Introduction
Nasopharyngeal carcinoma (NPC) has a high incidence
rate (27.2/100,000 males in 2003) in Southern China (1). Although the majority of primary NPC
cases can be successfully treated with radiotherapy, local
recurrence and metastatic NPC remain major problems in the
treatment of NPC (2). In a
preclinical cell model, Lun et al (3) recently demonstrated that a
subpopulation of Epstein-Barr virus (EBV) and cluster of
differentiation (CD)44-positive NPC cells are resistant to
chemotherapy and exhibit properties of cancer stem cells (CSCs).
The transmembrane glycoprotein CD44 is primarily considered a
multifunctional protein that participates in signaling pathways
involved in cancer dissemination (4). The versatility of CD44 in the
regulation of cell growth and migration is due to its interaction
with various cellular molecules, including ankyrin, ezrin, radixin
and moesin (4).
The developmental signaling pathways Wnt, Notch and
Hedgehog are known to be used by CSCs to regulate cell growth and
differentiation (5). In addition
to these stem cell-associated signaling pathways, previous studies
also indicate that microRNA (miRNA) (6,7) and
epigenetic mechanisms (8-10) serve an important role in the
regulation of CSC growth and differentiation. In NPC, it has been
recently identified that the CREB-binding protein (CBP)/catenin
antagonist and Wnt modulator ICG-001 could inhibit the growth of
EBV-positive NPC cells via downregulated expression of the tumor
suppressor/pro-differentiator miR-145 (11). It was also observed that ICG-001
reduced the population of cells expressing SRY-box
2Hi/CD44Hi (11). In the present study, the role of
miR-150 in the expression of CD44 in ICG-001-treated NPC cells was
further demonstrated.
Materials and methods
Cell culture
The EBV-positive C666-1 NPC cell line was maintained
in RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS;
Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin-streptomycin (P/S; Gibco; Thermo Fisher Scientific,
Inc.). The early passage of NPC xenograft-derived EBV-positive C17
cells (Professor Pierre Busson, Université Paris-Sud, Paris,
France) (12) were cultured in
RPMI-1640 medium supplemented with 7.5% FBS, 1% GlutaMAX (Gibco;
Thermo Fisher Scientific, Inc.), 0.2% Primocin (InvivoGen, San
Diego, CA, USA) and 7 µM Y-27632 (Cayman Chemical Company,
Ann Arbor, MI, USA), an inhibitor of Rho kinases I and II. The
EBV-negative HONE-1 NPC cell line was maintained in Dulbecco's
modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific,
Inc.) supplemented with 5% FBS and 5% newborn calf serum (Gibco;
Thermo Fisher Scientific, Inc.) with 1% P/S. The C666-1 and HONE-1
cell lines (13-16) were obtained from the Hong Kong NPC
AoE Research Tissue Bank and Cell Line Repository (Hong Kong,
China), and were authenticated using an AmpFLSTR Identifier PCR
Amplification kit (Thermo Fisher Scientific, Inc.), according to
the manufacturer's protocols. To further ensure the HONE-1 cells
used in the present study were free from HeLa cell contamination, a
single duplex detection polymerase chain reaction (PCR) assay
targeting a HeLa-specific L1 retrotransposon insertion, as
described by Rahbari et al (17), was conducted, which confirmed that
the cell line was not contaminated by HeLA cells. C666-1, C17, and
HONE-1 cells were treated with ICG-001 or dimethyl sulfoxide
(0.05%; vehicle control) for 3-7, 3-5 and 5 days, respectively. All
cell lines were cultured at 37°C in a 5% CO2 humidified
incubator. ICG-001 at 10 µM (1 µl 20 mM stock in 2 ml
RPMI-1640 medium (C666-1 and C17 cells) or DMEM (HONE-1 cells), or
same volume of DMSO was used for cell treatments unless otherwise
specified.
Cell transfection
C666-1 cells (3×105) were seeded onto
fibronectin-coated 35-mm culture dishes overnight at 37°C in a 5%
CO2 humidified incubator. Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) was then used in all
the transient transfection experiments, according to the
manufacturer's protocols. In all the siRNA or miRNA transfection
studies, 5 µl 20 µM oligonucleotide stocks was added
into the culture dishes containing 2 ml RPMI-1640 medium. To
investigate the knockdown effect of β-catenin or CD44, Ambion™
Silencer™ Pre-Designed small interfering (si)RNA targeting human
β-catenin (50 nM; Assay ID 146154; Thermo Fisher Scientific, Inc.)
or Silencer Select Pre-Designed & Validated siRNA targeting
human CD44 (50 nM; Assay ID s2681; Ambion; Thermo Fisher
Scientific, Inc.) was used in parallel with Silencer negative
control siRNA (5 0nM; cat. no. AM4611; Ambion; Thermo Fisher
Scientific, Inc.;). To investigate the effect of knocking down CBP,
ON-TARGETplus SMARTpool Human CBP siRNA (50 nM; cat. no.
L-003477-00-0005; GE Healthcare Dharmacon, Inc., Lafayette, CO,
USA) was used in parallel with ON-TARGETplus Non-targeting Control
siRNA #1 (50 nM; cat. no. D-001810-01-20; GE Healthcare Dharmacon,
Inc.). For miRNA precursor transfection, 50 nM Pre-miR miRNA
Precursor mimicking miR-150 precursor (pre-miR-150; Assay ID
PM10070; Ambion; Thermo Fisher Scientific, Inc.) was used in
parallel with 50 nM Pre-miR miRNA Precursor Negative Control #1
(Ambion; cat. no. AM17110; Thermo Fisher Scientific, Inc.). To
overexpress CD44, 200 ng expression vector pCMV3 with or without
the open reading frame of CD44 (cat. no. HG12211-UT; Sino
Biological Inc., Beijing, China) was transfected into the cells.
After 72 h, the cells were harvested and subjected to subsequent
assays.
Transwell migration assay
Transwell inserts (6.5 mm) with 8.0-µm pore
polycarbonate membranes (Corning Incorporated, Corning, NY, USA)
were used in the Transwell migration assay. The aforementioned
ICG-001-treated or transfected C666-1 cells (2×105) were
seeded in the upper chamber of the inserts containing RPMI-1640
medium, and the lower chamber contained RPMI-1640 medium
supplemented with 10% FBS. After 24 h of incubation at 37°C, the
cells remaining on the inserts were removed, while the migrated
cells at the bottom of the membrane were fixed in 4%
paraformaldehyde for 15 min at room temperature, permeabilized in
0.2% Triton-X for 10 min at room temperature and stained with DAPI
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for 30 min at room
temperature. The cells migrating across the membrane were then
visualized under a fluorescence microscope (magnification,
×200).
Western blotting
The aforementioned ICG-001-treated or transfected
C666-1 or aforementioned ICG-001-treated C17 cells were lysed in
lysis buffer [250 mM Tris (pH 8.0), 1% NP-40 and 150 mM NaCl]
containing 1% phosphatase inhibitor cocktail (Calbiochem; Merck
KGaA) and 0.25% protease inhibitors cocktail (Sigma-Aldrich; Merck
KGaA). Protein concentration was determined with a DC Protein Assay
kit (Bio-Rad Laboratories, Inc., Hercules, CA, USA), according to
the manufacturer's protocol. Cellular proteins were resolved in
SDS-PAGE (5% gel for CBP detection, and 7.5% gel for the detection
of β-catenin, CD44, ezrin and β-actin) and transferred to
polyvinylidene fluoride membranes (EMD Millipore, Billerica, MA,
USA). The membranes were blocked with 5% non-fat dry milk in TBS
with 0.1% Tween-20 (TBST) for 1 h at room temperature, followed by
incubation with primary antibodies against β-catenin (dilution
1:1,000; cat. no. 8480; Cell Signaling Technology, Inc., Danvers,
MA, USA), CBP (dilution 1:1,000; cat. no. sc-583; Santa Cruz
Biotechnology, Inc., Dallas, CA, USA), CD44 (dilution 1:1,000; cat.
no. 3570; Cell Signaling Technology, Inc.), ezrin (dilution
1:1,000; cat. no. 3145; Cell Signaling Technology, Inc.) and
β-actin (dilution 1:5,000; cat. no. A2228; Sigma-Aldrich; Merck
KGaA) for 3 h at room temperature. Subsequently, the membranes were
washed with TBST three times (15 min in total) and incubated with
corresponding horseradish peroxidase (HRP)-conjugated goat
anti-mouse IgG (H+L) secondary antibody (dilution 1:5,000; cat. no.
62-6520) or HRP-conjugated goat anti-rabbit IgG (H+L) secondary
antibody (dilution 1:5,000; cat. no. 65-6120 (both from Invitrogen;
Thermo Fisher Scientific, Inc.) at room temperature for 1 h.
Protein bands were detected with WESTSAVE Up (Western Blotting
Substrate) (Lab Frontier Co., Ltd., Seoul, Korea) and visualized on
X-ray films (FujiFilm Corporation, Tokyo, Japan) using Carestream
Kodak autoradiography GBX fixer/replenisher (Sigma-Aldrich; Merck
KGaA). β-actin was used as the internal control. Band intensities
were analyzed using ImageJ software (version 1.46; National
Institutes of Health, Bethesda, MD, USA).
Co-immunoprecipitation (Co-IP) assay
C666-1 cells subjected to Co-IP were lysed in the
aforementioned lysis buffer. An anti-CD44 antibody (1:100; cat. no.
3570; Cell Signaling Technology, Inc.) or a nonspecific IgG
antibody (1:100; cat. no. 5415; Cell Signaling Technology, Inc.)
was allowed to bind with protein G-sepharose (Sigma-Aldrich; Merck
KGaA) for 1 h at room temperature, then IP was performed on the
cell lysate with the sepharose-associated anti-CD44 or control
nonspecific IgG antibodies at 4°C overnight. The precipitates were
washed with aforementioned lysis buffer and eluted in SDS-sample
buffer [0.375 M Tris-HCl (pH 6.8), 12% SDS, 60% glycerol, 6%
2-mercaptoethanol and 0.025% bromophenol blue] at 95°C for 10 min.
Samples were then analyzed by western blotting as
aforementioned.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted using TRIzol®
reagent (Invitrogen; Thermo Fisher Scientific, Inc.), according to
the manufacturer's protocol. To detect CD44 mRNA expression, total
RNA of C666-1 cells was reverse transcribed to cDNA using Moloney
murine leukemia virus reverse transcriptase kit (cat. no.
28025-013; Invitrogen; Thermo Fisher Scientific, Inc.) with oligo
(dT)12-18 primer (cat. no. 18418012; Invitrogen; Thermo Fisher
Scientific, Inc.), according to manufacturer's protocols. qPCR was
then performed using Power SYBR® Green PCR Master mix
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. GAPDH was used as the internal control. The CD44 primer
sequences were as follows: Sense, 5′-TCAGAGGAGTAGGAGAGAGGAAAC-3′;
and antisense, 5′-GAAAAGTCAAAGTAACAATA ACAGTGG-3′ (18). The GAPDH primers were as follows:
Sense, 5′-GAAGGTGAAGGTCGGAGTC-3′; and antisense,
5′-GAAGATGGTGATGGGATTTC-3′ (19).
To detect miR-150 expression of aforementioned ICG-001-treated or
transfected C666-1, C17, HONE-1 cells and the C666-1 tumor spheres
prepared as subsequently mentioned, a TaqMan® MicroRNA
Reverse Transcription kit (Applied Biosystems; Thermo Fisher
Scientific, Inc.) was used for RT, according to the manufacturer's
protocol, while TaqMan 2X Universal PCR Master mix, no AmpERASE UNG
(Applied Biosystems; Thermo Fisher Scientific, Inc.) was used for
qPCR. All of the procedures were performed according to
manufacturer's protocols. Specific primers for miR-150 were
supplied by TaqMan MicroRNA Assays (Assay ID 000473; Applied
Biosystems; Thermo Fisher Scientific, Inc.). U6 small nuclear RNA
(Assay ID 001093; Applied Biosystems; Thermo Fisher Scientific,
Inc.) was used as the internal control. The relative expressions of
CD44 and miR-150 transcripts were calculated with the
2−ΔΔCq method (20).
Tumor sphere formation assay
A tumor sphere formation assay was performed as
previously described (8). Briefly,
C666-1 cells (2×103 cells/well) in DMEM/F12 (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 20 ng/ml
epidermal growth factor (Sigma-Aldrich; Merck KGaA), 20 ng/ml
fibroblast growth factor (Cell Signaling Technology, Inc.) and 20
ng/ml insulin-like growth factor (Cell Signaling Technology, Inc.)
were seeded onto 24-well ultra-low attachment culture plates
(Corning Incorporated) for 7 days. Growth factors were added to the
cultures every 2-3 days, and the cells were incubated at 37°C in a
5% CO2 humidified incubator. Following incubation, the
tumor spheres were observed under an inverted microscope
(magnification, ×50), the images were captured, and the size of
those tumor spheres measuring >20 µm was determined by
ImageJ software.
Target gene prediction for miRNA
Potential targets of miR-150 were predicted using
the online bioinformatics software TargetScan Human, version 6.2
(http://www.targetscan.org/vert_61/).
Luciferase reporter assay
To investigate the activity of Wnt signaling, C666-1
cells were transfected with the T-cell factor (TCF) reporter
plasmid M50 Super 8× TOPFlash (2 µg) for 24 h. An internal
control vector, pRL-TK (10 ng; Promega Corporation, Madison, WI,
USA), was co-transfected into the cells for normalization of the
transfection efficiency. Cells were then treated with or without
ICG-001 for 24 h. M51 Super 8× FOPFlash (a TOPFlash mutant with
mutated TCF sites) was used as a negative control. The firefly
luciferase activity of TOPFlash and FOPFlash, and the
Renilla luciferase activity of pRL-TK were measured using
Dual-Luciferase Reporter Assay system (Promega Corporation) in a
microplate luminometer (Tecan Group, Ltd., Mannedorf, Switzerland).
The luciferase reporter plasmids M50 Super 8× TOPFlash (cat. no.
12456) and M51 Super 8× FOPFlash (cat. no. 12457) were obtained
from Addgene, Inc. (Cambridge, MA, USA). All the transfections were
performed in the presence of Lipofectamine 2000, and the cells were
incubated at 37°C in a 5% CO2 humidified incubator.
To investigate the interaction between miR-150 and
the 3′-untranslated region (UTR) of CD44 gene transcript, CD44
(NM_000610) Human 3′ UTR Clone (wild-type 3′-UTR reporter clones
for CD44, wt-CD44 3′UTR) was purchased from OriGene Technologies,
Inc. (Rockville, MD, USA), while CD44 3′-UTR mutant constructs with
a mutated miR-150 seed region (mut-CD44 3′UTR) were generated using
a QuikChange Lightning Multi Site-Directed Mutagenesis kit (Agilent
Technologies, Inc., Santa Clara, CA, USA) with the primer
5′-AGATAAATAGCTTCACCCTTTGGGTGTGGGGGGG
AAGCATCTGAAAAATTTCTAGAGGGG-3′. The wild-type or mutant CD44 3′-UTR
luciferase reporter (50 ng) along with 200 nM miR-150 mimic
(Pre-miR-150; Ambion; Thermo Fisher Scientific, Inc.) or miRNA
mimic control (Pre-control; Ambion; Thermo Fisher Scientific, Inc.)
were transfected into C666-1 cells using Lipofectamine 2000 for 48
h. Prior to cell lysis with the Passive Lysis buffer provided by
the Luciferase Assay system (Promega Corporation), the signal of
the red fluorescent protein (RFP) transcribed by the vector was
determined with a fluorescence microscope (magnification, ×10).
Subsequently, luciferase activities were measured using the
Dual-Luciferase Reporter Assay system (Promega Corporation) in a
microplate luminometer and normalized to the signals of RFP.
Immunohistochemical (IHC) staining
A total of 8 female athymic BALB/c nu/nu mice (~15 g
per each) at 6-8 weeks were supplied by the Laboratory Animal Unit
of the University of Hong Kong (Hong Kong, China), and housed in
sterile rodent micro-isolator systems and given free access to
sterile water and food by the Department of Clinical Oncology of
Queen Elizabeth Hospital Hong Kong (Hong Kong, China). The animal
experiment was conducted under license from the Hong Kong
Department of Health and approved by the Committee on the Use of
Live Animals in Teaching and Research at the University of Hong
Kong. According to our previous study, mouse xenograft tumors were
generated by injecting C666-1 cells into the nude mice, and the
mice were sacrificed by cervical dislocation (11). The xenografts were removed and
fixed with 10% neutral buffered formalin at room temperature for 12
h, and then embedded in paraffin wax. The thickness of the sections
was 5 µm. Heat-induced epitope retrieval at 98°C was
performed in 10 mM sodium citrate buffer (Sigma-Aldrich; Merck
KGaA). The tissues were blocked with 10% goat serum (Sigma-Aldrich;
Merck KGaA) and 1% bovine serum albumin (Affymetrix; Thermo Fisher
Scientific, Inc.) in PBS for 30 min at room temperature, followed
by staining with an anti-CD44 antibody (overnight incubation at
room temperature; dilution, 1:100), polyclonal goat anti-mouse
biotinylated IgG (1.5 h incubation at room temperature; dilution
1:300; cat. no. E043301; Dako; Agilent Technologies, Inc.) and
streptavidin/HRP (45 min incubation at room temperature; dilution
1:300; cat. no. P039701-2; Dako; Agilent Technologies, Inc.). The
staining signals were visualized using 3,3′-diaminobenzidine
tetrahydrochloride hydrate (Sigma-Aldrich; Merck KGaA). The
sections were counterstained with hematoxylin (5 min at room
temperature) (Sigma-Aldrich; Merck KGaA) and the images were
captured under an inverted microscope (magnification, ×400). The
CD44 staining intensity was analyzed by Spectrum version 11.1.1.765
software (Aperio Technologies, Vista, CA, USA).
Statistical analysis
Data are presented as the mean ± standard deviation
of ≥3 independent experiments. The difference between control and
treatment groups was determined by one-way analysis of variance
followed by Holm-Sidak comparison method using SigmaPlot Version
12.0 (Systat Software, Inc., San Jose, CA, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
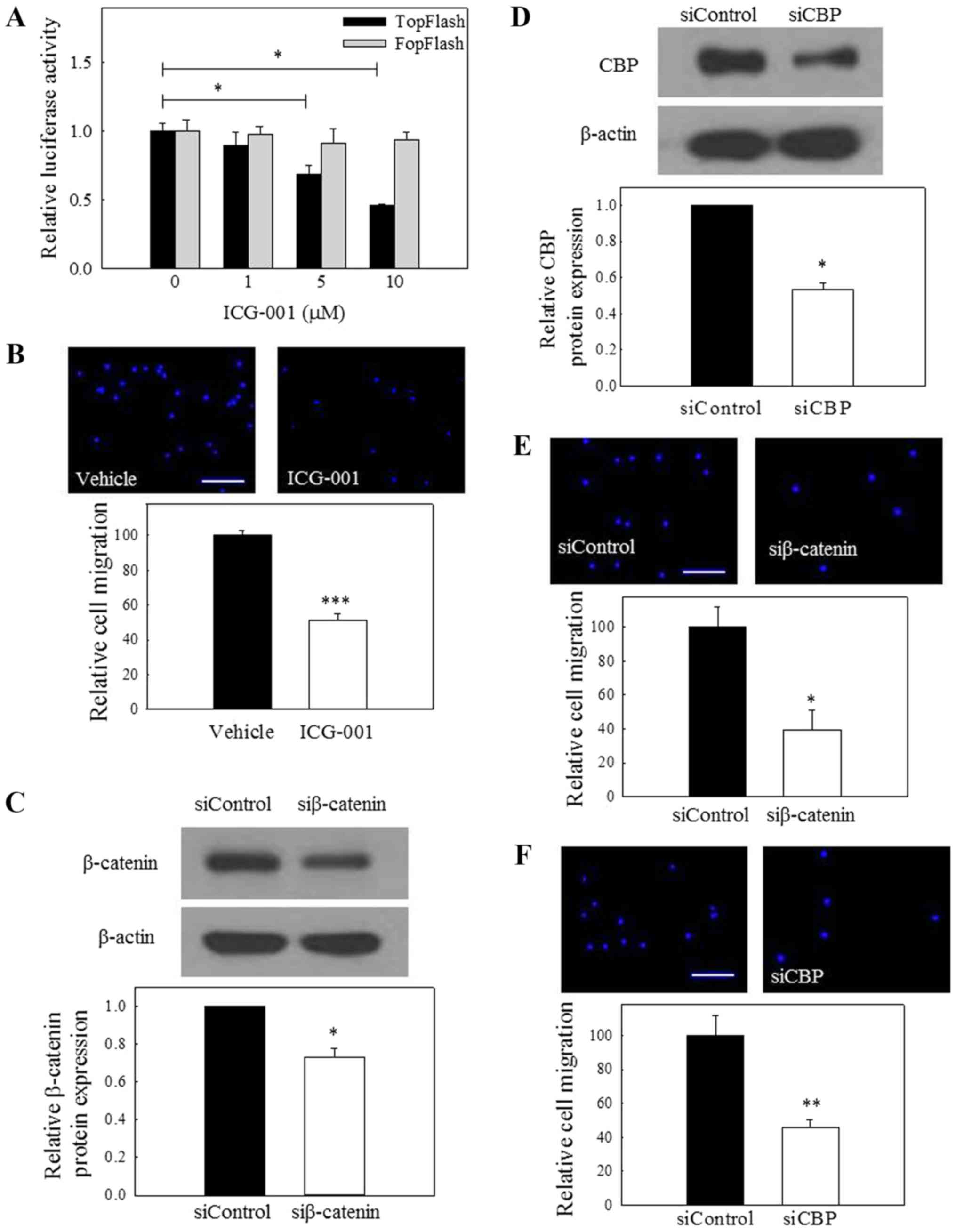
ICG-001 inhibits canonical Wnt signaling
in NPC cells
ICG-001 is a small molecule, CBP antagonist capable
of modulating Wnt-mediated β-catenin transcription. Thus, the
present study first confirmed the effect of ICG-001 on TCF reporter
activity in C666-1 NPC cells. The results depicted in Fig. 1A indicate that ICG-001 could
significantly reduce the luminescent signal of the TOPFlash
reporter, but not that of the FOPFlash reporter, which contains
mutated TCF sites. This observation indicated that ICG-001 could
antagonize β-catenin/TCF transcription in our cell model. Together
with a group of previously reported Wnt target genes that are
downregulated by ICG-001 in C666-1 cells (11), it was confirmed that ICG-001
specifically inhibited the canonical Wnt signaling pathway in NPC
cells.
ICG-001 inhibits the migration of NPC
cells
Our group previously demonstrated that ICG-001 could
restore the expression of the tumor suppressor miR-145 and inhibit
the growth of CSC-enriched NPC spheroid cells (11). The present study further revealed
that ICG-001 could significantly inhibit the migration of NPC cells
in a Transwell migration assay (Fig.
1B). Since ICG-001 is capable of interfering with the
β-catenin/CBP downstream signaling, it was hypothesized that siRNA
silencing of β-catenin or CBP expression could also inhibit the
migration of NPC cells. The present results demonstrated that siRNA
knockdown of the protein expression of β-catenin (Fig. 1C) or CBP (Fig. 1D) resulted in signifi-cantly
reduced migration of NPC cells (Fig.
1E and F). These observations indicate that β-catenin and CBP
are involved in the regulation of migration of NPC cells.
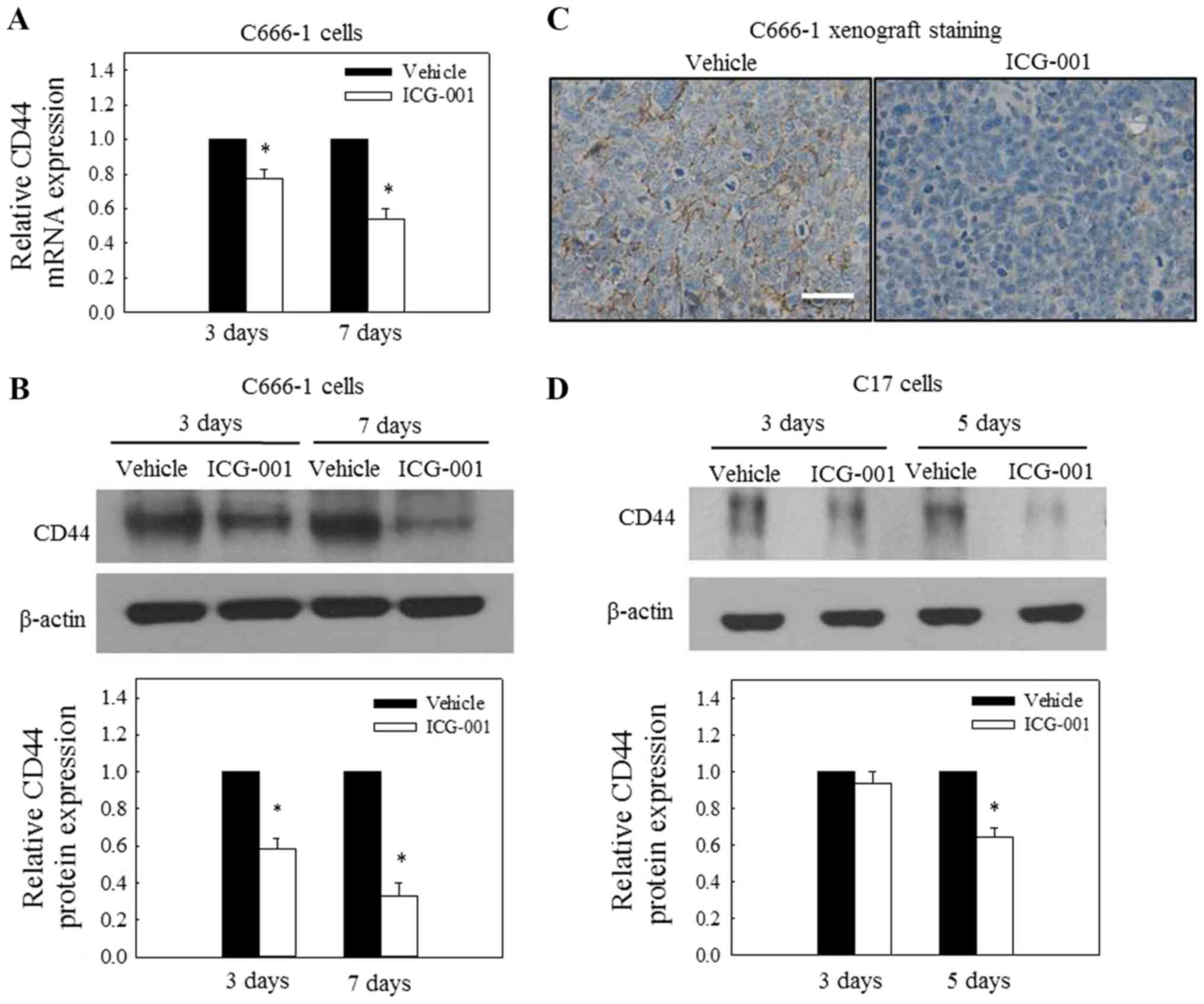
Involvement of CD44 in the migration of
NPC cells
The expression of CD44 has previously been
demonstrated to be regulated by Wnt/β-catenin signaling (21). In the present study, the expression
of CD44 was determined in ICG-001-treated NPC cells. The results
depicted in Fig. 2A and B indicate
that ICG-001 significantly inhibited the mRNA and protein
expression of CD44 in C666-1 cells. To further confirm the effect
of ICG-001 on the expression of CD44 in tumor tissues, IHC staining
of CD44 was performed on tumor tissues obtained from C666-1
tumor-bearing nude mice treated with ICG-001 or untreated. The
results from Fig. 2C indicated
reduced immunoreactivity of CD44 in the tumor tissues obtained from
ICG-001-treated animals. Additionally, the effect of ICG-001 on the
protein expression of CD44 was also significantly reduced in
EBV-positive NPC C17 cells (Fig.
2D). These results indicated that ICG-001 could downregulate
the expression of CD44 in various NPC cell lines.
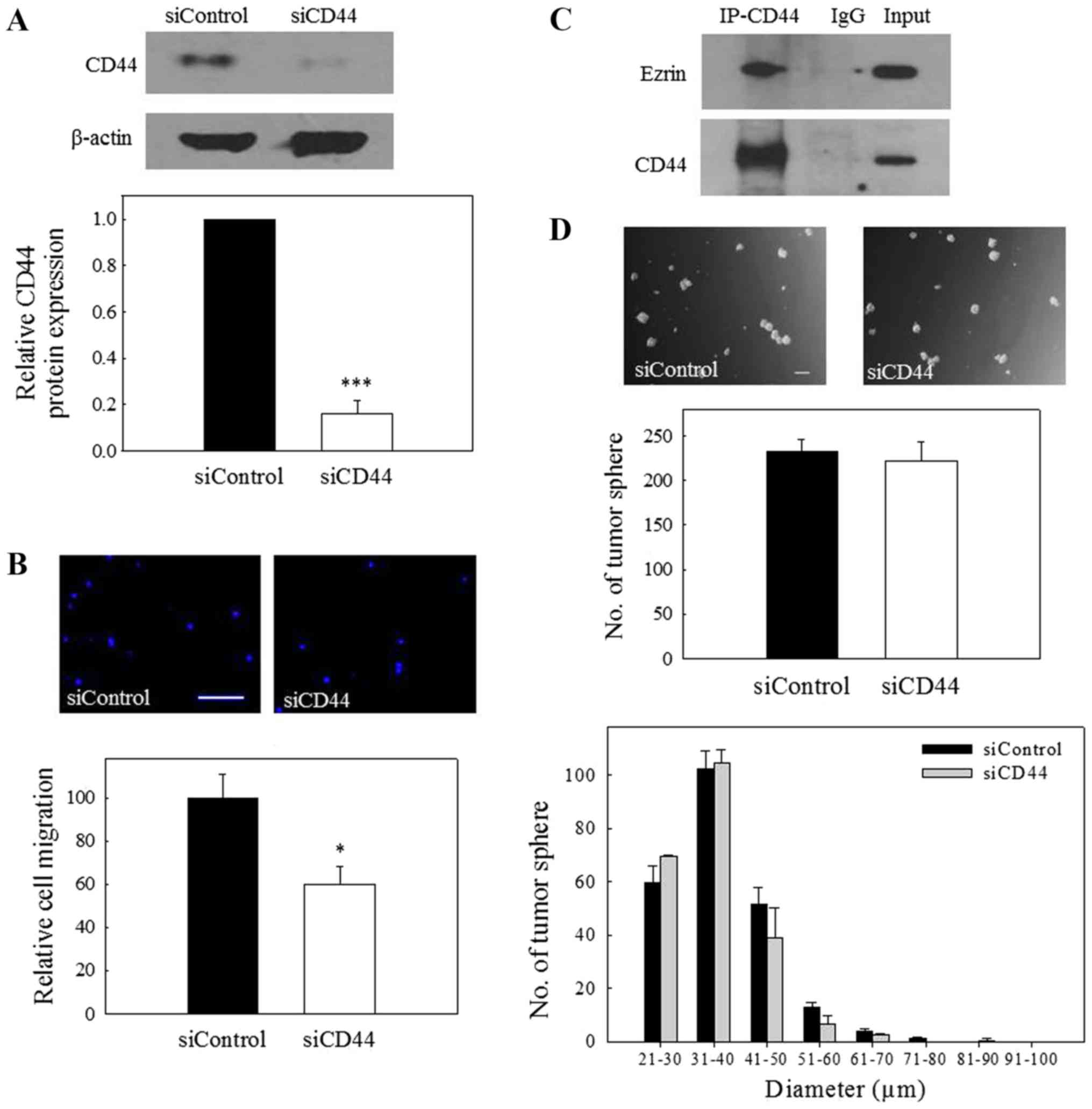
To confirm the involvement of CD44 in the migration
of NPC cells, the expression of CD44 was knocked down by
transfection of the NPC cells with CD44 siRNA. The results depicted
in Fig. 3A revealed that CD44
siRNA could significantly reduce the protein expression of CD44,
and this effect was accompanied by a significant reduction in the
migration of NPC cells (Fig. 3B).
Ezrin is a key molecule associating the plasma membrane components
with the cytoskeleton, and its association with CD44 is
particularly important in mediating cell migration (22,23).
To further confirm the interaction of CD44 with ezrin in NPC cells,
Co-IP was performed using total cell lysates. The results of
Fig. 3C revealed Co-IP between
CD44 and ezrin, indicating that CD44 interacts with the
aforementioned migration-regulatory components in NPC cells.
Notably, the capability of CD44 siRNA-transfected cells to form
tumor spheres was not significantly different to that of control
siRNA-transfected cells (Fig. 3D),
indicating that CD44 is not involved in the growth of C666-1 tumor
spheres. Collectively, the results from these experiments indicate
that CD44 is involved in the regulation of the migration of NPC
cells.
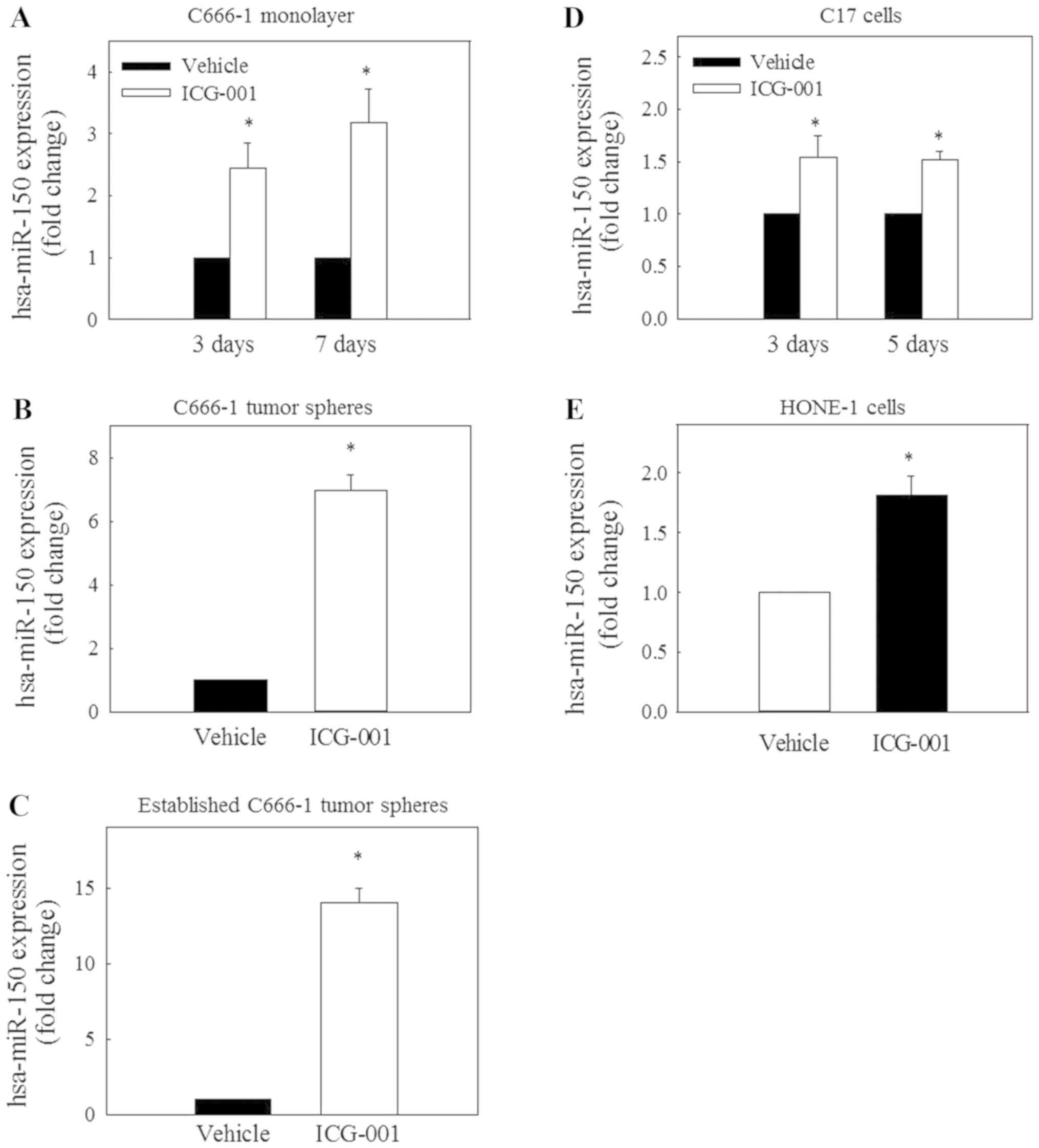
ICG-001 restores the expression of
miR-150
A recent study on miRNA expression demonstrated that
the expression level of miR-150 is reduced in NPC tissues (24). To evaluate the possible involvement
of miR-150 in the ICG-001-mediated inhibition of CD44 expression,
the expression of miR-150 was determined in C666-1 cells under
monolayer and spheroid culture conditions. The results in Fig. 4A-C revealed that ICG-001 could
significantly restore miR-150 expression under these culture
conditions. Significantly restored expression of miR-150 was also
observed in EBV-positive C17 (Fig.
4D) and EBV-negative HONE-1 (Fig.
4E) NPC cells.
It has been previously reported that the HONE-1 cell
line appears to be a part of the HeLa genomic sequence, indicating
that it may be a derivative of HeLa cervical carcinoma cells and
another cell line of unknown origin (25). To ensure that the HONE-1 cells used
in the present study were not contaminated by HeLa cells, a single
duplex detection PCR assay was performed. The results revealed that
no L1 insertion was detected in HONE-1 cells, indicating that the
HONE-1 cells used in the present study are not likely to be
cross-contaminated with HeLa cells (data not shown).
CD44 is a direct target of miR-150
In addition to the reduced expression of CD44 mRNA,
the present study sought to examine whether additional mechanisms
are also involved in the downregulated expression of CD44 in
ICG-001-treated NPC cells. To determine the possible involvement of
miRNA in the expression of CD44, the impact of overexpression of
miR-150 on the protein expression of CD44 was evaluated. Fig. 5A demonstrates that C666-1 cells
could be efficiently transfected with pre-miR-150. Subsequently, a
significant reduction in CD44 protein expression was observed in
pre-miR-150-transfected cells (Fig.
5B), indicating that miR-150 may be involved in the
ICG-001-induced downregulated expression of CD44 in NPC cells.
Based on the prediction results from TargetScan, miR-150
potentially targets the 3′-UTR of CD44 mRNA (Fig. 5C, upper panel). To further confirm
the specific action of miR-150, a 3′-UTR reporter assay was used to
verify the targeting of the 3′UTR of CD44 mRNA by miR-150. For that
purpose, C666-1 cells were transfected with a wild-type or mutant
CD44 3′-UTR reporter together with a pre-miR-150 (miR-150 mimic) or
a miRNA mimic control. The results in the lower panel of Fig. 5C revealed that miR-150 could
significantly reduce the wild-type 3′-UTR reporter activity,
whereas the effect of miR-150 on the mutant 3′-UTR was not
significant. These observations indicate that CD44 is a novel
target of miR-150 in NPC cells. In a functional study, transfection
of C666-1 cells with pre-miR-150 resulted in a significant
reduction in the migration of tumor cells (Fig. 5D). Furthermore, this inhibitory
effect could be attenuated when the cells were transfected with
pre-miR-150 and pCMV-CD44.
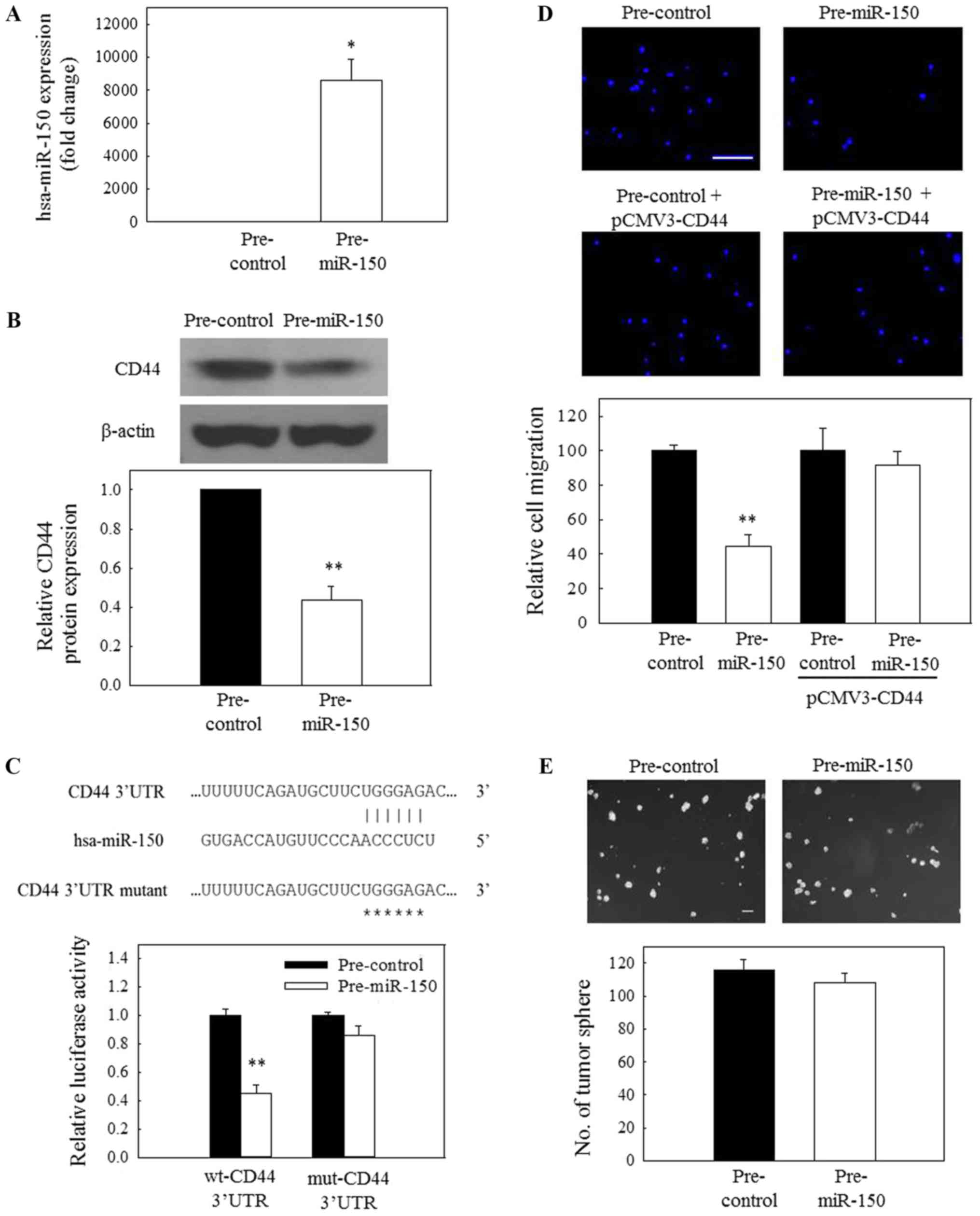 | Figure 5The miR-150/CD44 axis is involved in
the regulation of nasopharyngeal carcinoma cell migration. (A) The
transfection efficiency of pre-miR-150 at 50 nM in C666-1 cells was
confirmed by reverse transcription-quantitative polymerase chain
reaction. (B) Pre-miR-150 (50 nM) inhibited the protein expression
of CD44. (C) A putative binding site of miR-150 on CD44 mRNA 3′-UTR
was first predicted using TargetScan. Subsequently, C666-1 cells
were co-transfected with 200 nM pre-miR-150 or 200 nM pre-control
and 50 ng wt-CD44 3′-UTR reporter vector or 50 ng mut-CD44 3′-UTR
reporter vector. Luciferase activity was normalized to the red
fluorescence protein signal, and representative images are
depicted. (D) Overexpression of miR-150 (50 nM) resulted in the
inhibition of migration of C666-1 cells, but this inhibitory effect
was attenuated when the cells were treated with pre-miR-150 (50 nM)
and pCMV-CD44 (200 ng). Scale bar, 20 µm. (E) Treatment with
pre-miR-150 (50 nM) had no significant effect on the growth of
tumor spheres. Scale bar, 100 µm. *P<0.05 and
**P<0.01, compared with pre-control. miR, microRNA;
CD44, cluster of differentiation 44; pre-miR, precursor miR;
pre-control, precursor control miR; 3′-UTR, 3′-untranslated region;
wt, wild-type; mut, mutant. |
miR-150 overexpression inhibits tumor
cell migration but not spheroid growth
As aforementioned, siRNA knockdown of CD44 only
inhibited the migration but not the growth of NPC tumor spheres. In
the present study, two biological assays, spheroid formation and
tumor cell migration assays, were then used to further evaluate the
functional implication of the miR-150/CD44 axis in NPC cells. NPC
cells were transfected with the pre-miR-150. The results in
Fig. 5D revealed that the
migratory activity of pre-miR-150-transfected NPC cells was
significantly reduced, compared with the control-transfected cells.
However, exogenous miR-150 had no significant effect on the
formation of tumor spheres (Fig.
5E). Collectively, these data indicate that the miR-150/CD44
axis is involved in the regulation of NPC cell migration.
Discussion
CD44 is a cell surface membrane molecule involved in
the regulation of diverse functions, including migration, matrix
assembly, apoptosis resistance and drug resistance, in tumor cells
in a context-dependent manner (4).
It has previously been demonstrated that CD44 may interact with
hyaluronic acid and stimulate the proliferation of endothelial
cells (26). In colon cancer, CD44
has been indicated to regulate the in vitro and in
vivo growth of xenografts in animals (27). CD44 has also been implicated in the
self-renewal and maintenance of pluripotency (4). Another well-known function of CD44 is
the regulation of tumor cell migration via interaction with ezrin
(28). In NPC, an association
between the expression of CD44 and the characteristics of the
epithelial-mesenchymal transition (EMT) has previously been
reported (29). However, the
biological function of CD44 in NPC cells has not been fully
examined to date. In view of the importance of CD44 in the
regulation of tumor cell activities, the present study sought to
further examine the underlying mechanisms of ICG-001-induced
downregulation of CD44 expression in NPC cells.
An association between ICG-001-mediated inhibition
of in vitro migration of NPC cells and downregulated
expression of CD44 at the mRNA and protein level was observed.
ICG-001 is a CBP antagonist that has previously been demonstrated
to block the interaction between β-catenin and CBP, and to inhibit
the downstream transcription of a subset of Wnt target genes
(30). The reduced expression of
CD44 mRNA observed in the present study could be explained by the
fact that CD44 is a well-known Wnt downstream target gene (31). Notably, a significant increase in
the expression of miR-150 was also observed in ICG-001-treated NPC
cells. Previous studies demonstrated that miR-150 is one of the
miRNAs downregulated in NPC (24,32).
Further bioinformatics analysis using TargetScan indicated that
CD44 is a predicted target of miR-150. Using the CD44 3′-UTR
luciferase reporter assay, the present study confirmed that CD44 is
a novel target of miR-150. This observation indicated that the
restored expression of miR-150 may be an additional mechanism
contributing to the reduced protein expression of CD44 in
ICG-001-treated NPC cells.
Regarding the biological functions of miR-150 in
tumorigenesis, previous studies demonstrated that the expression of
miR-150 is significantly reduced in various tumor types, including
colon cancer (33), chronic
myeloid leukemia (34), acute
lymphocytic leukemia (35), mantle
cell lymphoma (36), Burkitt
lymphoma (BL) (37), gastric
cancer (38), esophageal squamous
cell carcinoma (39) and natural
killer (NK)/T-cell lymphoma (40).
In BL, re-expression of miR-150 inhibited tumor cell proliferation
and induced the differentiation of tumor cells by targeting c-Myb
(41). The involvement of the
miR-150/Myb axis in the regulation of tumor cell differentiation
was also demonstrated in myeloid leukemia (35). In NK/T-cell lymphoma,
overexpression of miR-150 resulted in the inhibition of tumor cell
proliferation and induction of apoptosis through downregulated
expression of Dyskeratosis congenita 1 and RAC-β
serine/threonine-protein kinase (40). In esophageal squamous cell
carcinoma, the tumor-suppressive activity of miR-150 was attributed
to its capacity to target the EMT inducer Zinc finger E-box-binding
homeobox 1 (39). These
observations indicate that miR-150 acts as a tumor suppressor in a
number of tumor types, and its effect is cellular
context-dependent. A previous study demonstrated that the reduced
level of miR-150 expression could be due to promoter methylation
(42). In the preliminary study,
the status of methylation of miR-150 promoter was examined.
However, a significant change of the methylation status of the
promoter was not observed following ICG-001 treatment. The
mechanistic action of miR-150 remains under investigation.
In the present study, a novel function of miR-150
was identified in its capacity to target CD44 and inhibit the
migration of NPC cells. This observation was further supported by
the results of the Co-IP/western blotting assay, which revealed
that CD44 co-precipitates with ezrin, an important cytoplasmic
component known to be associated with the migration-controlling
machinery in the cytoplasm (22,23).
This result is reminiscent of the previous observation by Endo
et al (43) that
overexpression of the EBV protein Latent membrane protein 1 (LMP-1)
in NPC cells could activate ezrin and the subsequent linking of
ezrin with CD44. Furthermore, IHC analysis of 200 NPC tissues
revealed that increased expression of ezrin was associated with an
increased rate of lymph node metastasis (44). A similar observation was also
previously reported in EBV-associated gastric carcinoma with
lymphoid stroma (GCLS), where high levels of ezrin in GCLS were
associated with lymph node metastasis (45). The importance of CD44 and ezrin has
also recently been demonstrated in breast cancer (23). Donatello et al (23) reported that CD44 and ezrin are
localized in different membrane locations in non-migrating cells.
Under migration-stimulating conditions, CD44 binds to ezrin and
regulates the migration of breast tumor cells (23). Collectively, these data indicate
that LMP-1/ezrin/CD44 serve an important role in the promotion of
NPC cell migration, and ICG-001 may have an anti-migratory function
through the restoration of the expression of the tumor suppressor
miR-150 in NPC cells.
ICG-001 is a CBP antagonist targeting the Wnt
signaling pathway (46). In the
present study, ICG-001 was observed to disrupt β-catenin/CBP/Wnt
signaling-mediated tumor cell migration via the miR-150/CD44 axis
in NPC cells. The present study also reported a novel function of
miR-150 in NPC cells. Together with our previous observations
(11), the present results
indicate that therapeutic intervention of the Wnt signaling pathway
with this CBP antagonist may be a strategy for inhibiting the
growth and dissemination of NPC tumor cells.
Funding
The present project was supported by the Research
Grants Council of the Hong Kong Special Administrative Region for
the NPC Area of Excellence (grant no. AoE/M 06/08 Center for
Nasopharyngeal Carcinoma Research) and the Strategic Development
Fund (grant no. HKBU SDF 15-1012-P04) of Hong Kong Baptist
University.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LSC, OYM, HHK, LC and KCC were involved in the in
vitro and in vivo studies. HLL, RKCN, RNSW, KWL, AWML,
GSWT, MK, MLL and NKM were involved in the project design and data
analysis. LSC and NKM wrote the manuscript. All authors have read
and approved the final manuscript.
Ethics approval and consent to
participate
For the C666-1 xenograft study, nude mice were
supplied by the Laboratory Animal Unit of the University of Hong
Kong (Hong Kong, China) and housed by the Department of Clinical
Oncology of Queen Elizabeth Hospital Hong Kong (Hong Kong, China).
The animal experiment was conducted under license from the Hong
Kong Department of Health and approved by the Committee on the Use
of Live Animals in Teaching and Research at the University of Hong
Kong.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Miss Carman Lo
(Department of Anatomical and Cellular Pathology, State Key
Laboratory in Oncology in South China, The Chinese University of
Hong Kong, Hong Kong, China) for the maintenance of NPC
xenograft-derived EBV-positive C17 NPC cells.
References
|
1
|
Cao SM, Simons MJ and Qian CN: The
prevalence and prevention of nasopharyngeal carcinoma in China.
Chin J Cancer. 30:114–119. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lee AW, Ma BB, Ng WT and Chan AT:
Management of nasopharyngeal carcinoma: Current practice and future
perspective. J Clin Oncol. 33:3356–3364. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lun SW, Cheung ST, Cheung PF, To KF, Woo
JK, Choy KW, Chow C, Cheung CC, Chung GT, Cheng AS, et al:
CD44+ cancer stem-like cells in EBV-associated
nasopharyngeal carcinoma. PLoS One. 7:e524262012. View Article : Google Scholar
|
|
4
|
Zöller M: CD44: Can a cancer-initiating
cell profit from an abundantly expressed molecule? Nat Rev Cancer.
11:254–267. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Takebe N, Miele L, Harris PJ, Jeong W,
Bando H, Kahn M, Yang SX and Ivy SP: Targeting Notch, Hedgehog, and
Wnt pathways in cancer stem cells: Clinical update. Nat Rev Clin
Oncol. 12:445–464. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Garofalo M and Croce CM: Role of microRNAs
in maintaining cancer stem cells. Adv Drug Deliv Rev. 81:53–61.
2015. View Article : Google Scholar :
|
|
7
|
Wang ZM, Du WJ, Piazza GA and Xi Y:
MicroRNAs are involved in the self-renewal and differentiation of
cancer stem cells. Acta Pharmacol Sin. 34:1374–1380. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ravasio R, Ceccacci E and Minucci S:
Self-renewal of tumor cells: Epigenetic determinants of the cancer
stem cell phenotype. Curr Opin Genet Dev. 36:92–99. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Avgustinova A and Benitah SA: The
epigenetics of tumour initiation: Cancer stem cells and their
chromatin. Curr Opin Genet Dev. 36:8–15. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Muñoz P, Iliou MS and Esteller M:
Epigenetic alterations involved in cancer stem cell reprogramming.
Mol Oncol. 6:620–636. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chan KC, Chan LS, Ip JC, Lo C, Yip TT,
Ngan RK, Wong RN, Lo KW, Ng WT, Lee AW, et al: Therapeutic
targeting of CBP/β-catenin signaling reduces cancer stem-like
population and synergistically suppresses growth of EBV-positive
nasopharyngeal carcinoma cells with cisplatin. Sci Rep. 5:99792015.
View Article : Google Scholar
|
|
12
|
Busson P, Ganem G, Flores P, Mugneret F,
Clausse B, Caillou B, Braham K, Wakasugi H, Lipinski M and Tursz T:
Establishment and characterization of three transplantable
EBV-containing nasopharyngeal carcinomas. Int J Cancer. 42:599–606.
1988. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Glaser R, Zhang HY, Yao KT, Zhu HC, Wang
FX, Li GY, Wen DS and Li YP: Two epithelial tumor cell lines (HNE-1
and HONE-1) latently infected with Epstein-Barr virus that were
derived from nasopharyngeal carcinomas. Proc Natl Acad Sci USA.
86:9524–9528. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yao KT, Zhang HY, Zhu HC, Wang FX, Li GY,
Wen DS, Li YP, Tsai CH and Glaser R: Establishment and
characterization of two epithelial tumor cell lines (HNE-1 and
HONE-1) latently infected with Epstein-Barr virus and derived from
nasopharyngeal carcinomas. Int J Cancer. 45:83–89. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cheng Y, Ho RL, Chan KC, Kan R, Tung E,
Lung HL, Yau WL, Cheung AK, Ko JM, Zhang ZF, et al: Anti-angiogenic
pathway associations of the 3p21.3 mapped BLU gene in
nasopharyngeal carcinoma. Oncogene. 34:4219–4228. 2015. View Article : Google Scholar
|
|
16
|
Chai AW, Cheung AK, Dai W, Ko JM, Ip JC,
Chan KW, Kwong DL, Ng WT, Lee AW, Ngan RK, et al:
Metastasis-suppressing NID2, an epigenetically-silenced gene, in
the pathogenesis of nasopharyngeal carcinoma and esophageal
squamous cell carcinoma. Oncotarget. 7:78859–78871. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rahbari R, Sheahan T, Modes V, Collier P,
Macfarlane C and Badge RM: A novel L1 retrotransposon marker for
HeLa cell line identification. Biotechniques. 46:277–284. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Peterson LF, Wang Y, Lo MC, Yan M, Kanbe E
and Zhang DE: The multi-functional cellular adhesion molecule CD44
is regulated by the 8;21 chromosomal translocation. Leukemia.
21:2010–2019. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lo MC, Yip TC, Ngan KC, Cheng WW, Law CK,
Chan PS, Chan KC, Wong CK, Wong RN, Lo KW, et al: Role of
MIF/CXCL8/CXCR2 signaling in the growth of nasopharyngeal carcinoma
tumor spheres. Cancer Lett. 335:81–92. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Wielenga VJ, Smits R, Korinek V, Smit L,
Kielman M, Fodde R, Clevers H and Pals ST: Expression of CD44 in
Apc and Tcf mutant mice implies regulation by the WNT pathway. Am J
Pathol. 154:515–523. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bretscher A, Reczek D and Berryman M:
Ezrin: A protein requiring conformational activation to link
microfilaments to the plasma membrane in the assembly of cell
surface structures. J Cell Sci. 110:3011–3018. 1997.
|
|
23
|
Donatello S, Babina IS, Hazelwood LD, Hill
AD, Nabi IR and Hopkins AM: Lipid raft association restricts
CD44-ezrin interaction and promotion of breast cancer cell
migration. Am J Pathol. 181:2172–2187. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang S, Mo Y, Midorikawa K, Zhang Z, Huang
G, Ma N, Zhao W, Hiraku Y, Oikawa S and Murata M: The potent tumor
suppressor miR-497 inhibits cancer phenotypes in nasopharyngeal
carcinoma by targeting ANLN and HSPA4L. Oncotarget. 6:35893–35907.
2015.PubMed/NCBI
|
|
25
|
Strong MJ, Baddoo M, Nanbo A, Xu M,
Puetter A and Lin Z: Comprehensive high-throughput RNA sequencing
analysis reveals contamination of multiple nasopharyngeal carcinoma
cell lines with HeLa cell genomes. J Virol. 88:10696–10704. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Murphy JF, Lennon F, Steele C, Kelleher D,
Fitzgerald D and Long AC: Engagement of CD44 modulates
cyclooxygenase induction, VEGF generation, and proliferation in
human vascular endothelial cells. FASEB J. 19:446–448. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Subramaniam V, Vincent IR, Gilakjan M and
Jothy S: Suppression of human colon cancer tumors in nude mice by
siRNA CD44 gene therapy. Exp Mol Pathol. 83:332–340. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Arpin M, Chirivino D, Naba A and
Zwaenepoel I: Emerging role for ERM proteins in cell adhesion and
migration. Cell Adhes Migr. 5:199–206. 2011. View Article : Google Scholar
|
|
29
|
Lin CH, Hung PH and Chen YJ: CD44 is
associated with the aggressive phenotype of nasopharyngeal
carcinoma through redox regulation. Int J Mol Sci. 14:13266–13281.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Takahashi-Yanaga F and Kahn M: Targeting
Wnt signaling: Can we safely eradicate cancer stem cells? Clin
Cancer Res. 16:3153–3162. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schmitt M, Metzger M, Gradl D, Davidson G
and Orian-Rousseau V: CD44 functions in Wnt signaling by regulating
LRP6 localization and activation. Cell Death Differ. 22:677–689.
2015. View Article : Google Scholar :
|
|
32
|
Li T, Chen JX, Fu XP, Yang S, Zhang Z,
Chen KhH and Li Y: microRNA expression profiling of nasopharyngeal
carcinoma. Oncol Rep. 25:1353–1363. 2011.PubMed/NCBI
|
|
33
|
Bao Y, Guo Y, Li Z, Fang W, Yang Y, Li X,
Li Z, Xiong B, Chen Z, Wang J, et al: MicroRNA profiling in Muc2
knockout mice of colitis-associated cancer model reveals epigenetic
alterations during chronic colitis malignant transformation. PLoS
One. 9:e991322014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Machová Poláková K, Lopotová T, Klamová H,
Burda P, Trněný M, Stopka T and Moravcová J: Expression patterns of
microRNAs associated with CML phases and their disease related
targets. Mol Cancer. 10:412011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Morris VA, Zhang A, Yang T, Stirewalt DL,
Ramamurthy R, Meshinchi S and Oehler VG: MicroRNA-150 expression
induces myeloid differentiation of human acute leukemia cells and
normal hematopoietic progenitors. PLoS One. 8:e758152013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhao JJ, Lin J, Lwin T, Yang H, Guo J,
Kong W, Dessureault S, Moscinski LC, Rezania D, Dalton WS, et al:
microRNA expression profile and identification of miR-29 as a
prognostic marker and pathogenetic factor by targeting CDK6 in
mantle cell lymphoma. Blood. 115:2630–2639. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang M, Yang W, Li M and Li Y: Low
expression of miR-150 in pediatric intestinal Burkitt lymphoma. Exp
Mol Pathol. 96:261–266. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Assumpção MB, Moreira FC, Hamoy IG,
Magalhães L, Vidal A, Pereira A, Burbano R, Khayat A, Silva A,
Santos S, et al: High-throughput miRNA sequencing reveals a field
effect in gastric cancer and suggests an epigenetic network
mechanism. Bioinform Biol Insights. 9:111–117. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yokobori T, Suzuki S, Tanaka N, Inose T,
Sohda M, Sano A, Sakai M, Nakajima M, Miyazaki T, Kato H, et al:
miR-150 is associated with poor prognosis in esophageal squamous
cell carcinoma via targeting the EMT inducer ZEB1. Cancer Sci.
104:48–54. 2013. View Article : Google Scholar
|
|
40
|
Watanabe A, Tagawa H, Yamashita J, Teshima
K, Nara M, Iwamoto K, Kume M, Kameoka Y, Takahashi N, Nakagawa T,
et al: The role of microRNA-150 as a tumor suppressor in malignant
lymphoma. Leukemia. 25:1324–1334. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen S, Wang Z, Dai X, Pan J, Ge J, Han X,
Wu Z, Zhou X and Zhao T: Re-expression of microRNA-150 induces
EBV-positive Burkitt lymphoma differentiation by modulating c-Myb
in vitro. Cancer Sci. 104:826–834. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jia Y, Ling M, Zhang L, Jiang S, Sha Y and
Zhao R: Downregulation of miR-150 expression by DNA
hypermeth-ylation is associated with high
2-hydroxy-(4-methylthio)butanoic acid-induced hepatic cholesterol
accumulation in nursery piglets. J Agric Food Chem. 64:7530–7539.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Endo K, Kondo S, Shackleford J, Horikawa
T, Kitagawa N, Yoshizaki T, Furukawa M, Zen Y and Pagano JS:
Phosphorylated ezrin is associated with EBV latent membrane protein
1 in naso-pharyngeal carcinoma and induces cell migration.
Oncogene. 28:1725–1735. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang L, Lin GN, Jiang XL and Lu Y:
Expression of ezrin correlates with poor prognosis of
nasopharyngeal carcinoma. Tumour Biol. 32:707–712. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tobo T, Hirahashi M, Yao T, Aishima S and
Oda Y: Ezrin expression and its phosphorylation in gastric
carcinoma with lymphoid stroma and Epstein-Barr virus infection.
Mol Clin Oncol. 1:220–224. 2013. View Article : Google Scholar
|
|
46
|
Teo JL and Kahn M: The Wnt signaling
pathway in cellular proliferation and differentiation: A tale of
two coactivators. Adv Drug Deliv Rev. 62:1149–1155. 2010.
View Article : Google Scholar : PubMed/NCBI
|