Introduction
Helicobacter pylori (H. pylori) is a
gram-negative, microaerophilic bacterium that colonizes the gastric
mucosa and infects ~50% of individuals worldwide. It has been
categorized as a group I carcinogen for gastric cancer (GC) by the
World Health Organization, and GC remains a major public health
problem worldwide (1,2). A previous study revealed that the
relative risk for the development of GC was 3.8-fold higher in
patients with H. pylori infection compared to those without
(3). In addition, the association
between H. pylori eradication and the reduced incidence of
GC has been demonstrated in a meta-analysis study (4). Nevertheless, with regards to the
prognosis of GC, several studies have suggested that H.
pylori infection is a favorable prognostic factor of GC in a
Chinese prospective cohort (5,6).
Conversely, other studies have indicated that H. pylori
infection has no association with prognosis in Chinese patients
(7,8). It was hypothesized that this
discrepancy may be due to the fact that GC with different H.
pylori genotypes exhibits different growth patterns and
pathobiological behavior, indicating different mechanisms of cancer
progression. Further knowledge regarding H. pylori-infected
GC may help to identify novel therapeutic targets.
Treatment of H. pylori infection is often
empiric and current guidelines recommend against the use of
standard triple therapy (clarithromycin, amoxicillin and proton
pump inhibitors) as first-line treatment (9). However, the prevalence of
amoxicillin-resistant H. pylori is very high in Africa
(65.6%) and Asia (11.6%) (9,10).
In H. pylori, resistance occurs through modifications in
penicillin-binding proteins (PBPs), leading to a decreased affinity
for the drug. These modifications include mutations and/or mosaics
in PBP2X and PBP2B, as well as in PBP1A for the highly resistant
isolates (11). To date, the
clinical significance of amoxicillin-resistant H. pylori
(PBP1A mutation-positive H. pylori, H.
pyloriP+) in GC, and the role of H.
pyloriP+ in GC cell malignant behavior and its
specific mechanism require further elucidation. Furthermore, the
H. pylori genome shows genetic diversity among distinct
isolates, and H. pylori pathogenicity is different in
distinct isolates. Clinically isolated H. pylori strains are
often subdivided into two types according to the
cytotoxin-associated gene (Cag) pathogenicity island-encoded CagA
protein (12). Notably,
CagA-positive H. pylori increases the risk for GC over the
risk associated with H. pylori infection alone (13). In addition, it has been reported
that infections involving H. pylori strains that possess the
virulence factor CagA have a worse clinical outcome than those
involving CagA-negative strains (14), and CagA is indispensable for H.
pylori-induced tumorigenesis in GC (15). Furthermore, CagA may promote
epithelial-mesenchymal transition (EMT), which indicates a strong
ability to induce invasion and metastasis of tumor cells, and to
stimulate gastric carcinogenesis (16). Therefore, in the present study, the
role of CagA- and PBP1A mutation-positive H. pylori (H.
pyloriCagA+/P+) was investigated in patients with GC
and in GC cells, in order to gain insights into the effects of
H. pylori during the development of GC.
Factors intrinsic to the host may also contribute to
the emergence of GC, and microRNAs (miRNAs/miRs) appear to serve an
important role in the etiology of this disease (17). miRNAs are noncoding RNAs that are
involved in post-translational regulation of gene expression
(18). Unique miRNAs are
associated with various histological subtypes, and the progression
and prognosis of GC (19). A
previous study demonstrated that H. pylori may induce
dysregulation of miRNAs, contributing to the etiology of H.
pylori-mediated GC cases (17). Therefore, the identification of
signature miRNAs associated with H. pylori infection may
provide novel insights into its carcinogenesis and the host
mechanisms that are involved in bacterial elimination. However,
whether miRNAs are involved in the effects of H.
pyloriCagA+/P+ on GC has received little
attention.
The present study investigated the clinical
significance of H. pyloriCagA+/P+ in patients
with GC, and explored the role of H.
pyloriCagA+/P+ in cell proliferation, invasion and
EMT of GC cells in vitro, in order to uncover the potential
mechanism underlying the effects of H.
pyloriCagA+/P+ on gastric carcinogenesis.
Furthermore, the involvement of potential miRNAs in the
pathogenesis of H. pyloriCagA+/P+-associated GC
was observed.
Materials and methods
Patients, H. pylori strains and culture
conditions. The present study was conducted between February 2014
and November 2017 at the Jiangsu Province Hospital of TCM (Nanjing,
China). Gastric biopsy specimens were collected and maintained in
selective tryptic soy broth as transport media for further
processing, as recommended by Siu et al (20). Only patients who were diagnosed
with GC, and were negative or positive for H. pylori were
recruited; patient characteristics are presented in Table I. A total of 184 pairs of GC
tissues and adjacent non-tumor tissues were collected during
surgery. None of the patients had received adjuvant chemotherapy,
radiotherapy or H. pylori eradication treatment prior to
surgery. The histological types, stages and types of treatment of
the patients with GC were recorded. Samples obtained from each
patient were collected for culture, and three clinical isolates
were recovered from GC tissues, as previously described (21), which were used in the present
study. Identification of H. pylori was determined by colony
morphology, Gram-staining, and biochemical profile reactions, such
as oxidase, catalase and urease tests (22). The molecular identification of
CagA-positive and CagA-negative strains was conducted as previously
described (23). The
amoxicillin-susceptible and amoxicillin-resistant H. pylori
strains examined in this study were isolated from GC tissues.
Amoxicillin resistance was identified by sub-culturing swabs of
bacteria from isolation plates to horse blood agar plates with and
without 0.5 mg/l amoxicillin (Merck KGaA, Darmstadt, Germany), as
previously described (24). A
PBP1A mutation in the C-terminal region (encoded by amino acids
320–660) was validated using reverse transcription-polymerase chain
reaction (RT-PCR) (22); the
primers used were as follows: Forward, 5′-GCGACATCTGGATGAAAAT-3′
and reverse, 5′-CCATTGTTCCAACATAATCA-3′ (22). All H. pylori strains were
cultured on chocolate blood agar, and were incubated at 37°C in a
humidified incubator containing 10% CO2 and 85%
N2 for 48 h.
 | Table IAssociation between the clinical
characteristics of the study subjects and the various H.
pylori strains. |
Table I
Association between the clinical
characteristics of the study subjects and the various H.
pylori strains.
| Clinical
characteristics | H.
pylori-negative (n=50) | HPCagA−
(n=48) |
HPCagA+/P− (n=55) |
HPCagA+/P+ (n=31) | Pa | Pb | Pc |
|---|
| Age, years (means ±
SD) | 57.70±9.64 | 54.31±7.82 | 52.96±8.95 | 55.08±11.15 | 0.38 | 0.22 | 0.56 |
| Sex | | | | | | | |
| Male | 35 | 27 | 30 | 19 | 0.23 | 0.98 | 0.70 |
| Female | 15 | 21 | 25 | 12 | | | |
| Location | | | | | 0.78 | 0.69 | 0.67 |
| Proximal | 9 | 11 | 16 | 7 | | | |
| Middle | 16 | 16 | 19 | 10 | | | |
| Distal | 25 | 21 | 20 | 14 | | | |
| Size | | | | | 0.90 | 0.032 | 0.028 |
| <5 cm | 38 | 36 | 23 | 5 | | | |
| ≥5 cm | 12 | 12 | 32 | 26 | | | |
| Histological
classification | | | | | 0.22 | 0.08 | 0.57 |
| Papillary
adenocarcinoma | 11 | 12 | 15 | 9 | | | |
| Tubular
adenocarcinoma | 26 | 31 | 24 | 17 | | | |
| Mucinous
adenocarcinoma | 5 | 1 | 5 | 2 | | | |
| Signetring cell
carcinoma | 8 | 4 | 11 | 3 | | | |
| Histological
differentiation | | | | | 0.19 | 0.25 | 0.14 |
| Well | 9 | 17 | 23 | 9 | | | |
| Moderately | 8 | 9 | 16 | 6 | | | |
| Poorly | 31 | 21 | 16 | 15 | | | |
| Others | 2 | 1 | 0 | 1 | | | |
| Invasion depth | | | | | 0.21 | 0.006 | <0.0001 |
| T1 | 13 | 20 | 10 | 4 | | | |
| T2 | 30 | 19 | 20 | 2 | | | |
| T3 | 5 | 5 | 19 | 9 | | | |
| T4 | 2 | 4 | 6 | 16 | | | |
| TNM stages | | | | | 0.62 | 0.015 | 0.019 |
| I | 22 | 18 | 11 | 6 | | | |
| II | 19 | 23 | 20 | 4 | | | |
| III | 7 | 4 | 20 | 13 | | | |
| IV | 2 | 3 | 4 | 8 | | | |
| Lymphatic
metastasis | | | | | 0.89 | 0.006 | 0.007 |
| No | 36 | 33 | 24 | 4 | | | |
| Yes | 14 | 15 | 31 | 27 | | | |
| Distant
metastasis | | | | | 0.27 | 0.008 | 0.009 |
| No | 33 | 37 | 21 | 3 | | | |
| Yes | 17 | 11 | 34 | 28 | | | |
Cell culture
The human gastric epithelial cell line (GES-1), and
GC cell lines, MGC-803, BGC-823, SGC-7901 and MKN45, were obtained
from the Cell Bank of the Chinese Academy of Sciences (Shanghai,
China) and maintained in Roswell Park Memorial Institute
(RPMI)-1640 medium (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA USA). 293T cells (Cell Bank of
Chinese Academy of Sciences) were cultured in Dulbecco's modified
Eagle's medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented
with 10% FBS. All cells were incubated at 37°C in a humidified
atmosphere containing 5% CO2.
H. pylori infection experiments. For cell
infection, a single colony of a 3-day old culture was cultured in 5
ml broth (tryptone 10 g/l; yeast extract 5 g/l; NaCl 10 g/l in
distilled water; pH 7.4) supplemented with 10% heat-inactivated FBS
(Gibco; Thermo Fisher Scientific, Inc.). The bacterial cultures
were incubated under microaerobic conditions generated by CampyGen
sachets at 37°C for 1–2 days with agitation at 150 rpm. When the
600 nm optical density of the bacterial culture in Brain Heart
Infusion broth (HiMedia Laboratories, Mumbai, India) reached 1.0,
the bacteria were harvested and incubated at 37°C in RPMI medium
for 30 min. SGC-7901 cells were microscopically enumerated using an
improved Neubauer counting chamber. Monolayer SGC-7901 cells in a
6-well plate (105/well) were incubated with H.
pylori at a multiplicity of infection of 50:1 for 12 h at 37°C
and 10% CO2, as previously described (25).
Cell transfection
miRNA mimic control (5′-UCACAACCUC
CUAGAAAGAGUAGA-3′), miR-134 mimics (sense
5′-UGUGACUGGUUGACCAGAGGGG-3′; antisense 5′-CCUCUGG
UCAACCAGUCACAUU-3′), inhibitor control (5′-UCACAA
CCUCCUAGAAAGAGUAGA-3′) and miR-134 inhibitors
(5′-CCCCUCUGGUCAACCAGUCACA-3′) were all purchased from Guangzhou
Ribobio Co., Ltd. (Guangzhou, China). Forkhead box M1
(FoxM1)-specific small interfering RNA (siRNA; si-FOXM1;
5′-UGAAUCUGCGUUUUC ACUCUC-3′) and negative control siRNA
(5′-UGCAAAUAAGGGUAAUC AUGC-3′) were synthesized by Shanghai
GenePharma Co., Ltd. (Shanghai, China). Lipofectamine®
2000 reagent (Invitrogen; Thermo Fisher Scientific, Inc.) was used
to transfect the aforementioned oligonucleotides into SGC-7901
cells according to the manufacturer's protocol. Briefly,
5×105 cells were seeded into each well of 6-well plates
18–24 h prior to transfection. The transfection medium (2 ml)
containing 1 µg/ml siRNAs or 100 nmol/l miRNA mimics/inhibitors
together with 6 µl Lipofectamine® 2000 was incubated at
room temperature for 15 min and was then transferred to each well
of the culture plates at 37°C; the cells were harvested for
analysis after 24 h. FoxM1 overexpression was achieved by
transfecting SGC-7901 cells with the FoxM1 (NM_202002) plasmid
(Shanghai GeneChem Co., Ltd., Shanghai, China). Attractene
transfection reagent (Qiagen GmbH, Hilden, Germany) was used to
transfect cells with the FoxM1 plasmid or an empty GV230 plasmid
(Shanghai GeneChem Co., Ltd.), in accordance with the
manufacturer's protocol.
miRNA microarray analysis
Five CagA-positive and PBP1a mutation-negative H.
pylori (H. pyloriCagA+/P−) and five H.
pyloriCagA+/P+ tumor tissue samples were selected
for miRNA microarray analysis. Total RNA was isolated using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) and miRNeasy mini kit (Qiagen GmbH), according to the
manufacturers' protocols. Total RNA was then labeled using the
miRCURY™ Array Power labeling kit (Exiqon; Qiagen GmbH) and
hybridization was performed using miRCURY™ LNA Array (Exiqon;
Qiagen GmbH). Fluorescent images were collected using a laser
scanner (GenePix 4000B; Molecular Devices, LLC, Sunnyvale, CA, USA)
and were digitized using Array-Pro image analysis software 6.3
(Media Cybernetics, Inc., Rockville, MD, USA). The SpotData Pro 3.0
software (CapitalBio Corporation, Beijing, China) was used for data
analysis. Hierarchical clustering was performed using Data Matching
Software MeV v4.8.1 (26).
RNA isolation and RT-quantitative (q)PCR
analysis
Total RNA was extracted from the treated cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). Total RNA was reverse transcribed using the cDNA Synthesis
kit (Takara Biotechnology Co., Ltd., Dalian, China), according to
the manufacturer's protocol, and quantification was performed using
SYBR Green (Takara Biotechnology Co., Ltd.) on a LightCycler480 PCR
system (Roche Diagnostics, Basel, Switzerland). Amplification of
target gene cDNA was normalized to GAPDH expression. The expression
levels of miR-134 were assessed using the Bulge-Loop™ miRNA qRT-PCR
Primer Set and the miRNA qRT-PCR Control Primer Set (Guangzhou
RiboBio Co., Ltd.), and U6 was used as an internal control. The
primer sequences used are listed in Table II. The thermocycling conditions
were as follows: 94°C for 10 min, followed by 40 cycles at 94°C for
10 sec, 60°C for 45 sec and 72°C for 60 sec, and a final extension
step at 72°C for 5 min. Each reaction was performed in triplicate.
Relative quantification of gene expression levels was expressed as
fold-change using the 2−ΔΔCq method (27). Each test was carried out in
triplicate.
| Table IIForward and reverse primers used for
RT-quantitative polymerase chain reaction. |
Table II
Forward and reverse primers used for
RT-quantitative polymerase chain reaction.
| Gene name | Sequence
(5′–3′) |
|---|
| Hsa-miR-134 | |
| RT primer |
GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCACTGGATACGTTGGTGTC |
| Forward |
ACACTCCAGCTGGGCACCAA |
| Hsa-miR-218 | |
| RT primer |
GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCACTGGATACGUGUAGAAA |
| Forward |
ACACTCCAGCTGGGTTGTGCTTGATCTAA |
| Hsa-miR-488 | |
| RT primer |
GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCACTGGATACGTTGAGAGT |
| Forward |
ACACTCCAGCTGGGCCCAGATAATGGCA |
| Hsa-miR-159 | |
| RT primer |
GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCACTGGATACGTCCTACCC |
| Forward |
ACACTCCAGCTGGGCAGACTTGGCCAT |
| U6 | |
| Forward |
CTCGCTTCGGCAGCACA |
| Reverse |
AACGCTTCACGAATTTGCGT |
| E-cadherin | |
| Forward |
AAAGGCCCATTTCCTAAAAACCT |
| Reverse |
TGCGTTCTCTATCCAGAGGCT |
| Vimentin | |
| Forward |
AGTCCACTGAGTACCGGAGAC |
| Reverse |
CATTTCACGCATCTGGCGTTC |
| α-SMA | |
| Forward |
CATTTCACGCATCTGGCGTTC |
| Reverse |
CCATCAGGCAGTTCGTAG |
| GAPDH | |
| Forward |
TGCACCACCAACTGCTTAGC |
| Reverse |
GGCATGGACTGTGGTCATGAG |
Western blot analysis
Radioimmunoprecipitation assay lysis buffer and
Bicinchoninic Acid Protein Quantification kit (Beijing Solarbio
Science & Technology Co., Ltd., Beijing, China) were employed
to extract total protein from SGC-7901 cells and determine protein
concentration, respectively. Equal amounts of protein (30 µg) were
separated by 10–12% SDS-PAGE, transferred to polyvinylidene
fluoride membranes. After blocking with 5% non-fat milk at room
temperature for 1 h, the membranes were incubated with E-cadherin
(cat. no. 14472, 1:1,000), Vimentin (cat. no. 5741, 1:1,000),
α-smooth muscle actin (α-SMA, cat. no. 48938, 1:1,000), FoxM1 (cat.
no. 20459, 1:1,000) and GAPDH (cat. no. 2118, 1:1,000; Cell
Signaling Technology, Inc., Danvers, MA, USA) antibodies overnight
at 4°C. The membranes were then incubated with goat anti-rabbit
immunoglobulin (Ig) G H&L (horseradish peroxidase) (1:5,000;
Abcam, Cambridge, MA, USA) for 1 h at room temperature.
Membrane-bound immune complexes were visualized using the enhanced
chemiluminescence detection reagent (Beyotime Institute of
Biotechnology, Haimen, China).
Cell proliferation and invasion
Cell proliferation was measured using the Cell
Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc.,
Kumamoto, Japan). At different time points, 10 µl CCK-8 solution
was added to each well, and the plates were incubated at 37°C for 2
h. Absorbance was measured at 570 nm using a Thermo Fisher
Scientific Microplate Reader (Thermo Fisher Scientific, Inc.). The
cell invasion assay was performed using Transwell chambers coated
with Matrigel (EMD Millipore, Billerica, MA, USA). At 48 h
post-transfection, 105 cells were added into the upper
chambers alongside serum-free medium. In the lower chambers, 500 µl
RPMI-1640 medium containing 10% FBS was added as a chemoattractant.
After 24 h, the Matrigel and cells on the upper surface were
removed by cotton swabs, and the cells that had migrated to the
lower surface were fixed in 4% paraformaldehyde (Sigma-Aldrich;
Merck KGaA) for 15 min at room temperature and stained with 0.1%
crystal violet (Sigma-Aldrich; Merck KGaA) for 15 min at room
temperature. Cells were visually counted in five randomly selected
fields under a light microscope (Axiovert 200 inverted microscope;
Zeiss AG, Oberkochen, Germany).
Luciferase assay and constructs
TargetScan (http://www.targetscan.org) and microRNA database
(http://www.microrna.org) bioinformatics software
were used to predict the downstream targets of miR-134. The FoxM1
promoter region was amplified from human genomic DNA (cat. no.
Roche-11691112001; Sigma-Aldrich; Merck KGaA) by PCR (forward,
5′-GGTCCGAGTAAAACAAGAGCG-3′; reverse, 5′-AGTGAGAGAGTATAGGAAGG-3′).
Mutated FoxM1, devoid of the miR-134 binding site, was generated
using the QuikChange II XL Site-Directed Mutagenesis kit (cat. no.
ST200521; Stratagene; Agilent Technologies, Inc., Santa Clara, CA,
USA). Wild type and mutated FoxM1 were sub-cloned into the
pGL3-basic luciferase vector (Promega Corporation, Madison, WI,
USA). 293T cells, cultured in 24-well plates (5×105),
were transfected with 2.4 µg/well wild type FoxM1-pGL3 plasmid or
mutant FoxM1-pGL3 plasmid, and were co-transfected with 20 nmol/l
miR-134 mimics or mimic control using 2 µl
Lipofectamine® 2000 transfection reagent (Invitrogen;
Thermo Fisher Scientific, Inc.), according to the manufacturer's
protocol. After 48 h of transfection, luciferase activity was
measured using the Bright-Glo™ Luciferase Assay system (Promega
Corporation) according to the manufacturer's protocol. Relative
percentages of luminescence intensity were calculated by comparison
with the mimic control-transfected cells.
Immunofluorescence analysis
After treatment, cells were fixed with 4%
paraformaldehyde (Sigma-Aldrich; Merck KGaA) for 15 min at room
temperature and blocked with 5% bovine serum albumin
(Sigma-Aldrich; Merck KGaA) for 30 min at room temperature. The
cells were then incubated with E-cadherin (1:400), α-SMA (1:500) or
FoxM1 (1:500) primary antibodies overnight at 4°C. After washing
with PBS-0.1% Tween, cells were incubated with Alexa
Fluor® 488 goat anti-rabbit IgG (cat. no. A-11034) or
Alexa Fluor® 594 goat anti-rabbit IgG (cat. no. B40925;
1:500; Invitrogen; Thermo Fisher Scientific, Inc.) for 30 min at
room temperature in the dark. DAPI (Sigma-Aldrich; Merck KGaA) was
used to identify the nuclei. Images were captured under a Nikon
Eclipse 800 epifluorescence microscope (Nikon Corporation, Tokyo,
Japan) with the appropriate filters.
Statistical analysis
All experiments were performed at least three times
and data are presented as the means ± standard error. Pearson's
χ2 test was used to investigate the associations between
H. pylori status and categorical variables. Student's t-test
was used to compare differences between two groups with continuous
variables. One-way analysis of variance and least significant
difference post hoc test was used to compare the differences
between more than two groups. Pearson's correlation was used to
analyze the relationship between miR-134 and FoxM1 expression.
Statistical analyses were performed using SPSS 20.0 (IBM Corp.,
Armonk, NY, USA) and GraphPad Prism 6.0 (GraphPad Software Inc., La
Jolla, CA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
H. pyloriCagA+/P+ infection is
associated with poor clinicopathological characteristics in
patients with GC
The study group consisted of 50 patients with H.
pylori-negative GC, 48 patients with CagA-negative H.
pylori (H. pyloriCagA−) GC, 55
patients with H. pyloriCagA+/P− GC,
and 31 patients with H. pyloriCagA+/P+ GC. As
shown in Table I, the demographic
and pathological characteristics of these patients were collected.
There were no significant differences among the groups with regards
to age, gender, tumor location, histological classification and
histological differentiation. Notably, there was a statistically
significant difference between H.
pyloriCagA+/P− infection and H.
pylori-negative groups with regards to tumor size, invasion
depth, TNM stages, lymphatic metastasis and distant metastasis.
Furthermore, patients with H. pyloriCagA+/P+
exhibited poor clinicopathological characteristics compared with
patients with H. pyloriCagA+/P−. These
results suggested that H. pyloriCagA+/P+ may have
an important role in the development of GC, which differs from
H. pyloriCagA+/P−.
H. pyloriCagA+/P+ induces
proliferation, invasion and EMT in SGC-7901 cells
The present study performed an in vitro assay
to further investigate the mechanisms underlying the relationship
between H. pyloriCagA+/P+ infection and poor
clinicopathological characteristics. To imitate the in vivo impact
of different H. pylori strains, SGC-7901 GC cells were
infected with H. pyloriCagA−, H.
pyloriCagA+/P− and H.
pyloriCagA+/P+. Notably, the CCK-8 assay (Fig. 1A) and Transwell assay (Fig. 1B) revealed that, when compared with
the H. pyloriCagA− group, H.
pyloriCagA+/P−-infected cells exhibited
significantly increased cell proliferation and invasion.
Furthermore, cell proliferation and invasion were further increased
in H. pyloriCagA+/P+-infected GC cells, as
compared to those infected with H.
pyloriCagA+/P−. In addition,
immunofluorescence staining revealed that H.
pyloriCagA+/P− and H.
pyloriCagA+/P+ infection reduced the fluorescence
intensity of E-cadherin, but increased the intensity of α-SMA
(Fig. 1C). Western blot analysis
indicated that H. pyloriCagA+/P−
infection induced EMT of SGC-7901 cells, whereas H.
pyloriCagA+/P+ exhibited more prominent EMT in GC
cells compared with the H.
pyloriCagA+/P− group (Fig. 1D). These findings may explain why
patients with H. pyloriCagA+/P+ and GC exhibited
advanced malignant behavior compared with patients with H.
pyloriCagA+/P− infection.
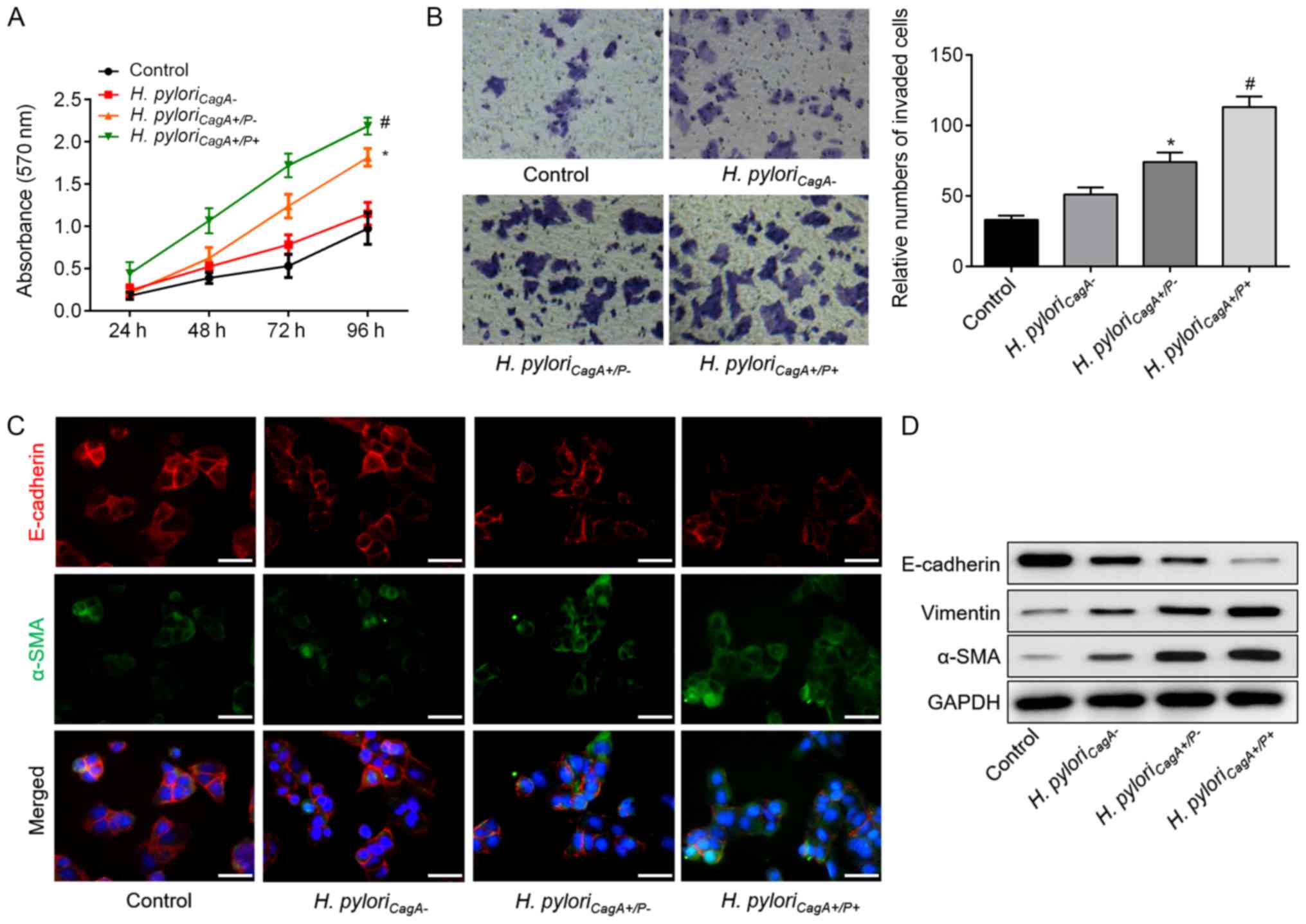 | Figure 1H. pyloriCagA+/P+
induces proliferation, invasion and EMT in SGC‐7901 gastric cancer
cells. (A) Growth curves for SGC‐7901 cells exposed to the
indicated treatments via Cell Counting Kit‐8 assay. (B) Transwell
assay was used to evaluate the invasion of SGC‐7901 cells infected
with H. pyloriCagA-, H.
pyloriCagA+/P− and H.
pyloriCagA+/P+. Untreated SGC‐7901 cells were used
as a control (magnification, ×100). (C) Double‐labeled
immunofluorescence staining was performed to examine the expression
of E‐cadherin (red) and α‐SMA (green) in SGC‐7901 cells from
different groups. Images merged with DAPI are presented. Scale
bar, 50 μm. (D) Protein expression levels of E‐cadherin, Vimentin
and α-SMA in SGC‐7901 cells infected with various H. pylori
strains. *P<0.05 vs. the control group; #P<0.05 vs. the H.
pyloriCagA+/P− group. α‐SMA, α‐smooth muscle actin;
CagA, cytotoxin‐associated gene A; H.
pyloriCagA‐, CagA‐negative H. pylori; H.
pyloriCagA+/P−, CagA‐positive and PBP1A
mutation‐negative H. pylori; H.
pyloriCagA+/P+, CagA‐and PBP1A mutation‐positive
H. pylori; H. pylori, Helicobacter pylori;
PBP1A, penicillin‐binding protein 1A |
miR-134 is downregulated in H.
pyloriCagA+/P+ GC tissues and cells
To determine the potential involvement of miRNAs in
gastric carcinogenesis induced by H.
pyloriCagA+/P- and H.
pyloriCagA+/P+, a miRNA microarray was performed on
five GC tissues, which were randomly selected from the H.
pyloriCagA+/P− and H.
pyloriCagA+/P+ groups. Notably, the results
identified nine upregulated miRNAs and 11 downregulated miRNAs in
the H. pyloriCagA+/P+ group compared with the
H. pyloriCagA+/P− group (Fig. 2A). The present study further
investigated the four most downregulated miRNAs, miR-134, miR-218,
miR-159 and miR-488, and validated the microarray results using
RT-qPCR. The expression levels of miR-134 were decreased in the
H. pyloriCagA− group compared with in
the control group, and were further decreased in the H.
pyloriCagA+/P+ group (Fig. 2B). Although miR-218 and miR488 were
decreased in H. pylori-infected GC tissues, there was no
significant difference between the different H. pylori
strains (Fig. 2C and D). In
addition, miR-159 was not altered in H. pylori-positive
tissues compared with in H. pylori-negative tissues.
Although miR-159 was downregulated in H.
pyloriCagA+/P− and H.
pyloriCagA+/P+ groups, when compared with the
control group, its expression did not differ between the H.
pyloriCagA+/P− and H.
pyloriCagA+/P+ groups (Fig. 2E). Based on these findings, the
present study focused on miR-134, with the aim of identifying its
regulatory role in H. pyloriCagA+/P+ GC
tissues.
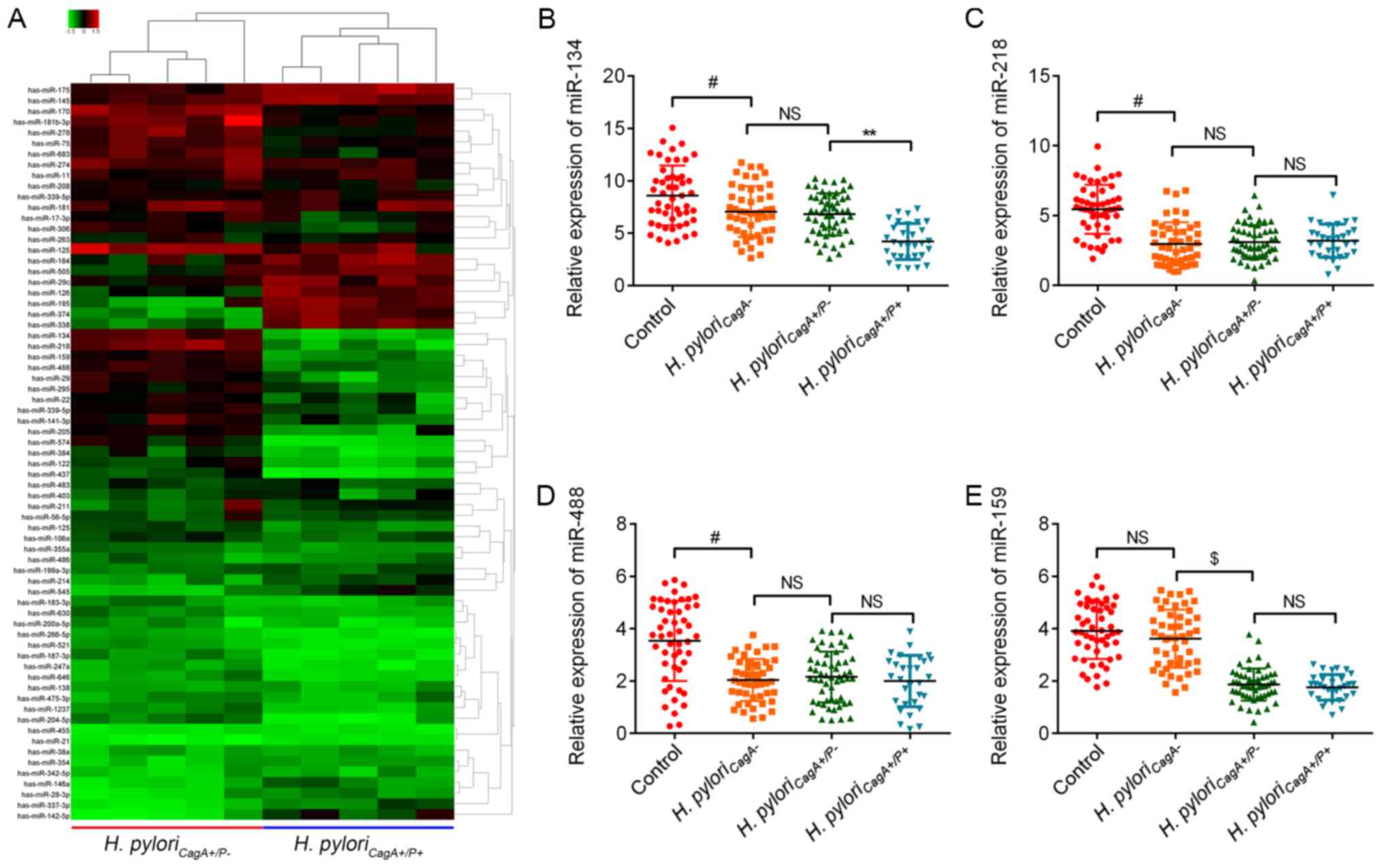 | Figure 2Aberrant miRNAs between H.
pyloriCagA+/P− and H.
pyloriCagA+/P+ GC tissues. (A) Heatmap of miRNA
expression between H. pyloriCagA+/P− and H.
pyloriCagA+/P+ GC tissues. The five tissues in each
group were selected at random. Green, downregulated; red,
upregulated. Relative expression levels of (B) miR‐134, (C)
miR‐218, (D) miR‐488 and (E) miR‐159 in GC tissues without H.
pylori infection, and in tissues infected with H.
pyloriCagA-, H. pyloriCagA+/P− and
H. pyloriCagA+/P+. #P<0.05 vs. the control
group; $P<0.05 vs. the H. pyloriCagA− group; **P<0.05
vs. H. pyloriCagA+/P− group. CagA,
cytotoxin‐associated gene A; H.
pyloriCagA−, CagA‐negative H.
pylori; H. pyloriCagA+/P−, CagA‐positive and
PBP1A mutation‐negative H. pylori; H.
pyloriCagA+/P+, CagA‐and PBP1A
mutation‐positive H. pylori; H. pylori,
Helicobacter pylori; miR, microRNA; NS, not significant;
PBP1A, penicillin‐binding protein 1A. |
miR-134 inhibits HP-induced cell
proliferation, invasion and reverses EMT
In order to elucidate the relationship between
miR-134 and H. pyloriCagA+/P+-induced malignant
behavior of GC cells, proliferation, invasion and EMT of SGC-7901
cells were analyzed. As shown in Fig.
3A, miR-134 expression differed between GES-1 cells and the
four GC cell lines. SGC-7901 cell exhibited the lowest miR-134
expression; therefore, it was then chosen for subsequent analyses.
As presented in Fig. 3B, SGC-7901
cells exposed to H. pylori infection exhibited reduced
miR-134 expression, and cells infected with H.
pyloriCagA+/P+ possessed the lowest expression
levels of miR-134; these findings were consistent with the results
from clinical samples. Therefore, SGC-7901 cells were transfected
with miR-134 mimics to induce miR-134 overexpression (Fig. 3C). Subsequently, CCK-8 and
Transwell assays demonstrated that miR-134 overexpression
significantly decreased H. pyloriCagA+/P+-induced
cell proliferation and invasion compared with in the control group
(Fig. 3D and E). As shown in
Fig. 3F, miR-134 overexpression
upregulated E-cadherin, and downregulated α-SMA expression. In
addition, western blot analysis revealed consistent alterations in
the aforementioned EMT markers in the H.
pyloriCagA+/P+ + miR-134 mimics groups compared with
in the H. pyloriCagA+/P+ group (Fig. 3G). These findings indicated that
miR-134 may act as a suppressor of proliferation, invasion and EMT
in H. pyloriCagA+/P+-infected SGC-7901 cells.
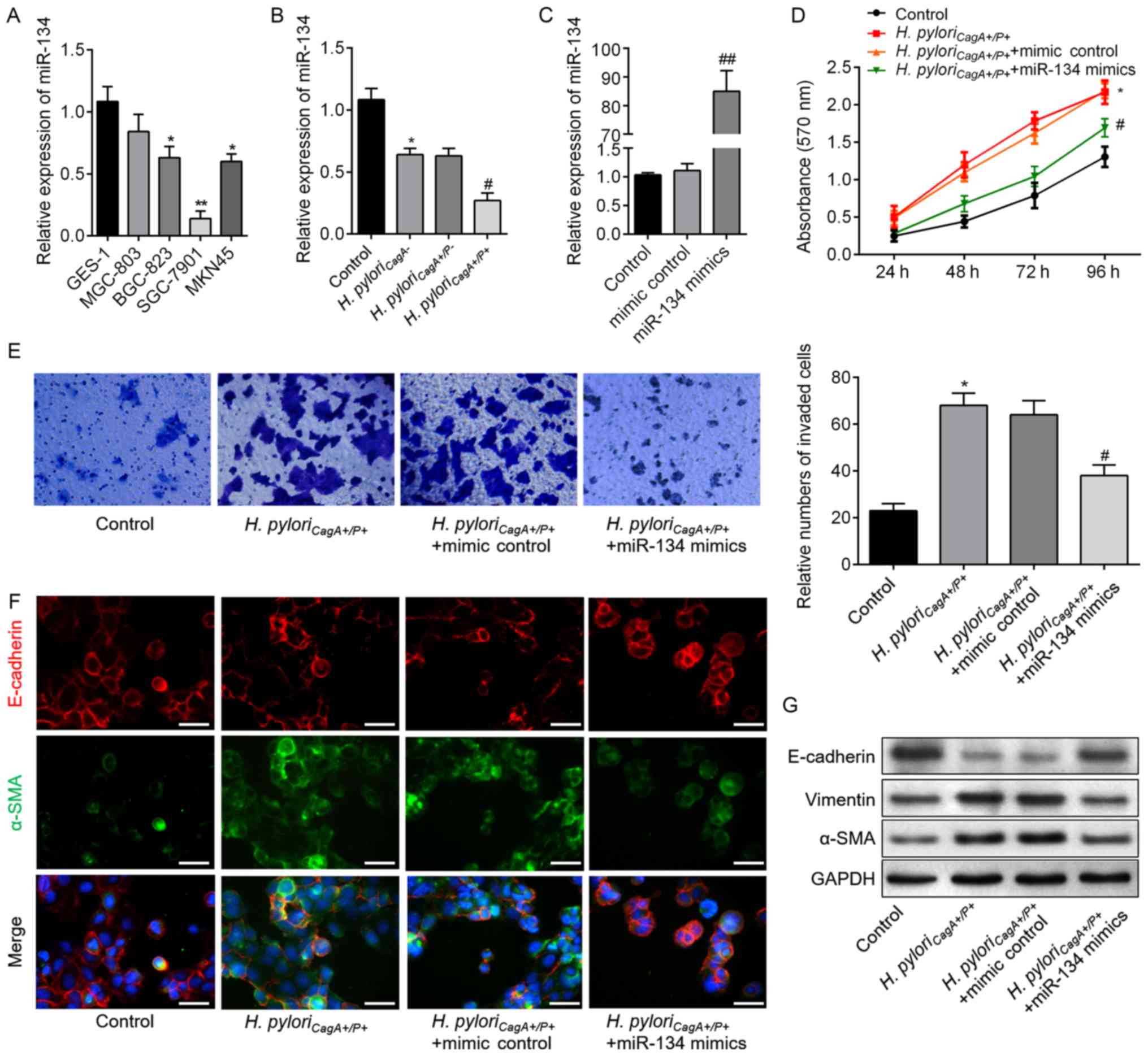 | Figure 3miR-134 inhibits proliferation,
invasion and EMT in H. pyloriCagA+/P+-infected
SGC-7901 cells. (A) miR-134 expression in a human gastric
epithelial cell (GES-1), and MGC803, BGC-823, SGC-7901 and MKN45
gastric cancer cell lines, as detected by reverse
transcription-qPCR analysis. *P<0.05,
**P<0.01 vs. GES-1 cells. (B) Relative expression
levels of miR-134 in SGC-7901 cells infected with H.
pyloriCagA-, H. pyloriCagA+/P- and
H. pyloriCagA+/P+. (C) Expression levels of
miR-134 in SGC-7901 cells transfected with mimic control or miR-134
mimics, as evaluated by reverse transcription-qPCR assay. (D)
SGC-7901 cells transfected with miR-134 mimics or mimic control
under H. pyloriCagA+/P+ infection were subjected
to Cell Counting Kit-8 analysis. (E) Invasive SGC-7901 cells were
stained with 0.1% crystal violet and counted 48 h post-transfection
(magnification, ×100). The number of invaded cells was determined
from three replicate wells and data are expressed as the means ±
standard deviation. (F) Double-labeled immunofluorescence staining
was performed to examine the expression of E-cadherin (red) and
α-SMA (green) in SGC-7901 cells from the various groups. Images
merged with DAPI are presented. Scale bar, 50 µm. (G) Protein
expression levels of E-cadherin, Vimentin and α-SMA in SGC-7901
cells treated as indicated. *P<0.05 vs. the control
group; #P<0.05 vs. the H.
pyloriCagA+/P+ + mimics control group. α-SMA,
α-smooth muscle actin; CagA, cytotoxin-associated gene A; H.
pyloriCagA-, CagA-negative H. pylori; H.
pyloriCagA+/P−, CagA-positive and PBP1A
mutation-negative H. pylori; H.
pyloriCagA+/P+, CagA- and PBP1A mutation-positive
H. pylori; H. pylori, Helicobacter pylori;
miR-134, microRNA-134; PBP1A, penicillin-binding protein 1A;
RT-qPCR, reverse transcription-quantitative polymerase chain
reaction. |
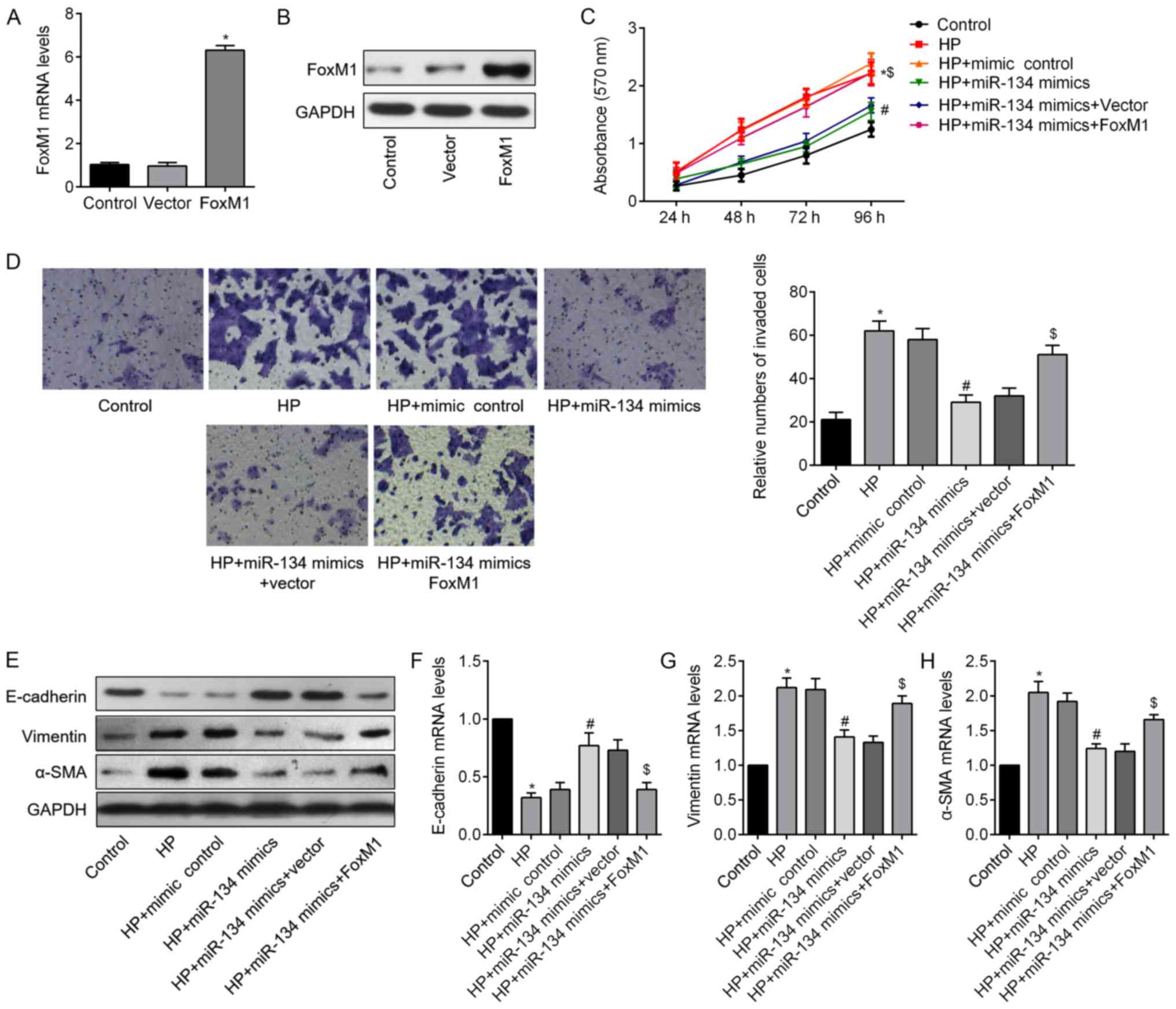
FoxM1 is identified as a direct target of
miR-134
To determine the target gene of miR-134 in H.
pyloriCagA+/P+-infected GC cells, candidate target
genes were searched using bioinformatics analysis. The analysis
revealed that FoxM1 possessed a putative miR-134-binding site
(Fig. 4A), which prompted further
validation of this relationship. FoxM1 expression was significantly
increased in H. pyloriCagA+/P+ GC tissues
compared with in H. pyloriCagA+/P- GC tissues
(Fig. 4B). Correlation analysis
revealed that FoxM1 expression was negatively correlated with
miR-134 expression (Fig. 4C).
Subsequently, knockdown of miR-134 was achieved via transfection
with miR-134 inhibitors; transfection efficiency was validated by
RT-qPCR (Fig. 4D). In response to
miR-134 overexpression, the mRNA and protein expression levels of
FoxM1 were significantly downregulated, whereas they were
upregulated by miR-134 knockdown in SGC-7901 cells (Fig. 4E and F). Dual luciferase reporter
assay revealed that miR-134 mimics significantly suppressed FoxM1
wild type luciferase activity compared with in the mimics control
group, but did not affect FoxM1 mutant type luciferase activity
(Fig. 4G). Furthermore,
immunofluorescence assay demonstrated that miR-134 mimics decreased
the expression of FoxM1 protein in H.
pyloriCagA+/P+-infected cells (Fig. 4H). These data suggested that FoxM1
was a direct target gene of miR-134 and was negatively regulated by
miR-134.
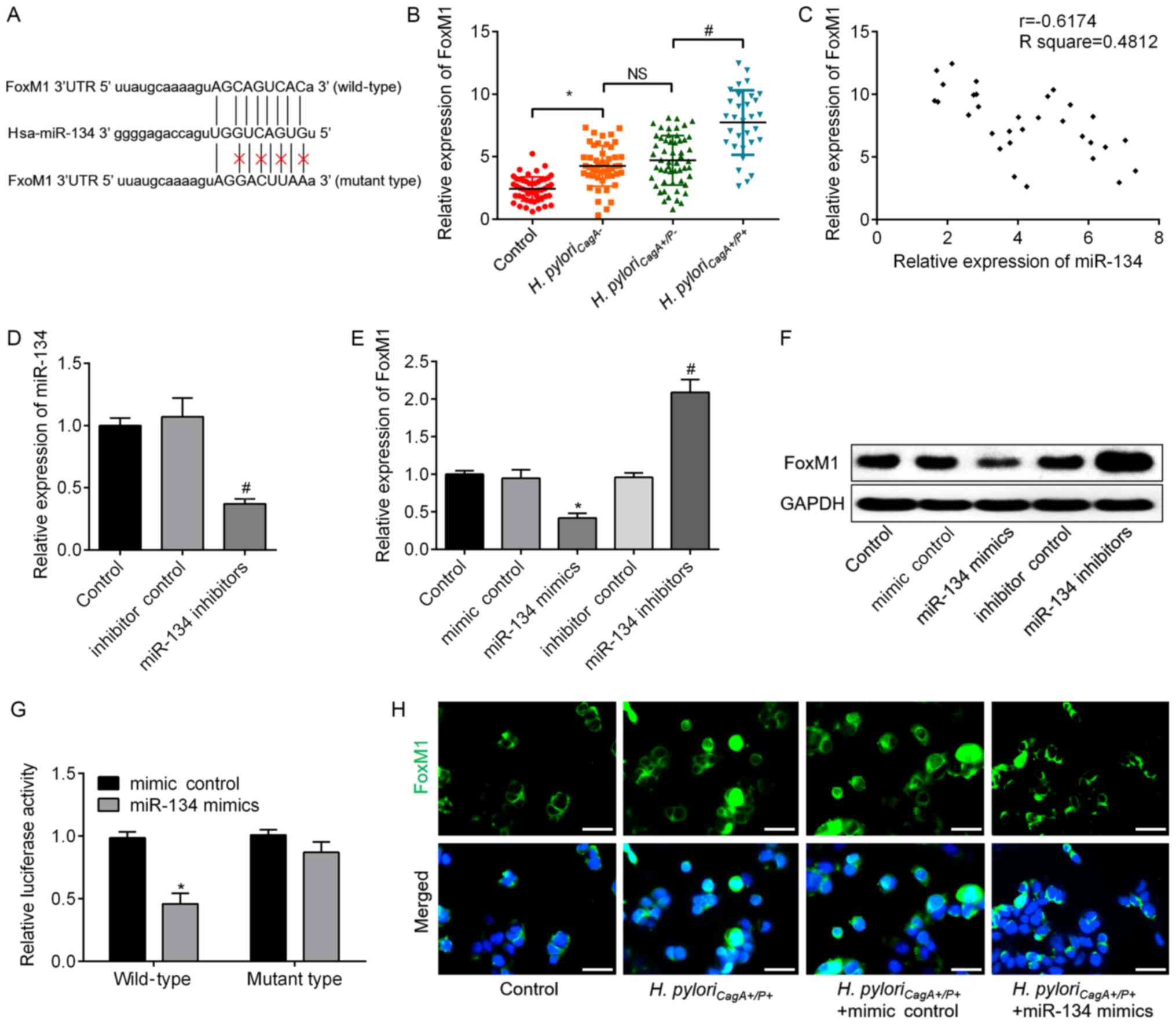 | Figure 4FoxM1 is a downstream target of
miR-134, which directly binds to the 3′UTR of FoxM1. (A) Putative
miR-134-binding sites in the 3′UTR of FoxM1 mRNA. A mutation was
generated in the FoxM1 3′UTR sequence in the complementary site for
the seed region of miR-134. (B) Relative expression levels of FoxM1
in GC tissues without H. pylori infection, and tissues
infected with H. pyloriCagA−, H.
pyloriCagA+/P− and H.
pyloriCagA+/P+. (C) A negative correlation was
observed between FoxM1 mRNA and miR-134 expression in GC tissues
infected with H. pyloriCagA+/P+. (D) Expression
levels of miR-134 in SGC-7901 cells transfected with inhibitor
control or miR-134 inhibitors, as determined by RT-qPCR analysis.
(E) mRNA expression levels of FoxM1 were analyzed by RT-qPCR
analysis in SGC-7901 cells transfected with mimic control, miR-134
mimics, inhibitor control or miR-134 inhibitors. GAPDH was used as
an internal control. (F) Protein expression levels of FoxM1 were
determined by western blotting in the groups. GAPDH was used as an
internal control. (G) Luciferase assay in 293T cells co-transfected
with miR-134 mimics or mimic control and a luciferase reporter
containing wild type or mutant FoxM1 3′UTR. (H) Immunofluorescence
analysis revealed the alteration of FoxM1 in H.
pyloriCagA+/P+-infected SGC-7901 cells transfected
with miR-134 mimics or mimic control. Images merged with DAPI are
presented. Scale bar, 50 µm. *P<0.05 vs. the mimic
control group; #P<0.05 vs. the inhibitor control
group. 3′UTR, 3′-untranslated region; CagA, cytotoxin-associated
gene A; H. pyloriCagA-, CagA-negative H.
pylori; H. pyloriCagA+/P−,
CagA-positive and PBP1A mutation-negative H. pylori; H.
pyloriCagA+/P+, CagA- and PBP1A mutation-positive
H. pylori; FoxM1, Forkhead box M1; H. pylori,
Helicobacter pylori; miR-134, microRNA-134; PBP1A,
penicillin-binding protein 1A; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction. |
Knockdown of FoxM1 impedes H.
pylori-induced EMT
Although elevated FoxM1 is identified as a
prognostic factor for GC, whether it is involved in EMT in H.
pyloriCagA+/P+-associated GC remains undefined.
Therefore, si-FoxM1 was used to knockdown FoxM1 in H.
pyloriCagA+/P+-infected SGC-7901 cells. The
transfection efficiency of si-FoxM1 was determined using RT-qPCR
and western blot analysis (Fig. 5A and
B). Subsequently, the role of FoxM1 in H.
pyloriCagA+/P+-induced EMT was determined. Western
blot analysis revealed that H. pylori-mediated EMT was
significantly reversed by knockdown of FoxM1 (Fig. 5C). Similarly, these findings were
validated at the mRNA level, as evidenced by the alterations in
EMT-associated genes (Fig. 5D–F).
These data revealed that H. pyloriCagA+/P+ may
trigger EMT of GC cells through regulation of FoxM1.
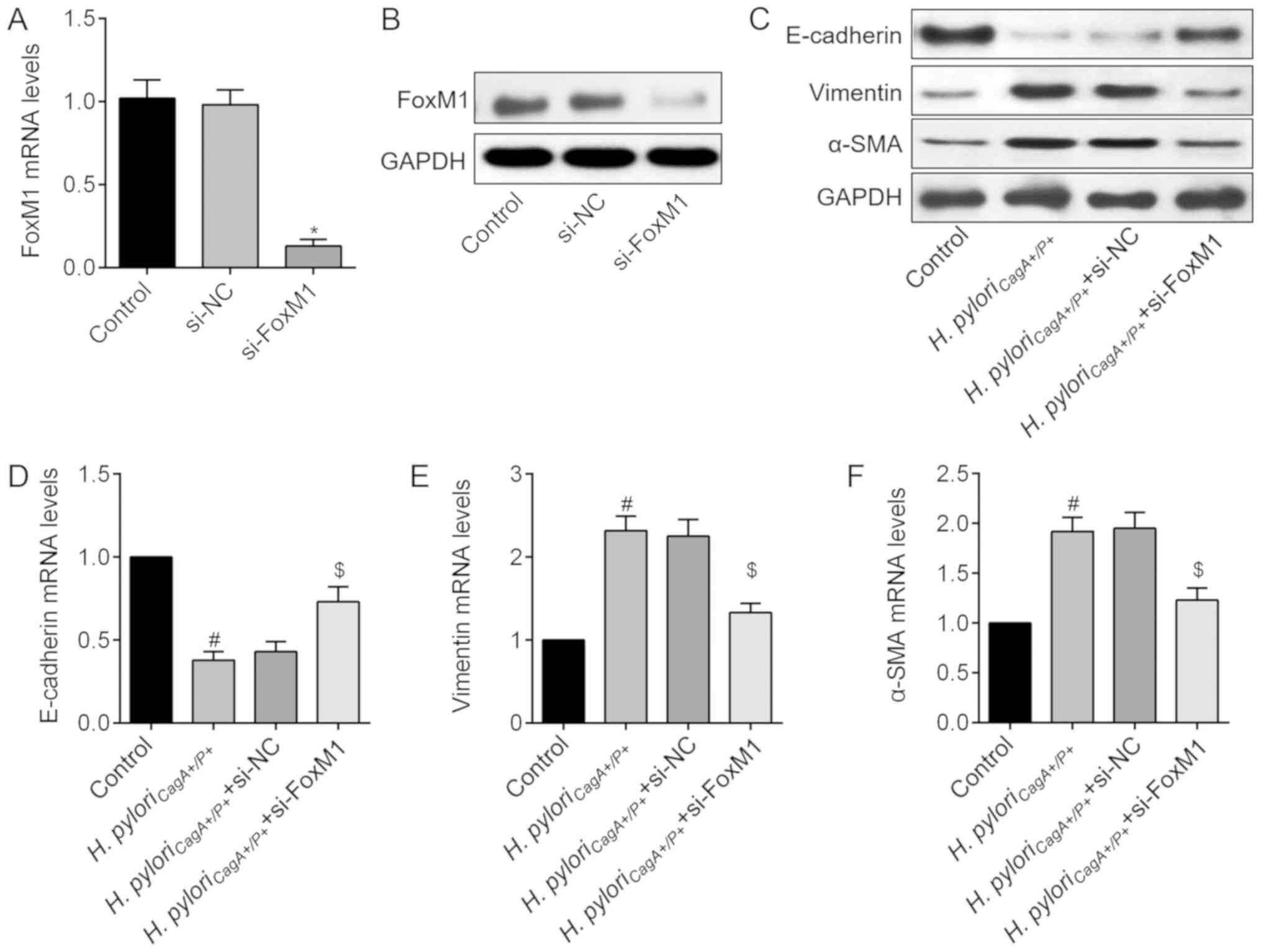 | Figure 5FoxM1 knockdown abolishes H.
pyloriCagA+/P+-induced EMT in SGC-7901 cells. (A)
FoxM1 mRNA expression levels in cells transfected with si-NC or
si-FoxM1. (B) FoxM1 protein expression was detected following
transfection with si-FoxM1. (C) Protein expression levels of EMT
markers, E-cadherin, Vimentin and α-SMA, in SGC-7901 cells
transfected with si-NC or si-FoxM1, and infected with H.
pyloriCagA+/P+. Relative mRNA expression levels of
(D) E-cadherin, (E) Vimentin and (F) α-SMA in SGC-7901 cells, as
determined by reverse transcription-quantitative polymerase chain
reaction analysis. *P<0.05 vs. the si-NC group;
#P<0.05 vs. the control group; $P<0.05.
vs. the H. pyloriCagA+/P+ group. α-SMA, α-smooth
muscle actin; CagA, cytotoxin-associated gene A; EMT,
epithelial-mesenchymal transition; H.
pyloriCagA−, CagA-negative H.
pylori; H. pyloriCagA+/P−,
CagA-positive and PBP1A mutation-negative H. pylori; H.
pyloriCagA+/P+, CagA- and PBP1A mutation-positive
H. pylori; FoxM1, Forkhead box M1; H. pylori,
Helicobacter pylori; NC, negative control; si, small
interfering RNA. |
miR-134 exerts its tumor suppressor
function through regulation of FoxM1
Having demonstrated that FoxM1 is a target gene of
miR-134, the present study further investigated whether it was
involved in the miR-134-mediated role in H.
pyloriCagA+/P+-infected GC cells. A FoxM1 plasmid
was transfected into SGC-7901 cells, and the mRNA and protein
expression levels of FoxM1 were significantly increased following
transfection (Fig. 6A and B). As
illustrated in Fig. 6C and D, the
inhibitory effects of miR-134 overexpression were reduced by
co-transfection with FoxM1. Furthermore, western blotting (Fig. 6E) and RT-qPCR analysis (Fig. 6F-H) demonstrated that
co-transfection with miR-134 mimics and FoxM1 markedly
downregulated E-cadherin, and upregulated Vimentin and α-SMA
compared with in the H. pyloriCagA+/P+ + miR-134
mimics group. These data suggested that miR-134 may inhibit H.
pyloriCagA+/P+-induced GC cell proliferation and
invasion, and reverse EMT by regulating FoxM1 in SGC-7901
cells.
Discussion
The present study demonstrated that H.
pyloriCagA+/P+ was associated with poor
clinicopathological characteristics in GC. In addition, H.
pyloriCagA+/P+ infection significantly promoted GC
cell proliferation and invasion, and induced EMT of GC cells. The
present results also demonstrated that H.
pyloriCagA+/P+ may promote GC cell malignant
behavior through regulation of the miR-134/FoxM1 regulatory
network. These data may help to further understand the role of
H. pyloriCagA+/P+ in gastric carcinogenesis.
Although the causal association between H.
pylori infection and GC incidence is well established, the
exact associations between H. pylori and the progression of
GC remain largely unknown (28).
The present study analyzed the association between H. pylori
infection and the clinical characteristics of GC in GC patients
without H. pylori infection, and in GC patients with H.
pyloriCagA−, H.
pyloriCagA+/P− and H.
pyloriCagA+/P+ infection. The results indicated that
H. pyloriCagA− infection was not
significantly associated with patient characteristics. This finding
was similar to the results of previous studies, which suggested
that H. pylori infection has no association with prognosis
in China, and the prognosis of H. pylori-negative GC is
equally poor (29). However, the
present study demonstrated that patients infected with H.
pyloriCagA+/P− possessed a higher proportion of
advanced characteristics than those with H.
pyloriCagA−. This result was also
supported by the findings that patients with H. pyloriCagA+
GC had a worse clinical outcome than those involving H.
pyloriCagA− strains (8). Furthermore, distinct characteristics
between H. pyloriCagA+/P− and H.
pyloriCagA+/P+ GC were observed. Therefore, the
present study focused on the role of PBP1A-mutated H. pylori
strains. In in vitro-selected amoxicillin-resistant H.
pylori strains, the resistance has been suggested to result
from alterations in PBP1A (30,31).
A single serine-to-arginine substitution in PBP1A induces
high-level amoxicillin resistance in H. pylori (32), which may explain the increase in
naturally occurring H. pylori strains with a high level of
amoxicillin resistance in recent years. Currently, amoxicillin
remains one of the first-line antibiotics used for prophylaxis and
treatment of H. pylori; however, the clinical significance
of H. pyloriCagA+/P+ in GC remains largely
unknown. The present study revealed that H.
pyloriCagA+/P+ infection was significantly
associated with various clinicopathological parameters, including
invasion depth, lymphatic metastasis and distant metastasis.
However, due to problems with follow-up, the 5 year overall or
disease-free survival rates among the different H. pylori
strain-infected cohorts were not analyzed. The present study
hypothesized that H. pyloriCagA+/P+ infection may
be associated with a poorer outcome for patients with GC as
compared to those with H.
pyloriCagA+/P− infection.
The present study further investigated the
mechanisms underlying the clinical significance of H.
pyloriCagA+/P+ infection. EMT is a
well-characterized embryological process that serves a critical
role in tumor metastasis. EMT is characterized by the loss of
epithelial markers (e.g., E-cadherin) and the acquisition of
mesenchymal markers (e.g., Vimentin and α-SMA) (33). The present results revealed that
H. pyloriCagA+/P+ induced a significant increase
in cell proliferation and invasion, a decrease in E-cadherin
expression, and an increase in Vimentin and α-SMA expression.
Furthermore, since emerging evidence has suggested that miRNAs
serve an important role in the control of H. pylori
infection-associated responses in GC (34), this study investigated whether
miRNAs were involved in H. pyloriCagA+/P+-induced
EMT in GC. To determine the miRNA expression profiles in H.
pyloriCagA+/P+-associated GC, a microarray chip was
used to measure miRNA expression levels in GC tissues with H.
pyloriCagA+/P+ or H.
pyloriCagA+/P− infection. Notably,
miR-134 was specifically downregulated in H.
pyloriCagA+/P+-infected tissues compared with those
with H. pyloriCagA+/P− infection.
Aberrant levels of miR-134 have been detected in
various malignancies, and may regulate tumor development,
differentiation, proliferation, invasion and metastasis (35,36).
Overexpression of miR-134 inhibits migration, invasion and EMT of
lung cancer cells by targeting integrin β1 mRNA (37). Similarly, Zha et al revealed
that miR-134 specifically targets integrin β1 to suppress the
invasion and metastasis of hepatocellular carcinoma cells in
vitro and in vivo (38). Additionally, forced expression of
miR-134 inhibits the migration and invasion of renal cell carcinoma
cells by blocking EMT (39). These
findings indicated the miR-134 may serve as a tumor suppressor in
human cancer (40); however, the
pathogenetic roles of miR-134 were obscure in GC, particularly in
GC associated with H. pylori infection. To the best of our
knowledge, the current study was the first to reveal that forced
overexpression of miR-134 could significantly reverse H.
pyloriCagA+/P+ infection-induced cell proliferation,
invasion and EMT. Notably, the molecular and modulated mechanisms
of miRNA are complex and variable (41), and a recent study revealed that
miR-134 has diverse target genes in cancer (40). Using bioinformatics analysis and
molecular experiments, the present study demonstrated that the
downregulation of miR-134 in H.
pyloriCagA+/P+-infected GC tissues was associated
with upregulated FoxM1 mRNA and protein expression. In agreement
with the sequence alignment, the luciferase reporter assays
confirmed the direct targeting of the FoxM1 3′-untranslated region
by miR-134, and suggested that a strong affinity may exist between
miR-134 and FoxM1 mRNA in patients with GC and H.
pyloriCagA+/P+ infection.
FoxM1 shares homology with the winged helix
DNA-binding domain and is predominantly expressed in fetal tissue.
It is maintained ubiquitously in all proliferating adult tissues
and some cancer cell lines, whereas its expression is absent from
differentiated cells (42). The
role of FoxM1 in the progression of GC has previously been
investigated. Promotion of gastric tumorigenesis by FoxM1 is
directly and significantly correlated with transactivation of
vascular endothelial growth factor expression and elevation of
angiogenesis (43). Increased
FOXM1 expression is also associated with invasive and metastatic
processes in GC, and is inversely associated with patient prognosis
(44). Mechanistically, FOXM1
promotes GC cell migration and invasion through inducing the
expression of Cath-D (45).
Furthermore, a negative correlation has been identified between
FoxM1 and E-cadherin expression in GC tissue (46). E-cadherin is associated with
epithelial phenotypes, which serve a critical role in the process
of EMT (47). In accordance with
the previous study, the present results indicated that knockdown of
FoxM1 significantly abolished H. pylori-induced EMT of
SGC-7901 cells. The functional studies revealed that the effects of
miR-134 on cell proliferation, invasion and EMT were reversed by
FoxM1 overexpression. Although a previous study revealed that FoxM1
is overexpressed in H. pyloriCagA+-induced gastric
carcinogenesis and is regulated by miR-370 (48), the present study is the first, to
the best of our knowledge, to suggest that FoxM1 was involved in
H. pyloriCagA+/P+-mediated GC cell EMT and was
negatively controlled by miR-134, delineating the mechanisms
governing FoxM1 regulation in GC.
The present study has numerous limitations. The
number of clinical samples used in this study was relatively small
and further studies are required to validate the role of H.
pyloriCagA+/P+ infection in patients with GC.
Additionally, H. pylori infections in many patients are
asymptomatic and only a few people develop clinical disease
(49). However, focusing on the
pathways implicated in H. pylori-induced tumorigenesis may
lead to novel therapeutic strategies for GC prevention. In
addition, with the growing proportion of amoxicillin-resistant
H. pylori, a kit for detection of PBP1A mutations would be
beneficial in clinical practice.
In conclusion, the present study revealed that
H. pyloriCagA+/P+ was associated with poor
clinicopathological characteristics in patients with GC. In
addition, H. pyloriCagA+/P+ induced significant
cell proliferation, invasion and EMT of GC cells. Mechanistically,
H. pyloriCagA+/P+ promoted EMT of GC cells
through suppressing miR-134, which targeted FoxM1 in GC.
Funding
The study was supported by Natural Science Funding
of China (grant nos. 31671869, 31471598, 31571852 and
31601487).
Availability of data and materials
All data generated or analyzed during this study
are included in this published article.
Authors' contributions
DDP designed the study and performed the
statistical analysis; LH and ZYW performed the experiments and data
correction; LH wrote the manuscript.
Ethics approval and consent to
participate
The present study was approved by the ethical board
of the Jiangsu Province Hospital of TCM (ethical no.
JSSZYY2014085). All studies performed involving human participants
were conducted in accordance with the Strengthening the Reporting
of Observational Studies in Epidemiology guidelines, and the 2013
Declaration of Helsinki. The patients, or their parents, provided
written informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
H. pylori
|
Helicobacter pylori
|
|
GC
|
gastric cancer
|
|
CagA
|
cytotoxin-associated gene A
|
|
PBP1A
|
penicillin-binding protein 1A
|
|
FoxM1
|
Forkhead box M1
|
|
miRNAs/miRs
|
microRNAs
|
|
EMT
|
epithelial-mesenchymal transition
|
Acknowledgments
Not applicable.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
2
|
Choi IJ, Kook MC, Kim YI, Cho SJ, Lee JY,
Kim CG, Park B and Nam BH: Helicobacter pylori Therapy for the
Prevention of Metachronous Gastric Cancer. N Engl J Med.
378:1085–1095. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Greenberg ER, Anderson GL, Morgan DR,
Torres J, Chey WD, Bravo LE, Dominguez RL, Ferreccio C, Herrero R,
Lazcano-Ponce EC, et al: 14-day triple, 5-day concomitant, and
10-day sequential therapies for Helicobacter pylori infection in
seven Latin American sites: A randomised trial. Lancet.
378:507–514. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee YC, Chiang TH, Chou CK, Tu YK, Liao
WC, Wu MS and Graham DY: Association Between Helicobacter pylori
Eradication and Gastric Cancer Incidence: A Systematic Review and
Meta-analysis. Gastroenterology. 150:1113–1124, e5. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang F, Sun GP, Zou YF, Zhong F, Ma T, Li
XQ and Wu D: Helicobacter pylori infection predicts favorable
outcome in patients with gastric cancer. Curr Oncol. 20:e388–e395.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang F, Sun G, Zou Y, Zhong F, Ma T and Li
X: Protective role of Helicobacter pylori infection in prognosis of
gastric cancer: Evidence from 2,454 patients with gastric cancer.
PLoS One. 8:e624402013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zheng F, Sun Z, Kan J, Yin J, Du F, Shen
G, Wang Z, Ren D, Bao X and Zhao J: Is It a Protective Factor of
Helicobacter pylori Infection in Overall Survival of All Gastric
Cancer? Evidence from Meta-Analysis. J Environ Pathol Toxicol
Oncol. 36:309–320. 2017. View Article : Google Scholar
|
|
8
|
Varga MG, Wang T, Cai H, Xiang YB, Gao YT,
Ji BT, Pawlita M, Waterboer T, Zheng W, Shu XO, et al: Helicobacter
pylori Blood Biomarkers and Gastric Cancer Survival in China.
Cancer Epidemiol Biomarkers Prev. 27:342–344. 2018. View Article : Google Scholar
|
|
9
|
Malfertheiner P, Megraud F, O'Morain CA,
Atherton J, Axon AT, Bazzoli F, Gensini GF, Gisbert JP, Graham DY,
Rokkas T, et al: European Helicobacter Study Group: Management of
Helicobacter pylori infection - the Maastricht IV/Florence
Consensus Report. Gut. 61:646–664. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Morgan DR, Torres J, Sexton R, Herrero R,
Salazar-Martínez E, Greenberg ER, Bravo LE, Dominguez RL, Ferreccio
C, Lazcano-Ponce EC, et al: Risk of recurrent Helicobacter pylori
infection 1 year after initial eradication therapy in 7 Latin
American communities. JAMA. 309:578–586. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nagai K, Davies TA, Jacobs MR and
Appelbaum PC: Effects of amino acid alterations in
penicillin-binding proteins (PBPs) 1a, 2b, and 2× on PBP affinities
of penicillin, ampicillin, amoxicillin, cefditoren, cefuroxime,
cefprozil, and cefaclor in 18 clinical isolates of
penicillin-susceptible, -intermediate, and -resistant pneumococci.
Antimicrob Agents Chemother. 46:1273–1280. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Polk DB and Peek RM Jr: Helicobacter
pylori: Gastric cancer and beyond. Nat Rev Cancer. 10:403–414.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Park JY, Forman D, Waskito LA, Yamaoka Y
and Crabtree JE: Epidemiology of Helicobacter pylori and
CagA-Positive Infections and Global Variations in Gastric Cancer.
Toxins (Basel). 10. pp. 102018, View Article : Google Scholar
|
|
14
|
Yong X, Tang B, Li BS, Xie R, Hu CJ, Luo
G, Qin Y, Dong H and Yang SM: Helicobacter pylori virulence factor
CagA promotes tumorigenesis of gastric cancer via multiple
signaling pathways. Cell Commun Signal. 13:302015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sokolova O, Borgmann M, Rieke C,
Schweitzer K, Rothkötter HJ and Naumann M: Helicobacter pylori
induces type 4 secretion system-dependent, but CagA-independent
activation of IκBs and NF-κB/RelA at early time points. Int J Med
Microbiol. 303:548–552. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yoon JH, Seo HS, Choi SS, Chae HS, Choi
WS, Kim O, Ashktorab H, Smoot DT, Nam SW, Lee JY, et al: Gastrokine
1 inhibits the carcinogenic potentials of Helicobacter pylori CagA.
Carcinogenesis. 35:2619–2629. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cadamuro AC, Rossi AF, Maniezzo NM and
Silva AE: Helicobacter pylori infection: Host immune response,
implications on gene expression and microRNAs. World J
Gastroenterol. 20:1424–1437. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Verma R and Sharma PC: Next generation
sequencing-based emerging trends in molecular biology of gastric
cancer. Am J Cancer Res. 8:207–225. 2018.PubMed/NCBI
|
|
19
|
Ueda T, Volinia S, Okumura H, Shimizu M,
Taccioli C, Rossi S, Alder H, Liu CG, Oue N, Yasui W, et al:
Relation between microRNA expression and progression and prognosis
of gastric cancer: A microRNA expression analysis. Lancet Oncol.
11:136–146. 2010. View Article : Google Scholar
|
|
20
|
Siu LK, Leung WK, Cheng AF, Sung JY, Ling
TK, Ling JM, Ng EK, Lau JY and Chung SC: Evaluation of a selective
transport medium for gastric biopsy specimens to be cultured for
Helicobacter pylor i. J Clin Microbiol. 36:3048–3050.
1998.PubMed/NCBI
|
|
21
|
Atrisco-Morales J, Martínez-Santos VI,
Román-Román A, Alarcón-Millán J, De Sampedro-Reyes J, Cruz-Del
Carmen I, Martínez-Carrillo DN and Fernández-Tilapa G: vacA s1m1
genotype and cagA EPIYA-ABC pattern are predominant among
Helicobacter pylori strains isolated from Mexican patients with
chronic gastritis. J Med Microbiol. 67:314–324. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kwon YH, Kim JY, Kim N, Park JH, Nam RH,
Lee SM, Kim JW, Kim JM, Park JY and Lee DH: Specific mutations of
penicillin-binding protein 1A in 77 clinically acquired
amoxicillin-resistant Helicobacter pylori strains in comparison
with 77 amoxicillin-susceptible strains. Helicobacter. 22:222017.
View Article : Google Scholar
|
|
23
|
Bisignano C, Filocamo A, La Camera E,
Zummo S, Fera MT and Mandalari G: Antibacterial activities of
almond skins on cagA-positive and-negative clinical isolates of
Helicobacter pylor i. BMC Microbiol. 13:1032013. View Article : Google Scholar
|
|
24
|
Qureshi NN, Gallaher B and Schiller NL:
Evolution of amoxicillin resistance of Helicobacter pylori in
vitro: Characterization of resistance mechanisms. Microb Drug
Resist. 20:509–516. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Al-Maleki AR, Loke MF, Lui SY, Ramli NSK,
Khosravi Y, Ng CG, Venkatraman G, Goh KL, Ho B and Vadivelu J:
Helicobacter pylori outer inflammatory protein A (OipA) suppresses
apoptosis of AGS gastric cells in vitro. Cell Microbiol. 19:192017.
View Article : Google Scholar
|
|
26
|
Yang J, Song H, Cao K, Song J and Zhou J:
Comprehensive analysis of Helicobacter pylori infection-associated
diseases based on miRNA-mRNA interaction network. Brief Bioinform.
Mar 20–2018.Epub ahead of print.
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
28
|
Yang Y, Li X, Du J, Yin Y and Li Y:
Involvement of microRNAs-MMPs-E-cadherin in the migration and
invasion of gastric cancer cells infected with Helicobacter pylor
i. Exp Cell Res. 367:196–204. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tsai KF, Liou JM, Chen MJ, Chen CC, Kuo
SH, Lai IR, Yeh KH, Lin MT, Wang HP, Cheng AL, et al: Taiwan
Gastrointestinal Disease and Helicobacter Consortium: Distinct
Clinicopathological Features and Prognosis of Helicobacter pylori
Negative Gastric Cancer. PLoS One. 12:e01709422017. View Article : Google Scholar
|
|
30
|
DeLoney CR and Schiller NL:
Characterization of an In vitro-selected amoxicillin-resistant
strain of Helicobacter pylor i. Antimicrob Agents Chemother.
44:3368–3373. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Paul R, Postius S, Melchers K and Schäfer
KP: Mutations of the Helicobacter pylori genes rdxA and pbp1 cause
resistance against metronidazole and amoxicillin. Antimicrob Agents
Chemother. 45:962–965. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gerrits MM, Schuijffel D, van Zwet AA,
Kuipers EJ, Vandenbroucke-Grauls CM and Kusters JG: Alterations in
penicillin-binding protein 1A confer resistance to beta-lactam
antibiotics in Helicobacter pylor i. Antimicrob Agents Chemother.
46:2229–2233. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liang L, Zeng M, Pan H, Liu H and He Y:
Nicotinamide N-methyltransferase promotes epithelial-mesenchymal
transition in gastric cancer cells by activating transforming
growth factor-β1 expression. Oncol Lett. 15:4592–4598.
2018.PubMed/NCBI
|
|
34
|
Polakovicova I, Jerez S, Wichmann IA,
Sandoval-Bórquez A, Carrasco-Véliz N and Corvalán AH: Role of
microRNAs and Exosomes in Helicobacter pylori and Epstein-Barr
Virus Associated Gastric Cancers. Front Microbiol. 9:6362018.
View Article : Google Scholar :
|
|
35
|
Li J, Wang Y, Luo J, Fu Z, Ying J, Yu Y
and Yu W: miR-134 inhibits epithelial to mesenchymal transition by
targeting FOXM1 in non-small cell lung cancer cells. FEBS Lett.
586:3761–3765. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu CJ, Shen WG, Peng SY, Cheng HW, Kao
SY, Lin SC and Chang KW: miR-134 induces oncogenicity and
metastasis in head and neck carcinoma through targeting WWOX gene.
Int J Cancer. 134:811–821. 2014. View Article : Google Scholar
|
|
37
|
Qin Q, Wei F, Zhang J and Li B: miR-134
suppresses the migration and invasion of non small cell lung cancer
by targeting ITGB1. Oncol Rep. 37:823–830. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zha R, Guo W, Zhang Z, Qiu Z, Wang Q, Ding
J, Huang S, Chen T, Gu J, Yao M, et al: Genome-wide screening
identified that miR-134 acts as a metastasis suppressor by
targeting integrin β1 in hepatocellular carcinoma. PLoS One.
9:e876652014. View Article : Google Scholar
|
|
39
|
Liu Y, Zhang M, Qian J, Bao M, Meng X,
Zhang S, Zhang L, Zhao R, Li S, Cao Q, et al: miR-134 functions as
a tumor suppressor in cell proliferation and
epithelial-to-mesenchymal Transition by targeting KRAS in renal
cell carcinoma cells. DNA Cell Biol. 34:429–436. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pan JY, Zhang F, Sun CC, Li SJ, Li G, Gong
FY, Bo T, He J, Hua RX and Hu WD: miR-134: A Human Cancer
Suppressor? Mol Ther Nucleic Acids. 6:140–149. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Korver W, Roose J and Clevers H: The
winged-helix transcription factor Trident is expressed in cycling
cells. Nucleic Acids Res. 25:1715–1719. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li Q, Zhang N, Jia Z, Le X, Dai B, Wei D,
Huang S, Tan D and Xie K: Critical role and regulation of
transcription factor FoxM1 in human gastric cancer angiogenesis and
progression. Cancer Res. 69:3501–3509. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ma J, Qi G, Xu J, Ni H, Xu W, Ru G, Zhao
Z, Xu W and He X: Overexpression of forkhead box M1 and
urokinase-type plasminogen activator in gastric cancer is
associated with cancer progression and poor prognosis. Oncol Lett.
14:7288–7296. 2017.
|
|
45
|
Yang L, Cui M, Zhang L and Song L: FOXM1
facilitates gastric cancer cell migration and invasion by inducing
Cathepsin D. Oncotarget. 8:68180–68190. 2017.PubMed/NCBI
|
|
46
|
Zhang J, Chen XY, Huang KJ, Wu WD, Jiang
T, Cao J, Zhou LS, Qiu ZJ and Huang C: Expression of FoxM1 and the
EMT-associated protein E-cadherin in gastric cancer and its
clinical significance. Oncol Lett. 12:2445–2450. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yu H, Shen Y, Hong J, Xia Q, Zhou F and
Liu X: The contribution of TGF-β in Epithelial-Mesenchymal
Transition (EMT): Down-regulation of E-cadherin via snail.
Neoplasma. 62:1–15. 2015. View Article : Google Scholar
|
|
48
|
Feng Y, Wang L, Zeng J, Shen L, Liang X,
Yu H, Liu S, Liu Z, Sun Y, Li W, et al: FoxM1 is overexpressed in
Helicobacter pylori-induced gastric carcinogenesis and is
negatively regulated by miR-370. Mol Cancer Res. 11:834–844. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gunawardhana N, Jang S, Choi YH, Hong YA,
Jeon YE, Kim A, Su H, Kim JH, Yoo YJ, Merrell DS, et al:
Helicobacter pylori-Induced HB-EGF Upregulates Gastrin Expression
via the EGF Receptor, C-Raf, Mek1, and Erk2 in the MAPK Pathway.
Front Cell Infect Microbiol. 7:5412018. View Article : Google Scholar :
|