Introduction
Glioblastoma or grade IV astrocytoma is the most
common form of glioma in adults, accounting for 60-70% of all
gliomas, as well as the most aggressive brain tumor (1). Glioblastoma treatment currently
involves extensive surgical resection followed by external-beam
radiation and concomitant temozolomide chemotherapy (2). Temozolomide is an alkylating agent
that forms O6-methylguanine in DNA, which miss-pairs
with thymine during the next DNA replication cycle (3). These critical lesions progress to
lethal DNA cross-links, which inhibit cell replication and result
in cell death (4,5). Despite this therapeutic effort,
numerous glioblastoma cases develop resistance to chemotherapy,
which results in a poor prognosis for the patients, who only have a
median survival time of 14.6 months after diagnosis (5). O6-methylguanine-DNA
methyltransferase (MGMT), a DNA repair enzyme, is responsible for
inducing temozolomide resistance (6,7).
MGMT removes alkyl groups from the O6 position of
guanine (8). MGMT promoter
methylation has been observed in 40-57% of glioblastoma cases;
additionally, when this occurs, MGMT is not transcribed and cannot
repair DNA damage caused by temozolomide. Left unrepaired, these
chemotherapy-induced lesions trigger cytotoxicity and apoptosis,
thus resulting in more efficient treatment (9). For this reason, MGMT promoter
methylation status can be considered as an important predictive
factor for good therapeutic response and hence survival of patients
with glioblastoma.
Histone deacetylases (HDACs) are primarily involved
in the deacetylation of histones, but a number of HDACs, including
HDAC6, can also affect the function of cytoplasmic non-histone
proteins, becoming key regulators of cancer signaling pathways
(10,11). A major substrate of HDAC6 is
acetylated α-tubulin (10), the
structural protein of the microtubules that form a whole variety of
cellular structures, including the primary cilium (12). HDAC6 overexpression, which has been
demonstrated to occur in numerous glioblastoma cases (13), rapidly deacetylates α-tubulin,
resulting in depolymerization of microtubules and disruption of the
primary cilium (12). Aberrant
ciliogenesis is a common defect, as determined in five glioblastoma
cells lines, which may contribute to the phenotype of these
malignant cells (14,15). Cilia have been implicated in
numerous signaling pathways important in embryonic development and
disease, including the Hedgehog (Hh) (16,17),
Wnt (18) and platelet-derived
growth factor (17) pathways.
In vertebrate cells, the sonic Hh pathway requires
primary cilia (19). A probable
mechanistic function of the cilium is to regulate the Hh pathway by
increasing the local concentration and bringing pathway components
together for key protein-protein interactions required for
regulation, including the presence of Smo in the primary cilium
being required for the genesis of Gli activated forms, while the
absence of Smo will generate Gli repressor forms (20-22).
Among Gli targets, a number of proteins, including cyclins D1 and
D2, insulin like growth factor binding protein-6, B-cell
lymphoma-2, GLI1 and Myc-N, can be observed (20). Furthermore, GLI1 has been
associated with increased expression of MGMT (23). Cyclopamine, an effective Smo
antagonist, competitively binds the Smo receptor and subsequently
inhibits the Hh pathway (23).
However, those cancer cells that lack cilia would not be responsive
to this type of Smo inhibitors, and instead would be required to be
treated with downstream inhibitors, such as the Gli antagonists
(21). Based on this, only
ciliated cells, or cells that had been previously exposed to cilia
formation promoters, such as tubastatin, would respond to
cyclopamine.
Epithelial-mesenchymal transition (EMT) is the
biological process by which cells lose their epithelial
characteristics and acquire a mesenchymal cell phenotype, which
includes enhanced migratory capacity, invasiveness, elevated
resistance to apoptosis and increased production of extracellular
matrix components (22,24-26)
(Table I). EMT is considered a
promoter of metastasis, due to the transformations through which
cells acquire motility (27). EMT
is induced by growth factors, including transforming growth factor
(TGF)-1, that decreases expression of E-cadherin (25,28,29),
but at the same time increases expression of HDAC6 (30). Following this reasoning, inhibition
of HDAC6 by a selective inhibitor, such as tubastatin A, would
reduce TGF-induced downregulation of E-cadherin, and therefore
EMT.
 | Table ICommon epithelial and mesenchymal
markers for epithelial-mesenchymal transition evaluation. |
Table I
Common epithelial and mesenchymal
markers for epithelial-mesenchymal transition evaluation.
| Types of proteins
or RNA | Epithelial
markers | Mesenchymal
markers |
|---|
| Cell-surface
proteins | E-cadherin
ZO-1 | N-cadherin |
| OB-cadherin |
| α5β1 integrin |
| Syndecan-1 |
| Cytoskeletal
markers | Cytokeratin | Vimentin |
| β-catenin |
| ECM proteins | α1 (IV)
collagen | α1 (I)
collagen |
| Laminin 1 | α1 (III)
collagen |
| Fibronectin |
| Laminin 5 |
| Transcriptional
factors | | Snail |
| Slug |
| ZEB1 |
| Twist |
| miRNAs | miR-200 family | miR-10b |
| miR-21 |
For the present study, two glioblastoma cell lines,
LN405 and T98G, were treated with a selective HDAC6 inhibitor,
tubastatin A, to determine whether this treatment modulates the
sonic Hh pathway, reduces tumor cell clonogenicity and migration
capacities, counteracts EMT and sensitizes glioblas-toma cells to
chemotherapy with temozolomide.
Materials and methods
Glioblastoma cell lines
The present study was performed using T98G and LN405
glioblastoma cell lines in all experiments conducted. T98G cells
were obtained from the European Collection of Cell Cultures
(Salisbury, UK) and are derived from a glioblastoma multiform tumor
from a 61-year-old Caucasian male. LN405 cells were purchased from
The Leibniz-Institute DSMZ (German Collection of Microorganisms and
Cell Cultures, Braunschweig, Germany) and correspond to
glioblastoma cells established from an astrocytoma tumor (grade IV)
of a 62-year-old female in 1986. The cell lines were cultured in
Gibco RPMI-1640 GlutaMAX™ medium, supplemented with 10% fetal
bovine serum (FBS) and 1% penicillin/streptomycin (all from Thermo
Fisher Scientific, Inc., Waltham, MA, USA). These cells were grown
as a monolayer in 75 cm2 flasks and maintained in an
incubator at 37°C in an atmosphere containing 5%
CO2.
MTT (thiazolyl blue tetrazolium bromide)
assay
The MTT tetrazolium reduction assay is frequently
used to estimate the number of viable eukaryotic cells and
calculate the median lethal dose (LD50) for screening collections
of compounds to determine if the test molecules exhibit direct
cytotoxic effects that eventually result in cell death. Viable
cells with active mitochondrial metabolism convert MTT substrate
into a purple colored formazan product with an absorbance maximum
~570 nm. When cells die, they lose the ability to convert MTT into
formazan, thus color formation serves as a useful and convenient
marker (presumably directly proportional) of only the viable cells.
This reaction generally involves NADH as a cofactor (31). This method requires the incubation
of the MTT substrate at a final concentration of 0.5 mg/ml for 1.5
h at 37°C, with a population of viable cells that have previously
been cultured in 96-well plates until reaching a confluence of
5,000 cells/well, and treated with each tested drug: For
cyclopamine and tubastatin A the concentrations used were 0, 5, 10,
15, 20, 25, 30, 35, 40, 45 and 50 µM; while for temozolomide
the concentrations used were 0, 50, 100, 150, 200, 250, 300, 350,
400, 450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950 and 1000
µM. After 1.5 h, MTT substrate was discarded to avoid
cytotoxicity due to the reagent [as the conversion to formazan by
cells in culture is time-dependent (31)] and dimethyl sulfoxide (DMSO) was
added to each sample. The resulting absorbance was monitored at 550
nm wavelength using the Multiskan EX reading spectrophotometer.
Following MTT tests, cyclopamine, tubastatin A and temozolomide
were used in LN405 cells at final concentrations of 27.5, 32.5 and
400 µM, respectively. In T98G cells, cyclopamine and
tubastatin A were used at final concentrations of 20 and 30
µM, respectively. Treatments were conducted at 37°C for 72
h. Temozolomide LD50 could not be achieved in T98G cells;
therefore, a final concentration of 400 µM was selected
(Table II).
| Table IIMutational status of PTEN and p53 in
the glioblastoma cell lines, and concentrations of cyclopamine,
tubastatin A and temozolomide. |
Table II
Mutational status of PTEN and p53 in
the glioblastoma cell lines, and concentrations of cyclopamine,
tubastatin A and temozolomide.
| Cell line | PTEN | p53 | Cyclopamine | Tubastatin A | Temozolomide |
|---|
| LN405 | MUT | MUT | 27.5 μM | 32.5 μM | 400 μM |
| T98G | WT | MUT | 20 μM | 30 μM | 400 μM |
2D colony formation assay
The aim of the 2D colony formation assay was to
investigate the attachment-dependent growth of cells when exposed
to different drugs. Cells were treated at 37°C for 72 h as follows:
LN405 with 27.5 µM cyclopamine, 32.5 µM tubastatin A
and 400 µM temozolomide; and T98G with 20 µM
cyclopamine, 30 µM tubastatin A and 400 µM
temozolomide. Concentrations and durations for the combination of
cyclopamine and tubastatin A, and the combination of temozolomide
and tubastatin A, were as for the single treatments. Additionally,
1% DMSO was used as a vehicle control. Subsequently, 300 cells/well
were cultured in six-well agarose plates, with 3 wells/condition at
37°C in a 5% CO2 incubator for 10 days. Subsequently,
cells were fixed with 4% paraformaldehyde for 40 min at room
temperature and stained with crystal violet (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) for 15 min at room temperature. Resulting
colonies were counted using a Suntex 560 Colony Counter (Gemini,
Apeldoorn, The Netherlands).
3D colony formation assay in soft
agar
The 3D colony formation assay was conducted to
investigate the attachment-independent growth capacity of the cell
lines when exposed to different drugs. Cells were treated at 37°C
for 72 h as follows: LN405 with 27.5 µM cyclopamine, 32.5
µM tubastatin A and 400 µM temozolomide; and T98G
with 20 µM cyclopamine, 30 µM tubastatin A and 400
µM temozolomide. Concentrations and durations for the
combination of cyclopamine and tubastatin A, and the combination of
temozolomide and tubastatin A, were as for the single treatments.
Additionally, 1% DMSO was used as a vehicle control. Subsequently,
10,000 cells/well were cultured in six-well agarose plates, with 3
wells/condition at 37°C in a 5% CO2 incubator for 2
weeks. Previously, 2 ml agarose 0.5% (cat. no. 8016; Pronadisa,
Laboratorios Conda, Torrejón de Ardoz, Madrid, Spain) with
Dulbecco’s modified Eagle’s medium (DMEM; Sigma-Aldrich; Merck
KGaA) were added to the wells. When this first layer had gelled,
10,000 cells contained in 2 ml agarose 0.2% and 1X DMEM were added
onto it. Once the top layer containing the cells had gelled, 2 ml
fresh medium (Gibco RPMI-1640 GlutaMAX™ medium supplemented with
10% FBS and 1% penicillin/streptomycin) were added, incubated at
37°C in a 5% CO2 incubator and changed every 3 days.
After 2 weeks, the medium was discarded, and the colonies were
stained with crystal violet for 5 min at room temperature. Samples
were washed 5 times with water to improve visualization of the
colonies, and then an image of each well was captured and analyzed
with the colony-forming unit free software OpenCFU (32).
Wound healing assay
This assay was performed to investigate the
migration capacity of the cells following treatment. Cells were
treated at 37°C for 72 h as follows: LN405 with 27.5 µM
cyclopamine, 32.5 µM tubastatin A and 400 µM
temozolomide; and T98G with 20 µM cyclopamine, 30 µM
tubastatin A and 400 µM temozolomide. Concentrations and
durations for the combination of cyclopamine and tubastatin A, and
the combination of temozolomide and tubastatin A, were as for the
single treatments. Additionally, 1% DMSO was used as a vehicle
control. Subsequently, cells were cultured at 37°C in a 5%
CO2 incubator in 24-well plates, at a concentration of
250,000 cells/well. After 24 h, a scratch was produced in the
middle of the well and medium was changed to one containing 2.5%
FBS in order to avoid proliferation and apoptosis of the cells.
Images were captured at 0, 8, 24, 32 and 48 h after scratching with
a Nikon SMZ18 light microscope, at ×10 magnification.
Cell death detection
ELISAPLUS
The Cell Death Detection ELISAPLUS (Roche
Diagnostics GmbH, Mannheim, Germany; cat. no. 11544675001) was used
in order to investigate the effect of different drugs on apoptosis.
This assay is based on a quantitative
sandwich-enzyme-immunoassay-principle using mouse monoclonal
antibodies directed against DNA and histones. This allows the
specific determination of mono- and oligonucleosomes in the
cytoplasmic fraction of cell lysates. For this experiment, cells
were cultured at 37°C in a 5% CO2 incubator in 96-well
plates at a concentration of 5,000 cells/well, with 4
wells/condition. After 24 h, treatments were added to each
corresponding well, and cells were treated at 37°C for 72 h as
follows: LN405 with 27.5 µM cyclopamine, 32.5 µM
tubastatin A and 400 µM temozolomide; and T98G with 20
µM cyclopamine, 30 µM tubastatin A and 400 µM
temozolomide. Concentrations and durations for the combination of
cyclopamine and tubastatin A, and the combination of temozolomide
and tubastatin A, were as for the single treatments. Additionally,
1% DMSO was used as a vehicle control. Apoptosis was measured at
24, 48 and 72 h following the manufacturer’s protocols.
Caspase-Glo 3/7 assay
The Caspase-Glo® 3/7 assay (Promega
Corporation, Madison, WI, USA) is a homogeneous, luminescent assay
that measures caspase-3 and caspase-7 activities. The kit provides
a luminogenic caspase-3/7 substrate, which contains the
tetrapeptide sequence DEVD. The addition of this reagent results in
cell lysis, followed by caspase cleavage of the substrate and
generation of a glow-type luminescent signal produced by
luciferase. Luminescence is proportional to the amount of caspase
activity present. Cells were cultured at 37°C for 72 h in Gibco
RPMI-1640 GlutaMAX™ medium, supplemented with 10% FBS and 1%
penicillin/streptomycin, in 96-well plates at a concentration of
5,000 cells/well, with 4 wells/condition. After 24 h, cells were
added to each corresponding well, that were treated at 37°C for 72
h as follows: LN405 with 27.5 µM cyclopamine, 32.5 µM
tubastatin A and 400 µM temozolomide; and T98G with 20
µM cyclopamine, 30 µM tubastatin A and 400 µM
temozolomide. Concentrations and durations for the combination of
cyclopamine and tubastatin A, and the combination of temozolomide
and tubastatin A, were as for the single treatments. Additionally,
1% DMSO was used as a vehicle control. Caspase-3/7 activation was
measured at 24, 48 and 72 h, following the manufacturer’s
protocols.
RNA extraction
Total RNA extraction from 72 h treated cells was
conducted following the TRIzol® reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) protocol, which allows sequential
precipitation of RNA, DNA and proteins from a single sample. Cells
were treated at 37°C for 72 h as follows: LN405 with 27.5 µM
cyclopamine, 32.5 µM tubastatin A and 400 µM
temozolomide; and T98G with 20 µM cyclopamine, 30 µM
tubastatin A and 400 µM temozolomide. Concentrations and
durations for the combination of cyclopamine and tubastatin A, and
the combination of temozolomide and tubastatin A, were as for the
single treatments. Additionally, 1% DMSO was used as a vehicle
control. Following homogenization of the samples with TRIzol
reagent, chloroform was added to separate (after 15 min
centrifugation at 12,000 × g and 4°C) into a clear upper aqueous
layer containing RNA, an interphase and a red lower organic layer
containing DNA and proteins. RNA precipitation was then achieved by
the addition of isopropanol and centrifugation for 30 min at 12,000
× g and 4°C. Subsequently, isopropanol was removed, and 1 ml 75%
ethanol in diethyl pyrocarbonate (DEPC) water was added, vortexed
and centrifuged for 5 min at 7,500 × g and 4°C. The process was
repeated once more. Subsequently, ethanol was removed, the RNA
pellet was left to dry, and resuspended in 15 µl DEPC water,
to finally be stored at −80°C for use in downstream applications.
Total RNA quantification in each sample was measured using
NanoDrop™ Microvolume Spectrophotometer (Thermo Fisher Scientific,
Inc.).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
For reverse transcription, 2 µg RNA were
mixed with 1 µl random primers (250 ng/µl) and 1
µl dNTPs mix (10 µM) in a final volume of 12
µl water. Random primers (cat. no. 48190011) were purchased
from Thermo Fisher Scientific, Inc.. This mixture was incubated for
5 min at 65°C. Subsequently, 4 µl first strand buffer and 2
µl DTT were added, and this was incubated for 2 min at 25°C.
Following this, 1 µl SuperScript II Reverse Transcriptase
(Invitrogen; Thermo Fisher Scientific, Inc.) was added for the
synthesis of cDNA and this final mixture was incubated for 10 min
at 25°C, 50 min at 42°C and finally 15 min at 72°C. Furthermore, 80
µl water were added and the cDNA was stored at −20°C.
RT-qPCR was used to analyze the expression of genes
associated with the sonic Hh pathway and EMT in six different
conditions (DMSO as control, tubastatin A, cyclopamine,
temozolomide, tubastatin A plus temozolomide and tubastatin A plus
cyclopamine). Cells were treated at 37°C for 72 h as follows: LN405
with 27.5 µM cyclopamine, 32.5 µM tubastatin A and
400 µM temozolomide; and T98G with 20 µM cyclopamine,
30 µM tubastatin A and 400 µM temozolomide.
Concentrations and durations for the combination of cyclopamine and
tubastatin A, and the combination of temozolomide and tubastatin A,
were as for the single treatments. Additionally, 1% DMSO was used
as a vehicle control. Amplification reactions were conducted in an
IQ5 multicolor real-time PCR detection system (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Subsequently, 1 µl
of each sample cDNA was used in a total volume of 20
µl/well, with a reaction mix containing 10 µl IQ™
SYBR® Green supermix (Bio-Rad Laboratories, Inc.). An
initial denaturation step at 95°C for 30 sec was followed by 40
cycles of amplification alternating between 95°C for 10 sec, the
corresponding annealing temperature for each gene for 30 sec and
72°C for another 30 sec (Table
III). Each sample was assayed in triplicate. Forward and
reverse primers were designed using Primer3Plus (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi).
Sequences and melting temperatures of each primer pair are depicted
in Table III. Ribosomal 18S gene
was used as a housekeeping reference gene for the relative
quantification of cDNA amount, using the comparative Cq method
(33), also known as the
2−∆∆Cq method.
| Table IIISequences and melting temperatures of
primers used for reverse transcription-quantitative polymerase
chain reaction. |
Table III
Sequences and melting temperatures of
primers used for reverse transcription-quantitative polymerase
chain reaction.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) | Temperature
(°C) |
|---|
| 18S |
GTAACCCGTTGAACCCATT |
CCATCCAATCGGTAGTAGCG | 63 |
| Gli1 |
AAGCGTGAGCCTGAATCTGT |
CAGCATGTACTGGGCTTTGA | 61 |
| PTCH1 |
AGTGTCGCACAGAACTCCACT |
GCATAGGCGAGCATGAGTAAG | 63 |
| Snail |
GGTTCTTCTGCGCTACTGCT |
TAGGGCTGCTGGAAGGTAAA | 63 |
| Slug |
CATTTCAACGCCTCCAAAA |
GGAATGGAGCAGCGGTAGT | 63 |
Protein extraction
Cells were treated at 37°C for 72 h as follows:
LN405 with 27.5 µM cyclopamine, 32.5 µM tubastatin A
and 400 µM temozolomide; and T98G with 20 µM
cyclopamine, 30 µM tubastatin A and 400 µM
temozolomide. Concentrations and durations for the combination of
cyclopamine and tubastatin A, and the combination of temozolomide
and tubastatin A, were as for the single treatments. Additionally,
1% DMSO was used as a vehicle control. Total protein extraction was
then conducted using radio immunoprecipitation assay lysis buffer
(50 mM Tris-hidroximetil-aminometano-HCl, pH 8.0, 150 mM NaCl, 0.5%
Triton® X-100 and 0.5% sodium deoxycholate).
Western blot analysis
A total of 20 µg of each bicinchoninic
acid-quantified protein sample were separated in 12% SDS-PAGE and
then transferred to a nitrocellulose membrane. Following blocking
with TBS-Tween 0.1% and 5% non-fat milk for 1 h at room
temperature, membranes were incubated overnight with the primary
antibody at 4°C. After three washes with TBS-Tween 0.1%, membranes
were incubated with the corresponding secondary antibodies at room
temperature for 1 h. To visualize the presence and quantity of
protein, Lumi-LightPLUS Western blotting substrate (Merck KGaA) was
used. The primary antibodies used in the present study were:
Acetylated α-tubulin (cat. no. T6793; 1:10,000; Merck KGaA) and
β-actin (cat. no. A5441; 1:10,000; Merck KGaA). The secondary
antibody used was horseradish peroxidase-conjugated anti-mouse
(cat. no. SC-516102; 1:10,000; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA).
Statistical analysis
Values are expressed as the mean ± standard
deviation or as the mean ± standard error of the mean. GraphPad 7.0
Software (GraphPad Software, Inc., La Jolla, CA, USA) was used to
analyze the statistics of the results obtained from the
experiments. The statistical tests used were the one-way analysis
of variance and Tukey’s multiple comparison test. P<0.05 was
considered to indicate a statistically significant difference.
Results
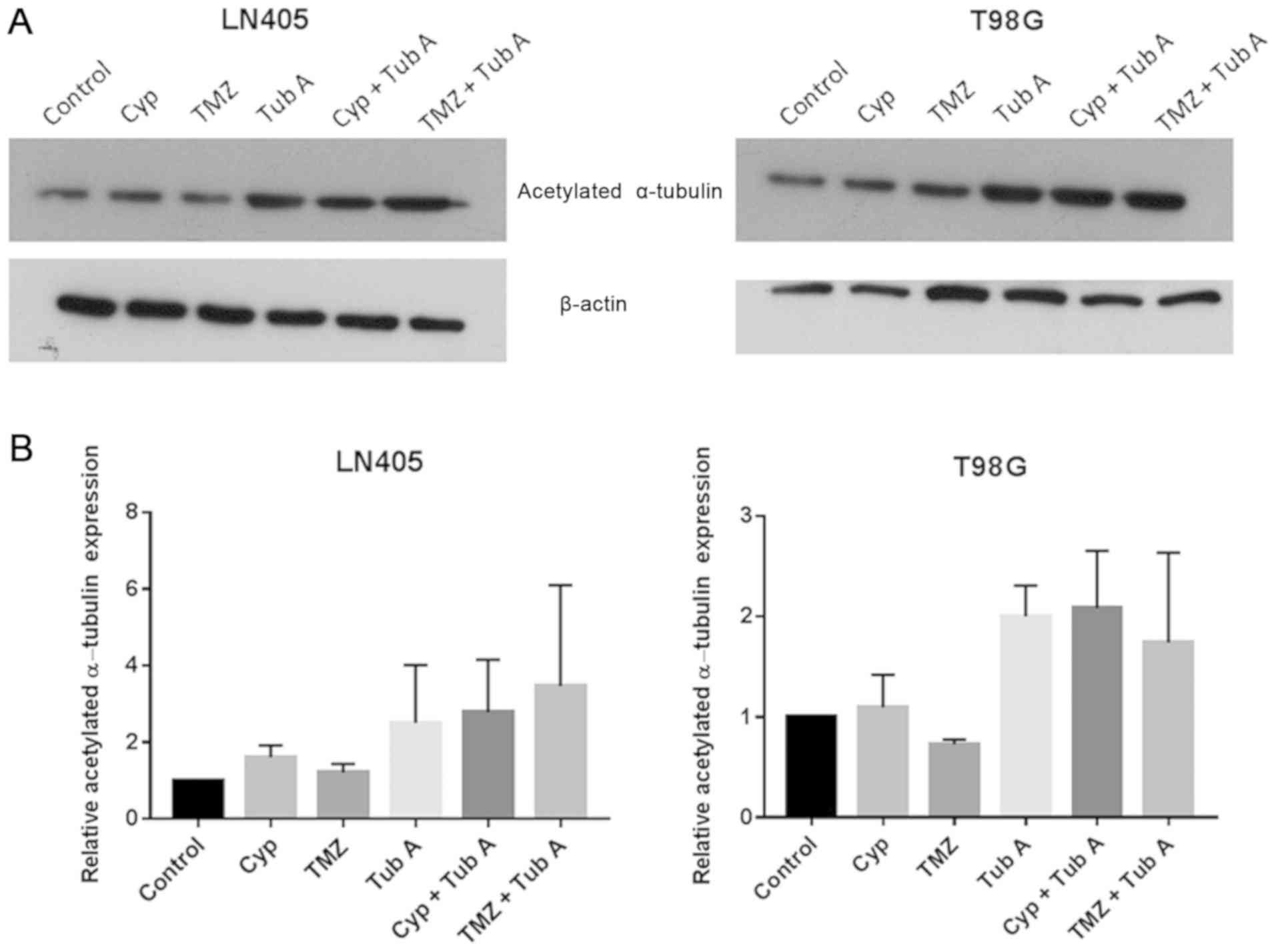
Tubastatin A increases acetylated
α-tubulin levels in glioblastoma cell lines
A western blot analysis for acetylated α-tubulin was
performed to ensure the effect of tubastatin A as an inhibitor of
HDAC6 (Fig. 1) in glioblastoma
cell lines. As expected, acetylated α-tubulin protein levels
increased in the groups treated with tubastatin A, both alone and
together with cyclopamine or temozolomide, compared with the
control group, confirming the indicated mechanism of action of this
drug. In the samples treated with cyclopamine or temozolomide
alone, no significant differences were observed.
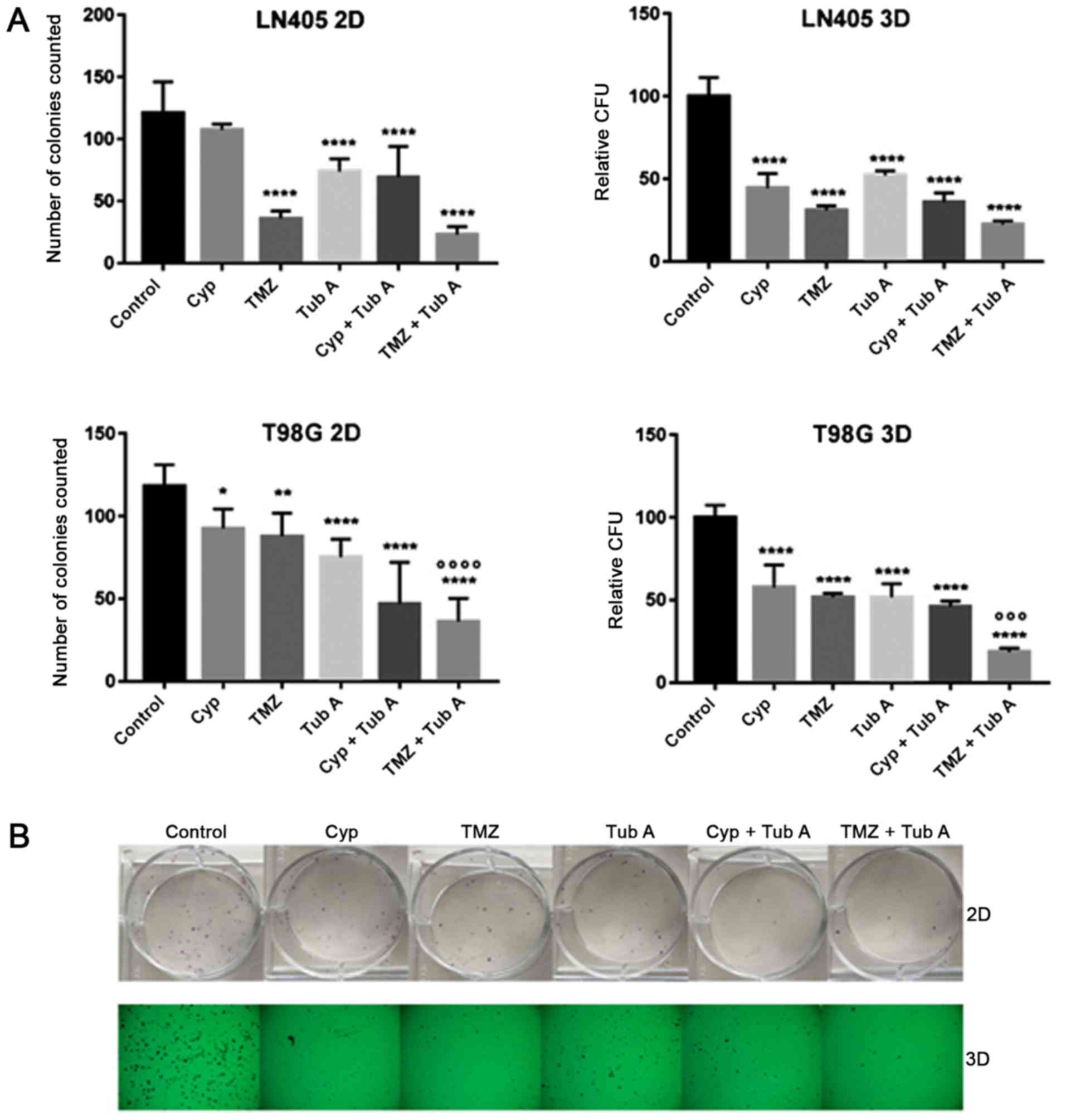
Tubastatin A, alone and combined with
temozolomide, reduces clonogenicity of glioblastoma cell lines
To evaluate the clonogenic capacity of T98G and
LN405 glioblastoma cells after treatment with cyclopamine,
temozolomide, tubastatin A, combination of cyclopamine with
tubastatin A, combination of temozolomide with tubastatin A, and
DMSO for 72 h, two different colony formation experiments were
performed: attachment-dependent (2D colonies); and
attachment-independent conditions (3D colonies) (Fig. 2). Tubastatin A single treatment,
and its combination with cyclopamine and temozolomide,
significantly reduced the number of colonies counted in both
experiments. The combination of temozolomide with tubastatin A was
the most efficient strategy for reducing clonogenicity of
glioblastoma cells, compared with the untreated group or cells
treated with temozolomide alone.
Tubastatin A decreases the migration
capacity of glioblastoma cell lines
A wound healing or scratching assay was then
conducted to analyze the migration capacity of T98G and LN405 cells
following treatment with cyclopamine, temozolomide, tubastatin A,
combination of cyclopamine with tubastatin A, and combination of
temozolomide with tubastatin A (Fig.
3). Even if all groups exhibited a closure of the scratch after
48 h, differences were evident among different treatments.
Cyclopamine alone had no significant effect on reducing cell
migration, compared with the control group. Tubastatin A induced a
reduced migration rate; however, a notable inhibition was observed
when both drugs were added together. The single treatment with
temozolomide reduced cell migration more, compared with the
individual treatment with cyclopamine. Tubastatin A produced
different results in the two cell lines. When tubastatin A was
combined with temozolomide, inhibition upon migration was enhanced,
as demonstrated by the inability of these cells to close the
gap.
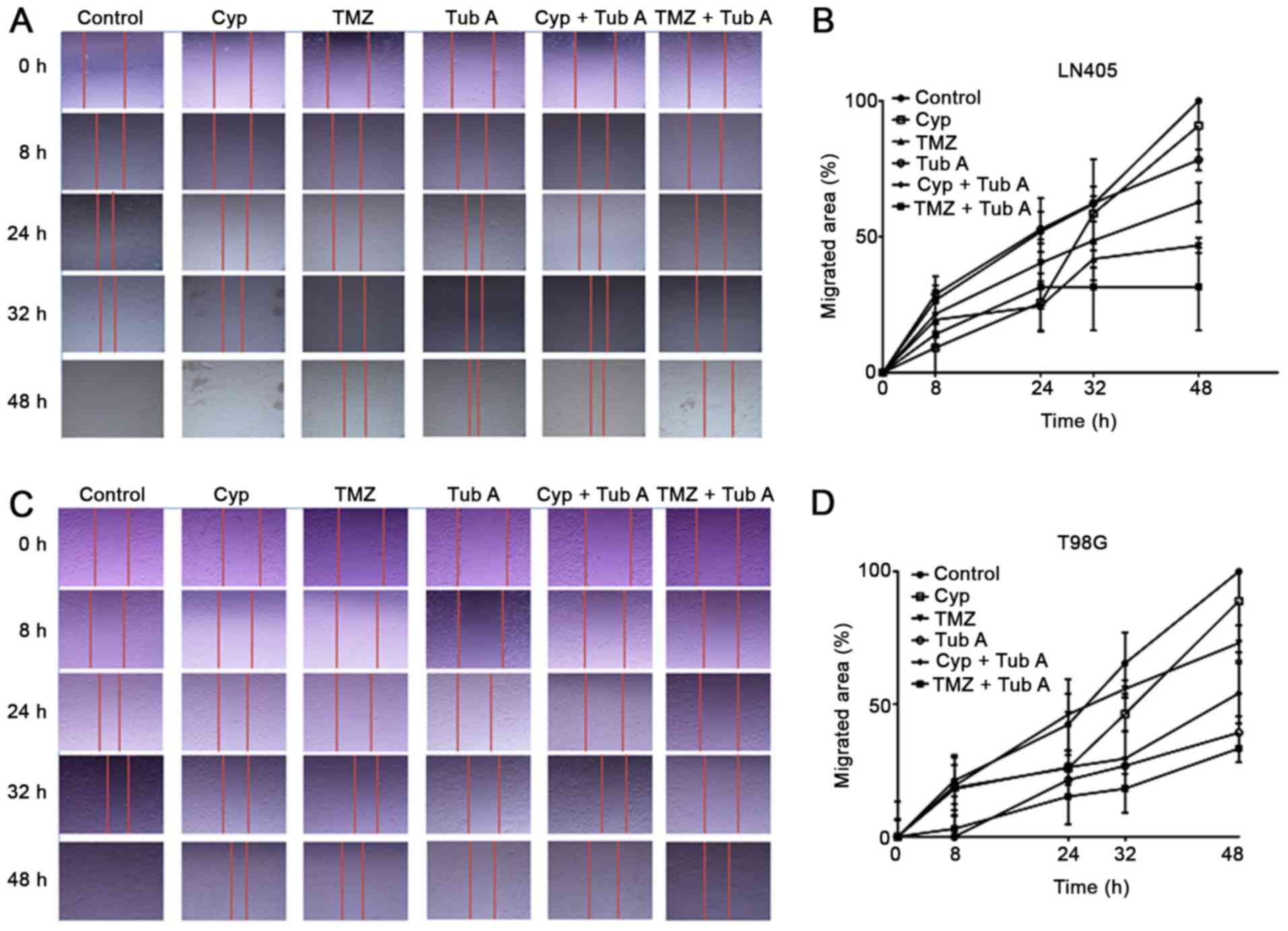 | Figure 3Changes in cell migration after
treatments of glioblastoma cells. Single treatment with TubA, and
combination with Cyp and TMZ, reduced cell migration with respect
to Cyp or TMZ alone in glioblastoma cells. (A) LN405 cells. Images
of the gap were captured at 0, 8, 24, 32 and 48 h following
scratching in every treatment condition. (B) LN405 cells. Graphs
representing the percentage of the migrated area in every condition
are incorporated. (C) T98G cells. Images of the gap were captured
at 0, 8, 24, 32 and 48 h following scratching in every treatment
condition. (D) T98G cells. Graphs representing the percentage of
the migrated area in every condition are incorporated. TubA,
tubastatin A; Cyp, cyclopamine; TMZ, temozolomide. |
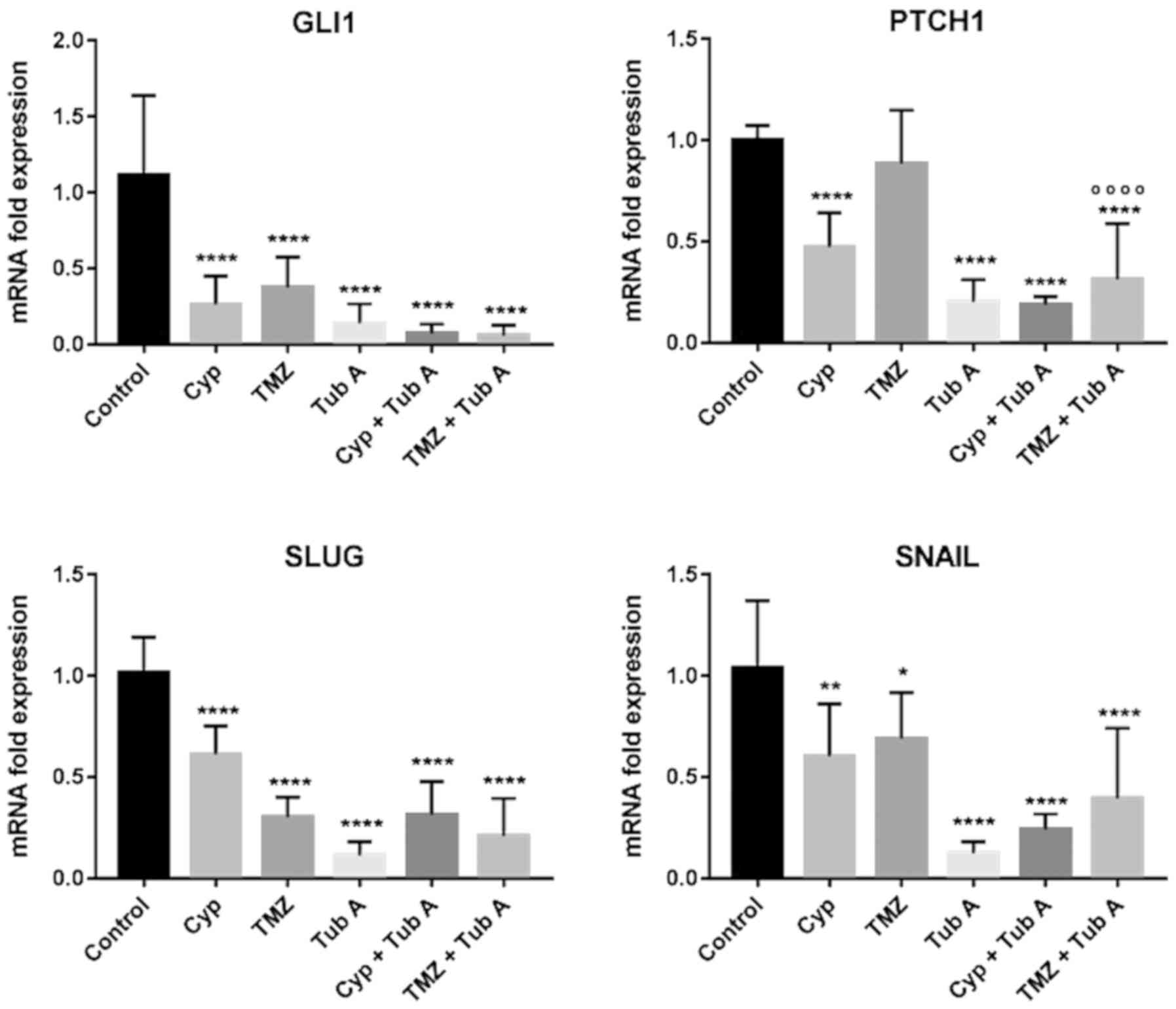
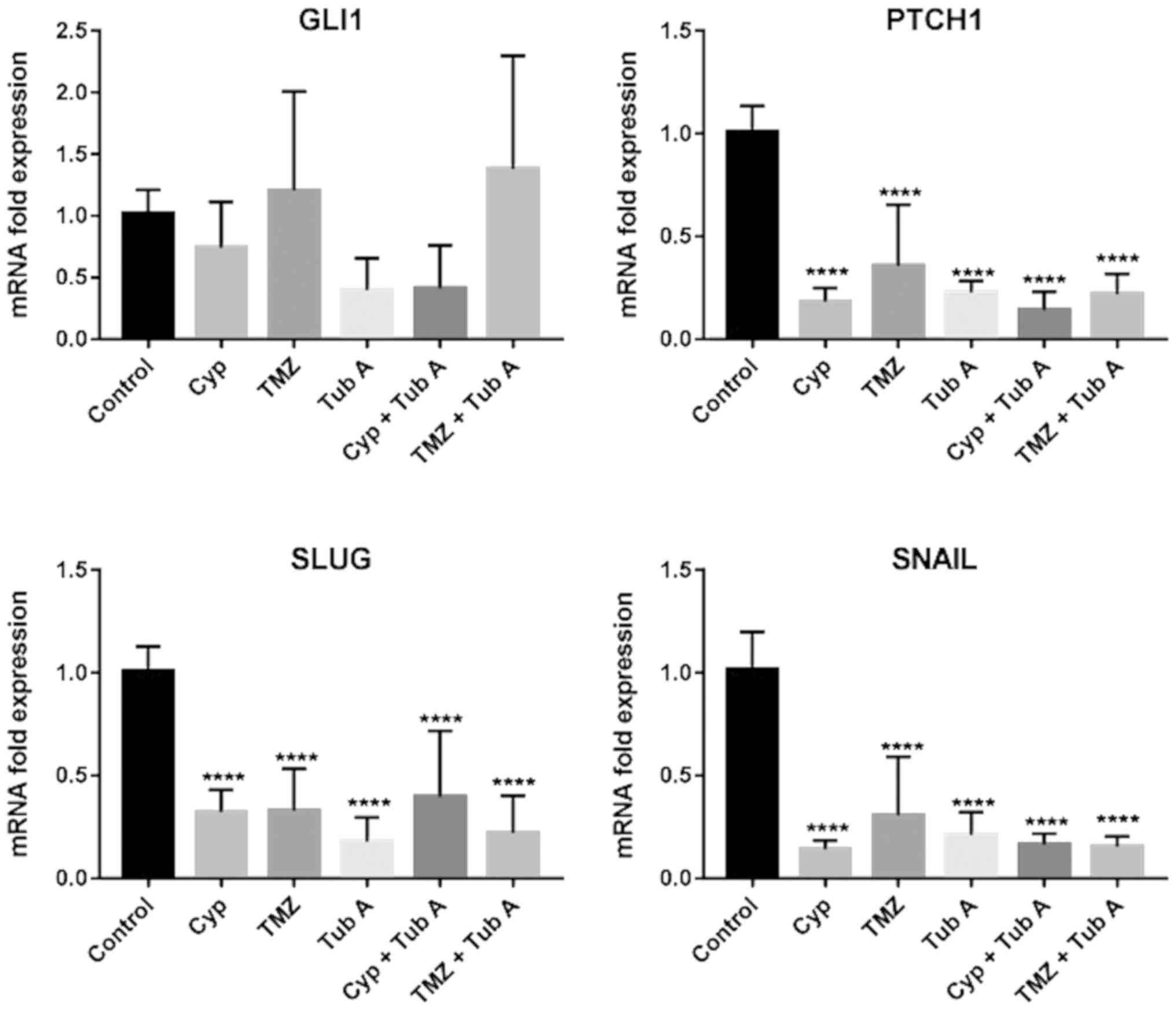
Tubastatin A downregulates the sonic Hh
pathway in glioblastoma cell lines
To observe whether tubastatin A treatment had any
effect on the regulation of the sonic Hh pathway, in LN405
(Fig. 4) and T98G (Fig. 5) glioblastoma cell lines, total RNA
was extracted from each experimental group and RT-qPCR (Table III) was performed following
reverse transcription for GLI1 and Patched 1 (PTCH1) genes.
Ribosomal 18S was used as a reference gene for the relative
quantification of these two genes using the comparative Cq method.
Treatment with cyclopamine had a significant effect on GLI1 and
PTCH1, both significantly reducing the expression following
treatment with these drugs. Nevertheless, in both cases, the most
significant decrease of PTCH1 expression was observed when cells
were treated both with cyclopamine and tubastatin A, indicating
that acetylation of α-tubulin and the possible restoration of
primary cilia may result in a downregulation of the sonic Hh
pathway. Additionally, the combined treatment of temozolomide and
tubastatin A decreased GLI and PTCH1 expression more, compared with
temozolomide alone.
Tubastatin A downregulates the expression
of mesenchymal markers in glioblastoma cell lines
EMT was investigated by RT-qPCR (Table III) for the mesenchymal markers
Snail and Snail family transcriptional repressor 2 (Slug) in LN405
(Fig. 4) and T98G (Fig. 5) glioblastoma cell lines. The
greatest reduction in expression of those markers was observed in
the tubastatin A single treatment. However, when tubastatin A did
not produce the greatest decay in expression of the markers, the
double treatments exhibited the greatest decay.
Tubastatin A accelerates temozolomide
action on apoptosis in glioblastoma cell lines
In order to investigate the effect of cyclopamine,
tubastatin A and temozolomide on apoptosis in T98G and LN405
glioblastoma cells, two different experiments were performed: Cell
death detection ELISAPLUS (Fig.
6); and Caspase-Glo 3/7 assay (Fig. 7). Evolution of both apoptosis
processes was monitored at 24, 48 and 72 h after treatment. Both
experiments demonstrated similar results, strengthening the
validation of these assays.
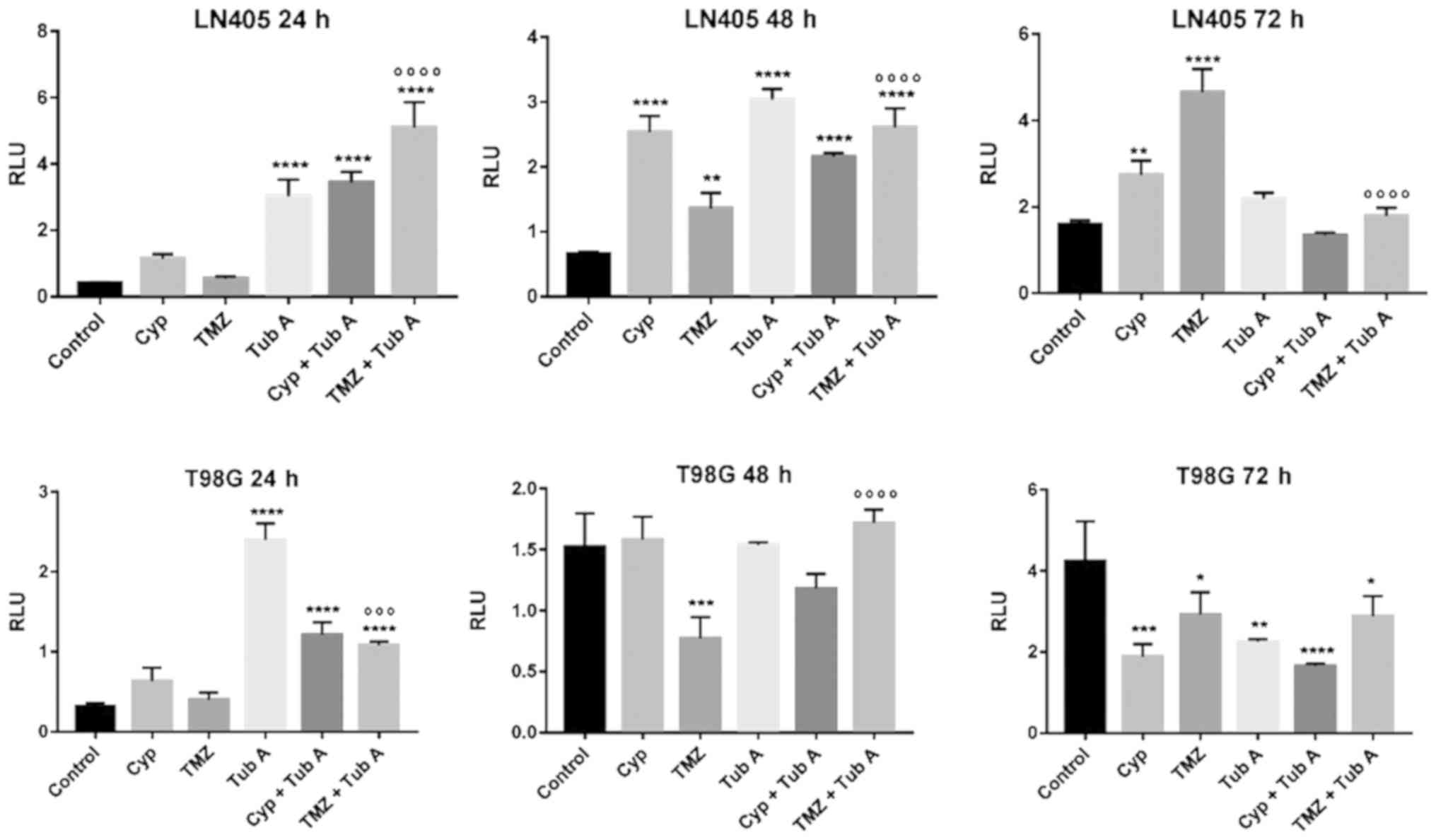 | Figure 7Changes in apoptosis, by caspase 3/7
activation, after treatments of LN405 and T98G glioblastoma cells.
Data are presented as the mean ± standard deviation.
*P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001, compared
with control; ºººP<0.001 and
ººººP<0.0001, compared with TMZ single treatment.
TubA, tubastatin A; Cyp, cyclopamine; TMZ, temozolomide; RLU,
relative light units. |
The treatment with cyclopamine had different effects
when inducing apoptosis in the two cell lines. In LN405 cells,
cyclopamine continued to induce apoptosis after 72 h of treatment,
whereas it did not have any effect in T98G cells after 48 h, when
the values were equalized with respect to its control. The
combination of cyclopamine with tubastatin A increased apoptosis at
24 h in both cell lines more than cyclopamine alone.
Tubastatin A notably induced apoptosis at 24 and 48
h with respect to the control, but this induction was attenuated
after reaching 72 h. The combination of tubastatin A with
temozolomide improved the increase of apoptosis induced by
temozolomide alone, at 24 and 48 h. However, at 72 h, temozolomide
alone produced the greatest increase in apoptosis, indicating that
the combination with tubastatin A accelerates the action of
temozolomide on apoptosis.
Discussion
Glioblastoma is the most common and the most
malignant form of brain tumor (1).
Despite the therapeutic effort conducted to treat this type of
glioma, the majority of patients develop resistance and have a poor
prognosis (2,34). The aim of the present study was to
investigate the effect of tubastatin A, a selective HDAC6
inhibitor, on glioblastoma cell lines. The cells used in the
present study, which lack the primary cilium (14), may restore it under treatment with
tubastatin A, and consequently, the cells may also reverse their
malignant phenotype and become sensitive to chemotherapy. The
present results demonstrated statistical differences encountered
following treating the glioblastoma cell lines with cyclopamine,
temozolomide, tubastatin A, combination of cyclopamine and
tubastatin A, combination of temozolomide with tubastatin A, or
DMSO, as a vehicle control.
With western blot analysis, an increase in
acetylated α-tubulin levels following treatment with tubastatin A,
alone or combined with cyclopamine or temozolomide, was
demonstrated (Fig. 1). This result
was expected, as inhibition of HDAC6 inhibits α-tubulin
deacetylation (15,35). The increase in acetylated α-tubulin
was greater when tubastatin A and temozolomide was used in the
LN405 cell line, while this increase with the combination treatment
was not observed in the T98G cell line. Therefore, it should be
considered that LN405 and T98G may produce different results, as
although they are glioblastoma cell lines, they are different at
the molecular level, as LN405 cells exhibit mutations in TP53 and
phosphatase and tensin homolog (PTEN), while T98G cell exhibit
mutations in TP53, but not in PTEN.
Both in vitro cell tumorigenicity
experiments, the 2D colony formation assay and the 3D soft agar
colony formation assay (Fig. 2),
revealed a significant decrease in clonogenicity of glioblastoma
cell lines following inhibition of HDAC6. A similar effect was
observed when tubastatin A was combined with cyclopamine in the 3D
assay. However, the combined therapy with temozolomide was the most
efficient treatment when it comes to reducing tumor growth, as
determined by the number of colonies counted. This indicates that
HDAC6 inhibition successfully reduces glioblastoma cell growth, as
occurs in other tumor types, including cholangiocarcinoma (36), and may sensitize them to
chemotherapy with temozolomide.
This observation was reinforced following analyzing
the results for the cell migration test in the wound healing assay
(Fig. 3). As occurs in the colony
formation assays, single treatment with cyclopamine or temozolomide
did not produce any significant difference, compared with the
control group. However, when tubastatin A was added, alone or
combined with any of the two other drugs, a significant inhibition
of the cell migration capacity was observed, which indicates
tubastatin A as a potential adjuvant drug to be administered
together with temozolomide.
Modulation of the sonic Hh pathway by tubastatin A
was also investigated. As depicted in Figs. 4 and 5, tubastatin A was the only drug that
achieved a decrease in the mRNA levels of GLI1 and PTCH1, and
therefore a downregulation of the sonic Hh pathway, when
administered alone. However, in the present study, a greater
decrease was expected when tubastatin A was combined with
cyclopamine, as restoration of the primary cilium caused by
tubastatin A would allow the action of cyclopamine on Smo
inhibition. This combined treatment also reduced GLI1 expression,
compared with untreated cells. These results may confirm the role
of the primary cilium in sonic Hh regulation and the efficiency of
the cyclopamine and tubastatin A treatment in modulating the
pathway when disrupted.
Finally, regarding the two genes associated with
EMT, the RT-qPCR experiments demonstrated a notable decrease in
expression of the mesenchymal genes Snail and Slug in all treated
groups, compared with the control group, even though these
differences were not statistically significant (Figs. 4 and 5). These observations indicated a
reversion of the EMT transition in these cells, as these markers
belong to a mesenchymal phenotype (37,38).
However, a report published by Stepanenko et al (39) demonstrated that a number of
glioblastoma cell lines treated with temozolomide increase vimentin
and Slug levels, although it was not the case for T98G cells.
Additionally, it was documented (40) that the loss of α-tubulin
acetylation acts as a mesenchymal marker for EMT; therefore,
increased expression of HDAC6 induces EMT. It can be considered
that treatment with tubastatin A is not only inducing the reversion
of EMT to a MET phenotype in glioblastoma cells, as it has been
demonstrated to increase acetylated α-tubulin levels, but also
represses TGF-1 induced EMT in cultured peritoneal mesothelial
cells, preventing peritoneal fibrosis (30).
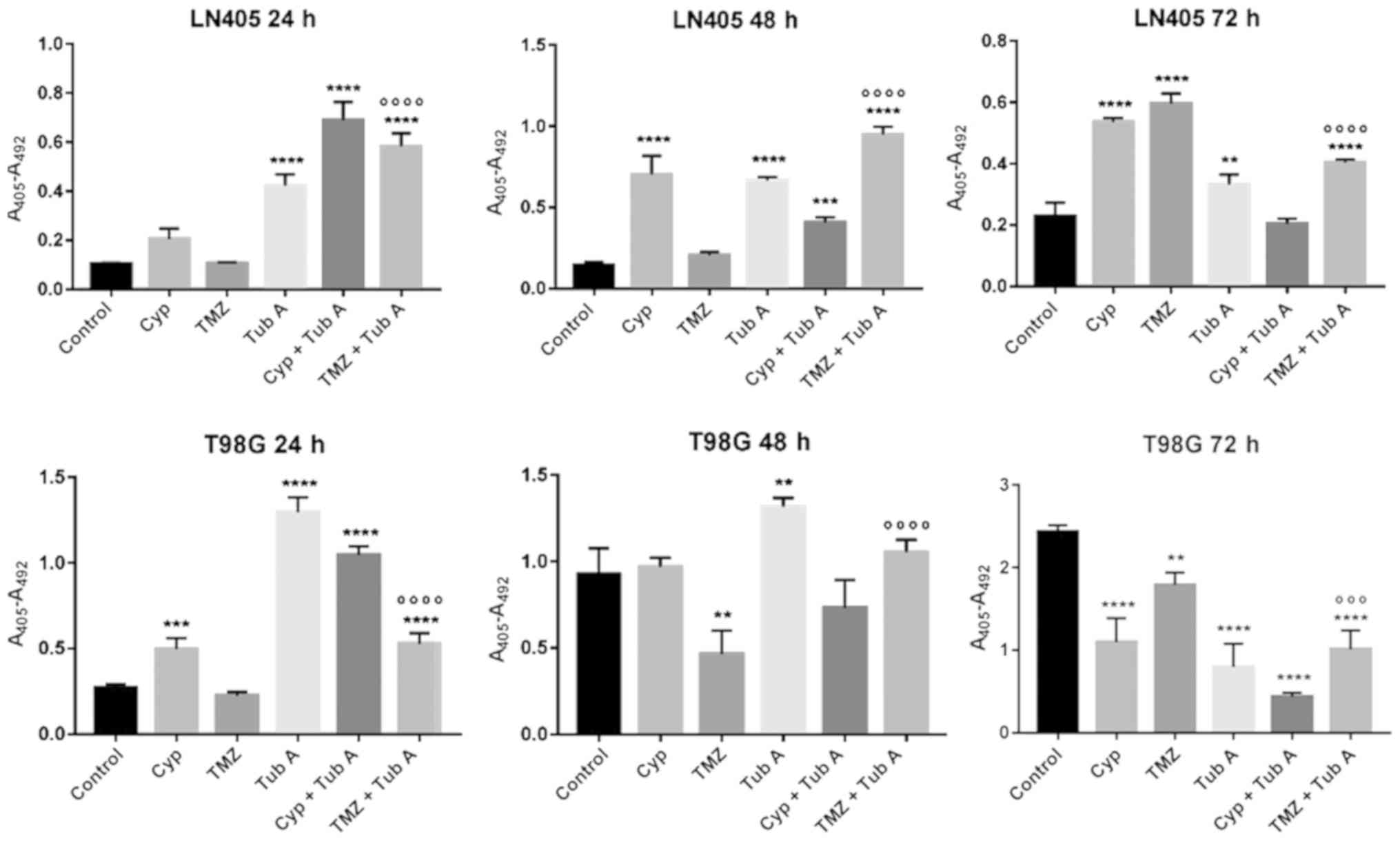
In the apoptosis assays (Fig. 6 and 7), cyclopamine increased cell death
during the first 24 h after treatment, but cannot be considered an
efficient drug when it comes to inducing apoptosis. Additionally,
the cyclopamine and tubastatin A combination treatment for 24 h
produced an increased effect, compared with cyclopamine alone.
Simultaneously, temozolomide single treatment did not produce
increased apoptosis, compared with the untreated group, which is a
poor result. Nevertheless, the most notable result from this
experiment is the highlighted effect of the inhibition of HDAC6 on
inducing apoptosis. Tubastatin A has been demonstrated to promote
cell death at a high rate not only in glioblastoma cells, but also
in other tumor types, including gastric cancer (41). Additionally, tubastatin A
accelerates temozolomide action, as the effect of single treatment
with temozolomide was already achieved 24 h earlier with the
combined treatment. This may be due to the fact that the inhibition
of HDAC6 favors the sensitization of the cells to temozolomide,
since tumors with overexpression of HDAC6 have an increased
resistance to temozolomide (42).
Furthermore, a novel mechanism for a possible explanation of the
increase of apoptosis by HDAC6 inhibition has recently been
determined (43), demonstrating
that HDAC6-selective inhibition is a novel epigenetic anticancer
therapeutic strategy targeting the p53-Hsp90 complex that can be
applied to wild-type p53-and mutated p53-bearing cancer, with
similar efficacy.
The present results indicated that the primary
cilium acts as a tumor suppressor in these glioblastoma cells, as
well as in other glioblastoma cell lines, including U87-MG, U-373G,
U-138MG or U-251MG (44), and
other tumor types, including cholangiocarcinoma (36), breast cancer (45), melanoma (46), sporadic clear cell renal cell
carcinoma (47), prostate cancer
(48) and lung cancer (49). However, there are studies that
indicate that cilia may function as an oncogene in other cases,
including pancreatic ductal carcinoma (50) and medulloblastoma (51), and other different glioblastoma
cell lines, due to the complicated heterogeneity of glioblastoma
attributed to the different responses of different cell lines or
patients (52). For this reason,
characterization of the patients is notable in order to classify
them as cilia positive or cilia negative, prior to choosing which
therapeutic strategy fits them best. However, a number of other
cellular tests and molecular experiments with more markers are
required with different glioblastoma cell lines prior to determine
the result, as the observations up to now are preliminary.
Funding
Financial support for this work was provided by a
grant from the Fundación Universidad de Navarra (Pamplona, Spain).
AU has been granted a predoctoral fellowship from the Asociación de
Amigos de la Universidad de Navarra (Pamplona, Spain).
Availability of data and materials
All data generated or analyzed during this study are
included within the manuscript.
Authors’ contributions
AU, EE, BM, JAR, MAI and JSC conceived and designed
the experiments. AU and EE performed the experiments. AU, EE and
JSC analyzed the data. EE and JSC drafted the paper. All authors
contributed to refine analysis of data and writing of the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
EMT
|
epithelial-mesenchymal transition
|
|
HDAC6
|
histone deacetylase 6
|
|
MGMT
|
O6-methylguanine-DNA
methyltransferase
|
Acknowledgments
Not applicable.
References
|
1
|
Jovčevska I, Kočevar N and Komel R: Glioma
and glioblastoma - how much do we (not) know? Mol Clin Oncol.
1:935–941. 2013.
|
|
2
|
Bleeker FE, Molenaar RJ and Leenstra S:
Recent advances in the molecular understanding of glioblastoma. J
Neurooncol. 108:11–27. 2012.
|
|
3
|
Thomas A, Tanaka M, Trepel J, Reinhold WC,
Rajapakse VN and Pommier Y: Temozolomide in the era of precision
medicine. Cancer Res. 77:823–826. 2017.
|
|
4
|
Perry J, Laperriere N, Zuraw L, Chambers
A, Spithoff K and Cairncross JG; Neuro-oncology Disease Site Group;
Cancer Care Ontario Program in Evidence-Based Care: Adjuvant
chemotherapy for adults with malignant glioma: A systematic review.
Can J Neurol Sci. 34:402–410. 2007.
|
|
5
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al European Organisation for Research and Treatment of Cancer
Brain Tumor and Radiotherapy Groups; National Cancer Institute of
Canada Clinical Trials Group: Radiotherapy plus concomitant and
adjuvant temozolomide for glioblastoma. N Engl J Med. 352:987–996.
2005.
|
|
6
|
Kitange GJ, Carlson BL, Schroeder MA,
Grogan PT, Lamont JD, Decker PA, Wu W, James CD and Sarkaria JN:
Induction of MGMT expression is associated with temozolomide
resistance in glioblastoma xenografts. Neuro-oncol. 11:281–291.
2009.
|
|
7
|
Qiu ZK, Shen D, Chen YS, Yang QY, Guo CC,
Feng BH and Chen ZP: Enhanced MGMT expression contributes to
temozolomide resistance in glioma stem-like cells. Chin J Cancer.
33:115–122. 2014.
|
|
8
|
Hegi ME, Diserens AC, Gorlia T, Hamou MF,
de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani
L, et al: MGMT gene silencing and benefit from temozolomide in
glioblastoma. N Engl J Med. 352:997–1003. 2005.
|
|
9
|
Kondo N, Takahashi A, Ono K and Ohnishi T:
DNA damage induced by alkylating agents and repair pathways. J
Nucleic Acids. 2010:5435312010.
|
|
10
|
Aldana-Masangkay GI and Sakamoto KM: The
role of HDAC6 in cancer. J Biomed Biotechnol. 2011:8758242011.
|
|
11
|
Seidel C, Schnekenburger M, Dicato M and
Diederich M: Histone deacetylase 6 in health and disease.
Epigenomics. 7:103–118. 2015.
|
|
12
|
Smith Q, Macklin B, Chan XY, Jones H,
Trempel M, Yoder MC and Gerecht S: Differential HDAC6 activity
modulates ciliogenesis and subsequent mechanosensing of endothelial
cells derived from pluripotent stem cells. Cell Rep. 24:895–908.e6.
2018.
|
|
13
|
Li S, Liu X, Chen X, Zhang L and Wang X:
Histone deacetylase 6 promotes growth of glioblastoma through
inhibition of SMAD2 signaling. Tumour Biol. 36:9661–9665. 2015.
|
|
14
|
Moser JJ, Fritzler MJ and Rattner JB:
Primary ciliogenesis defects are associated with human
astrocytoma/glioblastoma cells. BMC Cancer. 9:4482009.
|
|
15
|
Wang Z, Hu P, Tang F, Lian H, Chen X,
Zhang Y, He X, Liu W and Xie C: HDAC6 promotes cell proliferation
and confers resistance to temozolomide in glioblastoma. Cancer
Lett. 379:134–142. 2016.
|
|
16
|
Corbit KC, Aanstad P, Singla V, Norman AR,
Stainier DY and Reiter JF: Vertebrate Smoothened functions at the
primary cilium. Nature. 437:1018–1021. 2005.
|
|
17
|
Haycraft CJ, Banizs B, Aydin-Son Y, Zhang
Q, Michaud EJ and Yoder BK: Gli2 and Gli3 localize to cilia and
require the intraflagellar transport protein polaris for processing
and function. PLoS Genet. 1:e532005.
|
|
18
|
Simons M, Gloy J, Ganner A, Bullerkotte A,
Bashkurov M, Krönig C, Schermer B, Benzing T, Cabello OA, Jenny A,
et al: Inversin, the gene product mutated in nephronophthisis type
II, functions as a molecular switch between Wnt signaling pathways.
Nat Genet. 37:537–543. 2005.
|
|
19
|
Goetz SC and Anderson KV: The primary
cilium: A signalling centre during vertebrate development. Nat Rev
Genet. 11:331–344. 2010.
|
|
20
|
Braun S, Oppermann H, Mueller A, Renner C,
Hovhannisyan A, Baran-Schmidt R, Gebhardt R, Hipkiss A, Thiery J,
Meixensberger J, et al: Hedgehog signaling in glioblastoma
multiforme. Cancer Biol Ther. 13:487–495. 2012.
|
|
21
|
Hassounah NB, Bunch TA and McDermott KM:
Molecular pathways: The role of primary cilia in cancer progression
and therapeutics with a focus on Hedgehog signaling. Clin Cancer
Res. 18:2429–2435. 2012.
|
|
22
|
Pasca di Magliano M and Hebrok M: Hedgehog
signalling in cancer formation and maintenance. Nat Rev Cancer.
3:903–911. 2003.
|
|
23
|
Liu YJ, Ma YC, Zhang WJ, Yang ZZ, Liang
DS, Wu ZF and Qi XR: Combination therapy with micellarized
cyclopamine and temozolomide attenuate glioblastoma growth through
Gli1 down-regulation. Oncotarget. 8:42495–42509. 2017.
|
|
24
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009.
|
|
25
|
Skrypek N, Goossens S, De Smedt E,
Vandamme N and Berx G: Epithelial-to-mesenchymal transition:
Epigenetic reprogramming driving cellular plasticity. Trends Genet.
33:943–959. 2017.
|
|
26
|
Zeisberg M and Neilson EG: Biomarkers for
epithelial-mesenchymal transitions. J Clin Invest. 119:1429–1437.
2009.
|
|
27
|
Ramos FS, Wons L, Cavalli IJ and Ribeiro
EMSF: Epithelial-mesenchymal transition in cancer: An overview.
Integr Cancer Sci Ther. 4:1–5. 2017.
|
|
28
|
Iwadate Y: Epithelial-mesenchymal
transition in glioblastoma progression. Oncol Lett. 11:1615–1620.
2016.
|
|
29
|
Kahlert UD, Maciaczyk D, Doostkam S, Orr
BA, Simons B, Bogiel T, Reithmeier T, Prinz M, Schubert J,
Niedermann G, et al: Activation of canonical WNT/β-catenin
signaling enhances in vitro motility of glioblastoma cells by
activation of ZEB1 and other activators of
epithelial-to-mesenchymal transition. Cancer Lett. 325:42–53.
2012.
|
|
30
|
Xu L, Liu N, Gu H, Wang H, Shi Y, Ma X, Ma
S, Ni J, Tao M, Qiu A, et al: Histone deacetylase 6 inhibition
counteracts the epithelial-mesenchymal transition of peritoneal
mesothelial cells and prevents peritoneal fibrosis. Oncotarget.
8:88730–88750. 2017.
|
|
31
|
Dunigan DD, Waters SB and Owen TC: Aqueous
soluble tetrazolium/formazan MTS as an indicator of NADH- and
NADPH-dependent dehydrogenase activity. Biotechniques. 19:640–649.
1995.
|
|
32
|
Geissmann Q: OpenCFU, a new free and
open-source software to count cell colonies and other circular
objects. PLoS One. 8:e540722013.
|
|
33
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008.
|
|
34
|
Urdiciain A, Meléndez B, Rey JA, Idoate MA
and Castresana JS: Panobinostat potentiates temozolomide effects
and reverses epithelial–mesenchymal transition in glioblastoma
cells. Epigenomes. 2:52018.
|
|
35
|
Yang W, Liu Y, Gao R, Yu H and Sun T:
HDAC6 inhibition induces glioma stem cells differentiation and
enhances cellular radiation sensitivity through the SHH/Gli1
signaling pathway. Cancer Lett. 415:164–176. 2018.
|
|
36
|
Gradilone SA, Radtke BN, Bogert PS, Huang
BQ, Gajdos GB and LaRusso NF: HDAC6 inhibition restores ciliary
expression and decreases tumor growth. Cancer Res. 73:2259–2270.
2013.
|
|
37
|
Han SP, Kim JH, Han ME, Sim HE, Kim KS,
Yoon S, Baek SY, Kim BS and Oh SO: SNAI1 is involved in the
proliferation and migration of glioblastoma cells. Cell Mol
Neurobiol. 31:489–496. 2011.
|
|
38
|
Myung JK, Choi SA, Kim SK, Wang KC and
Park SH: Snail plays an oncogenic role in glioblastoma by promoting
epithelial mesenchymal transition. Int J Clin Exp Pathol.
7:1977–1987. 2014.
|
|
39
|
Stepanenko AA, Andreieva SV, Korets KV,
Mykytenko DO, Baklaushev VP, Huleyuk NL, Kovalova OA, Kotsarenko
KV, Chekhonin VP, Vassetzky YS, et al: Temozolomide promotes
genomic and phenotypic changes in glioblastoma cells. Cancer Cell
Int. 16:362016.
|
|
40
|
Gu S, Liu Y, Zhu B, Ding K, Yao TP, Chen
F, Zhan L, Xu P, Ehrlich M, Liang T, et al: Loss of α-tubulin
acetylation is associated with TGF-β-induced epithelial-mesenchymal
transition. J Biol Chem. 291:5396–5405. 2016.
|
|
41
|
Dong J, Zheng N, Wang X, Tang C, Yan P,
Zhou HB and Huang J: A novel HDAC6 inhibitor exerts an anti-cancer
effect by triggering cell cycle arrest and apoptosis in gastric
cancer. Eur J Pharmacol. 828:67–79. 2018.
|
|
42
|
Li ZY, Zhang C, Zhang Y, Chen L, Chen BD,
Li QZ, Zhang XJ and Li WP: A novel HDAC6 inhibitor Tubastatin A:
Controls HDAC6-p97/VCP-mediated ubiquitination-autophagy turnover
and reverses Temozolomide-induced ER stress-tolerance in GBM cells.
Cancer Lett. 391:89–99. 2017.
|
|
43
|
Ryu HW, Shin DH, Lee DH, Choi J, Han G,
Lee KY and Kwon SH: HDAC6 deacetylates p53 at lysines 381/382 and
differentially coordinates p53-induced apoptosis. Cancer Lett.
391:162–171. 2017.
|
|
44
|
Sarkisian MR, Siebzehnrubl D, Hoang-Minh
L, Deleyrolle L, Silver DJ, Siebzehnrubl FA, Guadiana SM,
Srivinasan G, Semple-Rowland S, Harrison JK, et al: Detection of
primary cilia in human glioblastoma. J Neurooncol. 117:15–24.
2014.
|
|
45
|
Menzl I, Lebeau L, Pandey R, Hassounah NB,
Li FW, Nagle R, Weihs K and McDermott KM: Loss of primary cilia
occurs early in breast cancer development. Cilia. 3:72014.
|
|
46
|
Kim J, Dabiri S and Seeley ES: Primary
cilium depletion typifies cutaneous melanoma in situ and malignant
melanoma. PLoS One. 6:e274102011.
|
|
47
|
Schraml P, Frew IJ, Thoma CR, Boysen G,
Struckmann K, Krek W and Moch H: Sporadic clear cell renal cell
carcinoma but not the papillary type is characterized by severely
reduced frequency of primary cilia. Mod Pathol. 22:31–36. 2009.
|
|
48
|
Liu Z, Rebowe RE, Wang Z, Li Y, Wang Z,
DePaolo JS, Guo J, Qian C and Liu W: KIF3a promotes proliferation
and invasion via Wnt signaling in advanced prostate cancer. Mol
Cancer Res. 12:491–503. 2014.
|
|
49
|
Kim M, Suh YA, Oh JH, Lee BR, Kim J and
Jang SJ: KIF3A binds to β-arrestin for suppressing Wnt/β-catenin
signalling independently of primary cilia in lung cancer. Sci Rep.
6:327702016.
|
|
50
|
Emoto K, Masugi Y, Yamazaki K, Effendi K,
Tsujikawa H, Tanabe M, Kitagawa Y and Sakamoto M: Presence of
primary cilia in cancer cells correlates with prognosis of
pancreatic ductal adenocarcinoma. Hum Pathol. 45:817–825. 2014.
|
|
51
|
Barakat MT, Humke EW and Scott MP: Kif3a
is necessary for initiation and maintenance of medulloblastoma.
Carcinogenesis. 34:1382–1392. 2013.
|
|
52
|
Lai SW, Huang BR, Liu YS, Lin HY, Chen CC,
Tsai CF, Lu DY and Lin C: Differential characterization of
temozolomide-resistant human glioma cells. Int J Mol Sci.
19:1272018.
|