Introduction
Gliomas pose as a serious issue in the field of
neurooncology as they represent ~30% of all brain tumors and 80% of
all malignant brain tumors (1).
Furthermore, gliomas tend to be incurable and the median survival
is dependent on the tumor type. Patients diagnosed with low-grade
tumors exhibit a median survival of >10 years (2). The outcome is notably different in
the case of high-grade gliomas, for which the median survival
varies between 3 years for anaplastic glioma and 15 months for
glioblastoma multiforme (3). In
addition, there are few therapeutic solutions for the treatment of
gliomas. One of the most favorable approach is the combination of
chemo- and radiotherapy with surgical intervention (4); however, none of the currently
available methods provide satisfying results. Additionally, as
described by Neeman and Ben-Eliyahu (5), surgery does not only enhance the
passage of malignant cells into the blood and lymphatic vessels,
but also inhibits the apoptosis of abnormal cells and increases
their invasiveness, which correlates with higher metastatic
ability. Furthermore, alterations in the immune and neuroendocrine
systems in the response to surgery have been associated with the
promotion of cancer progression (6).
For patients, one of the most notable symptom
associated with surgical glioma excision is pain. As a preventative
measure, local anesthetics, such as lidocaine can be used. In
addition to analgesia, these agents may possess other beneficial
properties. Lidocaine exhibits its activity by blocking
voltage-gated sodium channels (VGSC) on nociceptors, which leads to
the elimination of pain; however, these receptors are expressed on
the surface of some cancer cells (7-9). In
the case of cancer, increased expression of VGSC has been
demonstrated in some highly metastatic lines compared with
low-metastatic lines (10). In
addition, the blockage of these channels by chemical agents leads
to reduced invasiveness (11,12).
This indicates the importance of VGSC in the metastatic process.
Therefore, the effects of lidocaine on cancer cells requires
further investigation. As reported by numerous studies, lidocaine
and other local anesthetic drugs decrease the growth, invasiveness
and migration potential of non-small-cell lung carcinoma, breast
cancer or human hepatocellular carcinoma cells (13-16).
Recently, the anti-proliferative effects of lidocaine on glioma
cells was reported (17); however,
in this case lidocaine activity may be associated with the
inhibition of the melastatin-like transient receptor potential 7
(TRPM7) action.
Local anesthetics have been proposed to induce the
death of cancer cells via the apoptotic and necrotic pathways
(18-20); however, our current research also
indicates the occurrence of autophagy. In normal cells, autophagy
aims to degrade unnecessary or damaged organelles, and proteins
(21). In cancer cells, this
process serves different roles. In the early stages of cancer, when
the tumor is not yet vascularized, autophagy can serve the cell as
a way to obtain nutrition; thus, tumorigenesis is induced.
Conversely, mutations in the genes responsible for autophagy are
generally associated with enhanced progression of cancer (22-24).
There are two explanations for this. The first is that autophagy
inhibition leads to increased necrosis rate, which is associated
with the inflammatory response and thus cancer development. The
second may be rapid accumulation of mutations due to the lack of
counteracting the effects of increased oxidative stress by
autophagy (25); however, in all
cases, autophagy is be easily detected by observing characteristic
structures, including the autophagosome and autophagolysosomes
(26).
The present study aimed to determine the effects of
lidocaine on rat C6 glioma cells; this local anesthetic induces
protective autophagy. From a clinical point of view, the findings
of the present study suggests that the use of lidocaine in a
patient with glioma should be carefully considered. We also propose
to use other anesthetic agents preferentially.
Materials and methods
Cell culture and treatment
C6, a rat glioma cell line, was purchased from
American Type Culture Collection (CCL-107, Manassas, VA, USA). The
cells were cultured in tissue culture flasks or 12-well plates (BD
Biosciences, Franklin Lakes, NJ, USA) and grown as a monolayer at
37°C in a humidified CO2 incubator (5% CO2)
in Ham's F12 medium (Lonza Group, Ltd., Basel, Switzerland). The
medium was supplemented with 10% fetal bovine serum, 2 mM glutamine
and 50 µg/ml gentamycin (both from Sigma-Aldrich; Merck
KGaA, Darmastadt, Germany). C6 cells were treated for 24 h in 37°C
(incubator) with 0.25, 0.5, 1, 5, 10, 15 and 30 mM of lidocaine
(Sigma-Aldrich; Merck KGaA) for the MTT assay and 5, 10 and 15 mM
for other experiments. The control cells were grown under the same
conditions in the absence of lidocaine. In order to inhibit
autophagy, the cells were pretreated with 100 nM bafilomycin A1
(Baf A1; Sigma-Aldrich; Merck KGaA) for 4 h in 37°C and then
incubated in the presence or absence of lidocaine for 24 h in 37°C
The C6 culture was tested for Mycoplasma based on the rapid
staining with DAPI (Sigma-Aldrich; Merck KGaA). The staining
process was conducted for 10 min at room temperature. The tests
were negative. All in vitro studies were performed on ells
of low passage number (<5). Following 24 h of incubation with
lidocaine (37°C), the cells were observed using an inverted
microscope (magnification, x40; Nikon Corporation, Tokyo, Japan),
at least 5 number of fields per view, which provided the basis for
further analysis.
MTT assay
The cytotoxic effect of lidocaine on cell viability
was assessed using a colorimetric MTT metabolic activity assay. The
cells were cultured in 12-well plates (0.11×106) for 24
h and then treated with 0.25, 0.5, 1, 5, 10, 15 and 30 mM of local
anesthetic for another 24 h (37°C). The MTT stock solution was
prepared by dissolving MTT (Sigma-Aldrich; Merck KGaA) in 5 mg/ml
PBS. Following lidocaine treatment, the cells were washed with PBS
and incubated with MTT solution which was mixed with Dulbecco's
modified Eagle's medium without phenol red (Lonza Group, Ltd. in
the ratio 1:9 for 3 h at 37°C. MTT was reduced by metabolically
active cells to insoluble purple formazan crystals which were
dissolved in isopropanol (2 ml); cells were centrifuged at 15,717 ×
g for 2 min at room temperature. The absorbance was measured at the
wavelength of 570 nm using a spectrophotometer (Spectra Academy,
K-MAC, Daejeon, Korea). The viability of glioma cells was expressed
as the percentage relative to the control cells, which was assumed
as 100%. The viability of cells pretreated with Baf A1 also was
studied using an MTT assay. The experiment was conducted in the
same manner as for cells without Baf A1 pretreatment. After
analyzing the results 5, 10 and 15 mM lidocaine concentrations were
used for subsequent experiments.
Cell death analysis
The apoptosis kssay kit containing propidium iodide
(PI), Annexin V Alexa Fluor® 488 and Propidium Iodide
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) was
used to measure the percentage of viable, apoptotic and necrotic
cells by detecting phosphatidyl serine expression and membrane
permeability. On this basis the populations of cells were detected
as Annexin V-negative/PI-negative (live), Annexin
V-positive/PI-negative (early apoptosis), Annexin
V-positive/PI-positive (late apoptosis) or Annexin
V-negative/PI-positive (necrosis). The procedure was performed
according to the manufacturer's protocols. After 24 h incubation
(37°C) of C6 cells with lidocaine (5, 10 and 15 mM), the cells were
trypsinized (0.25% trypsin solution, 37°C, 5 min), centrifuged (500
× g, 8 min, room temperature) and suspended in Annexin binding
buffer included in the applied kit (ABB, 100 µl).
Subsequently, to each 100 µl of sample, 5 µl Annexin
V Alexa Fluor 488 was added and incubated in dark for 20 min at
room temperature. After this time, the cells were centrifuged (300
× g, 5 min at room temperature, resuspended in 100 µl ABB
again and 1 µl PI was added for 3 min to each sample at room
temperature. The data were analyzed using a Guava easyCyte 6HT-2L
Benchtop Flow Cytometer (Merck KGaA) and FlowJo vX0.7 software
(FlowJo LLC, Ashand, OR, USA). The cells pretreated with Baf A1
were also assessed using the Annexin V Alexa Fluor® 488
and Propidium Iodide assay and the experiment was conducted in the
same manner as for cells without Baf A1 pretreatment.
Cell cycle analysis
Alterations in the cell cycle of C6 cells after 24 h
treatment with lidocaine (5, 10 and 15 mM) were investigated using
Tali Cell Cycle Kit (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocols. Briefly, the treated
cells (0.11×106) were trypsinzed (0.25% trypsin
solution, 37°C, 5 min), centrifuged (500 × g, 8 min at room
temperature) and fixed in ice-cold 70% ethanol (-18°C, 24 h). The
next day, after washing with PBS and centrifugation (500 × g, 7 min
at room temperature), the cells were resuspended in the Tali Cell
Cycle Solution. After 30 min of incubation at room temperature, the
data were determined using Guava easyCyte 6HT-2L Benchtop Flow
Cytometer. The percentage of cells in each phase of the cell cycle
was assessed using FlowJo vX0.7 software.
Morphological and ultrastructural
analysis Light microscopy
For the morphological analysis of the C6 cells
following treatement with lidocaine the cells were seeded onto
12-well plate in density of 0.11×106. After 24 h the
cells were treated with appropriate doses of lidocaine (5, 10 and
15 mM at 37°C) or left untreated in the case of control group.
After another 24 h, hematoxylin staining was conducted. First, the
cells were fixed with 4% paraformaldehyde (pH 7.4; PA, SERVA
Electrophoresis GmbH, Heidelberg, Germany), rinsed three times with
PBS and washed with dH2O. For nuclear staining, Mayer's
hematoxylin was used (3 min at room temperature). Subsequently, the
cells were rinsed under tap water and washed with PBS. The
preparations were mounted using Aqua PolyMount (Polysciences, Inc.,
Warrington, PA, USA). The morphology of cell was observed using an
Eclipse E800 light microscope (Nikon Corporation; x40 magnification
with at least 10-20 fields per view analyzed) and NIS-Elements 4.0
software (Nikon Corproation).
Transmission electron microscopy
(TEM)
As an indicator of autophagy, vacuoles were detected
by TEM. For this reason the cells were seeded onto 6-well plate in
density of 0.4×106. After 24 h incubation with lidocaine, the C6
cells were fixed for 30 min with 3.6% (v/v) glutaraldehyde (6°C;
Polysciences, Inc.) in 0.1 M sodium cacodylate buffer (pH 7.4; Carl
Roth GmbH + Co. KG, Karlsruhe, Germany) and washed with 0.1 M
sodium cacodylate buffer. The cells were then entrapped within
fibrin clots at room temperature, which consist of bovine thrombin
(50 U/ml) (Biomed-Lublin, Lublin, Poland) and fibrinogen (3 mg/ml;
Sigma-Aldrich; Merck KGaA). Then, the cells were post-fixed with 1%
(w/v) osmium tetroxide (SERVA Electrophoresis GmbH) in 0.1 M sodium
cacodylate buffer for 1 h at room temperature. The samples were
rinsed with sodium cacodylate buffer and dehydrated via a graded
ethanol (30-90%) and acetone (90-100%) (both from Avantor
Performance Materials Poland S.A., Gliwice, Poland) series. The
studied material was then embedded in epoxy resin (Epon 812; Carl
Roth GmbH + Co. KG), in which polymerization was carried out at
37°C for 24 h and then at 65°C for 5 days. Following
polymerization, the selected part of material was cut into
ultrathin sections (65 nm) using a Reichert Om-U3 ultramicrotome
(Leica Microsystems, Inc., Buffalo Grove, IL, USA), placed on
copper grids and counterstained with 1% uranyl acetate (7 min at
room temperature; Ted Pella, Inc., Redding, CA, USA). The samples
were examined using a TEM JEM 100 CX (JEOL, Ltd., Tokyo, Japan); at
least 15-20 fields per view were analyzed.
Fluorescent staining Acridine orange (AO)
staining
To detect the typical structures for autophagy, such
as acidic vesicular organelles (AVOs), AO (Sigma-Aldrich; Merck
KGaA) was used. The C6 glioma cells were seeded on coverslips in
12-well plates (0.11×106 of cells) and treated with
lidocaine for 24 h. Subsequently, the cells were incubated in
medium containing 1 µg/ml AO for 15 min at 37°C. Then, the
medium was removed, the cells were rinsed with PBS at room
temperature and immediately examined with Nikon Eclipse E800
fluorescence microscope and NIS-Elements 4.0 software (Nikon
Corporation). Additionally, AVOs were studied using a Guava
easyCyte 6HT-2L Benchtop Flow Cytometer. Flow cytometry data were
analyzed using FlowJo vX0.7 software.
Immunofluorescence staining of
LC3-II
To detect microtubule-associated protein 1A/1B-light
chain 3 (LC3)-II protein, the cells were seeded on coverslips
(0.11×106 cells) and treated with lidocaine in the
concentrations of 5, 10 and 15 mM (37°C). After 24 h incubation
with lidocaine the cells were fixed with 4% paraformaldehyde for 20
min at room temperature, rinsed with PBS and permeabilized with
0.25% Triton X-100 (SERVA Electrophoresis GmbH). Then, to block
nonspecific binding sites, 1% (w/v) bovine serum albumin (BSA;
Sigma-Aldrich; Merck KGaA) in PBS was used (20 min at room
temperature). Then, the cells were incubated with rabbit
anti-LC3-II antibody (1:2,000; Thermo Fisher Scientific, Inc., cat.
no. PA1-46286) for 1 h at room temperature, and Alexa Fluor
488-labeled goat anti-rabbit secondary antibody (1:100; Invitrogen;
Thermo Fisher Scientific, Inc., cat. no. A27034) for 1 h at room
temperature. Following washing with PBS, the cell nuclei were
stained with DAPI (1:20,000; Sigma-Aldrich; Merck KGaA) for 10 min
at room temperature. Finally, the slides were mounted with
Aqua-Poly/Mount and examined using a Nikon Eclipse E800
fluorescence microscope (magnification, x40 in at least 10- 20
fields per view) and NIS-Elements 4.0 software.
Fluorescence staining of cytoskeletal
proteins (tubulin, vimentin and actin)
The C6 cells following treatment with lidocaine were
cultured on glass coverslips (24 h, 37°C). For β-tubulin staining,
the cells were prefixed with bifunctional protein crosslinking
reagent dithiobis(succinimidyl propionate) (DTSP; 1 mM
3,30-dithiodipropionic acid) diluted 1:50 in Hanks' Balanced Salt
solution (both from Sigma-Aldrich; Merck KGaA) for 10 min (RT).
Then, the cells were permeabilized in TSB (0.5% Triton X-100) SERVA
Electrophoresis GmbH] in microtubule stabilizing buffer (MTSB; 1 mM
EGTA, 10 mM PIPES, 4% poly(ethylene glycol); Sigma-Aldrich; Merck
KGaA) with the addition of DTSP (in dimethyl sulfoxide) for 10 min
and rinsed with TSB for 5 min. The cells were fixed with 4%
paraformaldehyde in MTSB for 15 min (room temperature), washed with
PBS and incubated with 1% BSA diluted in Tris-buffered saline for
15 min (room temperature). Tubulin was labeled using a mouse
monoclonal antibody against β-tubulin (Sigma-Aldrich; Merck KGaA,
cat. no. T5293) and anti-rabbit antibody-Alexa Fluor 488
(Invitrogen; Thermo Fisher Scientific, Inc., cat. no. A27034).
In the case of F-actin and vimentin labeling, the
cells were fixed with 4% paraformaldehyde in PBS, for 15 min,
treated with 0.25% Triton X-100 for 5 min and blocked in 1% (w/v)
BSA/PBS at room temperature. To stain F-actin,
phalloidin-tetramethylrhodamine (Sigma-Aldrich; Merck KGaA, cat.
no. P1951) was used and vimentin was detected by using mouse
anti-vimentin antibody (Sigma-Aldrich; Merck KGaA, cat. no. V6630)
and anti-mouse antibody-Alexa Fluor 488 (Invitrogen; Thermo Fisher
Scientific, Inc., cat. no. A27034). As aforementioned, cell nuclei
were stained with DAPI and the slides were mounted in
Aqua-Poly/Mount. All slides were examined using a Nikon Eclipse
E800 fluorescence microscope (magnification, x40, in at least 10-20
fields per view) and NIS-Elements 4.0 software.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA from C6 cells was extracted using the
Total RNA Mini Plus kit (A&A Biotechnology, Gdynia, Poland)
according to the manufacturer's protocols, followed by the
spectrophotometric determination of the concentration and purity of
the obtained RNA (BioSpectrometer basic; Eppendorf, Hamburg,
Germany). A one-step RT-qPCR was performed with LightCycler RNA
Master SYBR Green I kit (Roche Applied Science, Mannheim, Germany)
on a LightCycler 2.0 Instrument (Roche Applied Science). The total
reaction mixture (20 µl per single LightCycler capillary)
contained 100 ng of RNA and 0.2 µM of each primer in
addition to the LightCycler RNA Master SYBR-Green I kit components.
The sequences of primers were as follows: Beclin-1 (Becn1,
forward, 5′-AGCTGCCGTTATACTGTT CTG-3′ and reverse,
5′-ACTGCCTCCTGTGTCTTCAAT CTT-3′; LC3B, forward,
5′-GCCTTCTTCCTCCTGGTGAAT-3′ and reverse,
5′-TTTTTGCCTTGGTAGGGGCTT-3′; and GAPDH, forward,
5′-CACTGAGCATCTCCCTCACAA-3′ and reverse,
5′-TGGTATTCGAGAGAAGGGAGG-3′. The thermocycling conditions were as
follows: RT for 20 min at 61°C (one cycle); qPCR was conducted with
denaturation for 1 min at 95°C (one cycle) and 45 cycles of
denaturation for 5 sec at 95°C, followed by annealing and extension
for 20 sec at 57-61°C (depending on the melting temperature of the
primers) and 5 sec at 72°C, respectively. The data obtained from at
least three independent experiments were analyzed with LightCycler
Software Version 4.0 (Roche Applied Science). The relative gene
expression was normalized to that of GAPDH internal control and
assessed using the 2−ΔΔCq method (27).
Statistical analysis
The data were presented as the mean ± standard
deviation. GraphPad Prism 6.0 software (GraphPad Software, Inc., La
Jolla, CA, USA) was used for statistical analyses. A one sample
t-test was performed to analyze MTT and RT-qPCR data as it compares
the mean with a hypothetical value (100 or 1). A nonparametric
Kruskal-Wallis test with Dunn's post hoc test or the one-way
analysis of variance with Tukey's post hoc test (ANOVA, normal
distributions) were used to evaluate the differences in mean values
between cells treated with lidocaine compared with the control
(cells with and without Baf A1 pretreatment), independently (cell
death and RT-qPCR). In the case of multiple comparison the two-way
ANOVA was performed with Dunnett's multiple comparisons test (cell
cycle, fluorescence intensity of AO). P<0.05 was considered to
indicate a statistically significant difference.
Results
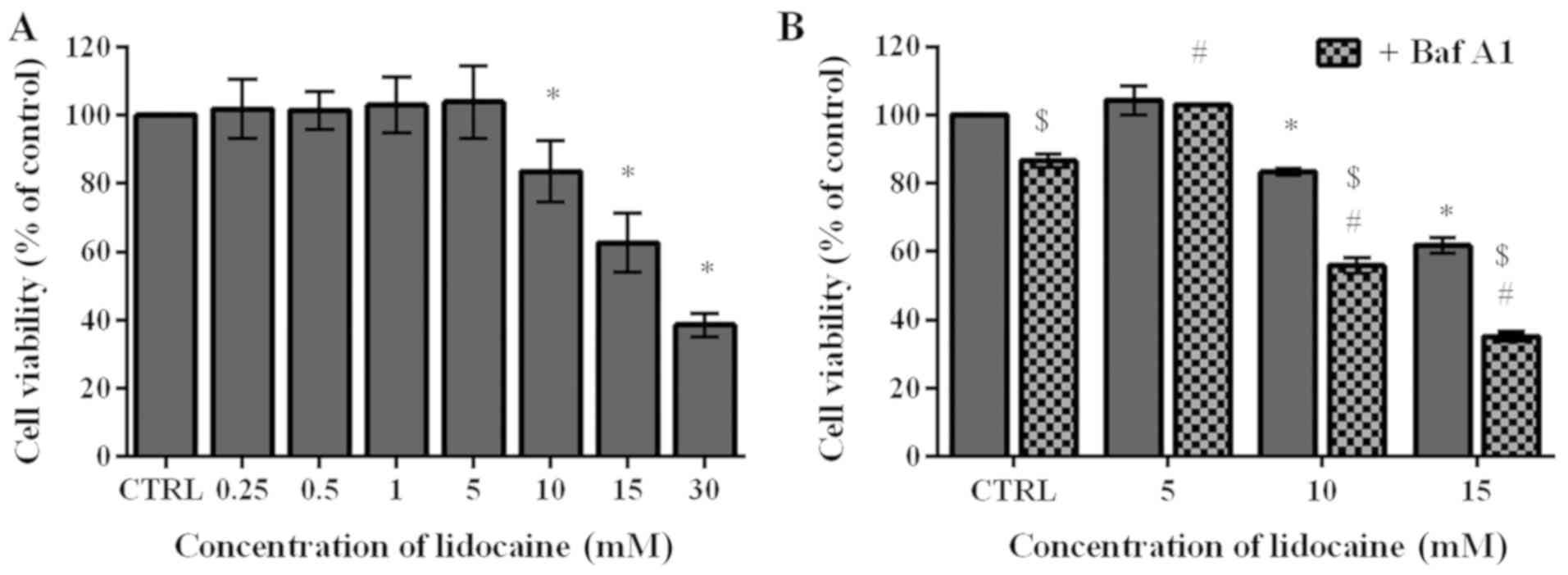
Lidocaine decreases the viability of
glioma C6 cells
To determine the cytotoxic effect of lidocaine on
the rat glioma cells, an MTT assay was conducted. During surgical
interventions, 5-20 mM lidocaine is applied (28); thus for the first step of the
investigation 0.25, 0.5, 1, 5, 10, 15 and 30 mM were selected in
the present study. The lowest four concentrations exhibited limited
impact on the C6 cells. The cell viability following treatment with
these low doses slightly increased by 1.72, 1.43, 2.97 and 3.95%,
respectively, compared with the control, which was assumed as 100%.
Conversely, treatment with higher doses (10, 15 and 30 mM) resulted
in 83.48, 62.63 and 38.59% cell viability, respectively, which was
significantly lower compared with the control (Fig. 1A). The MTT assay was also applied
to assess the viability of cells pretreated with Baf A1, which
inhibits autophagy by preventing the fusion of autophagosomes and
lysosomes (29). In cells
incubated with Baf A1 for 4 h, the viability significantly
decreased to 86.54% compared with the untreated cells. Following
treatment with the lowest dose of lidocaine (5 mM), significant
differences between untreated cells and those pretreated with Baf
A1 were not observed; however, pretreatment with the inhibitor of
autophagy followed by incubation with 10 and 15 mM lidocaine
induced significant decreases in cell viability compared with cells
treated with lidocaine alone; the percentage of live cells amounted
to 55.91 and 35.18%, respectively (Fig. 1B). Additionally, we observed
statistically significant differences between cells treated with
lidocaine and Baf A1, and the control (Fig. 1B).
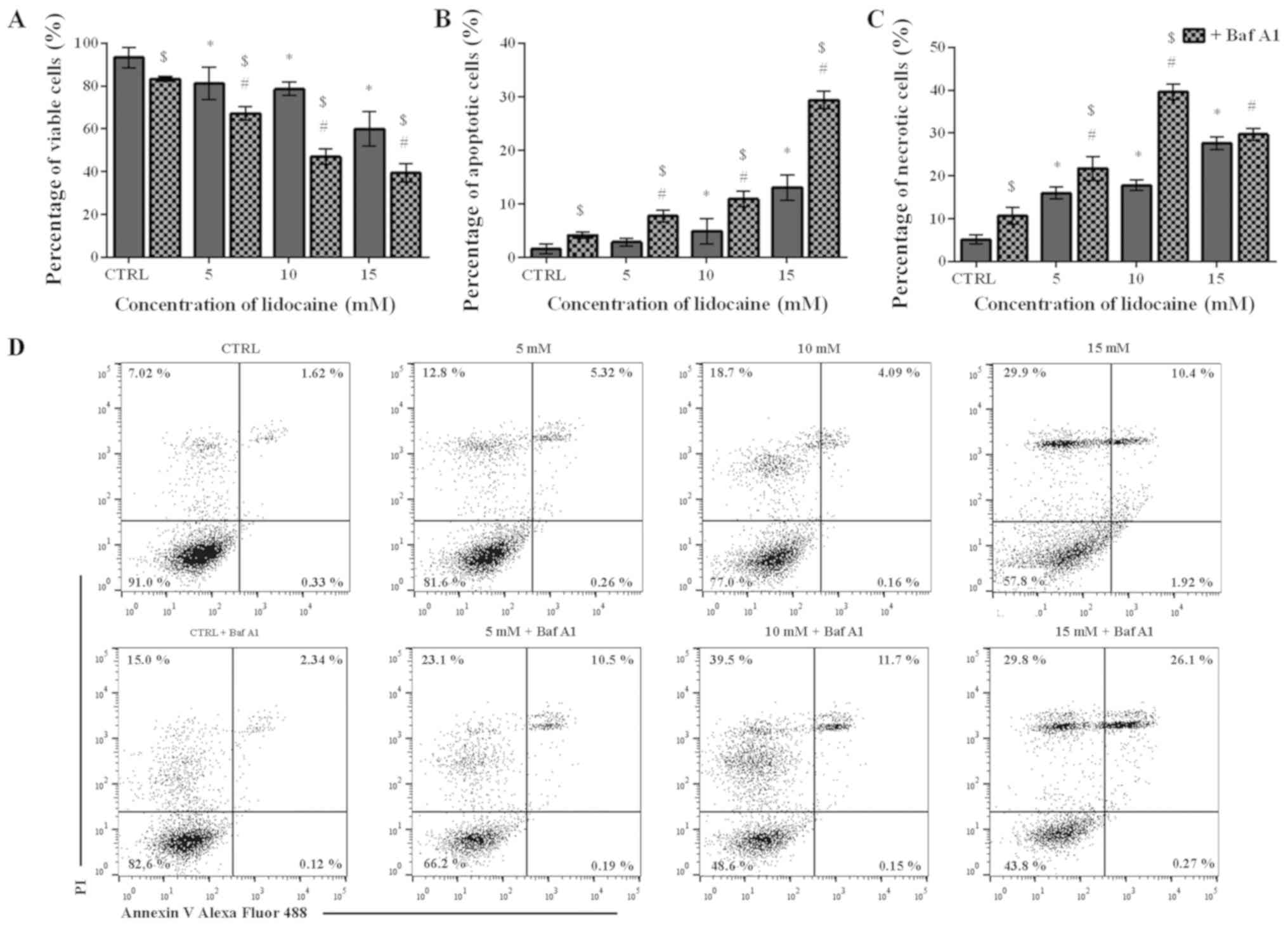
Lidocaine induces cell death mainly by
necrosis
To assess the mechanism underlying the cytotoxic
effect of lidocaine on the glioma cell line, Annexin V and PI
staining was applied. The percentage of live, apoptotic and
necrotic cells was analyzed in the populations treated only with
lidocaine and pretreated with Baf A1. Following treatment with
lidocaine alone, a dose-dependent decrease in the live cell
population was observed. In the control, the percentage of live
cells amounted to 93.26%, which significantly decreased to 81.16,
78.57 and 59.98% after treatment with 5, 10 and 15 mM lidocaine,
respectively (Fig. 2A). The number
of live cells were significantly lower when Baf A1 was applied.
Following pretreatment with the inhibitor of autophagy, 83.27% of
live cells was noted; however, when 5, 10 and 15 mM lidocaine were
added, the percentage of live cells was significantly lower
compared with lidocaine treatment alone and reached 67.24, 46.94
and 39.47%, respectively (Fig.
2A). In C6 cells treated with 5, 10 and 15 mM lidocaine, the
percentage of apoptotic cells increased from 1.58% in the control
to 2.88, 4.93 and 13.06%, respectively; however, only in the case
of 10 and 15 mM lidocaine, the differences were statistically
significant. Furthermore, the percentage of apoptotic cells was
increased in response to pretreatment with Baf A1. In control cells
treated only with inhibitor of autophagy, the percentage of
apoptotic cells amounted to 4.19%; following incubation with 5, 10
and 15 mM lidocaine the number of apoptotic cells significantly
increased to 7.78, 10.93 and 29.38%, respectively (Fig. 2B).
As presented in Fig.
2C, the population of necrotic cells increased in a
dose-dependent manner from 5.11% to 15.95, 17.77 and 27.62% after
treatment with 5, 10 and 15 mM lidocaine, respectively. In the case
of cells pretreated with Baf A1 alone, the number of necrotic cells
was significantly higher compared with the untreated control and
amounted to 10.66%; the number of necrotic cells significantly
increased to 21.74, 39.62 and 29.69% following Baf A1 pretreatment
and incubation with increasing lidocaine concentrations. However,
significant differences were not observed following treatment with
15 mM lidocaine in the presence or absence of Baf A1 (Fig. 2C). Representative plots are
presented in Fig. 2D.
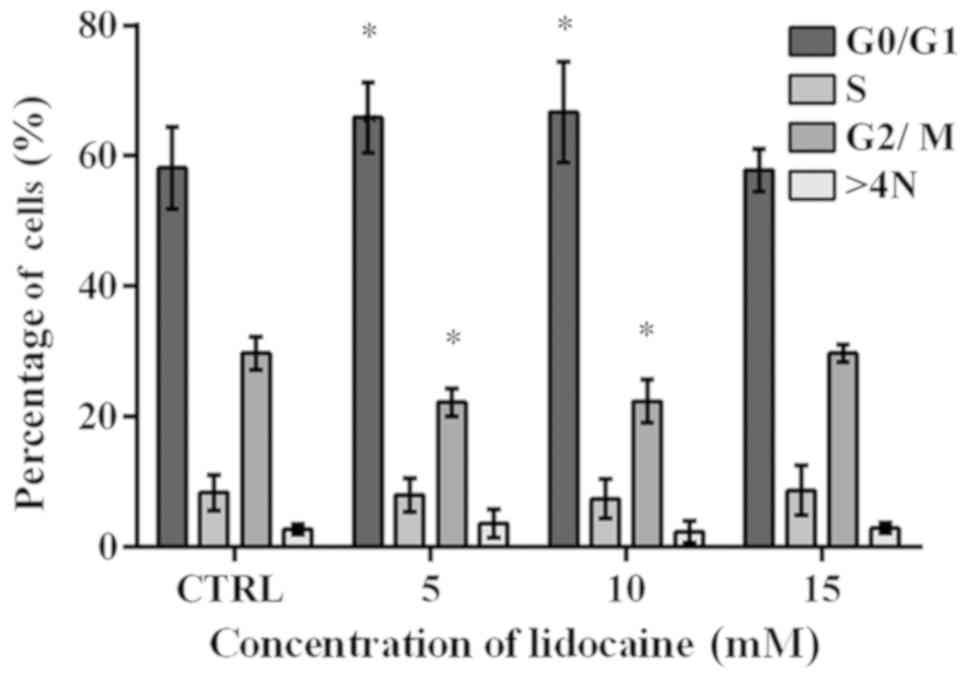
Lidocaine treatment induces alters the
cell cycle distribution
The cell cycle was analyzed using flow cytometry. A
significant increase from 58.15% in the control to 65.87 and 66.72%
in the percentage of cells with DNA content corresponding to G0/G1
phase was observed following treatment with 5 and 10 mM lidocaine,
respectively (Fig. 3). The results
obtained for the control cells were similar to that after treatment
with lidocaine (15 mM). The population of cells in S-phase reached
8.35, 8.02, 7.44 and 8.71% in the control and cells treated with 5,
10 and 15 mM lidocaine, respectively; however, the results were not
statistically significant (Fig.
3). Following incubation with 5 and 10 mM lidocaine, a
significant decrease in the number of cells in G2/M phase compared
with the control was noted (Fig.
3). In the control, the mean percentage of cells with DNA
content corresponding to G2/M phase amounted to 29.75%, and
following treatment with 5 and 10 mM lidocaine decreased to 22.20
and 22.40%, respectively. In turn, after incubation with the
highest dose of lidocaine, the population of cells in G2/M phase of
cell cycle was similar to the control cells. In addition, a slight
difference in the percentage of polyploid cells (2.78% in the
control cells, and 3.66, 2.37 and 2.92% following treatment with 5,
10 and 15 mM lidocaine, respectively) (Fig. 3).
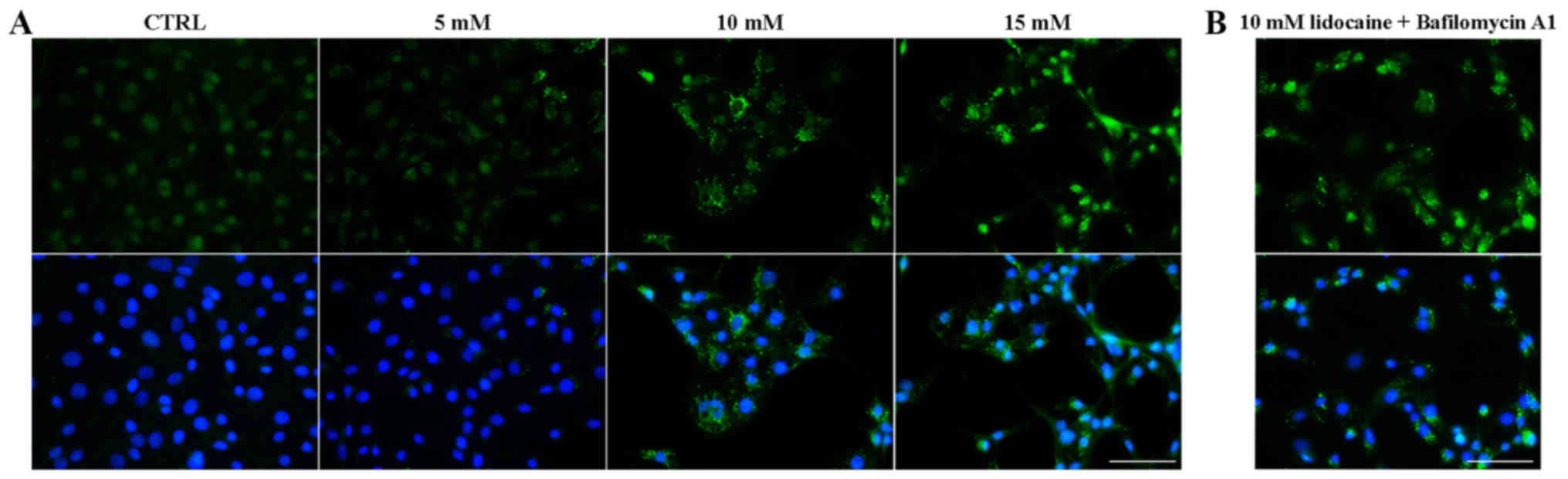
Lidocaine induces the formation of
autophagic vacuoles
Small vacuole-like structures were observed under an
inverted microscope following treatment with lidocaine. A
spindle-shaped morphology, which is typical for C6 cells, was
visible in the control and treatment with the lowest dose of
lidocaine, whereas following incubation with 10 and 15 mM of
lidocaine, numerous vacuoles in the cytoplasm of C6 cells had
formed (Fig. 4). The same results
were observed after hematoxylin staining. TEM was used to assess
cell morphology on the ultrastructural level. As presented in
Fig. 4, cells incubated with
lidocaine in 10 and 15 mM exhibited numerous vacuole-like
structures in the cytoplasm area compared with the control cells.
Furthermore, following treatment with the lowest dose of lidocaine,
fewer, small vacuoles, which were not visible via light microscopy,
were also observed by TEM. Some of these structures contained
amorphous materials and organelles in the various stages of
degeneration. Higher doses of lidocaine revealed larger vacuoles.
Additionally, the majority of them were enclosed by a single
membrane, which suggests autolysosome formation. The
ultrastructural feature of autophagy, swollen mitochondria with
disorganized cristae was also observed (Fig. 4).
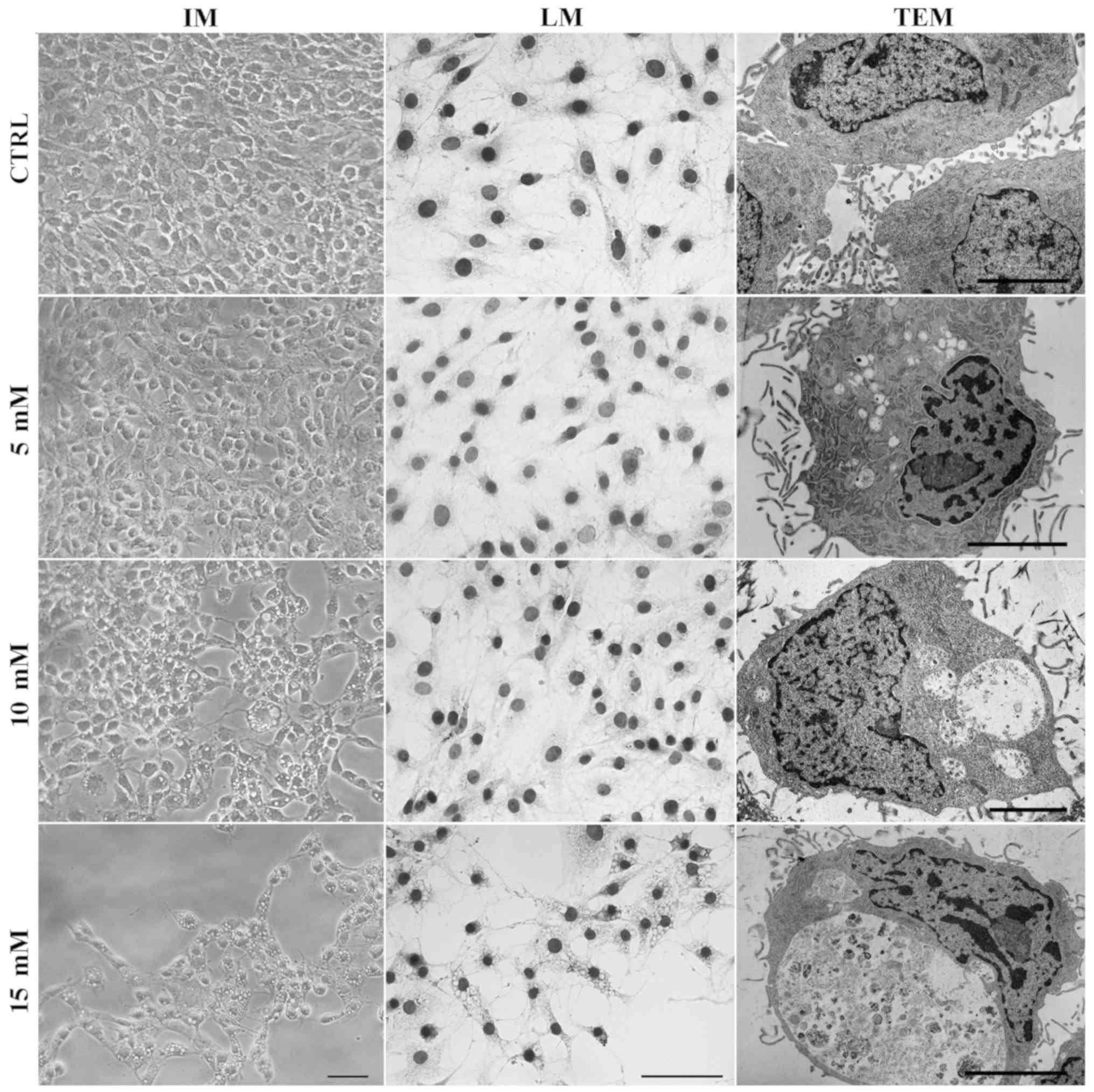 | Figure 4Lidocaine induces autophagic vacuole
formation in rat glioma cell line. The effects of lidocaine were
evaluated by IM in C6 cells following treatment with various
concentrations of lidocaine (5, 10 and 15 mM) for 24 h. Scale bar,
50 µm. Hematoxylin staining (LM) of CTRL cells, and cells
treated with 5, 10 and 15 mM lidocaine. Scale bar, 50 µm.
The detection of autophagic-like vacuoles using TEM in control
cells and incubated with 5, 10 and 15 mM lidocaine. Scale bar, 10
µm. CTRL, control; IM, inverted microscopy; LM, light
microscopy; TEM, transmission electron microscopy. |
Lidocaine induces autophagy
LC3 is a specific marker of autophagy and exists in
varied forms. LC3-I (18 kDa) is present in the cytosol, and
undergoes proteolysis and lipid modification for conversion into
LC3-II (16 kDa) (30). The shorter
form is located in the membrane of autophagosomes whereby the
expression levels of LC3-II correspond to the amount of these
structures (31). In control cells
and after treatment with the lowest dose of lidocaine, the
fluorescence staining of LC3-II was poor and diffuse, but following
treatment with higher doses of lidocaine the punctate pattern of
LC3-II expression was observed. Based on the images from
fluorescence microscopy, increased LC3-II staining occurred in
response to 10 mM lidocaine than with the highest dose (Fig. 5A). The punctate accumulation of
LC3-II following pretreatment of cells with Baf A1 and incubation
with 10 mM lidocaine was also noted, which is consistent with the
inhibition of autophagy (Fig.
5B).
In addition, lidocaine treatment led to a
significant dose-dependent increase in the transcript expression
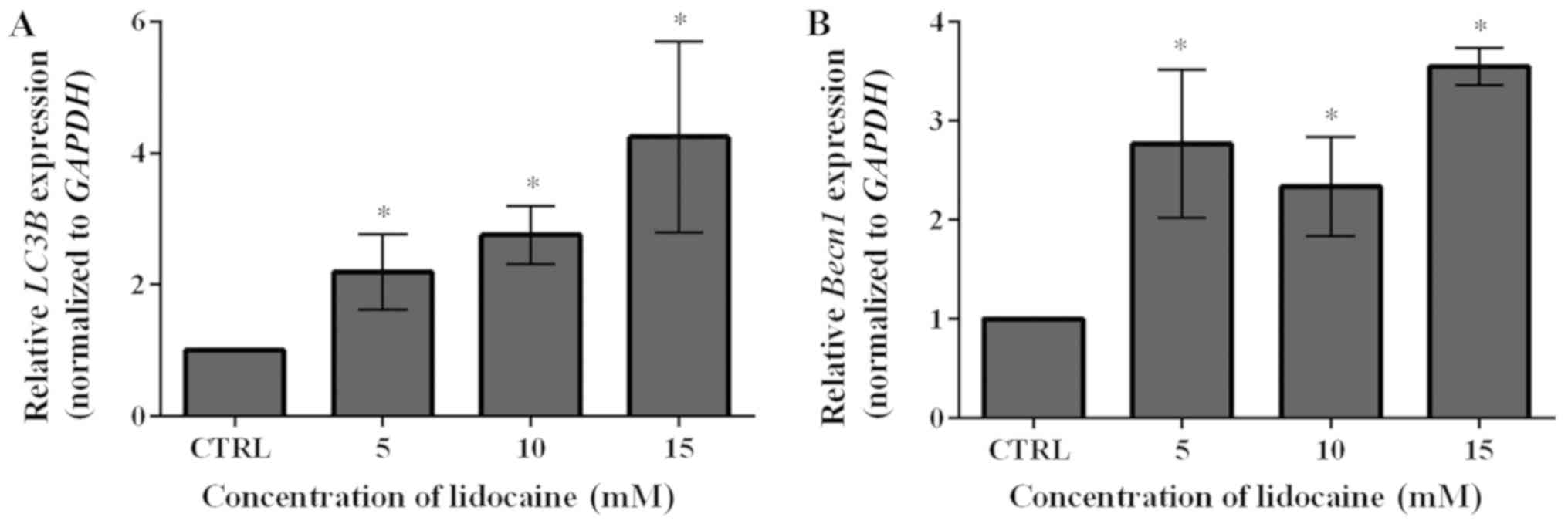
level of LC3B compared with the control (Fig. 6A). To further investigate the
occurrence of autophagy, the mRNA expression levels of another
autophagy marker, Becn1, were examined following treatment
with lidocaine. As presented in Fig.
6B, this agent significantly upregulated the mRNA expression of
Becn1; the expression levels were notably lower in response
to 10 mM lidocaine compared with the other treatment groups.
The second fluorescence method was staining with AO.
AO labels AVOs, such as autolysosomes and is also used in autophagy
assays. In AO-stained cells, the cytoplasm and nucleus fluoresced
bright green, whereas AVOs fluoresced bright red. The intensity of
red fluorescence is proportional to the degree of acidity and the
acidic compartment volume (32).
In control C6 glioma cells and cells treated with 5 mM lidocaine,
few AVOs (punctate fluorescence) were observed. Lidocaine (10 and
15 mM) promoted the formation of AVOs; however, more were detected
following treatment with 10 mM lidocaine compared with the highest
dose (Fig. 7A). Additionally, in
the cells pretreated with Baf A1 and incubated with 10 mM
lido-caine, the signal of red fluorescence was almost completely
blocked (Fig. 7B). The results
were confirmed by using flow cytometry. In addition, the mean
fluorescence intensity of AO was the highest in C6 cells treated
with 10 mM lidocaine; a significant increase in the mean
fluorescence intensity was also observed following treatment with
15 mM lidocaine compared with the control. Pretreatment with Baf A1
revealed significant decreases in the mean fluorescence intensity
compared with the untreated control (Fig. 7C).
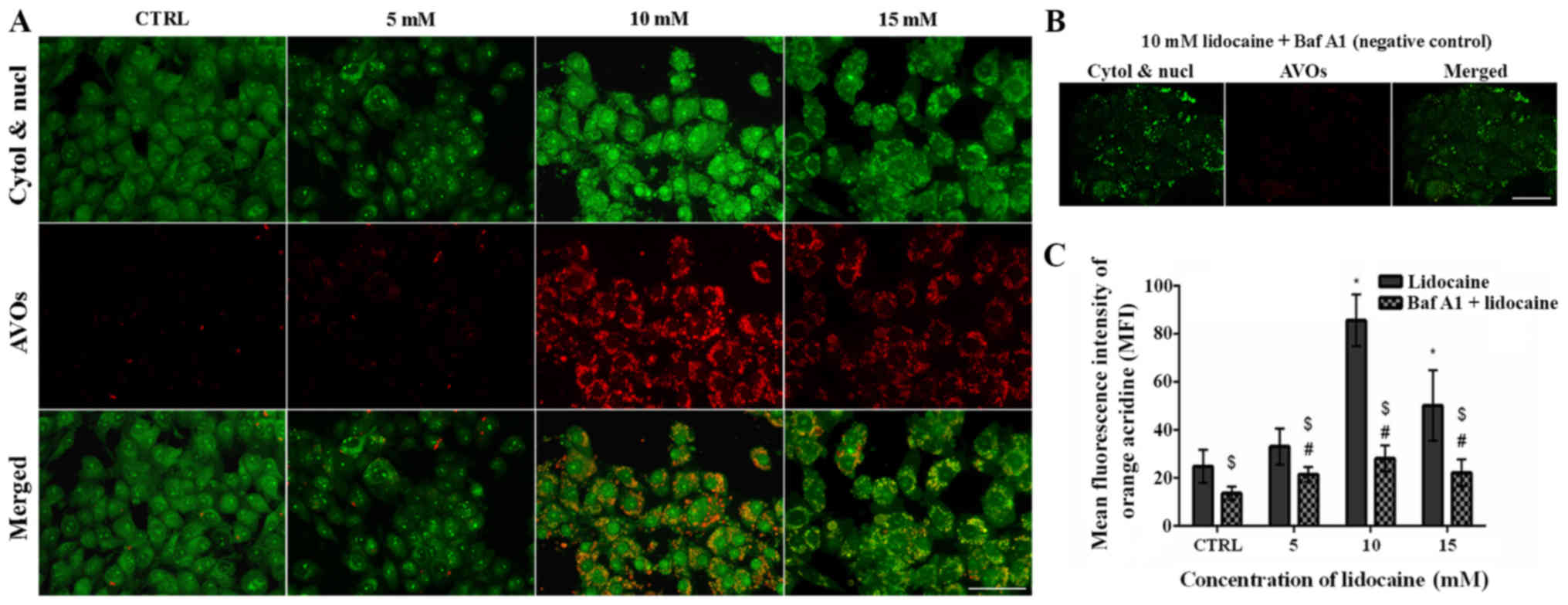 | Figure 7Detection of autophagy by acridine
orange staining. (A) Fluorescence staining of AVOs (red
fluorescence), as well as the cytoplasm and nucleus (green
fluorescence) in control cells, and cells treated with 5, 10 and 15
mM lidocaine. Scale bar, 50 µm. (B) Negative control
pretreated with Baf A1 (100 nM) and incubated with 10 mM lidocaine.
Scale bar, 50 µm. (C) MFI of AO was determined using flow
cytometer. *P<0.05 lidocaine doses vs. CTRL;
#P<0.05 lido-caine doses + Baf A1 vs. CTRL + Baf A1.
$P<0.05 cells pretreated with Baf A1 and then treated
with lidocaine vs. cells treated just with lidocaine. AO, acridine
orange, AVOs, acidic vesicular organelles; Baf A1, bafilomycin A1;
CTRL, control; cytol, cytoplasm; MFI, mean fluorescence intensity;
nucl, nucleus. |
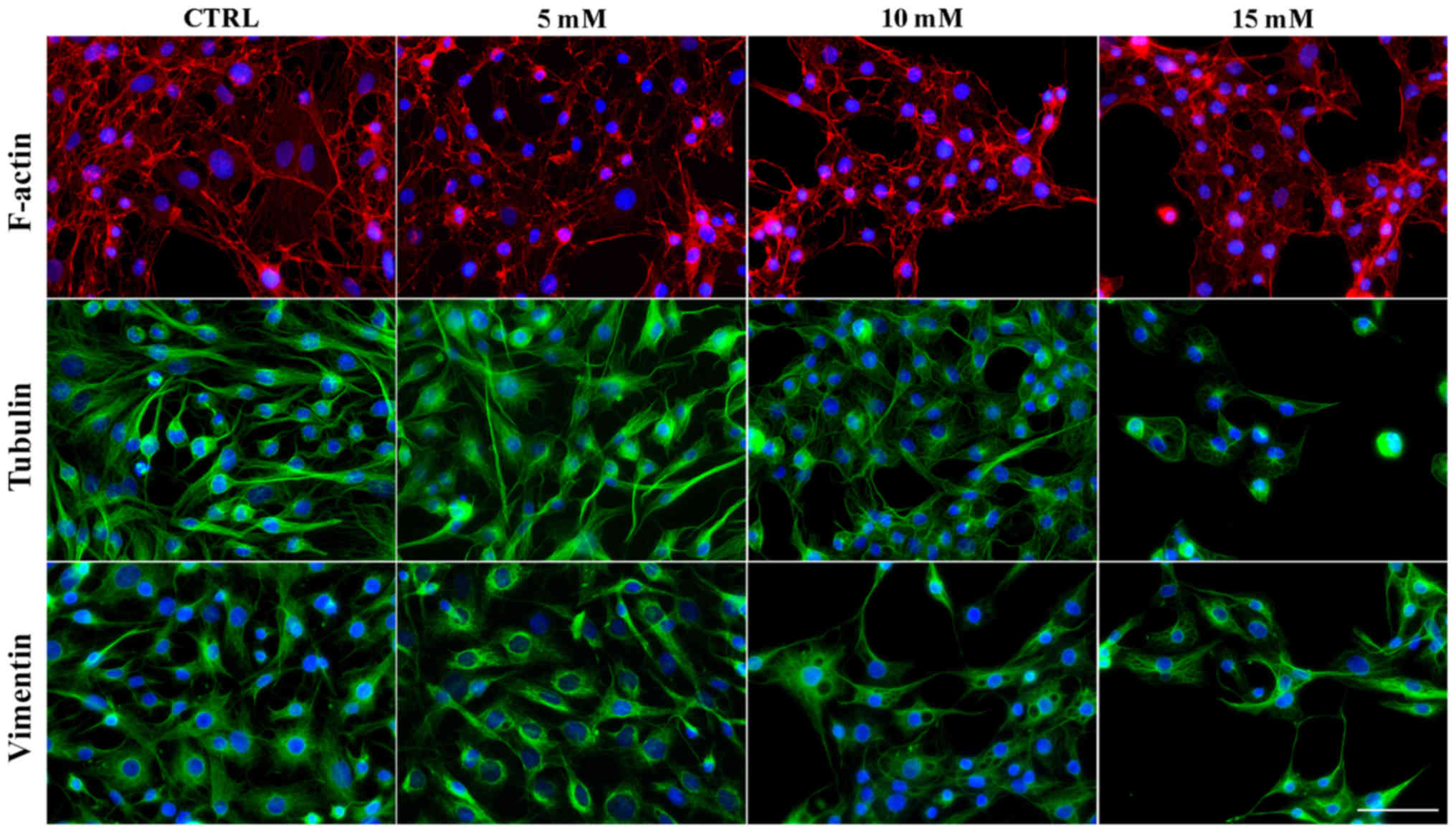
Lidocaine induces alterations in the
organization of the main cytoskeletal components
The cytoskeleton is a very important organelle in
the cell which undergoes reorganization during numerous cellular
processes (33,34). The results of the present study
revealed that lidocaine induced notable alterations in the
organization of cytoskeleton of glioma cells, which was associated
with autophagic structure formation. Control cells were
characterized by highly-developed F-actin with long stress fibers
in the cytoplasm, cytoplasmic protrusions and regions of cell-cell
junctions (Fig. 8). Following
treatment with the lowest dose of lidocaine, marked changes in
microfilament structure were not observed (Fig. 8). Treatment with 10 and 15 mM
lidocaine demonstrated the reorganization of actin filaments
(Fig. 8). These cells exhibited a
diffuse network of F-actin with short actin fibers and/or small,
punctate accumulation within the cytoplasm. The accumulation of
actin in the cortical region of cells following incubation with 10
mM lidocaine was also observed (Fig.
8). In small, shrunken cells the intensity of actin
fluorescence increased.
The fluorescence staining of β-tubulin also
presented marked lidocaine-induced alterations in the organization
of microtubules and mitotic spindle morphology (Fig. 8). In the control cells and cells
incubated with the lowest dose of lidocaine, β-tubulin was
organized in a regular and dense network of long microtubules,
which radiated from the microtubule-organizing centers (MTOCs)
(Fig. 8). Following treatment with
higher doses lidocaine, a less dense network of microtubules formed
compared with the control; however, the MTOCs were still visible
(Fig. 8). Additionally, in
shrunken cells a significantly higher fluorescence of tubulin was
noticed (Fig. 8).
The last analyzed element of the cytoskeleton was
vimentin. Analysis of actin and tubulin reorganization did not
reveal vacuole-like structures, but after vimentin staining,
autophagic vacuoles were detected. In control cells and cells
treated with 5 mM lidocaine, extended intermediate filament
networks in the cytoplasmic area and the accumulation of vimentin
near the nucleolemma were observed. Following treatment with 10 and
15 mM lidocaine, vacuole-like structures surrounded by vimentin
fibers were observed (Fig. 8).
Discussion
In the present study, the effects of lidocaine on
the rat glioma C6 cell line were investigated. Malignant gliomas
are primary brain tumors characterized by rapid and invasive growth
(35). The standard treatment is
based on radical brain tumor surgery, maximally safe radiation and
concomitant chemotherapy (36-38).
Despite the efforts of scientists and physicians to improve the
effectiveness of treatment, further developments are required.
Local anesthetics were reported as agents with the potential to
notably inhibit tumor growth (17). One of them is lidocaine, which can
be administered to local or regional tissue, and inhibits nerve
conduction. Several studies have demonstrated lidocaine to suppress
cell proliferation (28,39,40).
Jurj et al (28) reported
the antiproliferative effect of a clinical concentration of
lidocaine on human hepatocarcinoma cells (HepG2). Other scientists
revealed the antiproliferative, apoptotic and cytotoxic effect of
this agent on various types of cancer cells. Sakaguchi et al
(39) suggested that the
inhibition of epidermal growth factor receptor activity by
lidocaine is one way to decrease the proliferation of human tongue
cancer cells (39). In addition,
lidocaine enhances the therapeutic effect of drugs, including
mitomycin C, pirarubicin and Su Fu'ning lotion in BIU-87 bladder
cancer cells (40). Additionally,
the combination of lidocaine with mitomycin C in mice with
orthotopic bladder cancer resulted in prolonged survival and
reduced tumor size (40). The
antitumor effect of lidocaine on human breast cancer,
hepatocellular carcinoma cells, non-small cell lung cancer cells
and thyroid cancer cells was also observed (41-44).
Furthermore, lidocaine was reported to suppress glioma cell
proliferation (16,17). In the present study, a significant
decrease in cell viability after incubation with 10, 15 and 30 mM
lidocaine was observed compared with the control. Similar results
of Leng et al (17)
revealed the inhibition of proliferation of glioma cells (C6 rat
glioma cell line and A172 human glioblastoma cell line) and
indicated that lidocaine mediated this effect by decreasing the
expression of TRPM7 channels.
Decreases in cell proliferation following treatment
with cytostatic drugs or other agents are usually associated with
the induction of cell death. Apoptosis is the most desirable type
of cell death during cancer therapy. Lu et al (45) proposed that lidocaine induces
alterations in intracellular calcium ion concentration and
mitochondrial membrane potential in dose-dependent manners in
U87-MG glioma cells by activating the overexpression of
TRPV1 to induce intrinsic apoptosis via the mitochondrial
pathway (46). Cell death via the
mitochondrial pathway induced by lidocaine is not restricted to
glioma cells only, but is also activated in human hepatocellular
carcinoma (HepG2), Jurkat T-lymphoma and human thyroid carcinoma
(8505C) cells (16,44,47).
Additionally, local anesthetic agents induce apoptosis via
mitogen-activated protein kinase (13,44).
Furthermore, numerous studies have reported that low doses of
lidocaine induce the mitochondrial pathway of apoptosis (47-49).
Li and Han (48) proposed that
lidocaine in human neuroblastoma SH-SY5Y cells promoted endoplasmic
reticulum stress-mediated apoptosis. In the present study, the
apoptosis of C6 cells was noted, but the predominant type of cell
death was necrosis. A similar effect was observed by Kamiya et
al (50), in which lidocaine
induced apoptosis at low concentrations and necrosis at much higher
concentrations (>15 mM) in human histiocytic lymphoma cells. In
human SHEP neuroblastoma cells, a high concentration of lidocaine
(10 mM) promoted the transition from apoptosis to
caspase-independent necrosis (47). Despite the induction of apoptosis
and necrosis, the small vacuoles that were visible under the
inverted microscope attracted our attention in the present study.
Following hematoxylin staining of cells treated with 10 and 15 mM
lidocaine, small enclosed compartments were observed. The present
study proposed that these may have been autophagic vacuoles, and so
were analyzed at the ultrastructural level. Using TEM, few of these
structures contained amorphous materials and possibly the
organelles at the various stages of degeneration. In cells of the
other treatment groups, the vacuoles were empty and large.
Additionally, what was not visible under the light microscope
following treatment with the lowest dose of lidocaine, few, small
vacuoles were observed. Similar findings at the ultrastructural
were presented by Martinet et al (51), who described the protocol for
studying autophagic vacuoles by TEM. On the basis of this data, it
was assumed that lidocaine induced autophagy. Although the
potential disadvantages of TEM methods make it impossible to limit
research to only one method; the application of
immunocyto-chemistry or immunohistochemistry, and molecular
approaches for the unambiguous detection of autophagy are
recommended.
Autophagy is a highly dynamic and regulated process,
which is often accompanied by G0/G1 arrest; however, this is not
required for the induction of autophagy (52). In the present study, significant
alterations in the cell cycle distribution were reported in
response to 5 and 10 mM lidocaine. The results are consistent with
those of Xing et al (16),
in which 5 mM lidocaine induced cell cycle arrest in G0/G1 in HepG2
cells, even if the main process of cell death was apoptosis.
Additionally, autophagy is a very specific process, and can protect
cancer cells to sustain their growth and survival in unfavorable
conditions, such as the presence of cytostatic drugs; however,
autophagy may be an alternative form of cell death (53). Therefore, autophagy may be
exploited for cell survival under nutrient-restricted conditions,
hypoxia or the cytotoxic effect of drugs; however, autophagic type
II cell death may be therapeutically valuable and mediates the
cytotoxic effects of anticancer drugs leading to tumor cell death
(53,54). The autophagic process is associated
with the formation and clearance of autophagosomes (55,56).
The formation of these structures is directly associated with the
cytosolic form of LC3 (LC3-I), which is conjugated to
phosphatidylethanol-amine and converted into
LC3-phosphatidylethanolamine conjugate (LC3-II) (30,57).
LC3-II is located in autophagosomal membranes; following the fusion
of autophagosomes and lysosomes, the autophagosomal elements, such
as LC3-II are hydrolyzed (30).
Thus, LC3 protein and the formation of LC3-II are the main
indicators of autophagy (30,51).
In the present study, lidocaine treatment led to increased
transcript levels of LC3B, which were the highest in glioma
cells following treatment with 15 mM lidocaine. Similar results
were obtained using fluorescence methods, which confirmed the
findings of RT-qPCR analysis. The formation of autophagic vacuoles
in the present study was increased by lidocaine, and the highest
degree of LC3-II punctate staining was observed following treatment
with 10 and 15 mM lidocaine. Xiong et al (58), described the effects of local
anesthetic i.a. lidocaine on human neuroblastoma SH-SY5Y cells, and
indicated increases in the levels of LC3-II and the formation of
autolysosomes using a dual fluorescence-based LC3 punctuation assay
following the incubation of neuronal cells with lidocaine. Apart
from LC3, autophagy is regulated by a series of regulators, such as
Beclin-1, an essential initiator of autophagy (59,60).
Beclin-1 serves a key role in the recruitment of autophagic
proteins to the pre-autophagosomal structure, but also in the
formation of the core complex, comprising Beclin-1, vacuolar
protein sorting (Vps)34 and Vps15 (59). Of note, Beclin-1 is a factor which
determines whether the cells undergo autophagy or apoptosis. This
function is associated with interactions with anti-apoptotic
proteins B-cell lymphoma-2 (Bcl-2) or Bcl-xL via its BH3 domain
(59,61). This complex inhibits the assembly
of the pre-autophagosomal structure, thereby suppressing autophagy
(62). In autophagic cells, the
levels of Beclin-1 are increased and those of Bcl-2 are decreased.
In the present study, it was observed that lidocaine upregulated
the mRNA expression of Becn1. The importance of increased
Beclin-1 levels during autophagy was confirmed by Xiong et
al (58). Autophagy inhibition
was achieved by the transfection of cells with Becn1 small
interfering (si)RNA; knockdown in SH-SY5Y cells decreased LC3-II
content, thereby suppressing cell viability (58). Reduction of Beclin-1 by siRNA
resulted in defective autophagy and decreased the number of AVOs in
human glioblastoma U87 and esophageal squamous cell carcinoma
EC9706 cells (63,64).
The cytoskeleton is a very dynamic structure
comprising microtubules, microfilaments and intermediate filaments.
It is known that their organization and alterations are closely
associated with the cell state (65,66).
This organelle is involved in numerous cellular processes,
including mitosis, proliferation, migration and cell death
(67,68). In addition, the cytoskeleton is
also involved in the autophagic process (69). The formation of actin branches by
its reorganization is key for the biogenesis of autophagosomes.
Actomyosin-based transport is used to feed the growing phagophore;
finally, mature autophagosomes undergo intracellular transport and
fusion with lysosomes and endosomes, with the involvement of actin,
tubulin and signaling proteins (70).
Previously, the involvement of actin in autophagy
was first observed by Aplin et al (71); F-actin depolymerizing drugs
(cytochalasin D and latrunculin B) were reported to inhibit
autophagosome formation and autophagy (71). At present, numerous studies have
demonstrated that actin is involved in the autophagic process;
however, as well as actin assembly and reorganization, actin may
serve as a marker of autophagy (72,73).
Numerous scientific reports proposed the association between the
Arp 2//3 complex and actin polymerization during autophagy
(74-77). It has been reported that the Arp
2/3 complex is capable of forming branched actin networks, which
are important for all types of membrane remodeling activities in
cells, as wells as autophagy (76,77).
The present study proposed that actin filaments are important in
the first steps of autophagy due to its colocalization with early
omegasomes This suggests a role of actin in the initial steps of
autophagosome formation. Following the induction of autophagy,
actin networks bind the Arp 2/3 complex and polymerize inside the
phagophore. This process is responsible for generating the shape
(membrane curvature) of the autophagosomes and the fusion with
lysosomes to form the autolysosomes. In mature autophagosomes,
actin comet tails are formed (74,78).
Additionally, the fusion of autopha-gosomes with lysosomes depends
on the stability of actin filament branches, which is regulated by
the well-known stabilizer of actin comprising the Arp 2/3 complex
(71,78). The binding of the Arp 2/3 complex
with cortactin and the depletion of cortactin results in the loss
of the F-actin network, which inhibits autophagosome-lysosome
fusion (79-81). In the present study, the
reorganization of actin filaments was also observed.
Cells incubated with 10 mM lidocaine exhibited
diffuse networks of F-actin with short actin fibers and/or small,
punctate accumulations within the cytoplasm, and fibers of actin in
the cortical region in the present study. This unstable form of
actin may be associated with the continuous changes and dynamics of
the autophagy process. The short fibers that are formed in
autophagic cells are the components of the branched actin
network.
Microtubules also undergo reorganization. Our study
presents the importance of microtubule organization and
micro-tubule-based motors in the autophagic process. Following the
treatment of glioma cells with lidocaine, microtubules formed less
dense networks compared with the control; MTOCs were still visible.
This reorganization of the microtubule network may be associated
with the stimulation of autophagosome formation. Additionally, the
network of microtubules creates the pathway along which
pre-autophagosomal structures and autophagosomes are transported
(71). Destabilization of this
cytoskeleton element delays the arrival of autophagosomes near
lysosomes and thus, inhibits their fusion (69,82,83).
Clearly visible MTOCs following treatment with lidocaine were noted
in the present study. Jahreiss et al (83) reported that in rat kidney cells,
lysosomes are localized at the perinuclear region about MTOCs,
whereas autophagosomes form at the cortical region of the cells;
thus autophagosomes translocate to the lysosomes in a
microtubule-dependent manner (83). Similar results were obtained in
cervical cancer cells, in which the MTOC-directed movement of these
structures was associated with the microtubule network (84).
Intermediate filaments are also involved in the
autophagic process. In the study presented by Ruangjaroon et
al (85), in SH-SY5Y cells
following treatment with 50 µM fipronil, vimentin formed a
ring-like structure surrounding vacuoles. Identical localization of
this protein was observed in the present study, which suggests that
intermediate filaments serve an important role in the formation of
autophagic vacuoles. In addition, vimentin can inhibit autophagy
via a protein kinase B-dependent mechanism forming a complex with
14-3-3 protein and Beclin-1 (86).
However, confirmation regarding the nature of the
autophagy observed is required as the activation of this process
may be beneficial or harmful to cells (58). The pro-survival or pro-death
mechanism of this process requires further investigation as to
whether the increased rate of autophagy is due to the cellular
response or resistance to treatment (87). To inhibit autophagy and obtain a
negative control, the glioma cells were pretreated with Baf A1.
Fluorescence analysis (AO and LC3II staining) revealed that
lidocaine induced the autophagic process. Additionally, an MTT
assay indicated that the incubation of cells with Baf A1 prior to
lidocaine treatment (10 and 15 mM) significantly decreased the cell
viability compared with in cells treated only with lidocaine.
Furthermore, the addition of Baf A1 increased the number of
apoptotic cells in response to higher doses of lidocaine. This
suggests that lidocaine-induced autophagy may serve a protective
role. Similar results have been reported by Xiong et al in
which the protective mechanism of autophagy was suggested to be
directed against neurotoxity of local anesthetics in human
neuroblastoma cell line (58).
Autophagy was inhibited via downregulation of Beclin-1; however,
the same results were obtained via altering the expression of other
proteins, which may be directly or indirectly involved in the
process of autophagy (58,88,89).
In addition, critical autophagy regulators are also cytoskeletal
proteins involved in all stages of the process, and their
stabilization or destabilization may affect the course of autophagy
(71). There are numerous examples
in which prosurvival autophagy occurs in response to chemotherapy.
For instance, this process protects human breast cancer MCF-7 cells
from epirubicin-induced apoptosis, and esophageal squamous
carcinoma cells and colorectal cancer against the effects of 5-FU
(90-92). In addition, several drugs
indirectly associated with i.a. anesthetics may induce autophagy.
The results of the present study may improve understanding of the
effects of this process in different cancer cells, which may
contribute to developments of novel therapeutic strategies for
treatment of patients with glioma.
Funding
The present study was co-supported via research
tasks within the framework of statutory activities no. 294
(Nicolaus Copernicus University in Toruń, Faculty of Medicine,
Collegium Medicum in Bydgoszcz).
Availability of data and materials
The data and materials described in manuscript are
available upon request.
Authors' contributions
MI made substantial contributions to the design of
the resent study and supervised its quality. MI, WZ, MHW and AKW
wrote this paper and performed the experiments. MG and DG made
substantial contributions in collecting all the data and analyzed
the data in the study. MI, WZ and MHW critically revised the
manuscript for important intellectual content. All authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Goodenberger ML and Jenkins RB: Genetics
of adult glioma. Cancer Genet. 205:613–621. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ohgaki H and Kleihues P: Population-based
studies on incidence, survival rates, and genetic alterations in
astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol.
64:479–489. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bleeker FE, Molenaar RJ and Leenstra S:
Recent advances in the molecular understanding of glioblastoma. J
Neurooncol. 108:11–27. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Burdett S and Stewart L; Glioma
Meta-Analysis Trialists Group: Chemotherapy for high-grade glioma.
Neuroepidemiology. 22:3662003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Neeman E and Ben-Eliyahu S: Surgery and
stress promote cancer metastasis: New outlooks on perioperative
mediating mechanisms and immune involvement. Brain Behav Immun.
30(Suppl): S32–S40. 2013. View Article : Google Scholar
|
|
6
|
Gottschalk A, Sharma S, Ford J, Durieux ME
and Tiouririne M: Review article: The role of the perioperative
period in recurrence after cancer surgery. Anesth Analg.
110:1636–1643. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Roger S, Rollin J, Barascu A, Besson P,
Raynal PI, Iochmann S, Lei M, Bougnoux P, Gruel Y and Le Guennec
JY: Voltage-gated sodium channels potentiate the invasive
capacities of human non-small-cell lung cancer cell lines. Int J
Biochem Cell Biol. 39:774–786. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gao R, Shen Y, Cai J, Lei M and Wang Z:
Expression of voltage-gated sodium channel alpha subunit in human
ovarian cancer. Oncol Rep. 23:1293–1299. 2010.PubMed/NCBI
|
|
9
|
Diss JK, Archer SN, Hirano J, Fraser SP
and Djamgoz MB: Expression profiles of voltage-gated Na(+) channel
alpha-subunit genes in rat and human prostate cancer cell lines.
Prostate. 48:165–178. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang M, Kozminski DJ, Wold LA, Modak R,
Calhoun JD, Isom LL and Brackenbury WJ: Therapeutic potential for
phenytoin: Targeting Na(v)1.5 sodium channels to reduce migration
and invasion in metastatic breast cancer. Breast Cancer Res Treat.
134:603–615. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Grimes JA, Fraser SP, Stephens GJ, Downing
JEG, Laniado ME, Foster CS, Abel PD and Djamgoz MBA: Differential
expression of voltage-activated Na+ currents in two
prostatic tumour cell lines: Contribution to invasiveness in vitro.
FEBS Lett. 369:290–294. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Laniado ME, Lalani EN, Fraser SP, Grimes
JA, Bhangal G, Djamgoz MB and Abel PD: Expression and functional
analysis of voltage-activated Na+ channels in human
prostate cancer cell lines and their contribution to invasion in
vitro. Am J Pathol. 150:1213–1221. 1997.PubMed/NCBI
|
|
13
|
Wang HW, Wang LY, Jiang L, Tian SM, Zhong
TD and Fang XM: Amide-linked local anesthetics induce apoptosis in
human non-small cell lung cancer. J Thorac Dis. 8:2748–2757. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Piegeler T, Votta-Velis EG, Liu G, Place
AT, Schwartz DE, Beck-Schimmer B, Minshall RD and Borgeat A:
Antimetastatic potential of amide-linked local anesthetics:
Inhibition of lung adenocarcinoma cell migration and inflammatory
Src signaling independent of sodium channel blockade.
Anesthesiology. 117:548–559. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lirk P, Berger R, Hollmann MW and Fiegl H:
Lidocaine time- and dose-dependently demethylates deoxyribonucleic
acid in breast cancer cell lines in vitro. Br J Anaesth.
110:1652013. View Article : Google Scholar
|
|
16
|
Xing W, Chen DT, Pan JH, Chen YH, Yan Y,
Li Q, Xue RF, Yuan YF and Zeng WA: Lidocaine induces apoptosis and
suppresses tumor growth in human hepatocellular carcinoma cells in
vitro and in a xenograft model in vivo. Anesthesiology.
126:868–881. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Leng T, Lin S, Xiong Z and Lin J:
Lidocaine suppresses glioma cell proliferation by inhibiting TRPM7
channels. Int J Physiol Pathophysiol Pharmacol. 9:8–15.
2017.PubMed/NCBI
|
|
18
|
Johnson ME, Saenz JA, DaSilva AD, Uhl CB
and Gores GJ: Effect of local anesthetic on neuronal cytoplasmic
calcium and plasma membrane lysis (necrosis) in a cell culture
model. Anesthesiology. 97:1466–1476. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Johnson ME, Uhl CB, Spittler KH, Wang H
and Gores GJ: Mitochondrial injury and caspase activation by the
local anesthetic lidocaine. Anesthesiology. 101:1184–1194. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lirk P, Haller I, Hausott B, Ingorokva S,
Deibl M, Gerner P and Klimaschewski L: The neurotoxic effects of
amitriptyline are mediated by apoptosis and are effectively blocked
by inhibition of caspase activity. Anesth Analg. 102:1728–1733.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Glick D, Barth S and Macleod KF:
Autophagy: Cellular and molecular mechanisms. J Pathol. 221:3–12.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh
H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al:
Promotion of tumorigenesis by heterozygous disruption of the beclin
1 autophagy gene. J Clin Invest. 112:1809–1820. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yue Z, Jin S, Yang C, Levine AJ and Heintz
N: Beclin 1, an autophagy gene essential for early embryonic
development, is a haploinsufficient tumor suppressor. Proc Natl
Acad Sci USA. 100:15077–15082. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mariño G, Salvador-Montoliu N, Fueyo A,
Knecht E, Mizushima N and López-Otín C: Tissue-specific autophagy
alterations and increased tumorigenesis in mice deficient in Atg4C
autophagin-3. J Biol Chem. 282:18573–18583. 2007. View Article : Google Scholar
|
|
25
|
Ekiz HA, Can G and Baran Y: Role of
autophagy in the progression and suppression of leukemias. Crit Rev
Oncol Hematol. 81:275–285. 2012. View Article : Google Scholar
|
|
26
|
Izdebska M, Klimaszewska-Wiśniewska A,
Hałas M, Gagat M and Grzanka A: Green tea extract induces
protective autophagy in A549 non-small lung cancer cell line.
Postepy Hig Med Dosw (Online). 69:1478–1484. 2015.
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−ΔΔC(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
28
|
Jurj A, Tomuleasa C, Tat TT,
Berindan-Neagoe I, Vesa SV and Ionescu DC: Antiproliferative and
apoptotic effects of lidocaine on human hepatocarcinoma cells. A
preliminary study. J Gastrointestin Liver Dis. 26:45–50.
2017.PubMed/NCBI
|
|
29
|
Klionsky DJ, Elazar Z, Seglen PO and
Rubinsztein DC: Does bafilomycin A1 block the fusion of
autophagosomes with lysosomes? Autophagy. 4:849–850. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tanida I, Ueno T and Kominami E: LC3 and
autophagy. Methods Mol Biol. 445:77–88. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee YK and Lee JA: Role of the mammalian
ATG8/LC3 family in autophagy: Differential and compensatory roles
in the spatiotemporal regulation of autophagy. BMB Rep. 49:424–430.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Paglin S, Hollister T, Delohery T, Hackett
N, McMahill M, Sphicas E, Domingo D and Yahalom J: A novel response
of cancer cells to radiation involves autophagy and formation of
acidic vesicles. Cancer Res. 61:439–444. 2001.PubMed/NCBI
|
|
33
|
Izdebska M, Zielińska W, Grzanka D and
Gagat M: The role of actin dynamics and actin-binding proteins
expression in epithelial-to-mesenchymal transition and its
association with cancer progression and evaluation of possible
therapeutic targets. BioMed Res Int. 2018:45783732018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Grzanka D, Gagat M and Izdebska M:
Involvement of the SATB1/F-actin complex in chromatin
reorganization during active cell death. Int J Mol Med.
33:1441–1450. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sampetrean O and Saya H: Modeling
phenotypes of malignant gliomas. Cancer Sci. 109:6–14. 2018.
View Article : Google Scholar :
|
|
36
|
Nakada M, Nakada S, Demuth T, Tran NL,
Hoelzinger DB and Berens ME: Molecular targets of glioma invasion.
Cell Mol Life Sci. 64:458–478. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li G, Qin Z, Chen Z, Xie L, Wang R and
Zhao H: Tumor microenvironment in treatment of glioma. Open Med
(Wars). 12:247–251. 2017.
|
|
38
|
MostovenkoEVégváriÁRezeliMLichtiCFFenyöDWangQLangFFSulmanEPSahlinKBMarko-VargaGet
al: Large scale identification of variant proteins in glioma stem
cells. ACS Chem Neurosci. 9:73–79. 2018. View Article : Google Scholar :
|
|
39
|
Sakaguchi M, Kuroda Y and Hirose M: The
antiproliferative effect of lidocaine on human tongue cancer cells
with inhibition of the activity of epidermal growth factor
receptor. Anesth Analg. 102:1103–1107. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yang X, Zhao L, Li M, Yan L, Zhang S, Mi
Z, Ren L and Xu J: Lidocaine enhances the effects of
chemotherapeutic drugs against bladder cancer. Sci Rep. 8:5982018.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chamaraux-Tran TN, Mathelin C, Aprahamian
M, Joshi GP, Tomasetto C, Diemunsch P and Akladios C: Antitumor
effects of lidocaine on human breast cancer cells: An in vitro and
in vivo experimental trial. Anticancer Res. 38:95–105. 2018.
|
|
42
|
Le Gac G, Angenard G, Clément B, Laviolle
B, Coulouarn C and Beloeil H: Local anesthetics inhibit the growth
of human hepatocellular carcinoma cells. Anesth Analg.
125:1600–1609. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang L, Hu R, Cheng Y, Wu X, Xi S, Sun Y
and Jiang H: Lidocaine inhibits the proliferation of lung cancer by
regulating the expression of GOLT1A. Cell Prolif. 50:502017.
View Article : Google Scholar
|
|
44
|
Chang YC, Hsu YC, Liu CL, Huang SY, Hu MC
and Cheng SP: Local anesthetics induce apoptosis in human thyroid
cancer cells through the mitogen-activated protein kinase pathway.
PLoS One. 9:e895632014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lu J, Ju YT, Li C, Hua FZ, Xu GH and Hu
YH: Effect of TRPV1 combined with lidocaine on cell state and
apoptosis of U87-MG glioma cell lines. Asian Pac J Trop Med.
9:288–292. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Martinou JC and Youle RJ: Mitochondria in
apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev
Cell. 21:92–101. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Werdehausen R, Braun S, Essmann F,
Schulze-Osthoff K, Walczak H, Lipfert P and Stevens MF: Lidocaine
induces apoptosis via the mitochondrial pathway independently of
death receptor signaling. Anesthesiology. 107:136–143. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li K and Han X: Endoplasmic reticulum
stress is involved in the lidocaine-induced apoptosis in SH-SY5Y
neuroblastoma cells. J Mol Neurosci. 56:122–130. 2015. View Article : Google Scholar
|
|
49
|
Kawasaki C, Kawasaki T, Ogata M, Sata T
and Chaudry IH: Lidocaine enhances apoptosis and suppresses
mitochondrial functions of human neutrophil in vitro. J Trauma.
68:401–408. 2010. View Article : Google Scholar
|
|
50
|
Kamiya Y, Ohta K and Kaneko Y:
Lidocaine-induced apoptosis and necrosis in U937 cells depending on
its dosage. Biomed Res. 26:231–239. 2005. View Article : Google Scholar
|
|
51
|
Martinet W, Timmermans JP and De Meyer
GRY: Methods to assess autophagy in situ - transmission electron
microscopy versus immunohistochemistry. Methods Enzymol.
543:89–114. 2014. View Article : Google Scholar
|
|
52
|
Valentin M and Yang E: Autophagy is
activated, but is not required for the G0 function of BCL-2 or
BCL-xL. Cell Cycle. 7:2762–2768. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Reyjal J, Cormier K and Turcotte S:
Autophagy and cell death to target cancer cells: Exploiting
synthetic lethality as cancer therapies. Adv Exp Med Biol.
772:167–188. 2014. View Article : Google Scholar
|
|
54
|
Eskelinen EL: The dual role of autophagy
in cancer. Curr Opin Pharmacol. 11:294–300. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhou J, Hu SE, Tan SH, Cao R, Chen Y, Xia
D, Zhu X, Yang XF, Ong CN and Shen HM: Andrographolide sensitizes
cisplatin-induced apoptosis via suppression of
autophagosome-lysosome fusion in human cancer cells. Autophagy.
8:338–349. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chen G, Ke Z, Xu M, Liao M, Wang X, Qi Y,
Zhang T, Frank JA, Bower KA, Shi X, et al: Autophagy is a
protective response to ethanol neurotoxicity. Autophagy.
8:1577–1589. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yin Z, Pascual C and Klionsky DJ:
Autophagy: Machinery and regulation. Microb Cell. 3:588–596. 2016.
View Article : Google Scholar
|
|
58
|
Xiong J, Kong Q, Dai L, Ma H, Cao X, Liu L
and Ding Z: Autophagy activated by tuberin/mTOR/p70S6K suppression
is a protective mechanism against local anaesthetics neurotoxicity.
J Cell Mol Med. 21:579–587. 2017. View Article : Google Scholar :
|
|
59
|
Kang R, Zeh HJ, Lotze MT and Tang D: The
Beclin 1 network regulates autophagy and apoptosis. Cell Death
Differ. 18:571–580. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Wirawan E, Lippens S, Vanden Berghe T,
Romagnoli A, Fimia GM, Piacentini M and Vandenabeele P: Beclin1: A
role in membrane dynamics and beyond. Autophagy. 8:6–17. 6–17.
2012. View Article : Google Scholar
|
|
61
|
Decuypere JP, Parys JB and Bultynck G:
Regulation of the autophagic Bcl-2/Beclin 1 interaction. Cells.
1:284–312. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Marquez RT and Xu L: Bcl-2:Beclin 1
complex: multiple, mechanisms regulating autophagy/apoptosis toggle
switch. Am J Cancer Res. 2:214–221. 2012.PubMed/NCBI
|
|
63
|
Huang X, Qi Q, Hua X, Li X, Zhang W, Sun
H, Li S, Wang X and Li B: Beclin 1, an autophagy-related gene,
augments apoptosis in U87 glioblastoma cells. Oncol Rep.
31:1761–1767. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Du H, Che J, Shi M, Zhu L, Hang JB, Chen Z
and Li H: Beclin 1 expression is associated with the occurrence and
development of esophageal squamous cell carcinoma. Oncol Lett.
14:6823–6828. 2017.PubMed/NCBI
|
|
65
|
Pawlak G and Helfman DM: Cytoskeletal
changes in cell transformation and tumorigenesis. Curr Opin Genet
Dev. 11:41–47. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Pegoraro AF, Janmey P and Weitz DA:
Mechanical properties of the cytoskeleton and cells. Cold Spring
Harb Perspect Biol. 9:a0220382017. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Hall A: The cytoskeleton and cancer.
Cancer Metastasis Rev. 28:5–14. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Pollard TD: The cytoskeleton, cellular
motility and the reductionist agenda. Nature. 422:741–745. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Kast DJ and Dominguez R: The
cytoskeleton-autophagy connection. Curr Biol. 27:R318–R326. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Kruppa AJ, Kendrick-Jones J and Buss F:
Myosins, actin and autophagy. Traffic. 17:878–890. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Aplin A, Jasionowski T, Tuttle DL, Lenk SE
and Dunn WA Jr: Cytoskeletal elements are required for the
formation and maturation of autophagic vacuoles. J Cell Physiol.
152:458–466. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Aguilera MO, Berón W and Colombo MI: The
actin cytoskeleton participates in the early events of
autophagosome formation upon starvation induced autophagy.
Autophagy. 8:1590–1603. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Reggiori F, Monastyrska I, Shintani T and
Klionsky DJ: The actin cytoskeleton is required for selective types
of autophagy, but not nonspecific autophagy, in the yeast
Saccharomyces cerevisiae. Mol Biol Cell. 16:5843–5856. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Monastyrska I, He C, Geng J, Hoppe AD, Li
Z and Klionsky DJ: Arp2 links autophagic machinery with the actin
cytoskeleton. Mol Biol Cell. 19:1962–1975. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Monastyrska I, Rieter E, Klionsky DJ and
Reggiori F: Multiple roles of the cytoskeleton in autophagy. Biol
Rev Camb Philos Soc. 84:431–448. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Kast DJ, Zajac AL, Holzbaur EL, Ostap EM
and Dominguez R: WHAMM directs the Arp2 3 complex to the ER for
autophagosome biogenesis through an actin comet tail mechanism.
Curr Biol. 25:1791–1797. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Mi N, Chen Y, Wang S, Chen M, Zhao M, Yang
G, Ma M, Su Q, Luo S, Shi J, et al: CapZ regulates autophagosomal
membrane shaping by promoting actin assembly inside the isolation
membrane. Nat Cell Biol. 17:1112–1123. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Coutts AS and La Thangue NB: Regulation of
actin nucleation and autophagosome formation. Cell Mol Life Sci.
73:3249–3263. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Fehrenbacher K, Huckaba T, Yang HC,
Boldogh I and Pon L: Actin comet tails, endosomes and
endosymbionts. J Exp Biol. 206:1977–1984. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Taunton J, Rowning BA, Coughlin ML, Wu M,
Moon RT, Mitchison TJ and Larabell CA: Actin-dependent propulsion
of endosomes and lysosomes by recruitment of N-WASP. J Cell Biol.
148:519–530. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Lee JY, Koga H, Kawaguchi Y, Tang W, Wong
E, Gao YS, Pandey UB, Kaushik S, Tresse E, Lu J, et al: HDAC6
controls autophagosome maturation essential for ubiquitin-selective
quality-control autophagy. EMBO J. 29:969–980. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Mackeh R, Perdiz D, Lorin S, Codogno P and
Poüs C: Autophagy and microtubules - new story, old players. J Cell
Sci. 126:1071–1080. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Jahreiss L, Menzies FM and Rubinsztein DC:
The itinerary of autophagosomes: From peripheral formation to
kiss-and-run fusion with lysosomes. Traffic. 9:574–587. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Kimura S, Noda T and Yoshimori T:
Dynein-dependent movement of autophagosomes mediates efficient
encounters with lysosomes. Cell Struct Funct. 33:109–122. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Ruangjaroon T, Chokchaichamnankit D,
Srisomsap C, Svasti J and Paricharttanakul NM: Involvement of
vimentin in neurite outgrowth damage induced by fipronil in SH-SY5Y
cells. Biochem Biophys Res Commun. 486:652–658. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Wang RC, Wei Y, An Z, Zou Z, Xiao G,
Bhagat G, White M, Reichelt J and Levine B: Akt-mediated regulation
of autophagy and tumorigenesis through Beclin 1 phosphorylation.
Science. 338:956–959. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Sui X, Chen R, Wang Z, Huang Z, Kong N,
Zhang M, Han W, Lou F, Yang J, Zhang Q, et al: Autophagy and
chemotherapy resistance: A promising therapeutic target for cancer
treatment. Cell Death Dis. 4:e8382013. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Redmann M, Benavides GA, Berryhill TF,
Wani WY, Ouyang X, Johnson MS, Ravi S, Barnes S, Darley-Usmar VM
and Zhang J: Inhibition of autophagy with bafilomycin and
chloroquine decreases mitochondrial quality and bioenergetic
function in primary neurons. Redox Biol. 11:73–81. 2017. View Article : Google Scholar
|
|
89
|
Yang YP, Hu LF, Zheng HF, Mao CJ, Hu WD,
Xiong KP, Wang F and Liu CF: Application and interpretation of
current autophagy inhibitors and activators. Acta Pharmacol Sin.
34:625–635. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Sun WL, Chen J, Wang YP and Zheng H:
Autophagy protects breast cancer cells from epirubicin-induced
apoptosis and facilitates epirubicin-resistance development.
Autophagy. 7:1035–1044. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Liu D, Yang Y, Liu Q and Wang J:
Inhibition of autophagy by 3-MA potentiates cisplatin-induced
apoptosis in esophageal squamous cell carcinoma cells. Med Oncol.
28:105–111. 2011. View Article : Google Scholar
|
|
92
|
Li J, Hou N, Faried A, Tsutsumi S and
Kuwano H: Inhibition of autophagy augments 5-fluorouracil
chemotherapy in human colon cancer in vitro and in vivo model. Eur
J Cancer. 46:1900–1909. 2010. View Article : Google Scholar : PubMed/NCBI
|