Introduction
Ovarian cancer is one of the most lethal
gynecological malignancies (1-3). It
has an unfavorable prognosis, and numerous of patients are
diagnosed at an advanced stage, because of the absence of
representative symptoms and sensitive diagnostic approaches
(4). Cytoreductive surgery and
postoperative adjuvant chemotherapy using platinum-based compounds
and taxanes, as a single or combination treatment, have been
standard for ovarian cancer (5).
Therefore, chemotherapy is the inevitable therapeutic option for
ovarian cancer. However, chemoresistance is a major hindrance to
clinical trials for this disease. Furthermore, ~75% of patients who
are initially sensitive to the platinum/paclitaxel-based
chemotherapy relapse due to chemoresistance, which results in
therapeutic failure, causing >90% of related deaths (6). Therefore, it is highly necessary to
develop new treatment strategies against chemoresistant ovarian
cancers.
Metformin has been widely used for the treatment of
type 2 diabetes mellitus for decades. Metformin is a complex drug
with various mechanisms of action. Previous studies have reported
that metformin decreases glucose production in the liver (7,8) and
increases glucose utilization in the gut, altering the microbiome
in the intestine and increasing glucagon-like peptide 1 secretion
(9). Molecularly, the established
direct target of metformin is the mitochondrial complex I in the
electron transport chain, which metformin binds to and inhibits,
thereby decreasing mitochondrial respiration and ATP production
(10). In vivo and in
vitro studies have demonstrated that metformin-induced energy
depletion could activate AMP-activated protein kinase (AMPK) in the
liver and hepatocytes, respectively (11,12).
Because AMPK serves a critical role in regulating metabolism and
maintaining cellular energy homeostasis, it has been considered an
important therapeutic target for controlling human diseases,
including metabolic diseases and cancer (13).
Accumulating in vitro and in vivo
studies have suggested that metformin has anticancer properties
and, therefore, inhibits the growth of various types of cancer,
including gastric, esophageal, colon and breast cancers (14-18).
The primary mechanism of the antitumor effects of metformin is
activating the AMPK signaling pathway. Activated AMPK activates the
tumor suppressor tuberous sclerosis complex 1 and 2 (TSC1/2), which
then negatively regulates mammalian target of rapamycin (mTOR).
mTOR is a key mediator of phosphatidylinositol 3-kinase (PI3K)/AKT
signaling, which is one of the most frequently altered pathways in
human cancer (19,20). Additionally, previous studies have
demonstrated that metformin decreases the activation of AKT in
several cancer cells not only via AMPK-dependent but also
independent mechanisms (21-24).
Although several reports suggest that metformin downregulates the
PI3K/AKT pathway, many aspects of the regulatory mechanism remain
unclear.
AKT, a well-known serine/threonine protein kinase,
has important roles in cell survival, proliferation and tumor
development (25). A previous
report from our group has demonstrated that mitochondrial E3
ubiquitin protein ligase 1 (MUL1) negatively regulates AKT, through
the induction of K48-linked polyubiquitination at the K284 residue
(26). This polyubiquitination of
AKT by MUL1 subsequently leads to its proteasomal degradation
(26).
The present study demonstrated that metformin
inhibited the growth of chemoresistant cancer cell lines.
Furthermore, the current results revealed that metformin
downregulated AKT protein expression by upregulating MUL1 E3
ligase. These findings we suggest that MUL1 may have a key role in
the antitumor effects of metformin.
Materials and methods
Reagents and cell culture
Human ovarian cancer A2780 cells were purchased from
the European Collection of Authenticated Cell Cultures (Salisbury,
UK), while SKOV3 and paclitaxel-resistant SKOV3-TR cells were
kindly provided by Dr Anil K Sood (The University of Texas MD
Anderson Cancer Center, Houston, TX, USA). A2780/Cis cells were
kindly provided by Professor Jae Ho Lee (Cheil General Hospital and
Women’s Healthcare, Seoul, Republic of Korea). The cells were
maintained in RPMI-1640 medium (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 10% (v/v) fetal bovine serum
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) and 1% (v/v)
penicillin/streptomycin. All the cells were incubated in a
humidified atmosphere of 5% CO2 at 37°C. Metformin
(Sigma-Aldrich; Merck KGaA) was dissolved in phosphate-buffered
saline (PBS). Human AKT serine/threonine kinase 2 (AKT2) cDNA was
cloned into pcDNA3.1-Myc/His (Invitrogen; Thermo Fisher Scientific,
Inc.), as previously described (26). The HA-ubiquitin (HA-Ub) plasmid
pMT123 was kindly provided by Dr Dirk Bohmann (University of
Rochester, Rochester, NY, USA).
Cell viability
Cells were seeded in 96-well plates
(2×104 cells/well), and their viability was evaluated
using the water-soluble tetrazolium (WST)-1 assay (EZ-Cytox cell
viability assay kit; ITSBio, Seoul, Korea), according to the
manufacturer’s protocol. Briefly, the cells were treated with the
indicated concentrations of metformin for 72 h and then the WST-1
solution was added to each well. The absorbance of the reaction
solution was then measured at 450 nm with a reference wavelength of
655 nm using an iMark microplate reader (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). Proliferation was assessed using the BrdU
cell proliferation assay (Cell Signaling Technology, Inc., Danvers,
MA, USA), according to the manufacturer’s protocol.
Immunoblotting
The protein expression levels were determined using
western blot analysis. The cells were harvested and lysed in
radioimmunoprecipitation assay (RIPA) lysis buffer (Thermo Fisher
Scientific, Inc.), supplemented with EDTA-free protease inhibitor
cocktail (Roche Diagnostics, Indianapolis, IN, USA). Total protein
concentration was determined using a bicinchoninic acid (BCA)
protein assay kit (Thermo Fisher Scientific, Inc.), according to
the manufacturer’s instruction. Then, 5X SDS sample buffer was
added to each cell lysate sample, and 40 µg of proteins were
loaded into 8-12% SDS-PAGE gel and separated. Then, the proteins
were transferred onto a nitrocellulose membrane (Whatman; Thermo
Fisher Scientific, Inc.). The membrane was blocked with 5% skim
milk for 1 h and subsequently incubated with the indicated
antibodies overnight at 4°C. After washing with Tris-buffered
saline with 0.1% Tween-20 (TBST), the membranes were incubated with
a horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rabbit
secondary antibody (Cell Signaling Technology, Inc.). Proteins were
visualized using enhanced chemiluminescence (ECL) reagents (Bio-Rad
Laboratories, Inc.) and detected with the ChemiDoc Touch Imaging
system (Bio-Rad Laboratories, Inc.). Anti-AMPK (cat. no. 2532),
anti-phosphorylated (p-) AMPK (cat. no. 50081), anti-AKT (cat. no.
4691), anti-AKT serine/threonine kinase 1 (AKT1; cat. no. 2967),
anti-AKT2 (cat. no. 5239), anti- AKT serine/threonine kinase 3
(AKT3; cat. no. 4059), anti-p-AKT (S473; cat. no. 9271),
anti-glycogen synthase kinase 3β (GSK3β; cat. no. 9315),
anti-p-GSK3β (cat. no. 9323), anti-Cyclin D1 (cat. no. 2922),
anti-β-actin (cat. no. 4967) and anti-Myc-tag (cat. no. 2272)
antibodies were purchased from Cell Signaling Technology, Inc.
Anti-MUL1 (cat. no. HPA026837) antibody was purchased from
Sigma-Aldrich (Merck KGaA). Anti-HA-tag antibody (cat. no. SC-7392)
was purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX,
USA). The primary antibodies were diluted to 1:1,000 in TBST. The
secondary anti-mouse IgG (cat. no. 7076) and anti-rabbit IgG (cat.
no. 7074) were purchased from Cell Signaling Technology, Inc., and
diluted to 1:5,000 in TBST. The intensity of each protein band
(normalized to β-actin) was quantified using ImageJ software
(version 1.6.0; National Institute of Health, Bethesda, MD,
USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNAs were isolated the TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). Reverse transcription
was performed to synthesize cDNA with 1 µg of total RNA
using 1X First-Strand buffer, 10 mM DTT, 10 U/µl Moloney
Murine Lukemia Virus (M-MLV) Reverse Transcriptase, 2 U/µl
RNaseOUT Recombinant Ribonucleaase Inhibitor (all from Invitrogen;
Thermo Fisher Scientific, Inc.), 0.5 mM dNTP Mix (Takara Bio Inc,
Shiga, Japan), and 100 pmol oligo(dT) primer (Bionics, Seoul,
Republic of Korea). The reaction mixture (20 µl) was
incubated for 50 min at 37°C, 15 min at 70°C and then held at 4°C.
qPCR was performed using the StepOnePlus system (Thermo Fisher
Scientific, Inc.). Each reaction (20 µl) was performed using
EvaGreen dye-based 1X HOT FIREPol EvaGreen qPCR Mix Plus (Solis
BioDyne, Tartu, Estonia), 1 µl of RT product and 10
pmol/µl primers. The reaction was incubated at 12 min at
95°C, followed by 40 cycles at 95°C for 15 sec, 50°C for 30 sec and
72°C for 30 sec. Relative quantification of MUL1 expression was
calculated according to the 2−∆∆Cq method (27) and normalized by an endogenous
internal control (β-actin) expression. The primers used were as
follows: β-actin, 5′-GGA TTC CTA TGT GGG CGA CGA-3′ (forward) and
5′-CGC TCG GTG AGG ATC TTC ATG-3′ (reverse); and MUL1, 5′-CAC AAG
ATG GTG TGG AAT CG-3′ (forward) and 5′-TCA GCA TCT CCT CGG TCT
CT-3′ (reverse).
RNA interference (RNAi)
SKOV3-TR and A2780/Cis cells were transfected with
100 pmol of MUL1 small interfering RNA (siRNA; Bioneer, Corporation
Daejeon, Korea) using Lipofectamine RNAiMAX (Invitrogen; Thermo
Fisher Scientific, Inc.). The sense sequence of MUL1 siRNA was
5′-GGGAUUUUUAUCUCGAGGC-3′. RNAi targeting MUL1 was delivered to the
cells using a lentivirus encoding MUL1 short hairpin (sh) RNA as
previously described (26).
In vivo ubiquitination assay
In vivo ubiquitination assays were performed
as previously described (26).
SKOV3-TR and A2780/Cis cells were transfected with Myc/His-tagged
AKT2 and HA-tagged ubiquitin and treated with metformin for 48 h.
Then the cells were treated with proteasome inhibitor MG132 for 6 h
prior to cell lysis. The cells were gathered, washed and lysed in
200 µl of denaturing lysis buffer (50 mM Tris-HCl pH 7.4,
0.5% SDS and 70 mM β-mercaptoethanol) by vortexing and boiling for
15 min at 95°C. The lysates were diluted with 800 µl buffer
A (50 mM NaH2PO4, 300 mM NaCl, and 10 mM
imidazole, pH 8.0) containing protease inhibitor cocktail and
MG132. Diluted lysates were incubated overnight at 4°C with Ni-NTA
beads (Qiagen GmbH, Hilden, Germany), which have an affinity for
proteins carrying a His tag. The beads were washed five times with
buffer B (50 mM NaH2PO4, 300 mM NaCl, and 20
mM imidazole, pH 8.0). Bound proteins were eluted by boiling in
SDS-PAGE sample buffer. Eluted proteins were immunoblotted with
anti-HA antibody for determination of ubiquitination levels of
AKT2.
Cell cycle analysis
The cells were harvested with trypsin, fixed in 70%
cold ethanol overnight at 4°C, and then stained with propidium
iodide (PI) solution for 1 h in the dark at 37°C. The cell pellets
were washed with PBS, and the cellular DNA content was analyzed
using a BD FACSCalibur flow cytometry platform (BD Biosciences, San
Jose, CA, USA). Cell cycle fractions were quantified using the Cell
Quest software (BD Biosciences).
Clonogenic assay
The cells were seeded at 1.5×103
cells/well in six-well cell culture plates and incubated for 24 h.
After 72 h exposure to 20 mM metformin, the cells were washed and
the medium was replaced with fresh medium. Then, the cells were
incubated for another 14 days, and the cell colonies were stained
with 0.1% crystal violet solution. The colonies on random area of
each well were counted, and the results were quantified using Image
J software (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
All data are presented as mean ± standard deviation
from triplicate experiments. Results were analyzed for statistical
significance using GraphPad Prism version 5 (GraphPad software,
Inc., San Diego, CA, USA) with the Student’s t-test or one-way
ANOVA followed by Tukey’s test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Metformin has anticancer activity against
chemoresistant ovarian cancer cell lines
Previous studies have reported that metformin
inhibits chemoresistant cancer cell growth, including that of the
ovarian cancer cell lines, SKOV3-TR and A2780/cis (28,29).
The present study further examined the in vitro cell growth
inhibition and antiproliferative effects of metformin on parental
and chemoresistant ovarian cancer cell lines, in specific SKOV3 and
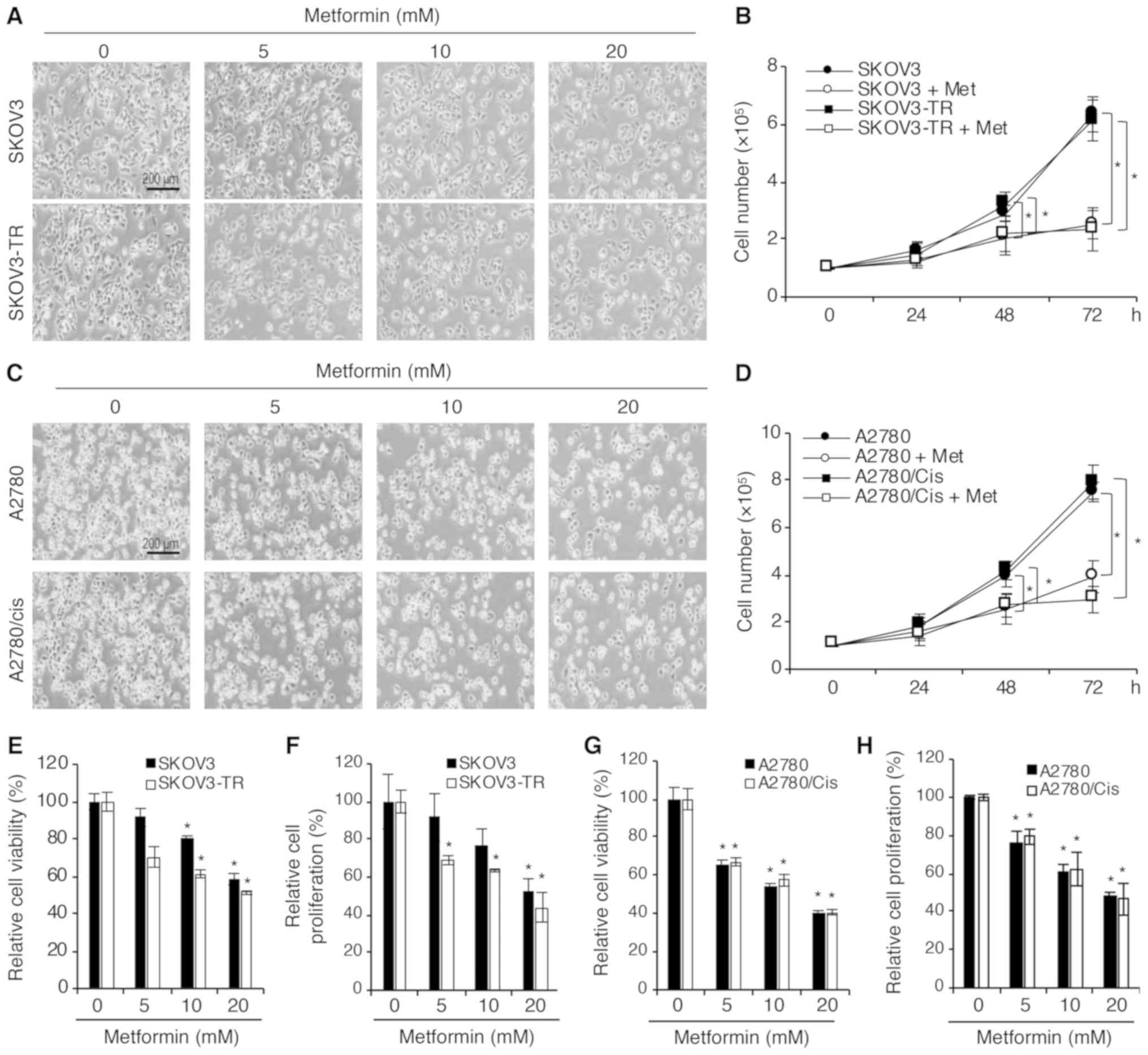
SKOV3-TR, and A2780 and A2780/cis. First, confluency changes
following metformin treatment were investigated. Consistent with
previous studies, metformin decreased cell confluency in all the
cell lines tested in a concentration-dependent manner (Fig. 1A and C). In addition, 20 mM
metformin significantly inhibited the growth of SKOV3 and SKOV3-TR
cells in a time-dependent manner (Fig.
1B). A similar result was observed in A2780 and A2780/cis cells
(Fig. 1D). The effect of metformin
on cell viability and proliferation was further evaluated. SKOV3,
SKOV3-TR, and A2780, A2780/cis cells were treated with various
concentrations of metformin for 48 h. The WST-1 assay demonstrated
that cell viability was significantly decreased in all cell lines
in a concentration-dependent manner (Fig. 1E and G). Furthermore, as shown in
Fig. 1F and H, the proliferation
of all cell lines was inhibited in a concentration-dependent manner
following exposure to metformin for 48 h. These data demonstrated
that metformin had anticancer activity not only on the parental but
also on the chemoresistant ovarian cancer cell lines. Thus, the
underlying mechanism of metformin was further investigated in the
present study using these two cell lines, SKOV3-TR and
A2780/cis.
Metformin decreases AKT expression in a
proteasome-dependent manner in parental and chemoresistant ovarian
cancer cell lines
The anticancer effect of metformin has been
previously reported to be mediated by regulation of AKT signaling
in various types of cancer (21-24).
Therefore, the present study sought to determine if metformin
regulated the activation of AKT in parental SKOV3 and A2780, and
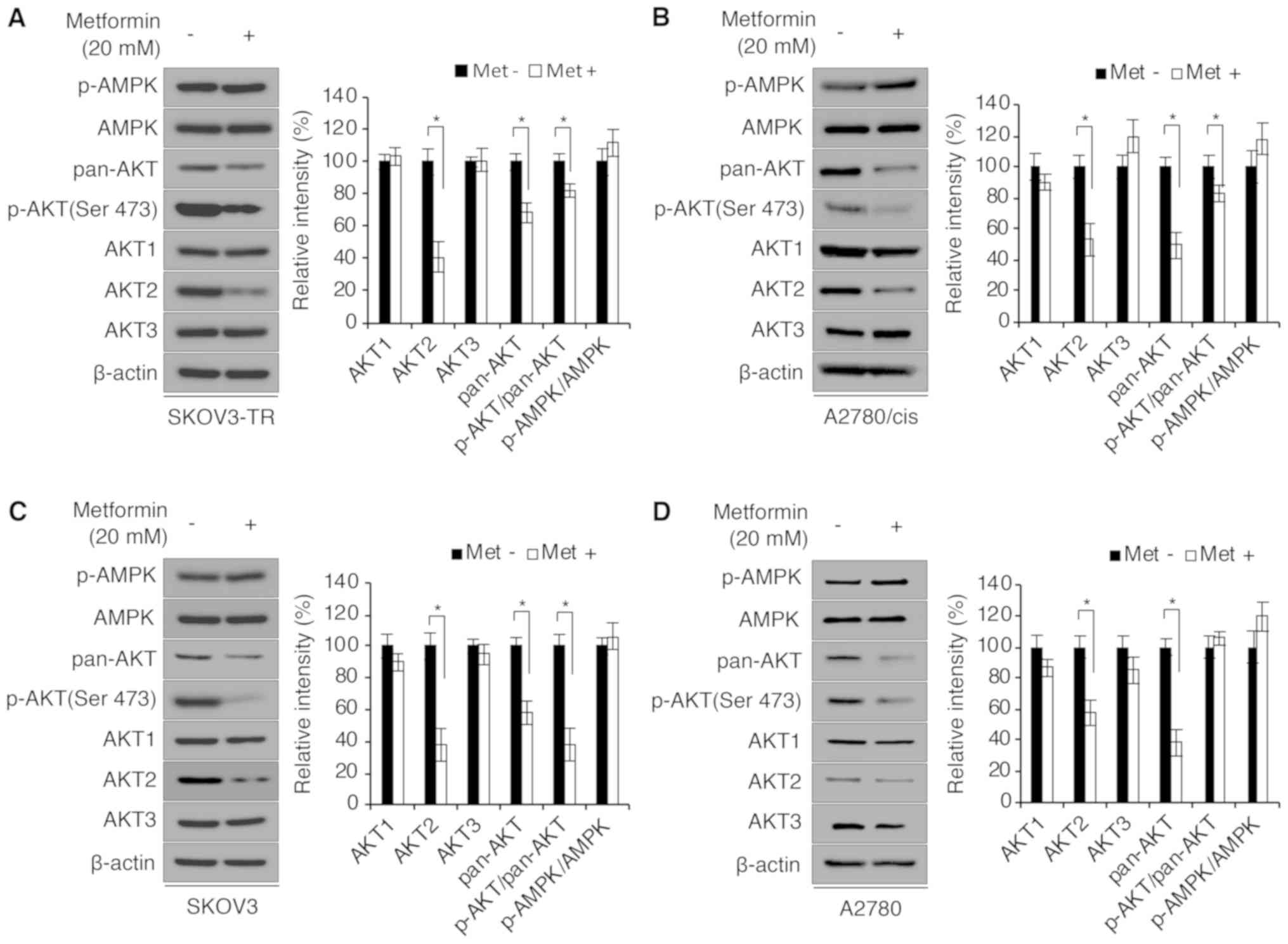
chemoresistant SKOV3-TR and A2780/cis cells. To this end, SKOV3-TR
and A2780/cis cells were treated with 20 mM metformin for 72 h. As
illustrated in Fig. 2A and B,
metformin significantly decreased p-AKT (Ser473) expression in both
cell lines. Although previous studies have demonstrated that
metformin increases the phosphorylation of AMPK and regulates the
PI3K/AKT pathway in an AMPK-dependent manner (21,30),
a significant difference in p-AMPK (Thr472) expression was not
observed in the present study. Thus, the mRNA and protein
expression levels of the AKT subfamily members, AKT1, AKT2 and
AKT3, were examined. Notably, among the AKT family of proteins, the
expression levels of AKT2 were significantly decreased following
metformin treatment (Fig. 2A and
B), but the mRNA expression levels of the AKT family
members were not changed (data not shown). Consistent with these
data, metformin was demonstrated to also decrease AKT2 protein
levels in parental SKOV3 and A2780 cells (Fig. 2C and D). Therefore, it was
hypothesized that metformin regulated AKT expression levels
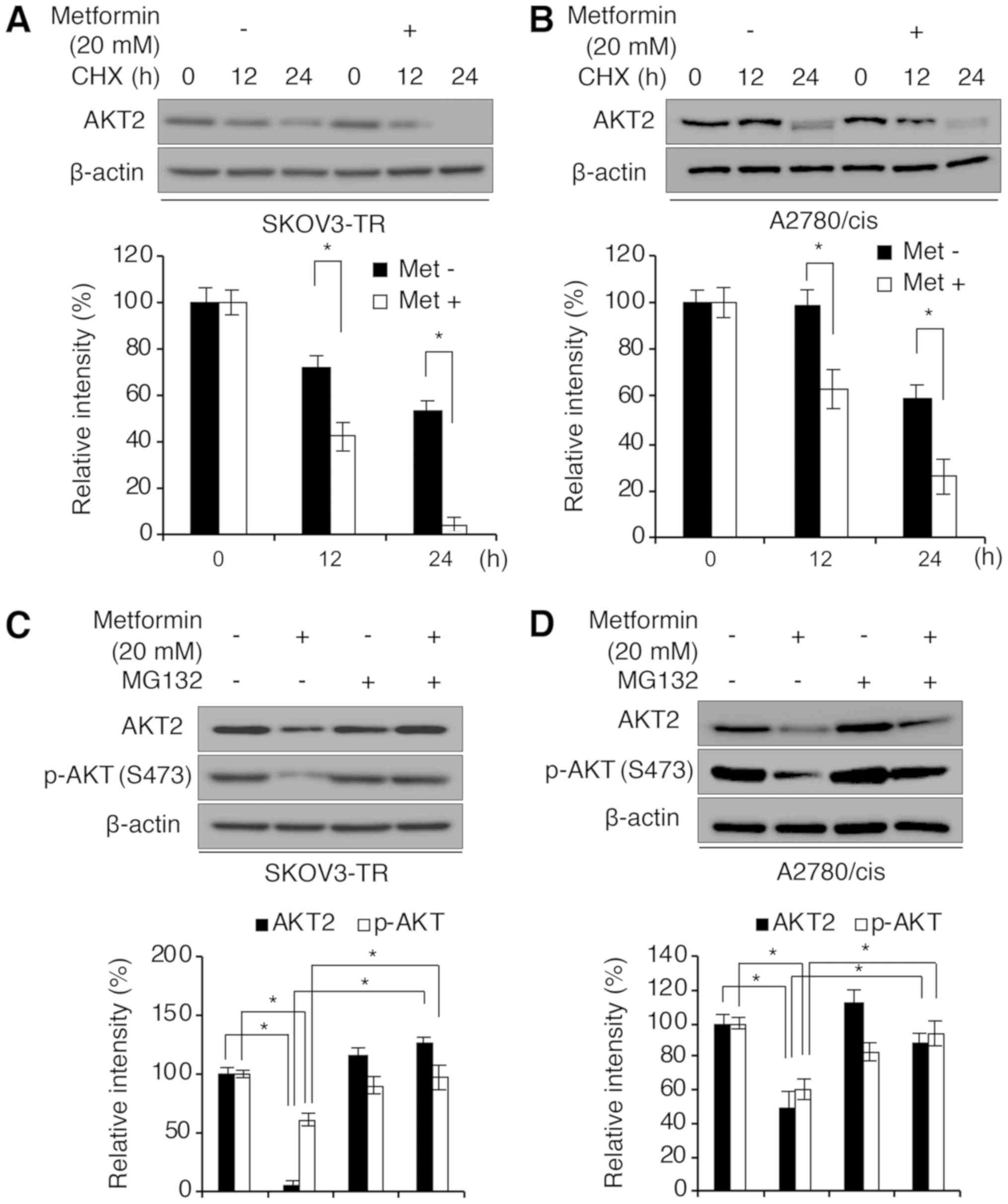
post-translationally. To investigate the difference in AKT
degradation following metformin exposure (effect on AKT2 protein
degradation by metformin), SKOV3-TR and A2780/cis cells were
treated with cycloheximide (CHX) to block de novo protein
synthesis following dimethyl sulfoxide (DMSO; vehicle control) or
20 mM metformin treatment. As illustrated in Fig. 3A and B, metformin treatment
significantly accelerated the protein degradation of AKT2 in both
SKOV3-TR and A2780/cis cell lines. To elucidate the mechanism of
metformin-induced AKT2 degradation, we then investigated whether
inhibition of the proteasome-dependent protein degradation pathway
could abrogate the effect of metformin on AKT2 protein stability.
SKOV3-TR and A2780/cis cells were treated with the peptide aldehyde
proteasome inhibitor MG132 or DMSO (vehicle control) for 12 h
following incubation with or without metformin for 48 h. MG132
treatment rescued the decreased protein expression of AKT2 and
p-AKT induced by metformin treatment in SKOV3-TR cells (Fig. 3C). Similar results were observed in
A2780/cis cells (Fig. 3D),
indicating that metformin decreased AKT2 and p-AKT protein
expression in a proteasome-dependent manner.
Metformin increases MUL1 expression
Previous studies have demonstrated that AKT could be
degraded by MUL1 and tetratricopeptide repeat domain 3 (TTC3) via
K48-linked ubiquitination in a proteasome-dependent manner
(26,31). Bae et al (26) have reported that MUL1 interacts
with AKT1 and AKT2 through a kinase domain of AKT and
preferentially degrades p-AKT. Western blot analysis revealed that
metformin particularly induced the degradation of AKT2 (and p-AKT)
among the three AKT isoforms (AKT1, AKT2 and AKT3; Fig. 2A and B). Therefore, the present
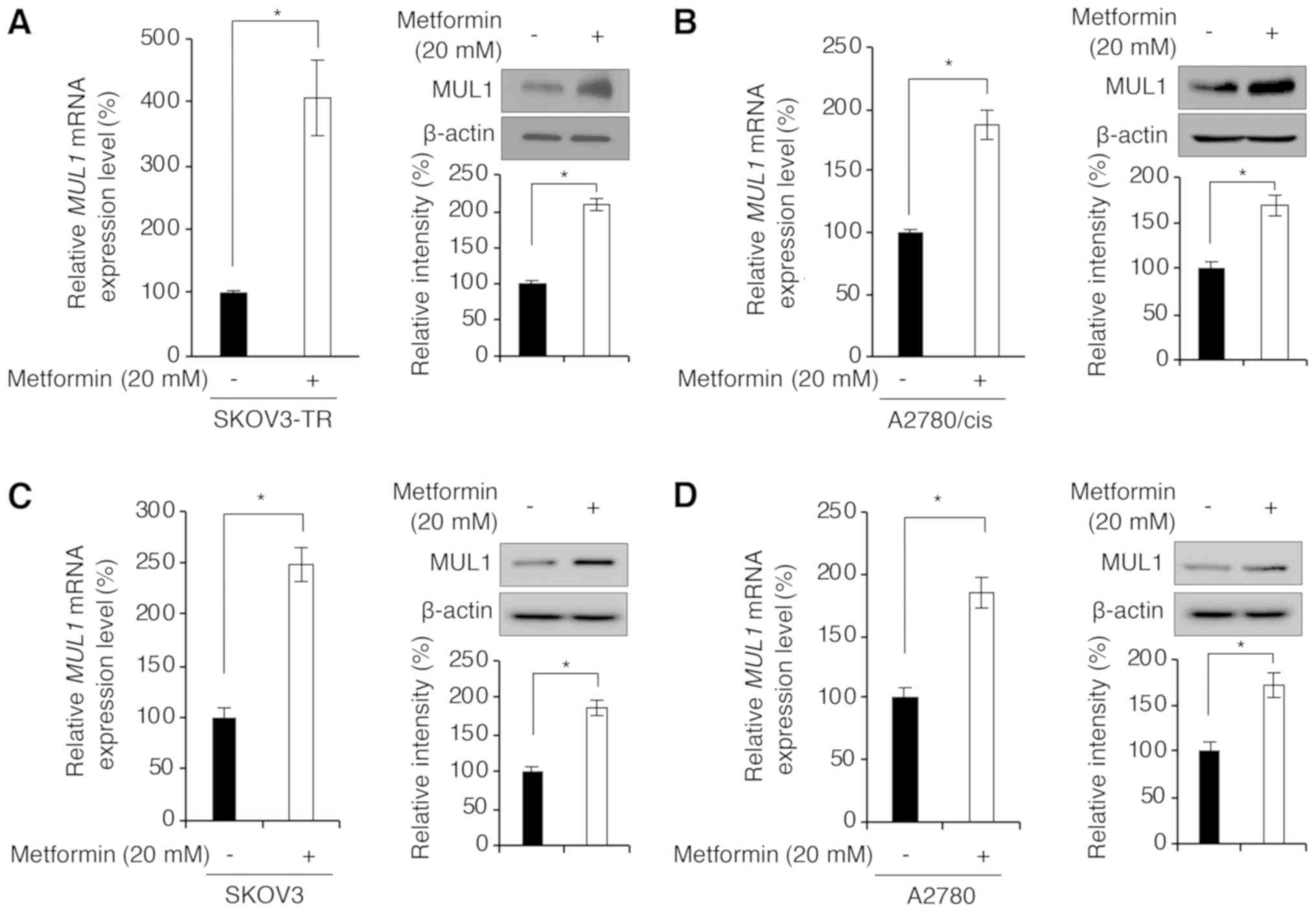
study investigated if metformin could increase MUL1 expression.
MUL1 mRNA expression levels in A2780/cis and SKOV3-TR cells treated
with metformin were measured using reverse
transcription-quantitative polymerase chain reaction (RT-qPCR). As
shown in Fig. 4A and B, metformin
treatment significantly increased MUL1 mRNA levels in A2780/cis and
SKOV3-TR cells. Similarly, the protein expression levels of MUL1
were also increased following metformin treatment (Fig. 4A and B). In addition, we examined
whether metformin-induced MUL1 expression was specific to the
chemoresistant cells. As illustrated in Fig. 4C and D, treatment with metformin
significantly upregulated MUL1 mRNA and protein expression in the
parental SKOV3 and A2780 cells. Together, these findings indicate
that metformin treatment enhanced both mRNA and protein expression
levels of MUL1.
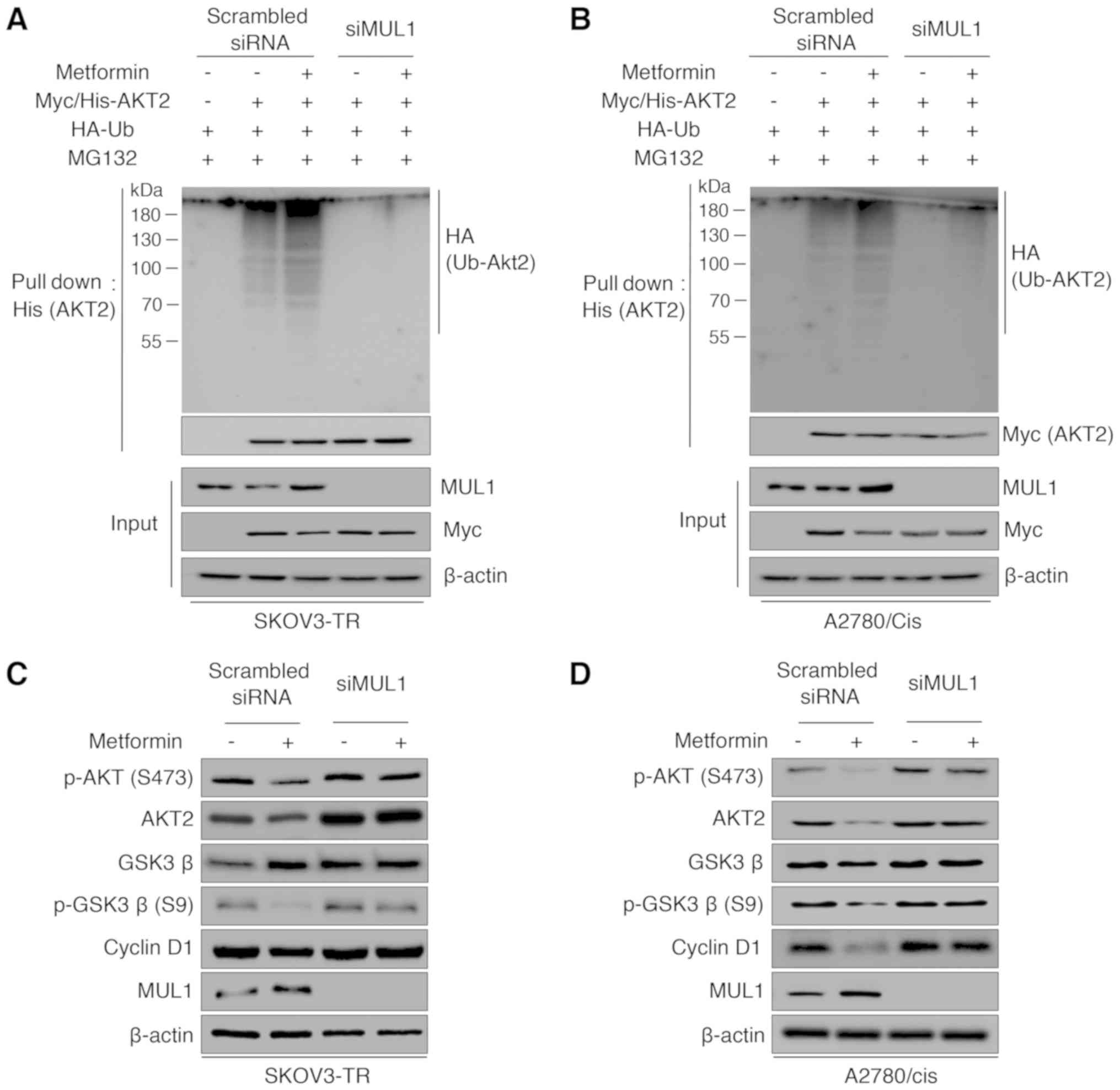
Metformin-induced MUL1 expression
promotes AKT degradation in a proteasome-dependent manner and
regulates the AKT downstream pathway
The aforementioned results led to the hypothsis that
metformin-induced AKT degradation may be mediated by MUL1. To test
this hypothesis, His-ubiquitin pull-down assays were performed. As
illustrated in Fig. 5A and B,
exposure to metformin induced polyubiquitination of AKT, and siRNA
directed against MUL1 abrogated metformin-induced AKT
ubiquitination in both SKOV3-TR and A2780/cis cell lines. As
metformin decreased the viability and proliferation of
chemoresistant ovarian cancer cell lines (Fig. 1), the effects of metformin on AKT
downstream genes associated with cell cycle progression and cell
growth were further examined. The GSK3β/cyclin D1 pathway is a
well-known downstream pathway of AKT associated with cell
proliferation and cycle progression. Several reports have suggested
that phosphorylation of GSK3β at Serine 9 by AKT decreases the
kinase activity of GSK3β for Thr286 of cyclin D1, which leads to
the cytoplasmic proteasomal degradation of cyclin D1 (32,33).
Thus, in the present study the protein expression levels of p-GSK3β
(Ser9) and cyclin D1 were determined following metformin treatment
using western blot analysis. As illustrated in Fig. 5C and D, metformin treatment
significantly inhibited GSK3β phosphorylation and cyclin D1
expression. However, knockdown of MUL1 using siRNA rescued the
protein expression levels of p-AKT, AKT2, p-GSK3β, and cyclin D1 in
both cell lines. Taken together, these findings suggest that the
increase in MUL1 expression induced by metformin regulated the AKT
downstream pathway.
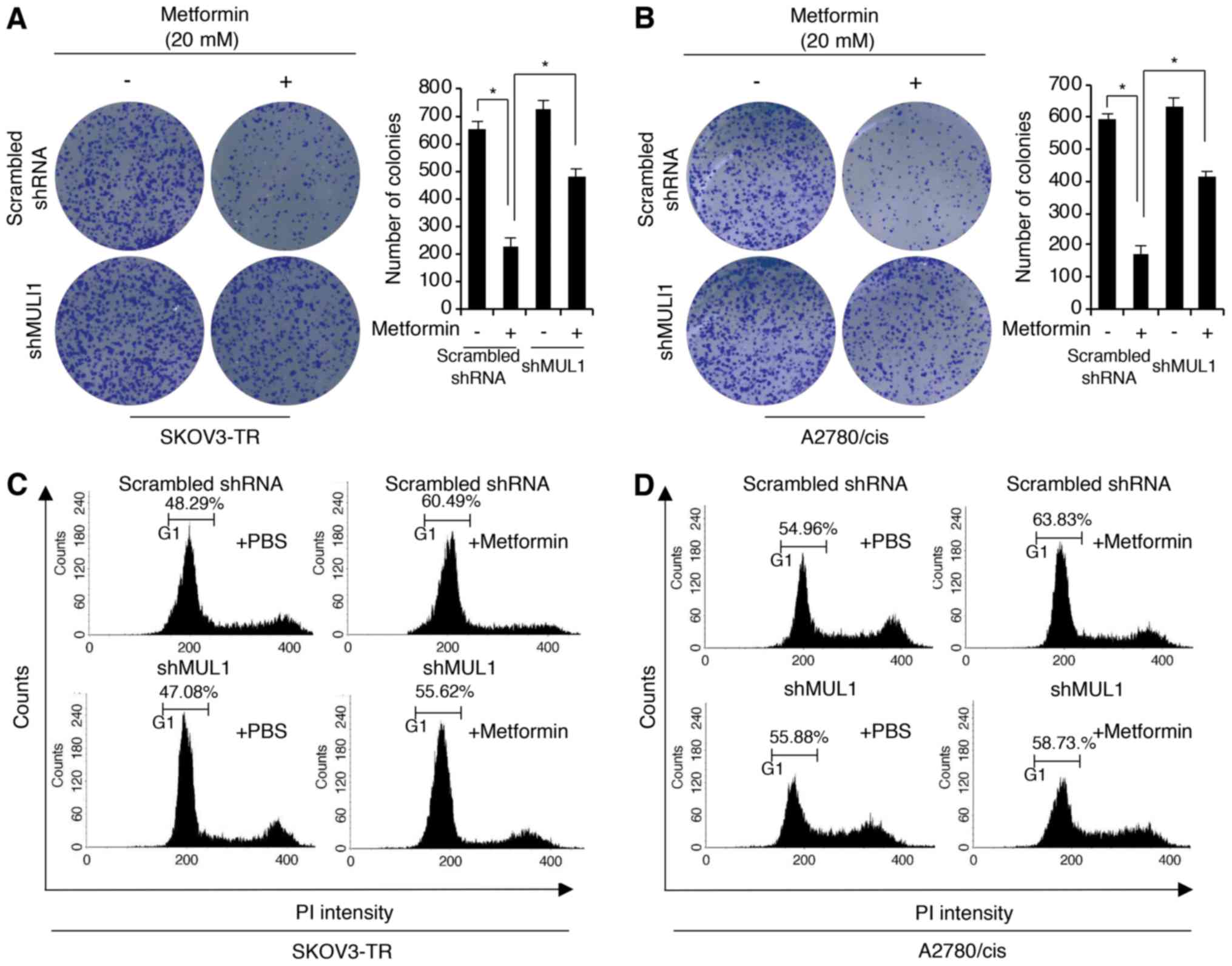
Antitumor effects of metformin are
regulated by MUL1
Next, the present study sought to determine whether
the increase in MUL1 expression was required for the antitumor
activity of metformin in the chemoresistant ovarian cancer cell
lines. To this end, MUL1 knockdown SKOV3-TR and A2780/cis cell
lines were generated, by expressing a shRNA construct targeting
MUL1 (shMUL1). First, the clonogenic growth ability was
investigated in the control and shMUL1 knockdown SKOV3-TR and
A2780/cis cells. The results revealed that metformin treatment
significantly inhibited clonogenic growth. However, the
metformin-mediated inhibition of clonogenic growth was partially
rescued in MUL1 stable knockdown cells, compared with the control
cells (Fig. 6A and B). Because the
results of Figs. 1 and 5 demonstrated that metformin treatment
decreased cell viability and proliferation and downregulated the
AKT/GSK3β/cyclin D1 pathway, which is associated with cell cycle
progression, further cell cycle analyses were conducted using flow
cytometry. As presented in Fig.
6C, there was a higher increase in the number of cells in the
G1-phase of the cell cycle in the metformin-treated control
SKOV3-TR cells compared with the metformin-treated shMUL1 SKOV3-TR
cells. Similar results were observed in A2780/cis cells (Fig. 6D). Together, these data suggest
that metformin-mediated MUL1 expression may be important for the
antitumor activity of metformin.
Discussion
Taxane (paclitaxel) and platinum drugs (such as
cisplatin) induce DNA damage and constitute the first-line
chemotherapy for ovarian cancer. Unfortunately, >70% of patients
with ovarian cancer who are prescribed paclitaxel show relapse and
develop chemoresistance (34).
This clinical therapeutic challenge is caused by several factors.
Currently, most ovarian cancers are left undiagnosed until they
reach an advanced stage because there are few reliable symptoms and
etiological factors in the early stages of ovarian cancer (35). Furthermore, chemoresistance is
reported to be responsible for 90% of deaths in patients with
advanced ovarian cancer (36).
This observation indicates that chemoresistance is the primary
factor in ovarian cancer relapse; however, the development of
strategies targeting these chemoresistant ovarian cancers remains a
fundamental challenge. These problems make ovarian cancer one of
the most lethal tumors with pernicious growth and progression,
frequent metastasis, and commonly acquired chemoresistance
(34).
The results of the present study demonstrated the
chemosensitizing effect of metformin on drug-resistant SKOV3-TR and
A2780/Cis cells. Biochemical assays revealed that metformin
significantly suppressed cell proliferation, viability, and cycle
progression in these cells. Notably, metformin increased both mRNA
and protein levels of MUL1 and promoted the degradation of AKT
protein in a proteasome-dependent manner. To further analyze this,
the effect of metformin on MUL1 and AKT expression was examined in
the parental cell lines, SKOV3 and A2780, because data in Fig. 1 demonstrated that metformin had
anticancer activity not only on the parental but also on the
chemoresistant ovarian cancer cell lines. The results demonstrated
that treatment with metformin decreased the level of AKT2 and
significantly upregulated MUL1 expression in those cell lines,
similar with the results from the SKOV3-TR and A2780/Cis resistant
lines. These findings indicate that metformin-induced MUL1
expression was not a result specific to chemoresistance. AKT is
known to be associated with the resistance of cancer cells to
various anticancer drugs (37-41),
including ovarian cancer cells (8). In addition, previous studies have
demonstrated that metformin decreases the expression of p-AKT in
several cancer cells. Hyperactivation of AKT, which is known to
stimulate cell survival and proliferation pathways, is frequently
observed in cancers. Furthermore, MUL1 has been previously
demonstrated to be an E3 ubiquitin ligase for AKT1 and AKT2
(26); therefore, the present data
further suggested that the chemosensitizing effect of metformin is
mediated by MUL1 expression in drug-resistant ovarian cancer cells.
This hypothesis was supported by the observation that silencing of
MUL1 expression suppressed metformin-mediated AKT degradation and
its downstream effects. Additionally, metformin significantly
decreased colony formation compared with control cells; however,
this inhibitory effect was suppressed by silencing MUL1 expression,
indicating that MUL1 regulated metformin-mediated AKT degradation
and anticancer effects in chemoresistant ovarian cancer cells. A
previous study has also reported that metformin exerts a
chemosensitizing effect on drug-resistant ovarian cancer cells
(29). Specifically, the authors
observed that metformin decreased proliferation levels with
downregulation of the inflammatory signaling pathway in
paclitaxel-resistant A2780 and cisplatin-resistant ACRP cell lines
(29). Further studies are
required to assess whether the effect of metformin is dependent on
inflammatory signaling, as there is presently no functional
evidence that MUL1 regulates inflammatory signaling in cancer
cells.
Metformin, a widely used drug for the treatment of
type 2 diabetes with relatively low side effects (7,8), has
attracted much attention in oncology owing to its anticancer
activities (14-18). Although the exact mechanisms
underlying the effects of metformin have not been completely
elucidated, the most well-known mechanism is activation and
phosphorylation of AMPK by inhibiting the activity of mitochondrial
complex I (11,12). The present study demonstrated that
treatment with metformin distinctly upregulated MUL1 expression and
downregulated AKT and its downstream targets GSK3β and cyclin D1.
However, the p-AMPK levels remained unchanged, suggesting that the
anticancer and chemosensitization effects of metformin are
independent of the AMPK-mediated pathway. Recent accumulating
evidence suggests that the anticancer activities of metformin are
mediated by not only AMPK-dependent but also -independent pathways.
Metformin decreases the expression levels of cyclin D1, which is an
important regulator of cell cycle progression, in the absence of
AMPK (42). In addition, the
antiproliferative effect of metformin is mediated by
AMPK-independent inhibition of mammalian target of rapamycin
complex 1 (mTORC1) signaling, which has been implicated in cancer
progression (43). Based on these
reports, the current findings suggest that AMPK activation is not
essential for the anticancer and chemosensitization effects of
metformin on drug-resistant ovarian cancer cells.
In summary, the present findings indicate that
metformin inhibited the growth and proliferation of drug-resistant
ovarian cancer cells, which was mediated by MUL1 expression and the
subsequent AKT degradation. Additionally, metformin promoted the
chemosensitization in a MUL1-dependent and AMPK-independent manner.
To the best of our knowledge, this is the first study to elucidate
the promoting effect and cellular mechanism of metformin and its
chemosensitizing potential in drug-resistant ovarian cancer
cells.
Funding
This study resulted from the Konkuk University
research support program.
Availability of data and materials
All data generated or analysed during this study are
included in this published article.
Authors’ contributions
JL, SA and SB performed the experiments and wrote
the manuscript. JHJ and KK participated in the design of the study
and performed the statistical analysis. JYK and ISA analyzed and
interpreted the data. SA and SB conceived the study, and
participated in its design and coordination and helped to draft
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
We thank all the members of our research group for
their support and advice during this study.
References
|
1
|
McGuire S: World Cancer Report 2014.
Geneva, Switzerland: World Health Organization, International
Agency for Research on Cancer, WHO Press. 2015 Adv Nutr. 7. pp.
418–419. 2016
|
|
2
|
Engel J, Eckel R, Schubert-Fritschle G,
Kerr J, Kuhn W, Diebold J, Kimmig R, Rehbock J and Hölzel D:
Moderate progress for ovarian cancer in the last 20 years:
Prolongation of survival, but no improvement in the cure rate. Eur
J Cancer. 38:2435–2445. 2002.
|
|
3
|
Beaufort CM, Helmijr JC, Piskorz AM,
Hoogstraat M, Ruigrok-Ritstier K, Besselink N, Murtaza M, van
IJcken WF, Heine AA, Smid M, et al: Ovarian cancer cell line panel
(OCCP): Clinical importance of in vitro morphological subtypes.
PLoS One. 9:e1039882014.
|
|
4
|
Banno K, Yanokura M, Iida M, Adachi M,
Nakamura K, Nogami Y, Umene K, Masuda K, Kisu I, Nomura H, et al:
Application of microRNA in diagnosis and treatment of ovarian
cancer. BioMed Res Int. 2014:2328172014.
|
|
5
|
Muggia FM: Relevance of chemotherapy dose
and schedule to outcomes in ovarian cancer. Semin Oncol. 31(Suppl
15): 19–24. 2004.
|
|
6
|
Agarwal R and Kaye SB: Ovarian cancer:
Strategies for overcoming resistance to chemotherapy. Nat Rev
Cancer. 3:502–516. 2003.
|
|
7
|
Hundal RS, Krssak M, Dufour S, Laurent D,
Lebon V, Chandramouli V, Inzucchi SE, Schumann WC, Petersen KF,
Landau BR, et al: Mechanism by which metformin reduces glucose
production in type 2 diabetes. Diabetes. 49:2063–2069. 2000.
|
|
8
|
Cao J, Meng S, Chang E, Beckwith-Fickas K,
Xiong L, Cole RN, Radovick S, Wondisford FE and He L: Low
concentrations of metformin suppress glucose production in
hepatocytes through AMP-activated protein kinase (AMPK). J Biol
Chem. 289:20435–20446. 2014.
|
|
9
|
McCreight LJ, Bailey CJ and Pearson ER:
Metformin and the gastrointestinal tract. Diabetologia. 59:426–435.
2016.
|
|
10
|
Wheaton WW, Weinberg SE, Hamanaka RB,
Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM,
Budigner GS, et al: Metformin inhibits mitochondrial complex I of
cancer cells to reduce tumorigenesis. eLife. 3:e022422014.
|
|
11
|
Musi N, Hirshman MF, Nygren J, Svanfeldt
M, Bavenholm P, Rooyackers O, Zhou G, Williamson JM, Ljunqvist O,
Efendic S, et al: Metformin increases AMP-activated protein kinase
activity in skeletal muscle of subjects with type 2 diabetes.
Diabetes. 51:2074–2081. 2002.
|
|
12
|
Stephenne X, Foretz M, Taleux N, van der
Zon GC, Sokal E, Hue L, Viollet B and Guigas B: Metformin activates
AMP-activated protein kinase in primary human hepatocytes by
decreasing cellular energy status. Diabetologia. 54:3101–3110.
2011.
|
|
13
|
Viollet B, Horman S, Leclerc J, Lantier L,
Foretz M, Billaud M, Giri S and Andreelli F: AMPK inhibition in
health and disease. Crit Rev Biochem Mol Biol. 45:276–295.
2010.
|
|
14
|
Kato K, Gong J, Iwama H, Kitanaka A, Tani
J, Miyoshi H, Nomura K, Mimura S, Kobayashi M, Aritomo Y, et al:
The anti-diabetic drug metformin inhibits gastric cancer cell
proliferation in vitro and in vivo. Mol Cancer Ther. 11:549–560.
2012.
|
|
15
|
Fujihara S, Kato K, Morishita A, Iwama H,
Nishioka T, Chiyo T, Nishiyama N, Miyoshi H, Kobayashi M, Kobara H,
et al: Antidiabetic drug metformin inhibits esophageal
adenocar-cinoma cell proliferation in vitro and in vivo. Int J
Oncol. 46:2172–2180. 2015.
|
|
16
|
Nangia-Makker P, Yu Y, Vasudevan A,
Farhana L, Rajendra SG, Levi E and Majumdar AP: Metformin: A
potential therapeutic agent for recurrent colon cancer. PLoS One.
9:e843692014.
|
|
17
|
Davies G, Lobanova L, Dawicki W, Groot G,
Gordon JR, Bowen M, Harkness T and Arnason T: Metformin inhibits
the development, and promotes the resensitization, of
treatment-resistant breast cancer. PLoS One. 12:e01871912017.
|
|
18
|
Dowling RJ, Niraula S, Stambolic V and
Goodwin PJ: Metformin in cancer: Translational challenges. J Mol
Endocrinol. 48:R31–R43. 2012.
|
|
19
|
Huang J and Manning BD: The TSC1-TSC2
complex: A molecular switchboard controlling cell growth. Biochem
J. 412:179–190. 2008.
|
|
20
|
Lien EC, Dibble CC and Toker A: PI3K
signaling in cancer: Beyond AKT. Curr Opin Cell Biol. 45:62–71.
2017.
|
|
21
|
Zakikhani M, Blouin MJ, Piura E and Pollak
MN: Metformin and rapamycin have distinct effects on the AKT
pathway and proliferation in breast cancer cells. Breast Cancer Res
Treat. 123:271–279. 2010.
|
|
22
|
Zhang J, Li G, Chen Y, Fang L, Guan C, Bai
F, Ma M, Lyu J and Meng QH: Metformin inhibits tumorigenesis and
tumor growth of breast cancer cells by upregulating miR-200c but
downregu-lating AKT2 expression. J Cancer. 8:1849–1864. 2017.
|
|
23
|
Liu Y, Zhang Y, Jia K, Dong Y and Ma W:
Metformin inhibits the proliferation of A431 cells by modulating
the PI3K/Akt signaling pathway. Exp Ther Med. 9:1401–1406.
2015.
|
|
24
|
Rattan R, Giri S, Hartmann LC and Shridhar
V: Metformin attenuates ovarian cancer cell growth in an AMP-kinase
dispensable manner. J Cell Mol Med. 15:166–178. 2011.
|
|
25
|
Fresno Vara JA, Casado E, de Castro J,
Cejas P, Belda-Iniesta C and González-Barón M: PI3K/Akt signalling
pathway and cancer. Cancer Treat Rev. 30:193–204. 2004.
|
|
26
|
Bae S, Kim SY, Jung JH, Yoon Y, Cha HJ,
Lee H, Kim K, Kim J, An IS, Kim J, et al: Akt is negatively
regulated by the MULAN E3 ligase. Cell Res. 22:873–885. 2012.
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-ΔΔC(T)) method. Methods. 25:402–408. 2001.
|
|
28
|
Kim NY, Lee HY and Lee C: Metformin
targets Axl and Tyro3 receptor tyrosine kinases to inhibit cell
proliferation and overcome chemoresistance in ovarian cancer cells.
Int J Oncol. 47:353–360. 2015.
|
|
29
|
Dos Santos Guimarães I, Ladislau-Magescky
T, Tessarollo NG, Dos Santos DZ, Gimba ER, Sternberg C, Silva IV
and Rangel LB: Chemosensitizing effects of metformin on cisplatin-
and paclitaxel-resistant ovarian cancer cell lines. Pharmacol Rep.
70:409–417. 2018.
|
|
30
|
Karnevi E, Said K, Andersson R and
Rosendahl AH: Metformin-mediated growth inhibition involves
suppression of the IGF-I receptor signalling pathway in human
pancreatic cancer cells. BMC Cancer. 13:2352013.
|
|
31
|
Suizu F, Hiramuki Y, Okumura F, Matsuda M,
Okumura AJ, Hirata N, Narita M, Kohno T, Yokota J, Bohgaki M, et
al: The E3 ligase TTC3 facilitates ubiquitination and degradation
of phos-phorylated Akt. Dev Cell. 17:800–810. 2009.
|
|
32
|
Luo J: Glycogen synthase kinase 3beta
(GSK3beta) in tumori-genesis and cancer chemotherapy. Cancer Lett.
273:194–200. 2009.
|
|
33
|
Cross DA, Alessi DR, Cohen P, Andjelkovich
M and Hemmings BA: Inhibition of glycogen synthase kinase-3 by
insulin mediated by protein kinase B. Nature. 378:785–789.
1995.
|
|
34
|
Giornelli GH: Management of relapsed
ovarian cancer: A review. Springerplus. 5:11972016.
|
|
35
|
Jelovac D and Armstrong DK: Recent
progress in the diagnosis and treatment of ovarian cancer. CA
Cancer J Clin. 61:183–203. 2011.
|
|
36
|
Sherman-Baust CA, Becker KG, Wood Iii WH,
Zhang Y and Morin PJ: Gene expression and pathway analysis of
ovarian cancer cells selected for resistance to cisplatin,
paclitaxel, or doxorubicin. J Ovarian Res. 4:212011.
|
|
37
|
Banno E, Togashi Y, de Velasco MA,
Mizukami T, Nakamura Y, Terashima M, Sakai K, Fujita Y, Kamata K,
Kitano M, et al: Clinical significance of Akt2 in advanced
pancreatic cancer treated with erlotinib. Int J Oncol.
50:2049–2058. 2017.
|
|
38
|
Cheung M and Testa JR: Diverse mechanisms
of AKT pathway activation in human malignancy. Curr Cancer Drug
Targets. 13:234–244. 2013.
|
|
39
|
Cassinelli G, Zuco V, Gatti L, Lanzi C,
Zaffaroni N, Colombo D and Perego P: Targeting the Akt kinase to
modulate survival, invasiveness and drug resistance of cancer
cells. Curr Med Chem. 20:1923–1945. 2013.
|
|
40
|
Guerrero-Zotano A, Mayer IA and Arteaga
CL: PI3K/AKT/ mTOR: Role in breast cancer progression, drug
resistance, and treatment. Cancer Metastasis Rev. 35:515–524.
2016.
|
|
41
|
Kim SH, Juhnn YS and Song YS: Akt
involvement in paclitaxel chemoresistance of human ovarian cancer
cells. Ann NY Acad Sci. 1095:82–89. 2007.
|
|
42
|
Ben Sahra I, Laurent K, Loubat A,
Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le
Marchand-Brustel Y and Bost F: The antidiabetic drug metformin
exerts an antitumoral effect in vitro and in vivo through a
decrease of cyclin D1 level. Oncogene. 27:3576–3586. 2008.
|
|
43
|
Kalender A, Selvaraj A, Kim SY, Gulati P,
Brûlé S, Viollet B, Kemp BE, Bardeesy N, Dennis P, Schlager JJ, et
al: Metformin, independent of AMPK, inhibits mTORC1 in a rag
GTPase-dependent manner. Cell Metab. 11:390–401. 2010.
|