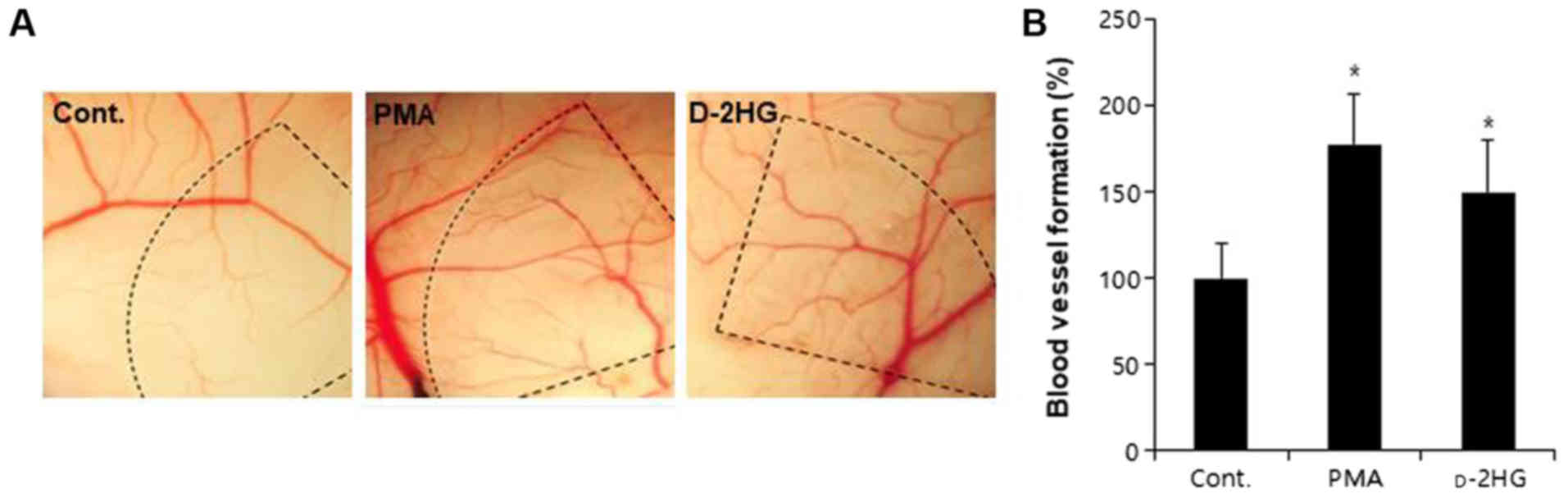
functional new blood vessels in CAMs ex vivo
To confirm the in vitro angiogenic activity
of d-2HG, we
performed ex vivo angiogenesis assays. d-2HG induced the formation of
neovessels from the large vessels in the CAM region, as shown in
Fig. 5A. Networks between new
vessels were very complex, and vessel branching points were
increased in the d-2HG-treated CAMs, similar to
those in the PMA-treated CAMs (Fig.
5B). Eggs exhibiting >30% neovessel formation were
considered positive, and the percentage of positive eggs was
calculated. The numbers of angiogenic CAMs were significantly
increased by treatment with d-2HG compared with that in the
controls (Fig. 5B).
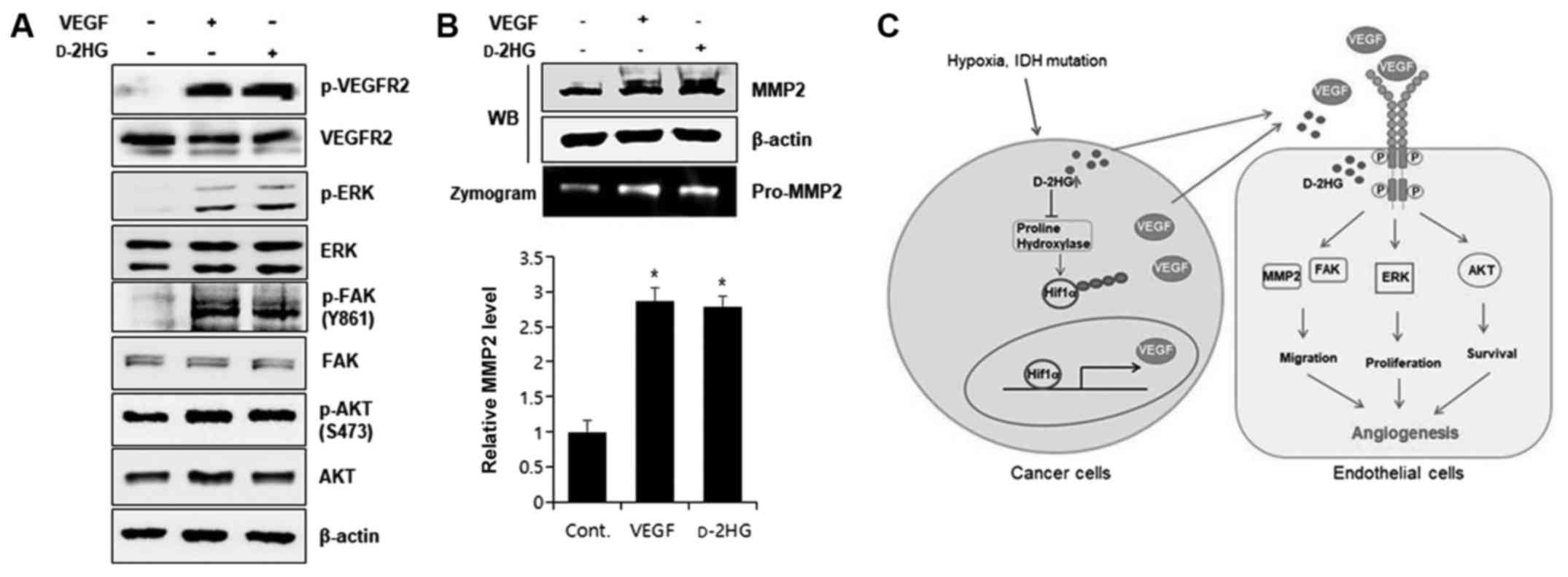
d-2HG activates VEGFR2 signaling
and matrix metalloproteinase (MMP)2 activity
VEGF/VEGFR2, the most prominent ligand-receptor
complex in the VEGF system, induces endothelial cell proliferation,
migration, survival, and new vessel formation associated with
angiogenesis (34). VEGFR2
activation promotes the expression of downstream signaling
molecules, such as AKT, p42/44 extracellular signal-regulated
kinase (ERK) and p38 mitogen-activated protein kinase (MAPK), and
focal adhesion kinase (FAK) (34).
In this study, we found that d-2HG clearly increased the
phosphorylation of VEGFR2 (Fig.
6A), suggesting that VEGFR2 expression and activation were
induced by d-2HG. The
activation of p42/44 ERK, AKT and FAK was observed following
treatment with d-2HG,
suggesting that d-2HG
increased angiogenic activity through the activation of VEGFR2
signaling.
MMP is activated by growth factor signaling, and its
activation is essential for matrix degradation and cell migration
and to release growth factors, such as VEGF, from the matrix
(35). Of note, d-2HG significantly enhanced MMP2
activation, as observed by gelatin zymography and western blot
analysis (Fig. 6B) to the same
level induced by VEGF, suggesting that d-2HG increased angiogenic
activity via the activation of MMP2. Taken together, these results
indicated that d-2HG
increased angiogenic capacity via VEGF secretion and activated
VEGFR2 signaling, including FAK, in vascular endothelial cells.
Discussion
In this study, we demonstrated that the d-form of HG, an oncometabolite,
increased angiogenesis. d-2HG is produced by mutated IDH
enzymes found in a subsets of leukemias and brain tumors (27). Additionally, d-2HG levels are increased
through glutamine anaplerosis, even in the absence of IDH mutation
(28). In this study, in IDH2R172K
mutant human breast cancer cells, 2HG levels and metabolites
involved in the glycolytic pathway were increased.
Standard methods for measuring metabolites by gas
chromatography mass spectrometry do not distinguish enantiomeric
species; thus, total 2HG measured in most assays includes both
d-2HG and
l-2HG. In this study,
we demonstrated the that d-2HG contents were increased in
the context of hypoxia and in IDH2 mutant cells. d-2HG exhibited cytotoxicity at
concentrations >5 mM in vascular endothelial cells; this
concentration was lower than that used in other reports, i.e., 50
mM, for the analysis of metabolites in cancer cells (14), suggesting that vascular endothelial
cells are more susceptible to higher concentrations of d-2HG. The serum levels of
d-2HG in patients
with IDH1/2-mutated biliary tract cancer have been shown to be
approximately 11 µM (median value) (36). Therefore, 250 µM
d-2HG was sufficient
to observe the angiogenic activity in endothelial cells.
The oncometabolite d-2HG increased endothelial cell
proliferation, migration and tube formation, and the angiogenic
activity of this compound was similar to that of VEGF. In addition,
VEGFR2 activation was observed following treatment with
d-2HG. d-2HG increased the activation of
VEGFR2 signaling. Phosphatidylinositol 3-kinase (PI3K)/AKT
signaling increases the survival of endothelial cells, whereas the
MAPK kinase/p42/44 ERK pathway is involved in endothelial cell
proliferation. The activation of FAK increases the migration
activity of endothelial cells. In addition to FAK activation, MMP2
increases matrix degradation and facilitates cell migration
(35,37). The functional activation of PI3K,
ERK and FAK by d-2HG
may increase angiogenic activity. MMP2 activation also increases
the levels of VEGF released from the matrix, promoting the binding
of VEGF to its receptors (35).
Therefore, d-2HG
increases both VEGF availability to bind with VEGFR2 and enhances
cell migration by modulation of MMP2 activity. However, the
mechanisms through which d-2HG regulates MMP2 activity are
still unclear.
VEGF expression can be enhanced by d-2HG through the activation of
the ERK/MAPK pathway to increase the expression of activating
protein (AP)-1, and AP-1 then increases VEGF gene
transcription. This event must occur very early as treatment with
d-2HG increased MAPK
expression, including p42/44 ERK expression, within 10 min. Since
it has been reported that hypoxia increases the formation of
l-2HG independent of
the IDH mutation, and elevated l-2HG stabilizes HIF-1α (30), in the presence of both metabolites
the angiogenic activity will be further increased. Therefore,
further studies are required to investigate whether l-2HG induces angiogenic activity
and to determine the differences between l-2HG and d-2HG.
Metabolites altered by IDH2R172K and IDH1R132H
mutations and by high-dose 2HG (30 mM) treatment in glioma cells
result in the elevation of numerous free amino acids, lipid
precursors, such as glycerol-phosphate, and the depletion of TCA
cycle intermediates, such as citrate; however, intermediates
related to the glycolytic pathway were not altered (14). In this study, increased metabolites
in IDH2R172K mutant cells overlapped with those induced by hypoxic
stress conditions in breast cancer cells, including 2HG. We found
that lipid precursors, such as glycerol-3 phosphate, and many
metabolites related to the glycolytic pathway, such as
glucose-6-phosphate, fructose-6-phosphate and lactic acid, were
elevated under hypoxic conditions and in IDH2R172K mutant cells.
Moreover, hypoxia and IDH2R172K mutation also increased lipid
precursors and numerous free amino acids, consistent with previous
findings (14), suggesting that
IDH2R172K mutation preferentially altered the metabolic machinery,
such as glycolysis and the PPP, to yield high levels of energy,
similar to hypoxia (2,14). The most markedly decreased
metabolite was the reduced form of glutathione both in hypoxic
conditions and in IDH2R172K mutant cells. As the reduced form of
glutathione is essential for scavenging oxidative stress by
providing electrons (33), the
IDH2R172K mutation may increase oxidative stress, as previously
reported (38).
The d-2HG-induced angiogenic activity
can be caused by alterations in various metabolites as 2HG
increases the levels of numerous metabolites in glioma cells
(14). In this study, lactate
levels were increased by hypoxia, and we found that as mutations
may play a role in increasing d-2HG-dependent angiogenesis
because lactate enhances hypoxia-induced responses, including
angiogenesis, independent of HIF via the N-Myc downstream regulated
gene-mediated ERK pathway (39).
Inhibition of glycolysis suppresses pathological angiogenesis
(40), suggesting that increased
d-2HG and glycolytic
metabolites cooperatively activate angiogenesis in tumors.
d-2HG treatment
induced VEGFR2 activation and downstream signaling. However, it is
unclear as to whether VEGFR2 and MMP2 levels are induced by
d-2HG in endothelial
cells. It is possible that tge induction of VEGFR2 and MMP2 by
d-2HG increases the
secretion or expression of VEGF transcription in endothelial cells
and VEGF also increased the transcription of these targets. It is
also possible that an epigenetic mechanism, such as increased
histone methylation of VEGFR2 or MMP2 gene promoters,
may be involved as d-2HG inhibits histone
demethylases as a gene inhibition mark (41). Collectively, these findings
demonstrated that d-2HG increased VEGF secretion in
cancer cells, but increased VEGFR2 activity and downstream
signaling in endothelial cells. Therefore, further studies are
warranted to determine whether VEGF is secreted in endothelial
cells to induce autocrine regulation and to determine the
mechanisms through which d-2HG activates VEGFR2 and
downstream kinases.
The discovery of the IDH1/2 mutation has led to
novel therapies for the restoration of normal IDH1/2 function or
blockage of the production or downstream effects of d-2HG (24). AGI-5198 and AGI-6780 are selective
inhibitors of the mutant IDH1 and IDH2 enzymes, respectively. They
normalize 2HG, reverse histone and DNA methylation and induce the
differentiation of TF-1 erythroleukemia cells, as well as primary
human acute myelogenous leukemia (AML) cells with IDH1/2 mutations
(13). As IDH1/2 mutations are
frequently reported in hematologic cancers and glioma, clinical
trials have been conducted to treat the patients with these IDH1/2
mutations (24). Therefore,
targeting IDH1/2 mutations has the advantage of developing specific
therapies to treat specific types of cancer only harboring IDH1/2
mutations (24).
By contrast, the targeting of VEGFR2 signaling
pathway is focused on the anti-angiogenic activity. Clinically, the
development of antibodies against VEGF and kinase inhibitors
targeting VEGFR2 is effective in modulating pathological
angiogenesis in diseases characterized by abnormal angiogenesis,
such as diabetic retinopathy and cancer (42). However, the anti-VEGF signaling
strategy can enhance active angiogenesis recurrence by the
increased HIF-1α owing to hypoxia (43). In addition, tge targeting of VEGFR2
downstream signaling pathway can be competing with pro-angiogenic
therapeutic strategies, such as wound healing (44).
VEGF can stimulate vasculogenesis in tumors, as
well as normal physiology by recruiting bone marrow-derived
hematopoietic progenitor cells (HPCs) and endothelial progenitor
cells (EPCs) (45,46). VEGF can increase the expression of
nitric oxide (NO), prostacyclins and other soluble mediators that
lead to vasodilation (47).
Therefore, VEGF targeted therapy will cause vascular constriction,
as well as the inhibition of physiological angiogenesis.
In conclusion, in this study, we provided insight
into the effects of d-2HG on angiogenesis in cancer
cells and demonstrated, for the first time, at least to the best of
our knowledge, that this molecule induced angiogenesis and may be a
good candidate for anti-angiogenic therapy. The findings of this
study suggest that enhancing our understanding of cancer metabolic
profiles and the functions of metabolites in cancer progression,
such as angiogenesis, may provide a good foundation for the
development of novel therapeutic agents in cancer research.
Therefore, the appropriate utilization of metabolic inhibitors
could be an effective clinical strategy.
Funding
This study was supported by the Korean government
(grant nos. NRF-2017R1A2B3002227 and NRF-2013R1A2A2 A01068868).
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
JS and SHY performed and analyzed the data from
culture studies, molecular works and in vitro as well as
in vivo angiogenic assay. SHL performed western blot
analysis. JHJ synthesized and provided the many chemicals for the
preliminary experiments and the conception of the study. YML
initiated the study, supervised the experiments, and the analyzed
data and wrote the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Dr Hai Yan (Duke
University) for kindly providing the pLenti6.2/V5 or
pLenti6.2/V5-IDH2 (R172K) plasmids.
References
|
1
|
McKeown SR: Defining normoxia, physoxia
and hypoxia in tumours-implications for treatment response. Br J
Radiol. 87:201306762014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bertout JA, Patel SA and Simon MC: The
impact of O2 availability on human cancer. Nat Rev
Cancer. 8:967–975. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Seok JK, Lee SH, Kim MJ and Lee YM:
MicroRNA-382 induced by HIF-1α is an angiogenic miR targeting the
tumor suppressor phosphatase and tensin homolog. Nucleic Acids Res.
42:8062–8072. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Folkman J: Role of angiogenesis in tumor
growth and metastasis. Semin Oncol. 29(Suppl 16): S15–S18. 2002.
View Article : Google Scholar
|
|
5
|
Ferrara N and Kerbel RS: Angiogenesis as a
therapeutic target. Nature. 438:967–974. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Warburg OPK and Negelein E: The metabolism
of tumors. Biochem Z. 152:319–344. 1924.In German.
|
|
8
|
Losman JA and Kaelin WG Jr: What a
difference a hydroxyl makes: Mutant IDH, (R)-2-hydroxyglutarate,
and cancer. Genes Dev. 27:836–852. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cairns RA, Harris IS and Mak TW:
Regulation of cancer cell metabolism. Nat Rev Cancer. 11:85–95.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Raimundo N, Baysal BE and Shadel GS:
Revisiting the TCA cycle: Signaling to tumor formation. Trends Mol
Med. 17:641–649. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wise DR, Ward PS, Shay JE, Cross JR,
Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC and
Thompson CB: Hypoxia promotes isocitrate dehydrogenase-dependent
carboxylation of α-ketoglutarate to citrate to support cell growth
and viability. Proc Natl Acad Sci USA. 108:19611–19616. 2011.
View Article : Google Scholar
|
|
13
|
Zhang C, Moore LM, Li X, Yung WK and Zhang
W: IDH1/2 mutations target a key hallmark of cancer by deregulating
cellular metabolism in glioma. Neuro-oncol. 15:1114–1126. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Reitman ZJ and Yan H: Isocitrate
dehydrogenase 1 and 2 mutations in cancer: Alterations at a
crossroads of cellular metabolism. J Natl Cancer Inst. 102:932–941.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Parsons DW, Jones S, Zhang X, Lin JC,
Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et
al: An integrated genomic analysis of human glioblastoma
multiforme. Science. 321:1807–1812. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mardis ER, Ding L, Dooling DJ, Larson DE,
McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath
SD, et al: Recurring mutations found by sequencing an acute myeloid
leukemia genome. N Engl J Med. 361:1058–1066. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Amary MF, Bacsi K, Maggiani F, Damato S,
Halai D, Berisha F, Pollock R, O'Donnell P, Grigoriadis A, Diss T,
et al: IDH1 and IDH2 mutations are frequent events in central
chondrosarcoma and central and periosteal chondromas but not in
other mesenchymal tumours. J Pathol. 224:334–343. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chan-On W, Nairismägi ML, Ong CK, Lim WK,
Dima S, Pairojkul C, Lim KH, McPherson JR, Cutcutache I, Heng HL,
et al: Exome sequencing identifies distinct mutational patterns in
liver fluke-related and non-infection-related bile duct cancers.
Nat Genet. 45:1474–1478. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cairns RA, Iqbal J, Lemonnier F, Kucuk C,
de Leval L, Jais JP, Parrens M, Martin A, Xerri L, Brousset P, et
al: IDH2 mutations are frequent in angioimmunoblastic T-cell
lymphoma. Blood. 119:1901–1903. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fassan M, Simbolo M, Bria E, Mafficini A,
Pilotto S, Capelli P, Bencivenga M, Pecori S, Luchini C, Neves D,
et al: High-throughput mutation profiling identifies novel
molecular dysregulation in high-grade intraepithelial neoplasia and
early gastric cancers. Gastric Cancer. 17:442–449. 2014. View Article : Google Scholar
|
|
21
|
Sjöblom T, Jones S, Wood LD, Parsons DW,
Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, et al:
The consensus coding sequences of human breast and colorectal
cancers. Science. 314:268–274. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kang MR, Kim MS, Oh JE, Kim YR, Song SY,
Seo SI, Lee JY, Yoo NJ and Lee SH: Mutational analysis of IDH1
codon 132 in glioblastomas and other common cancers. Int J Cancer.
125:353–355. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ye D, Ma S, Xiong Y and Guan KL:
R-2-hydroxyglutarate as the key effector of IDH mutations promoting
oncogenesis. Cancer Cell. 23:274–276. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mondesir J, Willekens C, Touat M and de
Botton S: IDH1 and IDH2 mutations as novel therapeutic targets:
Current perspectives. J Blood Med. 7:171–180. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim
SH, Ito S, Yang C, Wang P, Xiao MT, et al: Oncometabolite
2-hydroxyglutarate is a competitive inhibitor of
α-ketoglutarate-dependent dioxygenases. Cancer Cell. 19:17–30.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Molenaar RJ, Radivoyevitch T, Maciejewski
JP, van Noorden CJ and Bleeker FE: The driver and passenger effects
of isocitrate dehydrogenase 1 and 2 mutations in oncogenesis and
survival prolongation. Biochim Biophys Acta. 1846:326–341.
2014.PubMed/NCBI
|
|
27
|
Dang L, Jin S and Su SM: IDH mutations in
glioma and acute myeloid leukemia. Trends Mol Med. 16:387–397.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Colvin H, Nishida N, Konno M, Haraguchi N,
Takahashi H, Nishimura J, Hata T, Kawamoto K, Asai A, Tsunekuni K,
et al: Oncometabolite D-2-Hydroxyglurate directly induces
epithelial-mesenchymal transition and is associated with distant
metastasis in colorectal cancer. Sci Rep. 6:362892016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Koivunen P, Lee S, Duncan CG, Lopez G, Lu
G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, et al:
Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked
to EGLN activation. Nature. 483:484–488. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Intlekofer AM, Dematteo RG, Venneti S,
Finley LW, Lu C, Judkins AR, Rustenburg AS, Grinaway PB, Chodera
JD, Cross JR, et al: Hypoxia induces production of
L-2-hydroxyglutarate. Cell Metab. 22:304–311. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hur W, Ryu JY, Kim HU, Hong SW, Lee EB,
Lee SY and Yoon SK: Systems approach to characterize the metabolism
of liver cancer stem cells expressing CD133. Sci Rep. 7:455572017.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee SH, Jung YD, Choi YS and Lee YM:
Targeting of RUNX3 by miR-130a and miR-495 cooperatively increases
cell proliferation and tumor angiogenesis in gastric cancer cells.
Oncotarget. 6:33269–33278. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pompella A, Visvikis A, Paolicchi A, De
Tata V and Casini AF: The changing faces of glutathione, a cellular
protagonist. Biochem Pharmacol. 66:1499–1503. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Olsson AK, Dimberg A, Kreuger J and
Claesson-Welsh L: VEGF receptor signalling - in control of vascular
function. Nat Rev Mol Cell Biol. 7:359–371. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sternlicht MD and Werb Z: How matrix
metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol.
17:463–516. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Delahousse J, Verlingue L, Broutin S,
Legoupil C, Touat M, Doucet L, Ammari S, Lacroix L, Ducreux M,
Scoazec JY, et al: Circulating oncometabolite D-2-hydroxyglutarate
enantiomer is a surrogate marker of isocitrate
dehydrogenase-mutated intrahepatic cholangiocarcinomas. Eur J
Cancer. 90:83–91. 2018. View Article : Google Scholar
|
|
37
|
Ferrara N, Gerber HP and LeCouter J: The
biology of VEGF and its receptors. Nat Med. 9:669–676. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li S, Chou AP, Chen W, Chen R, Deng Y,
Phillips HS, Selfridge J, Zurayk M, Lou JJ, Everson RG, et al:
Overexpression of isocitrate dehydrogenase mutant proteins renders
glioma cells more sensitive to radiation. Neuro-oncol. 15:57–68.
2013. View Article : Google Scholar :
|
|
39
|
Lee DC, Sohn HA, Park ZY, Oh S, Kang YK,
Lee KM, Kang M, Jang YJ, Yang SJ, Hong YK, et al: A lactate-induced
response to hypoxia. Cell. 161:595–609. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Schoors S, De Bock K, Cantelmo AR,
Georgiadou M, Ghesquière B, Cauwenberghs S, Kuchnio A, Wong BW,
Quaegebeur A, Goveia J, et al: Partial and transient reduction of
glycolysis by PFKFB3 blockade reduces pathological angiogenesis.
Cell Metab. 19:37–48. 2014. View Article : Google Scholar
|
|
41
|
Lu C, Ward PS, Kapoor GS, Rohle D, Turcan
S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, et
al: IDH mutation impairs histone demethylation and results in a
block to cell differentiation. Nature. 483:474–478. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ferrara N and Adamis AP: Ten years of
anti-vascular endothelial growth factor therapy. Nat Rev Drug
Discov. 15:385–403. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Franco M, Man S, Chen L, Emmenegger U,
Shaked Y, Cheung AM, Brown AS, Hicklin DJ, Foster FS and Kerbel RS:
Targeted anti-vascular endothelial growth factor receptor-2 therapy
leads to short-term and long-term impairment of vascular function
and increase in tumor hypoxia. Cancer Res. 66:3639–3648. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bazzazi H, Isenberg JS and Popel AS:
Inhibition of VEGFR2 activation and its downstream signaling to
ERK1/2 and calcium by thrombospondin-1 (TSP1): In silico
investigation. Front Physiol. 8:482017.
|
|
45
|
Rafii S, Lyden D, Benezra R, Hattori K and
Heissig B: Vascular and haematopoietic stem cells: Novel targets
for anti-angiogenesis therapy? Nat Rev Cancer. 2:826–835. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bertolini F, Shaked Y, Mancuso P and
Kerbel RS: The multifaceted circulating endothelial cell in cancer:
Towards marker and target identification. Nat Rev Cancer.
6:835–845. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Curiel TJ, Cheng P, Mottram P, Alvarez X,
Moons L, Evdemon-Hogan M, Wei S, Zou L, Kryczek I, Hoyle G, et al:
Dendritic cell subsets differentially regulate angiogenesis in
human ovarian cancer. Cancer Res. 64:5535–5538. 2004. View Article : Google Scholar : PubMed/NCBI
|