Introduction
Chronic myelogenous leukemia (CML) is caused by the
constitutively active BCR/ABL kinase, which derives from chromosome
translocation t(9;22) (q34;q11.2) in hematopoietic stem cells
(1). The majority of patients with
CML in the chronic phase (CP) have no symptoms and without
treatment the disease develops into fatal blast crisis (BC)
(2). Starting with imatinib, the
development of tyrosine kinase inhibitors (TKIs) has markedly
improved treatment of patients with CML; however, some patients
progress to BC even with TKIs treatment (2). Survival following transition to BC is
~0.5-1 year (2) and novel
treatments for these patients are urgently required.
In general, in the transformation to malignant
cells, the activation of kinase signaling and inactivation of
phosphatases is required (3).
Inhibition of protein phosphatase 2A (PP2A) has been reported
essential in CML progression (4).
The PP2A complex consists of subunits A (structural subunit), B
(regulatory subunit), and C (catalytic subunit) (5). PP2A suppresses cell survival and
proliferation by dephosphorylating regulatory molecules, such as
protein kinase B (Akt) and extracellular signal-regulated kinase
(4,5). In CML, BCR/ABL presence increases the
expression of SET nuclear proto-oncoprotein (SET), an inhibitor of
PP2A, causing PP2A suppression and promoting cell proliferation
(4). Activation of PP2A causes
dephosphorylation of BCR/ABL by binding and activating tyrosine
phosphatase Src homology region 2 domain-containing phosphatase-1
and suppressing leukemogenesis (4).
Cytotoxic T cells recognize peptides bound to human
leukocyte antigen (HLA) class I molecules on the malignant cells
(6). These peptides are generally
derived from intrinsic proteins, such as tyrosinase and
melanoma-associated antigen recognized by T cells, through
degradation in proteasomes (6). In
the current study, the HLA-A*24:02 gene was introduced into
K562, a cell line established from CML-BC lacking HLA expression.
It was determined by mass spectrometry that peptides from
T-lymphokine-activated killer cell-originated protein
kinase/PDZ-binding-kinase (TOPK/PBK) were interacting with the
introduced HLA. TOPK is a serine/threonine protein kinase belonging
to the mitogen-activated protein kinase kinase family (7). It consists of 322 amino acids and is
known as cancer/testis antigen (8). TOPK is scarcely expressed in human
normal tissues except for the testis and high TOPK expression is
reported in various cancers, including lung, oral, esophageal and
gastric cancers, and in hematopoietic tumor cell lines,
particularly with high-grade malignancies (9-13).
TOPK is phosphorylated by cyclin-dependent kinase 1 (CDK1)/cyclin
B1 during the mitotic phase of the cell cycle (7). Phosphorylated TOPK activates certain
proteins associated with cytokinesis of tumor cells, including
histone H3, phosphatase and tensin homolog and p97 (14-16).
The current study focused on the association between CML
oncogenesis and TOPK. Findings of the current study suggest that
TOPK was induced by BCR/ABL and regulated by PP2A. TOPK may be a
therapeutic target for patients with BCR/ABL leukemia.
Materials and methods
Plasmids
HLA*A24 in pcDNA3.1 was obtained from the
RIKEN BioResource Research Center (Ibaraki, Japan). The coding
region for HLA*A24 was inserted into the BamHI and
EcoRI sites of retroviral vector pQCXIN (Clontech
Laboratories, Inc., Mountainview, CA, USA) yielding
HLA-A*24-pQCXIN. The HLA-A*24-FLAG sequence was generated from
HLA-A*24-pQCXIN using the pQC (forward, 5′-ACGCCA
TCCACGCTGTTTTGACCT-3′) and FLAG (reverse, 5′-AAG
AATTCTACTTATCGTCGTCATCCTTGTAATCCACTTTA CAAGCTGT-3′) primers. All
polymerase chain reaction (PCR) procedures were performed using LA
TaqDNA polymerase (Takara Biotechnology Co., Ltd., Dalian, China)
at 94°C for 5 min, followed by 30 cycles with 94°C for 30 sec, 30
sec at 55°C and 72°C for 1 min and a final extension at 72°C for 10
min. PCR products were cloned into pGEM-T easy vector (Promega
Corporation, Madison, WI, USA) by insertion into the BamHI
and EcoRI cloning sites of pQCXIN
(HLA-A*24-FLAG-pQCXIN).
Total RNA was isolated from cultured K562 using
TRIzol reagent (Thermo Fisher Scientific, Inc., Waltham, MA, USA).
cDNA was synthesized using SuperScript II Reverse Transcriptase
(Thermo Fisher Scientific, Inc.) at 42°C for 50 min. The TOPK
sequence from K562 cDNA was amplified using TOPK v2 F (forward,
5′-GGACTGAGAGTGGCTTTCAC-3′) and TOPK v2 R (reverse,
5′-CAAGCCACACTTCAGCTGAG-3′) primers as described above. PCR
products were subcloned into pGEM-T easy and the fragment was
subsequently inserted into the EcoRI site of pcDNA3 (Thermo
Fisher Scientific, Inc.) yielding TOPK-pcDNA3. Generation of the
TOPK-FLAG sequence was achieved using TOPK-pcDNA3 with TOPK v2 F
and FLAG primers. PCR products were subcloned into the EcoRI
site of pcDNA3 or pQCXIN generating TOPK-FLAG pcDNA3 and TOPK-FLAG
pQCXIN, respectively.
V245 pCEP4-HA B56α (#14532), V246 pCEP4-HA B56β
(#14533), V247 pCEP4-HA B56γ1 (#14534), V248 pCEP4-HA B56γ3
(#14535), V249 pCEP4-HA B56δ (#14536) and V250 pCEP4-HA B56ε
(#14536; Addgene, Inc., Cambridge, MA, USA), plasmids containing
various PP2A subunits were gifts from Dr David M. Virshup (Duke-NUS
Medical School, Singapore, Republic of Singapore) (17).
Patients with CML
A total of 16 patients diagnosed with CML according
to clinical criteria and laboratory features were recruited between
November 2002 and December 2015 at the Tokyo Medical and Dental
University Hospital (Tokyo, Japan) (18). Patients were >20 years, with a
median age of 63 years (range, 22-89 years) and comprised 11 males
and 5 females. Only CML patients with BCR/ABL were included.
Peripheral blood mononuclear cells (PBMCs) or bone marrow
mononuclear cells (BMMCs) of patients were obtained at the time of
diagnosis of CML as CP (2 PBMCs and 9 BMMCs) or BC (5 PBMCs and 3
BMMCs) according to European LeukemiaNet criteria (18). Three additional samples were
obtained during TKIs therapy, two for patients with CML-CP treated
with imatinib and dasatinib and one for a patient with CML-BC
treated with nilotinib. These patients were resistant to TKI
treatment and samples were obtained prior to switching TKIs. The
clinical course of the patient with CML-BC treated with nilotinib
is further described. All other samples were obtained from patients
not receiving TKI treatment at time of collection. PBMCs form 4
healthy volunteers (2 males and 2 females; age, 21-51 years) and
BMMCs from 3 patients with stage I-IIA malignant lymphoma without
BM involvement (2 males and 1 female; age, 42-81 years) were used
as control. PBMCs and BMMCs were stored as detailed below.
Cells and reagents
PBMCs and BMMCs of patients or healthy donors were
isolated from fresh samples (peripheral blood, 10-20 ml or bone
marrow fluid, 2-5 ml) by density gradient centrifugation (760 × g;
15 min; 20°C) using Separate-L (Muto Pure Chemicals Co., Ltd.,
Tokyo, Japan). Mononuclear cells were preserved using the
Cellbanker-1 (Nippon Zenyaku Kogyo Co., Tokyo, Japan) at -80°C.
MD901 lymphoma cells (19), and TMD2 (20) and TMD5 (21) leukemic cells were established and
provided by Tokyo Medical and Dental University (Tokyo, Japan).
TL-Oml (22) and ILT-M1 (23) T cell leukemic cells were
established and provided by Dr Mari Kannagi (Tokyo Medical and
Dental University). TonB210, a clone of murine interleukin (mIL)
3-dependent BaF3 pro-B cells transfected with a BCR/ABL cDNA under
the control of a tetracycline-inducible promoter, was provided by
Dr George Q. Daley (24). 32D,
32D/p210 myeloblasts (32D transfected with BCR/ABL) and
TonB210/T315I pro-B cells (inducible transfectant of Ba/F3 with
BCR/ABL T315I mutant) were established in our laboratory as
previously described (25,26). PLAT-A, an amphotropic virus
packaging embryonic kidney cell line (27) was kindly provided by Dr Toshio
Kitamura (The Institute of Medical Sciences, Tokyo University,
Tokyo, Japan). K562 leukemic cells were obtained from RIKEN
BioResource Center (Tsukuba, Japan). RPMI-8226 myeloma cells, EW36
and Raji B cell lymphoma cells, MOLM1 and MV4-11 leukemic cells,
and 293T cells were purchased from the American Type Culture
Collection (Manassas, VA, USA). Cells were cultured as follows:
RPMI-1640 (Wako Pure Chemical Industries, Ltd., Osaka, Japan) for
K562, MD901, Raji, TMD2, RPMI-8226, EW36 and MOLM1; Iscove’s
modified Dulbecco’s medium (IMDM; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) for MV4-11; α modified Eagle’s minimum
essential medium (α-MEM; Sigma-Aldrich; Merck KGaA) for TMD5;
RPMI-1640 with 3 U/ml recombinant mIL-3 (PeproTech, Inc., Rocky
Hill, NJ, USA) for 32D, 32D/p210, TonB210 and TonB210/T315I;
Dulbecco’s modified Eagle’s medium (DMEM) for 293T and PLAT-A; and
RPMI-1640 with 30 U/ml of IL-2 (PeproTech, Inc.) for TL-Oml and
ILT-M1. Each medium contained 10% fetal calf serum (FCS; Nichirei
Biosciences Inc., Tokyo, Japan). All cells were grown and treated
in a humidified atmosphere at 37°C with 5% CO2.
TOPK inhibitor OTS514 (purity, 99.97%) was provided
by OncoTherapy Science, Inc. (Kanagawa, Japan). Okadaic acid was
purchased from Merck KGaA (#459620). FTY720 was purchased from
Cayman Chemical Co., Ltd. (#10006292; Ann Arbor, MI, USA).
Doxycycline (DOX) was purchased from Sigma-Aldrich (#D9891; Merck
KGaA). Nocodazole was purchased from Merck KGaA (#487928). Imatinib
was provided by Novartis Pharma K.K. (Tokyo, Japan).
K562 cells were treated with 300 ng/ml nocodazole
for 0, 6, 12, 21 and 24 h to check the time-dependent effects of
nocodazole. K562 cells were further treated with nocodazole for 21
h followed by treatment with 20 nM okadaic acid or 0, 5, 10 and 15
µM FTY720 for 4 h at 37°C prior to harvesting. K562 cells
expressing FLAG-tagged TOPK (K562 TOPK-F pQCXIN) were treated with
or without 300 ng/ml nocodazole for 21 h to check the interaction
between TOPK and PP2A. Stock solutions were prepared using dimethyl
sulfoxide and solutions were diluted ≥1/1000 for treatment.
TonB210 cells were treated with 1 µg/ml of
DOX for 5 h at 37°C to induce BCR/ABL. Cells were then incubated
with imatinib (10 µM) for 24 h at 37°C. To check effects of
OTS514 on survival, TonB210 and TonB210/T315I were cultured in the
presence of 1 µg/ml of DOX in the absence of IL-3 for 48 h
prior to treatment with 20 nM OTS514 for 48 h at 37°C.
RT-quantitative (q) PCR
PBMCs or BMMCs from patients with CML and PBMCs from
healthy donors were used in these experiments. Total RNA was
extracted using from these samples using TRIzol reagent (Gibco;
Thermo Fisher Scientific, Inc.).
RT was performed using oligo(dT) primers and
SuperScript™ II reverse transcriptase (Thermo Fisher Scientific,
Inc.) to synthesize cDNA at 42°C for 50 min. cDNA amplification was
performed using the following primers (Sigma-Aldrich; Merck KGaA):
PBK/TOPK v2, forward, 5′-GGACTGAGA GTGGCTTTCAC-3′ and
reverse, 5′-CAAGCCACACTTCA GCTGAG-3′; and GAPDH, forward,
5′-CTGACTTCAACAGC GACACC-3′ and reverse,
5′-TCCTCTTGTGCTCTTGCTGG-3′. qPCR was performed using LightCycler
480 Probes Master kit and LightCycler 480 system software version
1.5.1 (Roche Diagnostics, Indianapolis, IN, USA) using TaqMan Gene
Expression assays (#4331182; PBK/TOPK, Hs00902990_m1 and GAPDH,
Hs0286624_g1; Thermo Fisher Scientific, Inc.). Relative fold
changes of gene expression were assessed using the
2−ΔΔCq method (28).
Transfection and infection
For transient expression, 293T cells
(3×105) were transfected with indicated plasmids using
Lipofectamine 3000 (Thermo Fisher Scientific, Inc.) according to
the manufacturer’s instructions. A total of 2 µg DNA was
used for the transfections, comprising 1 µg V245 pCEP4-HA
B56α, V246 pCEP4-HA B56β, V247 pCEP4-HA B56γ1, V248 pCEP4-HA B56γ3,
V249 pCEP4-HA B56δ or V250 pCEP4-HA B56ε and 1 µg TOPK-FLAG
pcDNA3. Additionally, a TOPK-FLAG (1 µg) plus pcDNA3 (1
µg) sample was established and a control using 2 µg
pcDNA3. Transfection efficiency was determined by western blot
analysis. As endogenous TOPK expression in 293T was very low
(16), no exogenous TOPK
expression was observed for the control sample at the applied
exposure time.
To obtain K562 cells stably expressing HLA-A*24,
PLAT-A cells (1×106) were transfected with 2 µg
of HLA-A*24-FLAG-pQCXIN using Lipofectamine PLUS reagent (Thermo
Fisher Scientific, Inc.). At 72 h of transfection, 2 ml of the
culture supernatant containing recombinant retrovirus was
harvested. K562 cells (1.2×106) were infected with the
obtained recombinant retrovirus Following 48 h of infection, K562
cells were cultured in the presence of G418 (1 mg/ml; Wako Pure
Chemical Industries, Ltd.) for one week and high expressing cells
were selected by fluorescence-activated cell sorting (FACS) using
flow cytometer (FACSAria II; Becton-Dickinson and Co., Franklin
Lakes, NJ, USA). Cells (3×105) were incubated with
fluorescein isothiocyanate (FITC)-conjugated anti-HLA-ABC antibody
(W6/32; 100 ng; #11-9983-41; Thermo Fisher Scientific, Inc.) at 4°C
for 30 min. Data obtained from flow cytometry were analyzed using
FlowJo 6.3.3 (FlowJo LLC, Ashland, OR, USA). Two independent K562
clones stably expressing HLA-A*24 were selected for the
immunoprecipitation experiment to identify the peptides binding to
HLA.
K562 cells expressing FLAG-tagged TOPK (K562 TOPK-F
pQCXIN) were obtained by the same procedure. Following one week of
selection with G418, cells of interest were isolated by limiting
dilution cloning. The clone that exhibited the highest exogenous
expression level by western blot analysis was selected for the
further analysis.
Clonogenic assay
CD34-positive cells were isolated from BMMCs
obtained from 3 patients with CML and 3 patients with stage I-IIA
malignant lymphoma without BM involvement as control using a
Dynabeads CD34 Positive Isolation kit (Thermo Fisher Scientific,
Inc.). BMMCs (4×107) were incubated with 100 µl
of washed Dynabeads CD34 at 4°C for 30 min with gentle rotation.
Following isolation with a magnet, CD34-positive cells were
released using 100 µl DETACHaBEAD solution at room
temperature for 45 min. CD34-positive cells (1×103) were
cultured in MethoCult medium (Stemcell Technologies, Inc.,
Vancouver, BC, Canada) for 14 days at 37°C in the presence or
absence of OTS514 (20 nM). Each treatment was assessed in
triplicate and colony numbers were counted by inverted microscopy
(magnification, ×100). The number of visible colonies was
determined as the ratio of OTS514-treated to untreated samples.
Western blot analysis
Cells were lysed in lysis buffer containing 50 mM
Tris-HCl (pH 7.5), 1% Triton X-100, 150 mM NaCl, 50 mM NaF, 5 mM
ethylenediaminetetraacetate, 40 mM β-glycerophosphate, 1 mM sodium
orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 10 mg/ml
aprotinin and 10 mg/ml leupeptin (Sigma-Aldrich; Merck KGaA) and
incubated on ice for 30 min. Lysates were centrifuged (17,400 × g;
4°C; 15 min), proteins were obtained in the supernatant and
quantified using the DC protein assay kit (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). Proteins (20 µg) were boiled with
an equal amount of 2X SDS buffer (0.1 M Tris-HCl pH 6.8, 4% SDS,
12% 2-mercaptoethanol, 20% glycerol, 1% bromophenol blue) at 100°C
for 5 min, separated on 10% SDS-PAGE gels and transferred to
polyvinylidene difluoride membranes. Membranes were blocked with
blocking buffer (5% non-fat milk or 5% BSA in 20 mM Tris-HCl pH
7.5, 150 mM NaCl, 0.1% Tween-20) and probed with primary antibodies
overnight at 4°C followed by secondary antibodies at room
temperature for 1 h. The following antibodies were used:
Horseradish peroxidase (HRP)-conjugated sheep anti-mouse IgG
(1:5,000; cat. no. NA9310), HRP-conjugated donkey anti-rabbit IgG
(1:5,000; cat. no. NA934; GE Healthcare, Chicago, IL, USA),
HRP-conjugated mouse anti-goat IgG (1:5,000; cat. no. sc-2354;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA), rabbit
anti-PBK/TOPK (1:5,000; cat. no. #4942), rabbit anti-phosphorylated
(p)-PBK/TOPK (Thr9; 1:5,000; cat. no. #4941), rabbit anti-HA-tag
(C29F4; 1:5,000; cat. no. #3724), rabbit anti-p-c-ABL (Tyr245;
1:5,000; cat. no. #2861) (all from Cell Signaling Technology, Inc.,
Danvers, MA, USA), mouse anti-PP2A-Aα (C-20; 1:1,000; cat. no.
sc-6112), rabbit anti-c-ABL (K-12; 1:1,000; cat. no. sc-131) (both
from Santa Cruz Biotechnology, Inc.) and mouse anti-β-actin
(1:10,000; cat. no. A1978; Sigma-Aldrich; Merck KGaA). Blots were
visualized using Pierce electrochemiluminescence western blotting
substrate (Pierce; Thermo Fisher Scientific, Inc.). Densitometric
analyses were performed using ImageJ 1.48 (National Institutes of
Health, Bethesda, MD, USA).
Immunoprecipitation
K562 cells expressing FLAG-tagged TOPK (K562 TOPK-F
pQCXIN) or 293T cells transiently transfected with TOPK-FLAG and
PP2A subunits-HA were lysed in lysis buffer as described above and
obtained supernatants were cleared with Protein A/G PLUS-Agarose
Immunoprecipitation reagent (Santa Cruz Biotechnology, Inc.) at 4°C
for 1 h. The obtained supernatant was treated with 30 µl
anti-FLAG M2 Affinity gel (#A2220; Sigma-Aldrich; Merck KGaA) at
4°C overnight. An equal amount of supernatant was incubated with 10
µg mouse IgG (#0107-01; SouthernBiotech, Birmingham, AL,
USA) on ice for 1 h and subsequently treated with 30 µl
Protein A/G PLUS-Agarose Immunoprecipitation reagent at 4°C
overnight as negative controls. Proteins were then separated by
SDS-PAGE as described and subjected to western bot analysis. Total
cell lysates (TCL) were analyzed prior to immunoprecipitation.
Cell-surface FLAG-tagged HLA molecules from K562
cells stably expressing HLA-A*24 (1×108 cells; two
independent clones) were solubilized in buffered detergent solution
(150 mM NaCl, 20 mM Tris-HCl pH 8.0, 1% CHAPS) at 4°C for 1 h
(29,30), reacted with anti-FLAG M2 antibody
(2 µl/ml; #F3165; Sigma-Aldrich, Merck KGaA) at 4°C for 1 h
and then incubated with Protein A/G PLUS-Agarose
Immunoprecipitation reagent according to the manufacturer’s
protocols. Bound HLA molecules were dissociated from the beads
through the addition of FLAG peptides (150 µg; DYKDDDDK
peptide; #044-30953; Wako Pure Chemical Industries, Ltd.). Samples
were further immunoprecipitated using anti-pan HLA-class I antibody
(2 µl; #14-9983-80; Thermo Fisher Scientific, Inc.) at 4°C
for 1 h and treated with Protein A/G PLUS-Agarose
Immunoprecipitation reagent as described above.
Measurement of living cells
Cell viability was determined using flow cytometer.
Following treatment with or without 20 nM OTS514 for 48 h in K562,
MOLM1, TMD5, MV4-11, TonB210, TonB210/T315I and K562 cells treated
with or without 10 nM OTS514 and/or 0.5 µM imatinib for 48 h
were stained with 3,3′-dehexyloxacarbocyamine iodine (40 nM;
DiOC6), which is transported into the mitochondria of living cells,
and propidium iodide (10 µg/ml; PI; both Molecular Probes;
Thermo Fisher Scientific, Inc.), which permeates into dead cells
through damaged cell membranes at 37°C for 15 min. In living cells,
DiOC6 is positive and PI is negative. The total number of cells was
counted using a flow cytometer. Data were analyzed using FlowJo
6.3.3.
Mass spectrometry
Peptides bound to HLA molecules were eluted in 0.2 M
acetic acid solution for 10 min at 4°C (30-32)
and dried in a vacuum evaporator (Centrifugal Concentration CC-101;
Tomy Seiko Co., Ltd., Tokyo, Japan) for 3 h at room temperature.
Subsequently, peptide samples were dissolved in 0.5%
trifluoroacetic acid (TFA) in 5% aqueous acetonitrile (ACN),
purified and concentrated using Pierce C18 Spin Columns (#89870;
Thermo Fisher Scientific, Inc.) according to the manufacturer’s
protocol. The final peptide elution was performed using 50%
ACN.
Aqueous peptide samples (2 µl) were separated
by nano-flow liquid chromatography (nLC) using the Easy-nLC II
(Thermo Fisher Scientific, Inc.) at room temperature. Separation
was performed using a NTCC-360 column (75 µm × 150 mm; 3
µm; Nikkyo Technology Co., Ltd., Hong Kong, China) with 0.1%
TFA in water as solvent A and 0.1% TFA in 70% ACN as solvent B at a
flow rate of 300 nl/min. Peptide samples that eluted from the
column were fragmented for amino acid sequencing using
collision-induced dissociation (CID) in tandem mass spectrometer
(LTQ Orbitrap velos; Thermo Fisher Scientific, Inc.) equipped with
a nano electrospray ion source (positive ion mode). Acquired CID
spectra were analyzed against a protein database (Swiss-Prot;
https://www.uniprot.org/downloads) using
proteomics software PEAKS Studio 6.0 (Bioinformatics Solutions,
Inc., Waterloo, ON, Canada). The PEAKS peptide score (-10lgP) was
calculated for every peptide-spectrum match. The score was derived
from the P-value that indicates the statistical significance of the
peptide-spectrum match (33).
Statistical analysis
For statistical analysis, Student’s t-tests were
performed in pairwise comparisons using EZR 3.0.2 (Saitama Medical
Center, Saitama, Japan), a graphical user interface for R (The R
Foundation for Statistical Computing, Vienna, Austria) and
Kruskal-Wallis followed by Dunn’s multiple comparisons test or
one-way analysis followed by Turkey’s post-hoc test were performed
for multiple group comparisons using GraphPad Prism 6 (GraphPad
Software, Inc., La Jolla, CA, USA). Data are presented as the mean
± standard error representative of three repeats. P<0.05 was
considered to indicate a statistically significant difference.
Results
Peptides derived from TOPK are binding to
HLA-A*24 in HLA-A*24-overexpressing K562 cells
To reveal novel molecular targets for hematopoietic
malignancies, FLAG-tagged HLA-A*24 cDNA was introduced into
HLA-deficient K562 cells. The introduced HLA molecule was isolated
through two cycles of immunoprecipitation using anti-FLAG and
anti-pan HLA class I antibodies. Peptides bound to HLA molecules
were eluted and analyzed by mass spectrometry. A total of >200
sequences ranging from 9-11 amino acids were identified. These
sequences exhibited an expected reduced amino acid complexity at
anchor residue positions for HLA-A*24 (data not shown) (34).
The most promising ten peptides with the highest
PEAKS peptide score (-10lgP) were selected based on assays using
two independent clones (Table I).
Peptides derived from TOPK were selected in further analysis, as
TOPK has previously been reported as a cancer/testis antigen that
serves a role in regulation of survival and proliferation of
malignant cells (14).
 | Table IPeptides interacting with HLA in
HLA-A*24:02 transduced in K562 cells were analyzed by mass
spectrometry. |
Table I
Peptides interacting with HLA in
HLA-A*24:02 transduced in K562 cells were analyzed by mass
spectrometry.
| Amino acid
sequence | −10lgP | Predicted protein
of origin (gene symbol) |
|---|
| RYFDPANGKF | 79.14 | Elongation factor 2
(EEF2) |
| KFIDTTSKF | 64.75 | 60S ribosomal
protein L3 (RPL3), L3-like (RPL3L) |
| EYPDRIMNTF | 64.03 | Tubulin β-2A, 2B,
3, 4B chain (TUBB2A, 2B, 3, 4B) |
| SYQKVIELF | 58.93 | PDZ-binding
kinase/lymphokine-activated killer T-cell-originated protein kinase
(PBK/TOPK) |
| KYIHSANVL | 54.66 | Mitogen-activated
protein kinase 1, 3, 4, 6 (MAPK1, 3, 4, 6) |
| NYARGHYTI | 52.05 | Tubulin α-1A, 1B,
1C, 3C/D, 3E, 4A chain (TUBA1A, 1B, 1C, 3C/D, 3E, 4A) |
| RYTDVSTRY | 41.7 | Thioredoxin-related
transmembrane protein 2 (TMX2) |
| RYQKSTELL | 36.58 | Histone H3.3
(H3F3A) |
| LDKLRFGFKK | 28.85 | Dynein heavy chain
17, axonemal (DNAH17) |
| AVLGRGHF | 20.34 |
Serine/threonine-protein kinase N3
(PKN3) |
The nature of the peptides binding to HLA dependents
on active signaling pathways, as these define the sources of the
metabolized proteins (29,35). It is speculated that TOPK is
actively metabolized in the CML derived cell line assessed in the
current study and TOPK expression in hematopoietic tumor cell
lines, including K562 and clinical samples from patients with CML
was further assessed.
TOPK is highly expressed in hematopoietic
tumor cell lines
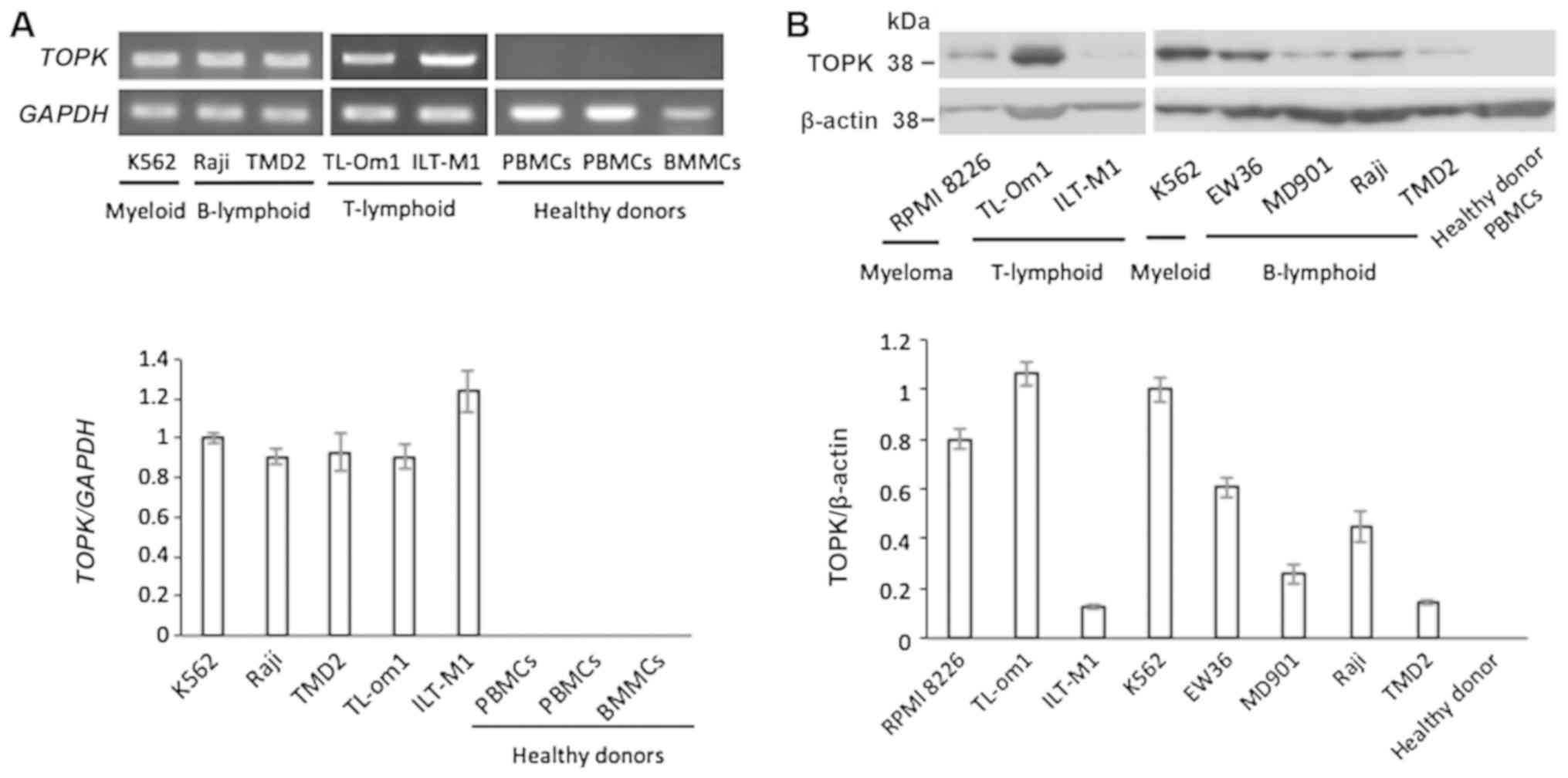
TOPK mRNA expression was determined by RT-qPCR in
various hematopoietic tumor cell lines, in addition to PBMCs or
BMMCs isolated from healthy donors. TOPK was expressed in all
analyzed cell lines, including K562, Raji, TMD2, TL-om1 and ILT-M1,
and expression was not detected in samples collected from healthy
donors (Fig. 1A). Consistently,
TOPK protein expression detected by western blotting was only
observed in hematopoietic tumor cell lines, including RPMI-8226,
TL-om1, ILT-M1, K562, EW36, MD901, Raji and TMD2, and not in
samples from healthy donors (Fig.
1B).
TOPK expression is increased in primary
CML cells
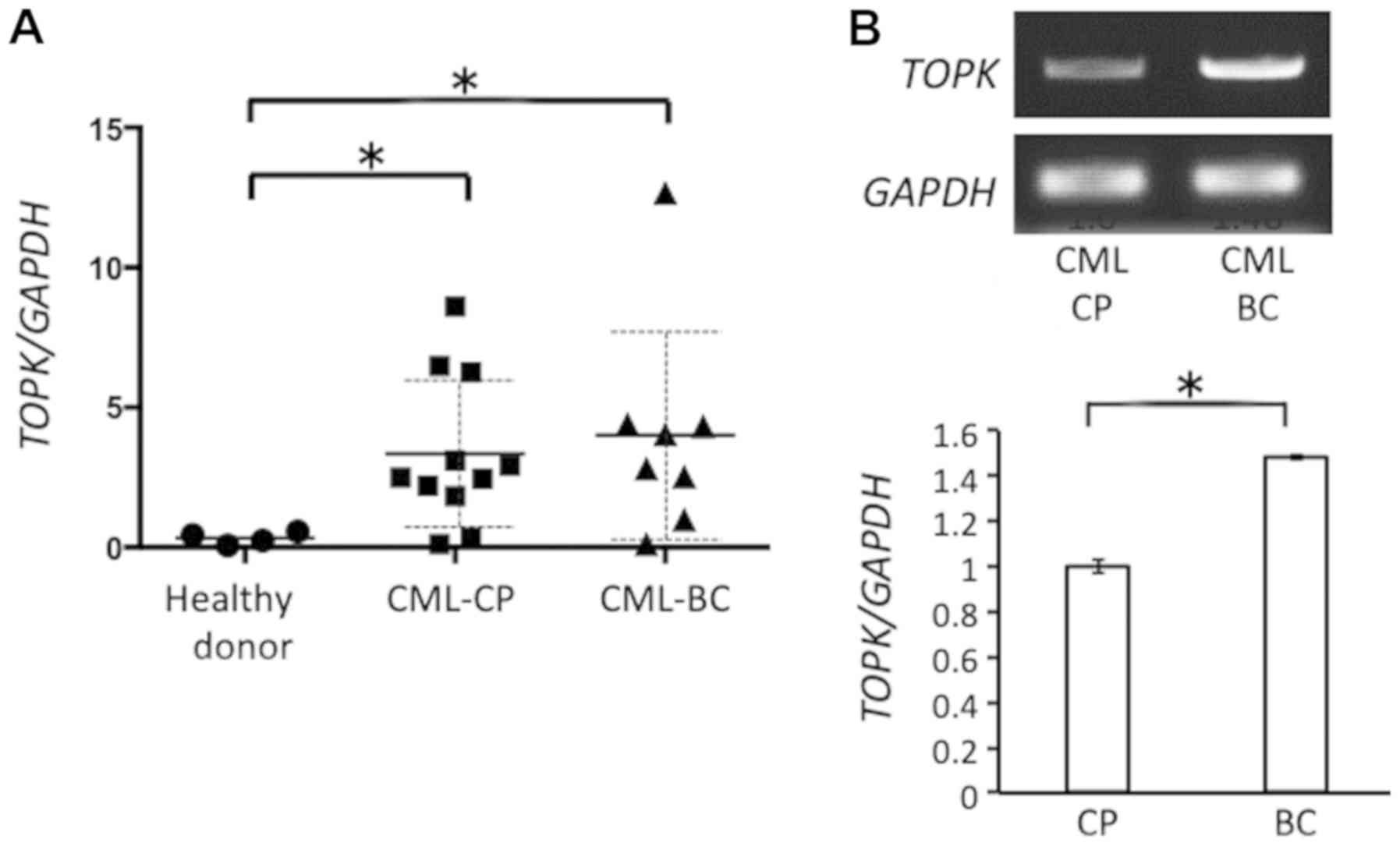
TOPK expression was analyzed in clinical samples
from patients with CML-CP or CML-BC and compared with samples from
healthy donors. TOPK expression was not significantly different in
PBMCs and BMMCs isolated from one patient with CML-CP (data not
shown). TOPK expression was significantly increased in CML-CP (2
PBMCs and 9 BMMCs) and CML-BC (5 PBMCs and 3 BMMCs) samples
compared with healthy donor samples (4 PBMCs; P<0.05 and
P<0.01, respectively; Fig. 2A).
No significant difference in TOPK mRNA expression was observed
between CML-CP and CML-BC samples (P>0.05). Expression of TOPK
mRNA was further compared in CML-CP and CML-BC samples obtained
from the same patients. In two out of three patients who provided
clinical samples at CML-CP and CML-BC, TOPK expression was
upregulated in CML-BC compared with CML-CP (Fig. 2B).
TOPK expression induces the kinase
activity of BCR/ABL
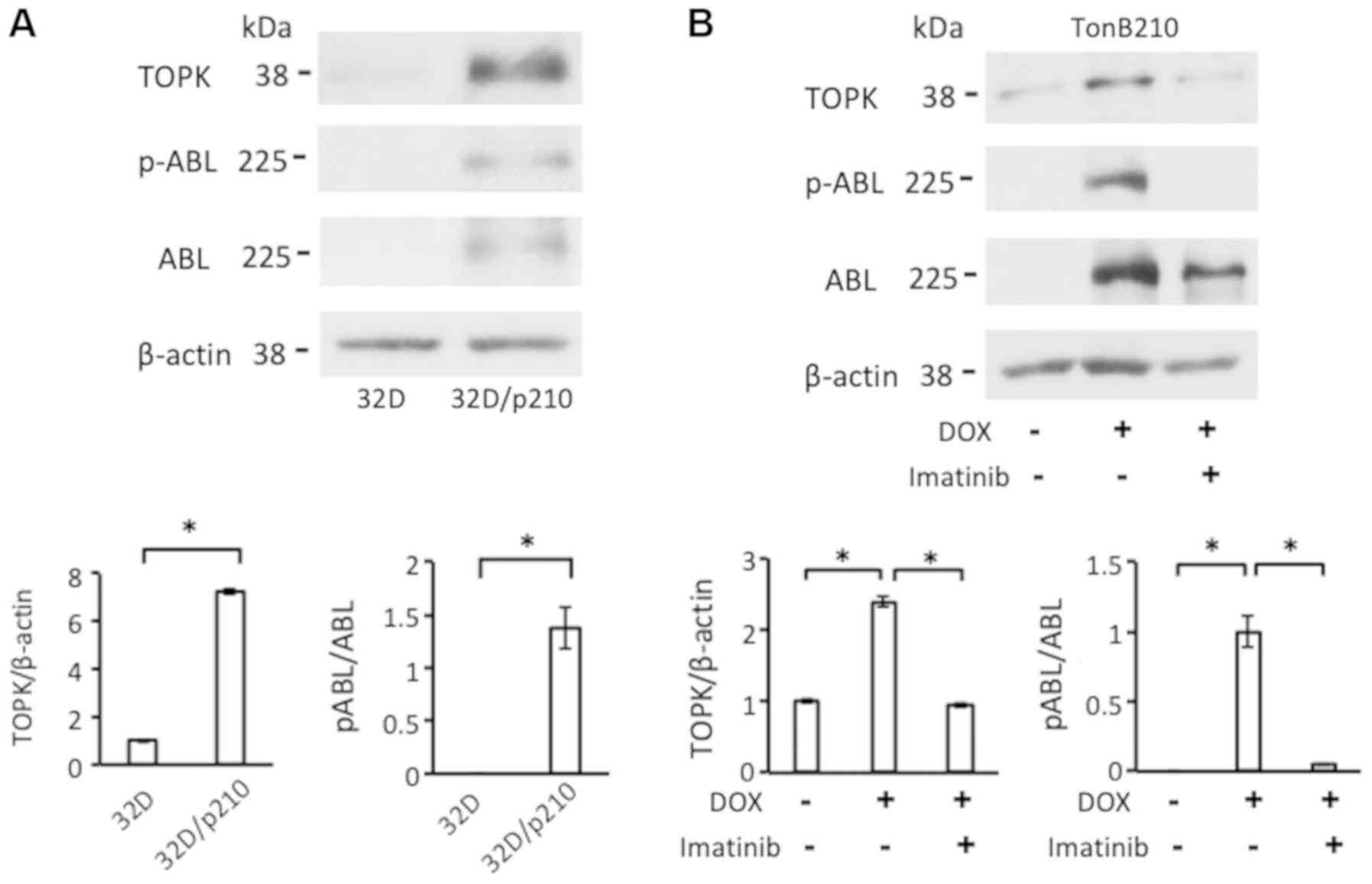
32D/p210 cells expressing BCR/ABL and the parental
cell line 32D not expressing BCR/ABL (26), were compared to evaluate an
association between TOPK expression and BCR/ABL. TOPK protein
expression was significantly higher in 32D/p210 compared with the
parental 32D (P>0.05; Fig. 3A).
An association between BCR/ABL and TOPK expression was further
analyzed using TonB210 cells, in which the addition of DOX induced
BCR/ABL expression and BCR/ABL autophosphorylation. TOPK protein
expression was significantly increased in TonB210 cells in the
presence of DOX compared with the untreated cells (P<0.01;
Fig. 3B). In addition, induction
of TOPK in TonB210 using DOX was significantly reversed when cells
were treated with imatinib (P<0.01; Fig. 3B) inhibiting BCR/ABL kinase
activity and autophosphorylation of BCR/ABL. The data suggested
that BCR/ABL may induce TOPK expression.
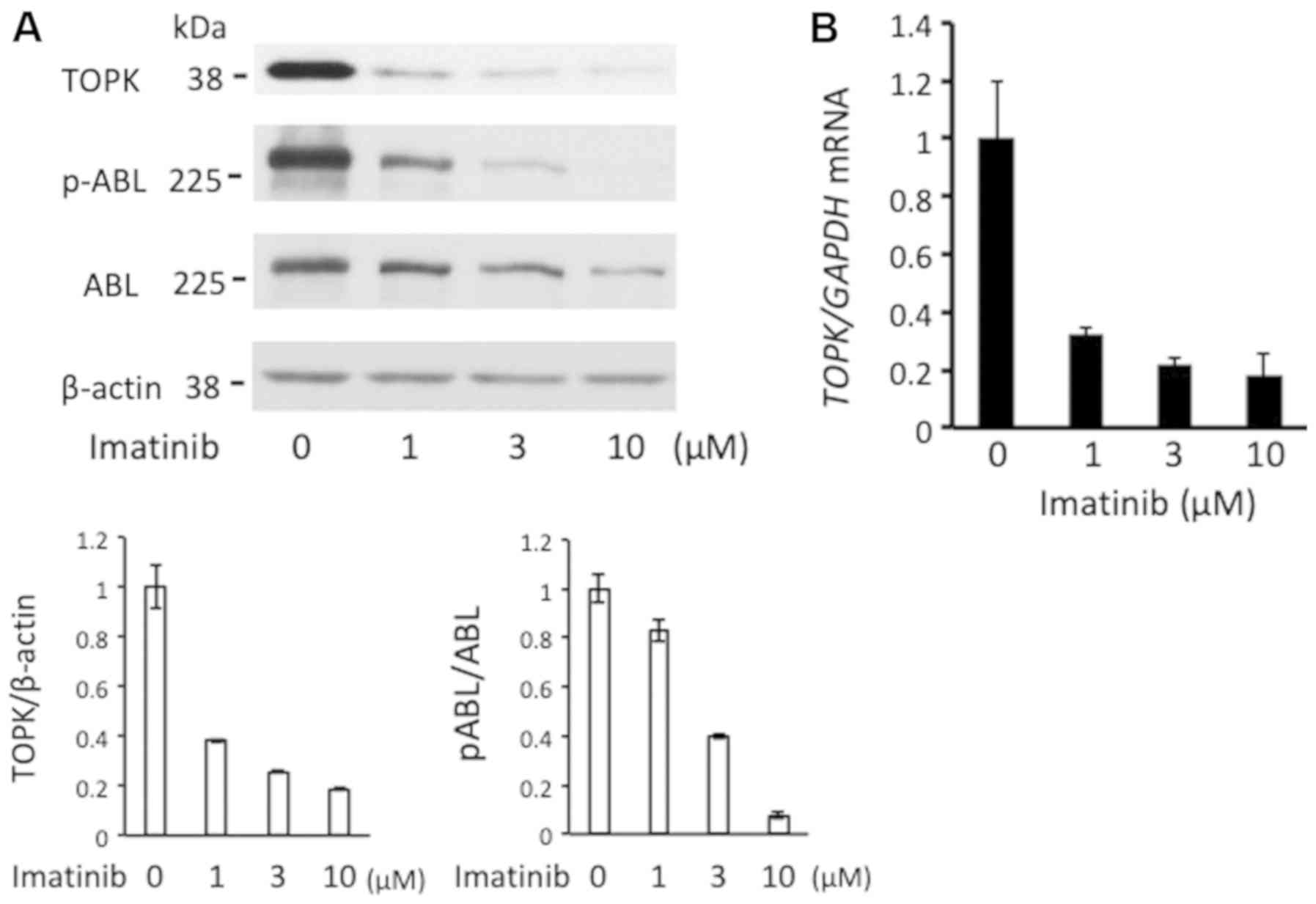
To further elaborate on this hypothesis and to
clarify the association between imatinib and ABL activation and
TOPK expression, imatinib was added to K652 cells at different
doses. As presented in Fig. 4A,
with increasing imatinib concentration, levels of TOPK and
phosphorylated BCR/ABL decreased in a potential dose-dependent
manner. Additionally, imatinib decreased expression levels of TOPK
mRNA in a potential dose-dependent manner (Fig. 4B). Similar results regarding
protein expression and phosphorylation were obtained using MOLM1
and TMD5 cells (data not shown). The results indicate an
association between TOPK expression and autophosphorylaiton of
BCR/ABL.
Mitotic arrest induced phosphorylation of
TOPK
Next, TOPK phosphorylation was assessed. TOPK is
phosphorylated at Thr9 by CDK1/cyclin B1 in the mitotic phase
(7,15). A crucial role of TOPK-Thr9 in cell
cycle progression of solid tumors has been reported previously
(14,36). Nocodazole induces mitotic arrest of
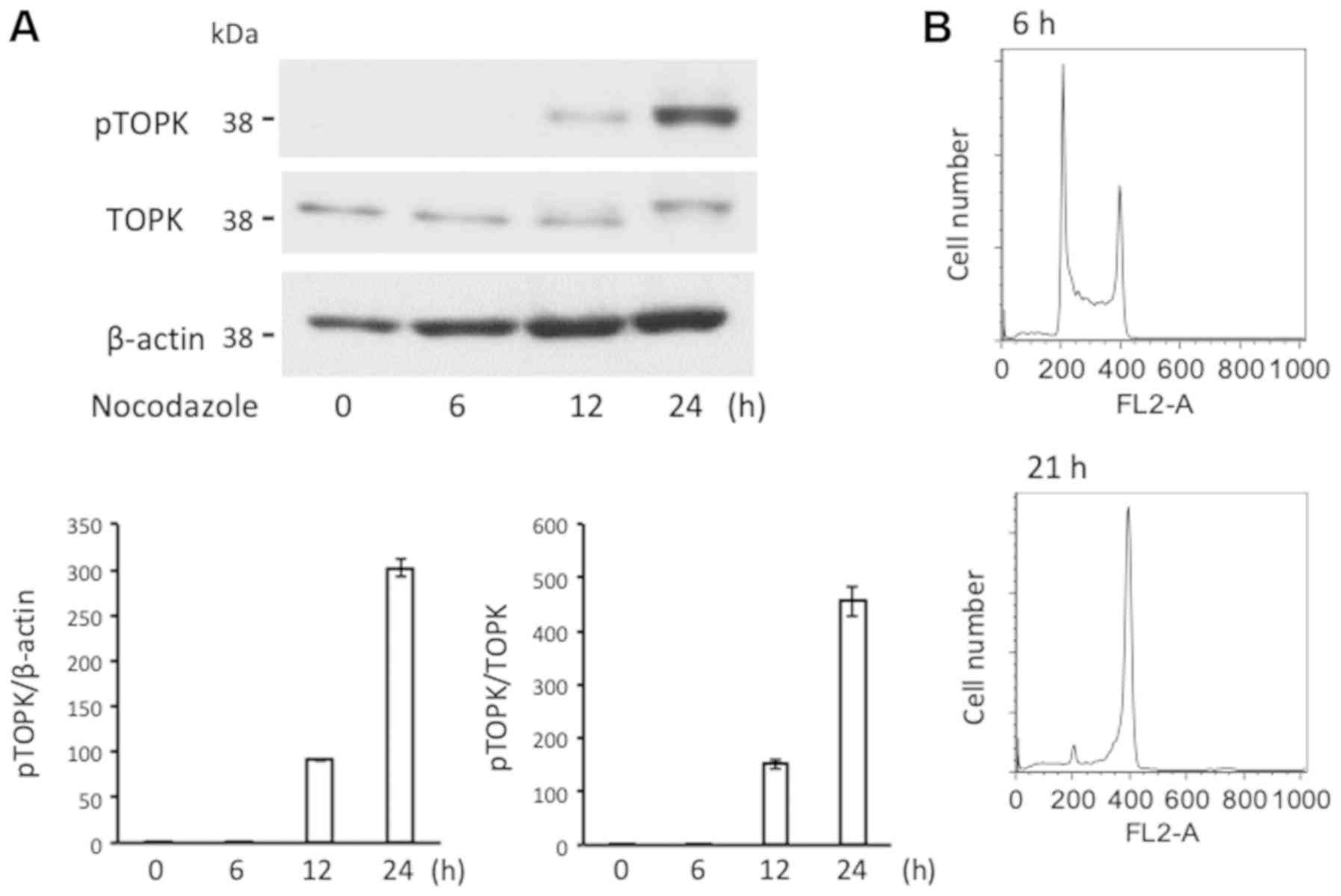
the cell cycle by microtubule depolymerization (37). In the current study, treatment with
nocodazole (300 ng/ml) enhanced the phosphorylation of TOPK in K562
cells a time-dependent manner (Fig.
5). Cell cycle analysis prior to and following 24 h treatment
with nocodazole suggested an accumulation of cells in the mitotic
phase (Fig. 5). At 24 h, the TOPK
band appeared with an increased molecular weight on the western
blot analysis, potentially due to mobility retardation caused by
phosphorylation, as reported previously (14).
PP2A is associated with and
dephosphorylates TOPK
PP2A is a serine/threonine phosphatase that acts as
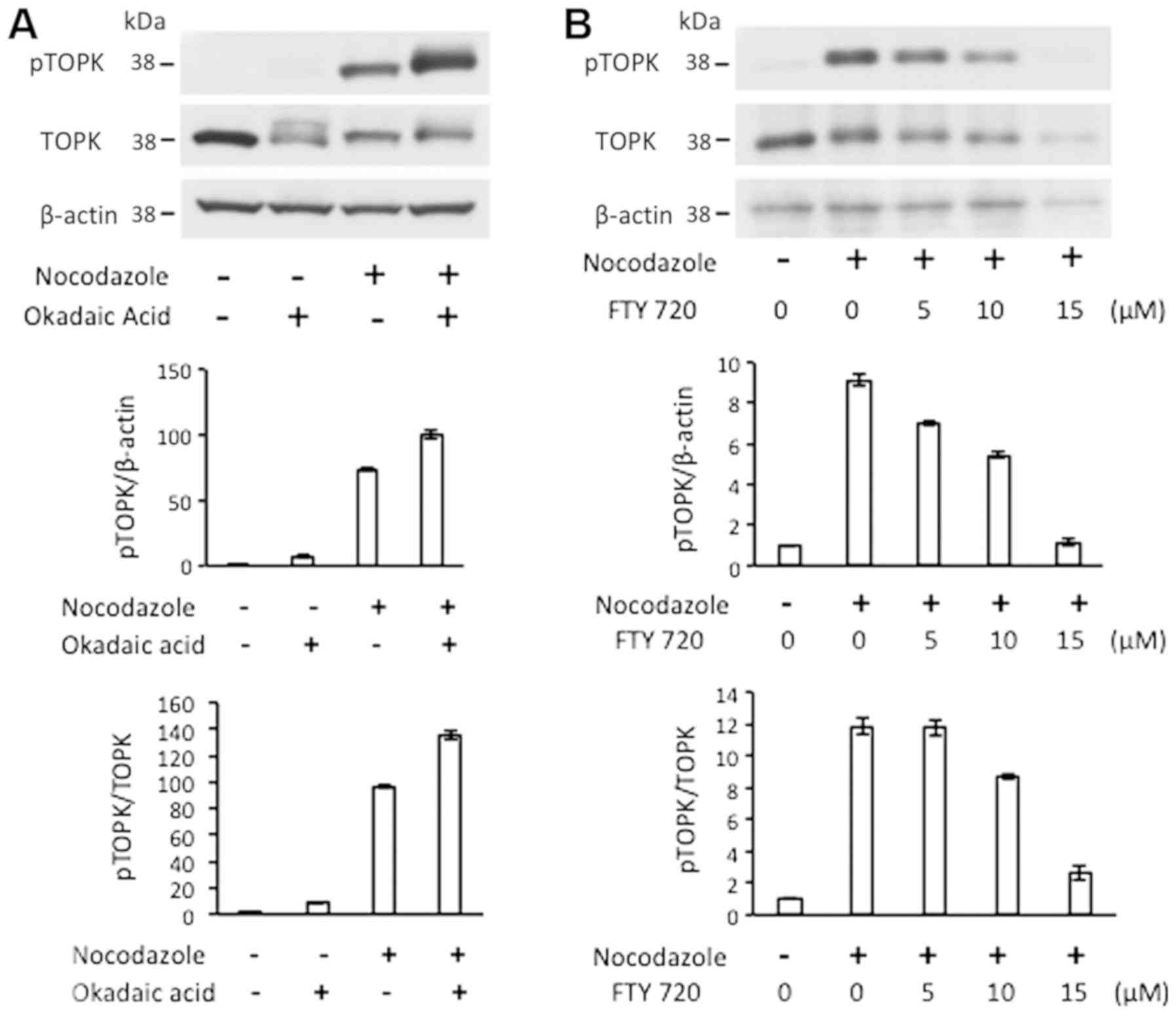
a tumor suppressor in BCR/ABL-positive cells (4). Okadaic acid is an inhibitor of
protein phosphatase 1 and PP2A, and FTY 720 is an activator of
PP2A. In K562 cells treated with nocodazole (300 ng/ml),
phosphorylation of TOPK was enhanced in the presence of okadaic
acid (200 nM; Fig. 6A) and
phosphorylation was reversed by FTY 720 (5, 10 and 15 µM) in
a dose-dependent manner (Fig. 6B).
Treatment with okadaic acid in the absence of nocodazole revealed a
TOPK band with a changed migration pattern that not detected using
the p-TOPK-Thr9 antibody (Fig.
6A). This data suggested a potential phosphorylation of TOPK at
a different amino acid.
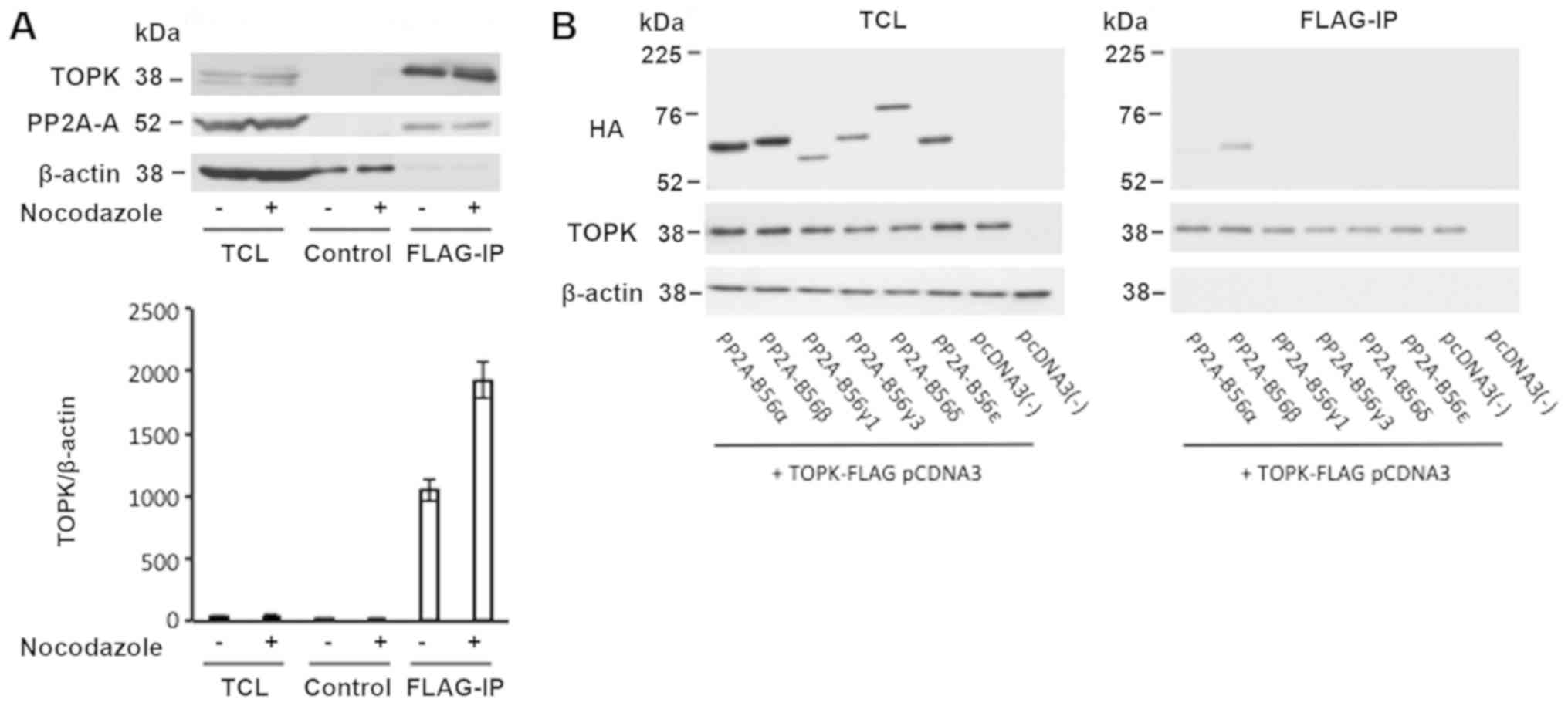
To further evaluate this potential connection, the
association between TOPK and PP2A was studied. K562 cells were
transfected with a FLAG-tagged TOPK overexpression vector. Western
blot analysis of TCL suggested the presence of PP2A-A and TOPK in
the cells. Although control mouse IgG was unable to precipitate
TOPK and PP2A-A, immunoprecipitation of TOPK with anti-FLAG beads
caused co-precipitation of PP2A-A in the presence and absence of
nocodazole (Fig. 7A). This data
suggested potential intermolecular interactions between TOPK and
the PP2A complex. PP2A-B is a regulatory subunit and in combination
with the PP2A-A and C dimer, it functions as PP2A phosphatase
(5). To elucidate which PP2A-B
subunit was involved in the interaction between PP2A and TOPK, 293T
cells were transfected with overexpression vectors for FLAG-tagged
TOPK and various PP2A-B56 subunits (α, β, γ1, γ3, δ and ε).
Expression was determined in the culture supernatant to confirm
transfection efficiency. Immunoprecipitation of TOPK using
anti-FLAG beads suggested that PP2A-HA-B56β and, to a lesser
extent, PP2A-HA-B56α interacted with TOPK (Fig. 7B). As β-actin was not detected in
the immunoprecipitated samples, interference from non-specific
precipitates was regarded as negligible. This result indicated that
TOPK and specific PP2A-B subunits may interact.
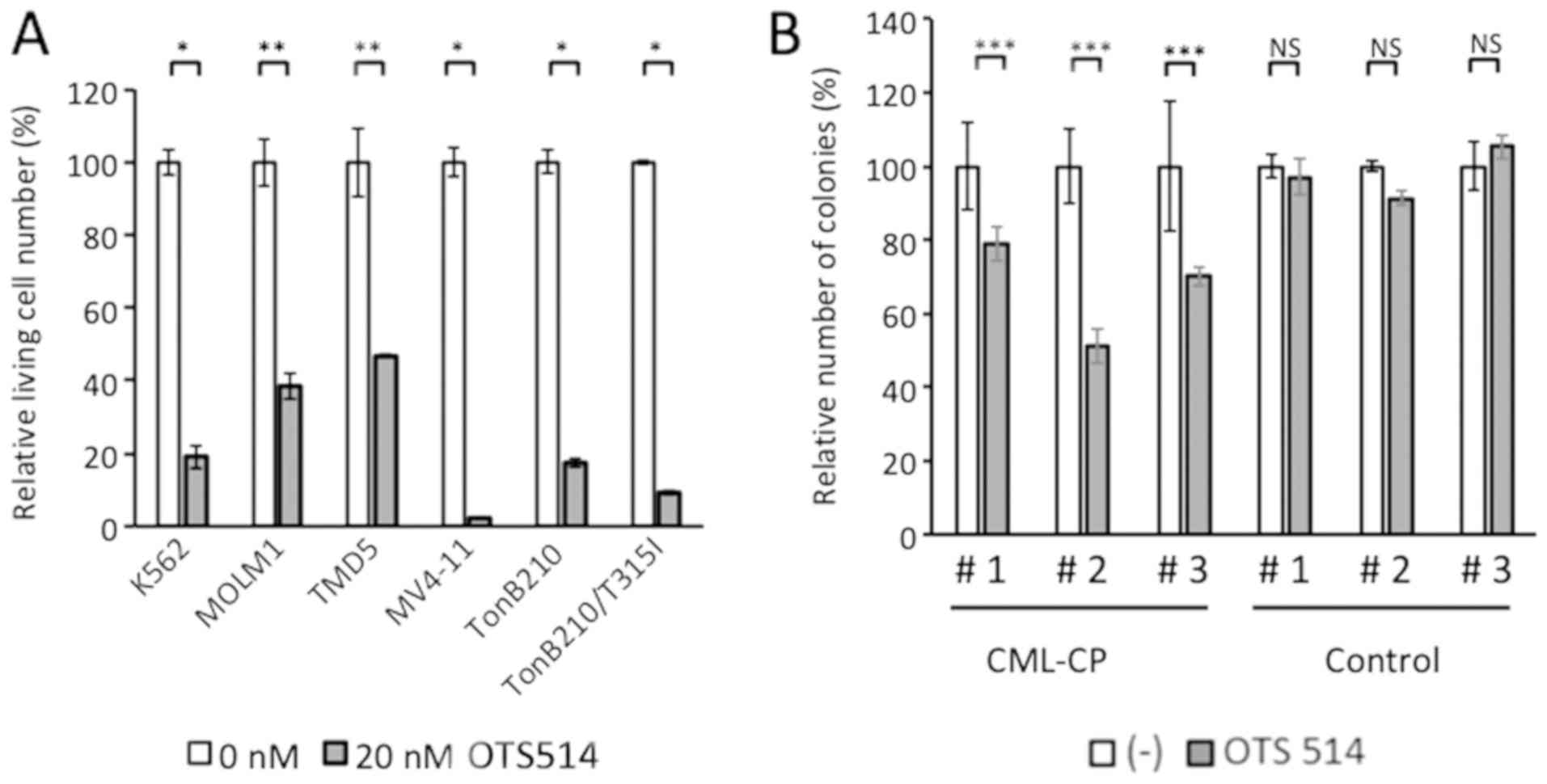
TOPK inhibitor suppresses proliferation
of BCR/ABL-positive cells
To elaborate on the effect of TOPK in
BCR/ABL-positive cells, experiments were performed using TOPK
inhibitor OTS514. OTS514 inhibits cell growth in several cell lines
in which TOPK is expressed at high levels (38). Suppression of proliferation by
OTS514 was assessed in various BCR/ABL-positive hematopoietic cell
lines (K562, MOLM1, TMD5, TonB210 and TonB210/T315I). Cells were
treated with 20 nM OTS514 for 48 h and evaluated by FACS analysis.
In each cell line, proliferation was suppressed significantly in
the inhibitor treated cells compared with the untreated controls
(P<0.001; Fig. 8A).
Additionally, it was examined whether OTS514 affects
CD34-positive primary cells. CD34-positive BMMCs from three
patients with CML-CP and three patients with stage I-IIA malignant
lymphoma without BM involvement were used in clonogenic assays.
Colony numbers were assessed at 14 days of culturing. As presented
in Fig. 8B, OTS514 significantly
decreased the number of colonies of the three samples from patients
with CML-CP (P<0.05) compared with the untreated control. No
significant differences were observed when treating the primary
cells isolated the control patients. The data suggested that an
inhibitory effect of OTS514 on the clonogenic capacity may be
specific for CML-CP cells.
OTS514 and imatinib exhibit combinatory
effects
To examine effects of the combination of OTS514 and
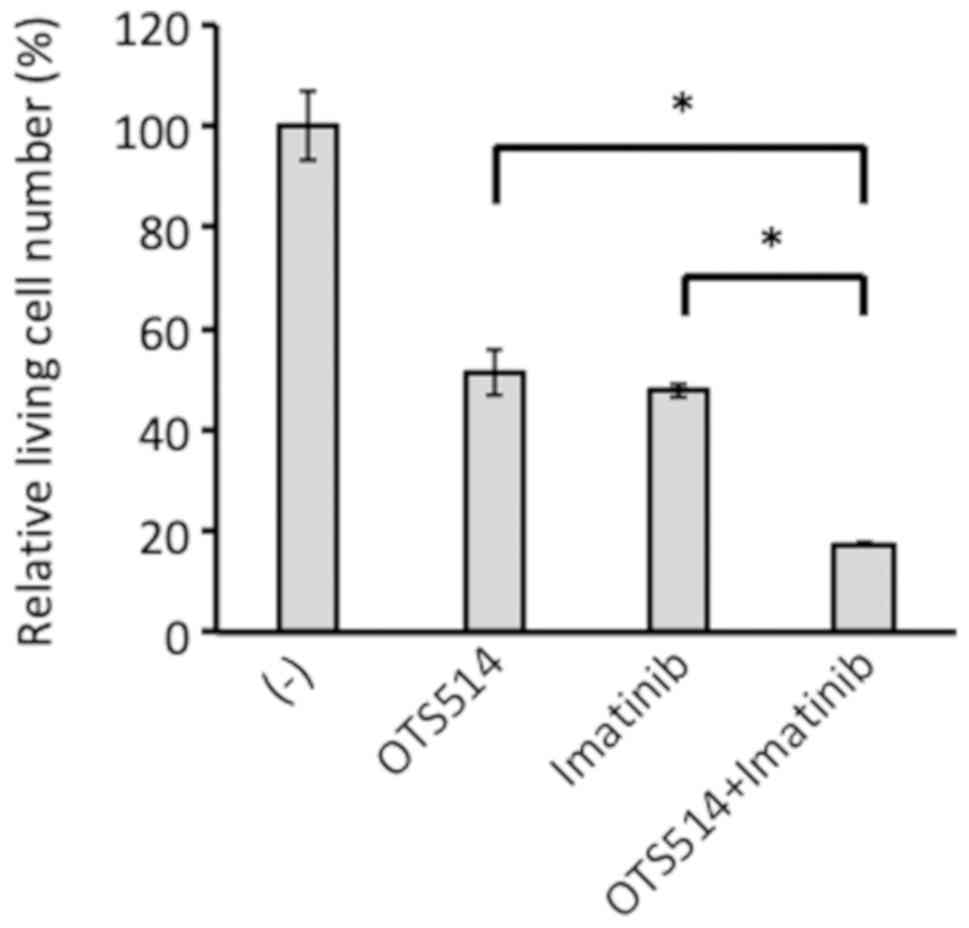
imatinib, K562 cells were treated with OTS514 (10 nM), imatinib
(0.5 µM) and a combination of both, and viability was
assessed by FACS analysis. OTS514 and imatinib single treatments
reduced the number of viable cells to a similar extent. Combined
treatment further significantly reduced the number of viable cells
compared with the single treatments (P<0.0001; Fig. 9). These results suggested that
inhibition of TOPK and BCR/ABL may affect the viability of CML
cells in an additive manner.
Discussion
In the present study, it was observed that peptides
describing TOPK-breakdown products were interacting with HLA-A in
K562 cells. Future experiments may focus on anti-TOPK CTL using by
this peptide in in vitro analyses. Difficulties may arise
from TOPK expression being associated with T cell activation and
anti-TOPK T cells may be hard to detect.
It was described that TOPK was induced by BCR-ABL as
a downstream target. Data suggested that expression of TOPK was
induced in BCR/ABL overexpressing cells and reduced when BCR-ABL
was inhibited using imatinib. Future studies evaluating the
knockdown or knockout of BCR/ABL are required to confirm this
mechanism and additional CML cell lines may be assessed. The
present data indicated that BCR/ABL induced TOPK transcription,
although certain cell lines, including TMD2, exhibited low TOPK
protein levels and abundant TOPK mRNA, implying a potential
presence of post-transcriptional regulators. The mechanism of this
transcriptional induction of TOPK by BCR/ABL requires further
elucidation. Previously, it was reported that transcription factor
forkhead box protein M1 (FoxM1) regulates TOPK expression (39). As FoxM1 is activated by BCR/ABL
(39,40), it may further be associated with
TOPK upregulation by BCR-ABL.
TOPK expression was significantly increased in
samples from patients with CML compared with healthy donors,
suggesting that TOPK may be associated with the development of CML.
Mechanisms leading to BC may vary on a case to case basis;
explaining for the results reported here, where no significant
differences were observed in patients with BC and CP. In two of
three patients that provided clinical samples in CP and BC, TOPK
expression was increased in BC compared with CP. The case presented
in more detail, was the patient exhibiting the highest overall
recorded expression of TOPK. The patient initially presented with
major molecular response to nilotinib (BCR/ABL international scale
[IS] <0.1%), prior to occurrence of sudden myeloid BC with E255K
mutation (IS=136.55%) at 11 months following diagnosis. Disease
progressed rapidly and the patient succumbed 3 weeks following BC
diagnosis. High TOPK expression may have contributed to the rapid
progression, as TOPK expression is associated with aggressive
development in other malignancies (9-13).
The samples size used in the current study was small and samples
for PBMCs and BMMCs were combined for the analysis. Further
analysis using an increased number of clinical samples is required
to elucidate CML development further.
In addition, TOPK is associated with PP2A, a key
tumor inhibitor of BCR/ABL-positive cells (4). PP2A suppresses cell survival and
proliferation by dephosphorylation regulatory markers (4,5). It
was suggested here that PP2A may interact with TOPK. PP2A is able
to accept substrates following binding of the B subunit to the
dimer formed by subunits A and C (41). The B subunit defines the potential
target molecule and is classified into four subfamilies (B/B55,
B′/B56, B″/B72 and B′″/PR93) (42). The present study revealed that
PP2A-B56β interacted most strongly with TOPK. It has been reported
that PP2A-B56β mediates the interaction of the PP2A core enzyme
complex with Akt (43). A
downstream target of Akt, CDC-like kinase 2, was reported to
phosphorylate B56β, which led to dephosphorylation of Akt by PP2A.
Akt is serine/threonine kinase regulating proliferation and
survival of cells and interacts with B56β (44). Based on the results presented in
the current study it was hypothesized that TOPK may utilize a
similar mechanism as Akt to regulate dephosphorylation by PP2A.
However, further studies are required to evaluate these claims.
Suppression of cell proliferation using a TOPK
inhibitor has been reported for various cell lines expressing TOPK
at high levels (38). The current
study confirmed that TOPK inhibitor OTS514 suppressed proliferation
in hematopoietic cell lines expressing BCR/ABL with or without the
T315I mutation, which confers resistance to most TKIs currently in
clinical use (45). Additionally,
OTS514 significantly enhanced the effect of imatinib on cell
viability. TOPK inhibitors may describe novel therapeutic agents
for ABL-mutated and -unmutated cases, to be used with or without
TKIs. Further studies are required to examine effects of the TOPK
inhibitor in various imatinib-resistant cell lines exhibiting
BCR/ABL-independent and -dependent molecular mechanisms.
Treatment of patients with CML has dramatically
improved since the introduction of TKIs, and the current goal is to
eradicate CML stem cells and to prevent relapse following TKI
treatment completion. The TOPK inhibitor OTS514 suppressed colony
formation in CD34-positive cells from patients with CML patients
but not in cells isolated from controls. Further studies are
required to examine whether the TOPK inhibitor may promote
elimination of CML stem cells.
The current study suggested that TOPK was induced by
BCR/ABL and was dephosphorylated by PP2A. Further studies are
warranted to evaluate the mechanisms of action and to elucidate the
significance of TOPK in BCR/ABL-positive and its potential as a
novel therapeutic.
Funding
This current study was supported by the Ministry of
Education, Culture, Sports, Science and Technology of Japan (grant
no. 15K09467).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors’ contributions
EU, SS, OM and TF contributed to project conception
and the design of experiments. EU, SS, RY, KW and TK performed the
experiments and analyzed the results. EU, SS, OM and TF wrote the
paper with contributions from all of the other coauthors. All
authors read and approved the final version of the manuscript.
Ethical approval and consent to
participate
Consent was obtained in compliance with the
Declaration of Helsinki and all patients provided written informed
consent. Approval was obtained from the Ethics Committee of the
Tokyo Medical and Dental University (Tokyo, Japan).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Ms. Kaori Okada, Ms.
Miyu Kato, Ms. Megumi Iida, Ms. Ikumi Suzuki, Ms. Miho Ohnishi and
Dr Ayaka Usui for technical assistance. The authors would further
like to thank Dr Suyoun Chung, Dr David M. Virshup, Dr George Q.
Daley, Dr Mari Kannagi, Dr Toshio Kitamura and Dr Shuji Tohda for
gifting experimental materials.
References
|
1
|
Deininger MW, Goldman JM and Melo JV: The
molecular biology of chronic myeloid leukemia. Blood. 96:3343–3356.
2000.
|
|
2
|
Hehlmann R, Saussele S, Voskanyan A and
Silver RT: Management of CML-blast crisis. Best Pract Res Clin
Haematol. 29:295–307. 2016.
|
|
3
|
Westermarck J: Targeted therapies don’t
work for a reason; the neglected tumor suppressor phosphatase PP2A
strikes back. FEBS J. 285:4139–4145. 2018.
|
|
4
|
Neviani P, Santhanam R, Trotta R, Notari
M, Blaser BW, Liu S, Mao H, Chang JS, Galietta A, Uttam A, et al:
The tumor suppressor PP2A is functionally inactivated in blast
crisis CML through the inhibitory activity of the BCR/ABL-regulated
SET protein. Cancer Cell. 8:355–368. 2005.
|
|
5
|
Schönthal AH: Role of serine/threonine
protein phosphatase 2A in cancer. Cancer Lett. 170:1–13. 2001.
|
|
6
|
Chang CC, Campoli M and Ferrone S: HLA
class I antigen expression in malignant cells: Why does it not
always correlate with CTL-mediated lysis? Curr Opin Immunol.
16:644–650. 2004.
|
|
7
|
Matsumoto S, Abe Y, Fujibuchi T, Takeuchi
T, Kito K, Ueda N, Shigemoto K and Gyo K: Characterization of a
MAPKK-like protein kinase TOPK. Biochem Biophys Res Commun.
325:997–1004. 2004.
|
|
8
|
Gaudet S, Branton D and Lue RA:
Characterization of PDZ-binding kinase, a mitotic kinase. Proc Natl
Acad Sci USA. 97:5167–5172. 2000.
|
|
9
|
Nandi A, Tidwell M, Karp J and Rapoport
AP: Protein expression of PDZ-binding kinase is up-regulated in
hematologic malignancies and strongly down-regulated during
terminal differentiation of HL-60 leukemic cells. Blood Cells Mol
Dis. 32:240–245. 2004.
|
|
10
|
Wei DC, Yeh YC, Hung JJ, Chou TY, Wu YC,
Lu PJ, Cheng HC, Hsu YL, Kuo YL, Chen KY, et al: Overexpression of
T-LAK cell-originated protein kinase predicts poor prognosis in
patients with stage I lung adenocarcinoma. Cancer Sci. 103:731–738.
2012.
|
|
11
|
Ohashi T, Komatsu S, Ichikawa D, Miyamae
M, Okajima W, Imamura T, Kiuchi J, Nishibeppu K, Kosuga T, Konishi
H, et al: Overexpression of PBK/TOPK contributes to tumor
development and poor outcome of esophageal squamous cell carcinoma.
Anticancer Res. 36:6457–6466. 2016.
|
|
12
|
Ohashi T, Komatsu S, Ichikawa D, Miyamae
M, Okajima W, Imamura T, Kiuchi J, Kosuga T, Konishi H, Shiozaki A,
et al: Overexpression of PBK/TOPK relates to tumour malignant
potential and poor outcome of gastric carcinoma. Br J Cancer.
116:218–226. 2017.
|
|
13
|
Chang CF, Chen SL, Sung WW, Hsieh MJ, Hsu
HT, Chen LH, Chen MK, Ko JL, Chen CJ and Chou MC: PBK/TOPK
expression predicts prognosis in oral cancer. Int J Mol Sci.
17:172016.
|
|
14
|
Park JH, Lin ML, Nishidate T, Nakamura Y
and Katagiri T: PDZ-binding kinase/T-LAK cell-originated protein
kinase, a putative cancer/testis antigen with an oncogenic activity
in breast cancer. Cancer Res. 66:9186–9195. 2006.
|
|
15
|
Abe Y, Takeuchi T, Kagawa-Miki L, Ueda N,
Shigemoto K, Yasukawa M and Kito K: A mitotic kinase TOPK enhances
Cdk1/cyclin B1-dependent phosphorylation of PRC1 and promotes
cytokinesis. J Mol Biol. 370:231–245. 2007.
|
|
16
|
Park JH, Nishidate T, Nakamura Y and
Katagiri T: Critical roles of T-LAK cell-originated protein kinase
in cytokinesis. Cancer Sci. 101:403–411. 2010.
|
|
17
|
Seeling JM, Miller JR, Gil R, Moon RT,
White R and Virshup DM: Regulation of beta-catenin signaling by the
B56 subunit of protein phosphatase 2A. Science. 283:2089–2091.
1999.
|
|
18
|
Baccarani M, Deininger MW, Rosti G,
Hochhaus A, Soverini S, Apperley JF, Cervantes F, Clark RE, Cortes
JE, Guilhot F, et al: European LeukemiaNet recommendations for the
management of chronic myeloid leukemia: 2013. Blood. 122:872–884.
2013.
|
|
19
|
Miki T, Kawamata N, Arai A, Ohashi K,
Nakamura Y, Kato A, Hirosawa S and Aoki N: Molecular cloning of the
breakpoint for 3q27 translocation in B-cell lymphomas and
leukemias. Blood. 83:217–222. 1994.
|
|
20
|
Tohda S, Nara N, Murohashi I and Aoki N:
Establishment of an interleukin-3-dependent leukemic cell line from
a patient with chronic lymphocytic leukemia in the acute phase.
Blood. 78:1789–1794. 1991.
|
|
21
|
Tohda S, Sakashita C, Fukuda T, Murakami N
and Nara N: Establishment of a double Philadelphia
chromosome-positive acute lymphoblastic leukemia-derived cell line,
TMD5: Effects of cytokines and differentiation inducers on growth
of the cells. Leuk Res. 23:255–261. 1999.
|
|
22
|
Sugamura K, Fujii M, Kannagi M, Sakitani
M, Takeuchi M and Hinuma Y: Cell surface phenotypes and expression
of viral antigens of various human cell lines carrying human T-cell
leukemia virus. Int J Cancer. 34:221–228. 1984.
|
|
23
|
Murakami Y, Hasegawa A, Ando S, Tanaka R,
Masuda T, Tanaka Y and Kannagi M: A novel mother-to-child human
T-cell leukaemia virus type-1 (HTLV-1) transmission model for
investigating the role of maternal anti-HTLV-1 antibodies using
orally infected mother rats. J Gen Virol. 98:835–846. 2017.
|
|
24
|
Klucher KM, Lopez DV and Daley GQ:
Secondary mutation maintains the transformed state in BaF3 cells
with inducible BCR/ABL expression. Blood. 91:3927–3934. 1998.
|
|
25
|
Kurosu T, Tsuji K, Kida A, Koyama T,
Yamamoto M and Miura O: Rottlerin synergistically enhances
imatinib-induced apoptosis of BCR/ABL-expressing cells through its
mitochondrial uncoupling effect independent of protein kinase
C-delta. Oncogene. 26:2975–2987. 2007.
|
|
26
|
Kurosu T, Ohki M, Wu N, Kagechika H and
Miura O: Sorafenib induces apoptosis specifically in cells
expressing BCR/ABL by inhibiting its kinase activity to activate
the intrinsic mitochondrial pathway. Cancer Res. 69:3927–3936.
2009.
|
|
27
|
Morita S, Kojima T and Kitamura T: Plat-E:
An efficient and stable system for transient packaging of
retroviruses. Gene Ther. 7:1063–1066. 2000.
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
|
|
29
|
Bassani-Sternberg M, Pletscher-Frankild S,
Jensen LJ and Mann M: Mass spectrometry of human leukocyte antigen
class I peptidomes reveals strong effects of protein abundance and
turnover on antigen presentation. Mol Cell Proteomics. 14:658–673.
2015.
|
|
30
|
Hofmann S, Glückmann M, Kausche S, Schmidt
A, Corvey C, Lichtenfels R, Huber C, Albrecht C, Karas M and Herr
W: Rapid and sensitive identification of major histocompatibility
complex class I-associated tumor peptides by Nano-LC MALDI MS/MS.
Mol Cell Proteomics. 4:1888–1897. 2005.
|
|
31
|
Hunt DF, Henderson RA, Shabanowitz J,
Sakaguchi K, Michel H, Sevilir N, Cox AL, Appella E and Engelhard
VH: Characterization of peptides bound to the class I MHC molecule
HLA-A2.1 by mass spectrometry. Science. 255:1261–1263. 1992.
|
|
32
|
Purcell AW and Gorman JJ:
Immunoproteomics: Mass spectrometry-based methods to study the
targets of the immune response. Mol Cell Proteomics. 3:193–208.
2004.
|
|
33
|
Zhang J, Xin L, Shan B, Chen W, Xie M,
Yuen D, Zhang W, Zhang Z, Lajoie GA and Ma B: PEAKS DB: de novo
sequencing assisted database search for sensitive and accurate
peptide identification. Mol Cell Proteomics.
11:M111.0105872012.
|
|
34
|
Liu J, Wu P, Gao F, Qi J, Kawana-Tachikawa
A, Xie J, Vavricka CJ, Iwamoto A, Li T and Gao GF: Novel
immunodominant peptide presentation strategy: A featured
HLA-A*2402-restricted cytotoxic T-lymphocyte epitope stabilized by
intrachain hydrogen bonds from severe acute respiratory syndrome
coronavirus nucleocapsid protein. J Virol. 84:11849–11857.
2010.
|
|
35
|
Khodadoust MS, Olsson N, Wagar LE, Haabeth
OA, Chen B, Swaminathan K, Rawson K, Liu CL, Steiner D, Lund P, et
al: Antigen presentation profiling reveals recognition of lymphoma
immunoglobulin neoantigens. Nature. 543:723–727. 2017.
|
|
36
|
Dougherty JD, Garcia AD, Nakano I,
Livingstone M, Norris B, Polakiewicz R, Wexler EM, Sofroniew MV,
Kornblum HI and Geschwind DH: PBK/TOPK, a proliferating neural
progenitor-specific mitogen-activated protein kinase kinase. J
Neurosci. 25:10773–10785. 2005.
|
|
37
|
Head J, Lee LL, Field DJ and Lee JC:
Equilibrium and rapid kinetic studies on nocodazole-tubulin
interaction. J Biol Chem. 260:11060–11066. 1985.
|
|
38
|
Matsuo Y, Park JH, Miyamoto T, Yamamoto S,
Hisada S, Alachkar H and Nakamura Y: TOPK inhibitor induces
complete tumor regression in xenograft models of human cancer
through inhibition of cytokinesis. Sci Transl Med.
6:259ra1452014.
|
|
39
|
Yang YF, Pan YH, Cao Y, Fu J, Yang X,
Zhang MF and Tian QH: PDZ binding kinase, regulated by FoxM1,
enhances malignant phenotype via activation of β-Catenin signaling
in hepatocellular carcinoma. Oncotarget. 8:47195–47205. 2017.
|
|
40
|
Mancini M, Castagnetti F, Soverini S, Leo
E, De Benedittis C, Gugliotta G, Rosti G, Bavaro L, De Santis S,
Monaldi C, et al: FOXM1 transcription factor: A new component of
chronic myeloid leukemia stem cell proliferation advantage. J Cell
Biochem. 118:3968–3975. 2017.
|
|
41
|
Mumby M: The 3D structure of protein
phosphatase 2A: New insights into a ubiquitous regulator of cell
signaling. ACS Chem Biol. 2:99–103. 2007.
|
|
42
|
Eichhorn PJ, Creyghton MP and Bernards R:
Protein phosphatase 2A regulatory subunits and cancer. Biochim
Biophys Acta. 1795:1–15. 2009.
|
|
43
|
Rodgers JT, Vogel RO and Puigserver P:
Clk2 and B56β mediate insulin-regulated assembly of the PP2A
phosphatase holoenzyme complex on Akt. Mol Cell. 41:471–479.
2011.
|
|
44
|
Testa JR and Tsichlis PN: AKT signaling in
normal and malignant cells. Oncogene. 24:7391–7393. 2005.
|
|
45
|
Redaelli S, Piazza R, Rostagno R,
Magistroni V, Perini P, Marega M, Gambacorti-Passerini C and
Boschelli F: Activity of bosutinib, dasatinib, and nilotinib
against 18 imatinib-resistant BCR/ABL mutants. J Clin Oncol.
27:469–471. 2009.
|