Introduction
Osteosarcoma (OS) is a common primary malignancy in
adolescents and children, and it is associated with a high
incidence rate, rapid progression and distant metastasis (1). The treatment of early OS is mainly
surgical amputation. The 5-year survival rate of patients OS who
only undergo amputation is approximately 16%. However, this type of
surgery is highly traumatic and is associated with a high
disability rate (2). The
application of neoadjuvant chemotherapy has significantly improved
the 5-year survival rate of patients with OS; however, some
patients are still insensitive to chemotherapy, and their treatment
would also be affected by recurrence and metastasis of the tumor
(3,4). Although the occurrence of OS is
relatively rare compared with other types of tumors, the basic
research and clinical treatment strategies for OS have drawn much
attention in the medical field, as it is associated with a high
mortality and disability rate (1,5).
Therefore, it is important to identify effective and safe
strategies with which to inhibit the progression of OS.
MicroRNAs (miRNAs or miRs) are a non-coding single
chain RNA molecules of approximately 22 nucleotides in length, and
they are widely distributed in plants, nematodes and human cells
(6,7). In 1993, scientists discovered miRNAs
for the first time in Caenorhabditis elegans (8,9). The
number of miRNAs recognized in mammals has now reached tens of
thousands. Researchers have found that miRNAs play a pivotal role
in, for example, the biological processes of cell growth,
differentiation, apoptosis and embryo development (10-13).
The association between miRNAs and the occurrence and development
of tumors has also attracted increasing attention in life science
research (14). Researchers have
found that miRNAs can regulate the progression of OS (15-17).
miR-9 is a member of the miRNA family, and it has been found to be
abnormally expressed in a number of types of tumor cells, such as
breast cancer, lung cancer, gastric cancer and OS (18-22).
Nevertheless, the specific role and targets of miR-9 in OS have not
yet been fully elucidated.
Mitogen-activated protein kinases (MAPKs) are a
class of intracellular threonine tyrosine protein kinases, and
signal transduction is composed of cascade 3 cascade reactions
(23,24). Studies have confirmed that MAPKs
exist in the majority of cells, and are associated with the
proliferation, differentiation and apoptosis of various cells
(25-27). Three different MAPK signaling
pathways, including p38 MAPK, c-Jun N-terminal kinase (JNK) and
extracellular signal-regulated kinase (ERK) (28,29),
have been found in mammals. A number of studies have confirmed that
these 3 signals can regulate the progression of various types of
tumors, for example, OS, prostate cancer and glioma (30-32).
In the present study, a target gene for miR-9 was
predicted in OS according to the MicroRNA.org
web site. We also examined the effects of miR-9 on the
proliferation and cell cycle progression of human OS cells
(Saos-2), and further analyzed the underlying molecular
mechanisms.
Materials and methods
Tissue collection
From May, 2016 to June, 2017, 25 OS tissues and
adjacent normal tissues were collected from patients with OS who
were underwent treatment at Henan Provincial People’s Hospital
(Zhengzhou, China). All patients signed informed consent forms,
allowing their tissues to be used in this study. The Ethics
Committee of Henan Provincial People’s Hospital authorized this
research.
Cells and cell culture
Human OS cell lines (Saos-2) and 293 cells were
obtained from JiningShiye (Shanghai, China). The cells were
cultured in Dulbecco’s modified Eagle’s medium (DMEM; Solarbio,
Beijing, China) containing 10% fetal bovine serum (FBS; MRC,
Jiangsu, China) and 100X penicillin-streptomycin mixed solution
(Leagene, Beijing, China) in an incubator at 37°C with 95%
humidified and 5% CO2 (SR80G, Sheyanyiqi, Shanghai,
China).
Cell transfection and grouping
hsa-miR-9 mimics, hsa-miR-9 inhibitors and miRNA
negative control (50 nM) were purchased from GenePharma (Shanghai,
China). For p16 interference, siRNA p16 and unspecific scrambled
siRNA (control siNC) were purchased from GenePharma. The sequences
of selected regions to be targeted by siRNAs for p16 were as
follows:
5′-TGCTGTTAGCTCTGCTCTTGGGATTGGTTTTGGCCACTGACTGACCAATCCCAAGCAGAGCTAA-3′
(sense),
5′-CCTGTTAGCTCTGCTTGGGATTGGTCAGTCAGTGGCCAAAACCAATCCCAAGAGCAGAGCTAAC-3′
(antisense). All transfections of the Saos-2 cells were conducted
with the cells at 50-60% confluence using Lipofectamine™ 2000
(Thermo Fisher Scientific, Beijing, China) at 37°C for 48 h.
This experiment established 10 different groups as
follows: i) Negative control (Saos-2 cells were transfected with
miRNA negative control); ii) hsa-miR-9 mimics (Saos-2 cells were
transfected with hsa-miR-9 mimics); iii) hsa-miR-9 inhibitors
(Saos-2 cells were transfected with hsa-miR-9 inhibitors); iv)
control + p16-3′UTR (293 cells were transfected with miRNA negative
control and p16-3′UTR); v) hsa-miR-9 + p16-3′UTR (293 cells were
transfected with hsa-miR-9 mimics and p16-3′UTR); vi) hsa-miR-9 +
p16-3′UTR-mut (293 cells were transfected with hsa-miR-9 mimics and
p16-3′UTR-mutant); vii) si-p16 (Saos-2 cells were transfected with
siRNA p16); viii) control + si-NC (Saos-2 cells were transfected
with unspecific scrambled siRNA); ix) hsa-miR-9 inhibitors + si-NC
(Saos-2 cells were transfected with hsa-miR-9 inhibitors and
unspecific scrambled siRNA); and x) hsa-miR-9 inhibitors + si-p16
(Saos-2 cells were transfected with hsa-miR-9 inhibitors and siRNA
p16). These cell groups were used in different assays as
needed.
Luciferase reporter assay
The MicroRNA.org
website (http://www.microrna.org/microrna/getMirnaForm.do)
was used to predict the target gene of miR-9, and the dual
luciferase reporter assay was used to confirm the findings.
Luciferase activity was detected using the double luciferase report
system detection kit (Promega Corp., Madison, WI, USA). Firstly,
hsa-miR-9 mimics and miRNA negative control were co-transfected
with p16-3′UTR or p16-3′UTR mutant plasmids (400 ng) into 293 cells
using Lipofectamine™ 2000 for 48 h. The cells were then lysed using
1X passive lysis buffer (PLB) at 37°C for 15 min. The cell
suspension was transferred to a black enzyme plate. LARII and
stop&Glo® reagent (Promega Corp.) was then added to
the plate in a drop-wise manner. Luciferase activity was measured
using a GloMax® Discover Multimode Microplate Reader
(GM3000; Promega Corp.). pRL-TK (Promega Corp.) was used as an
internal control.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from the cells and tissues
using an RNA extraction kit (Takara, Beijing, China). cDNA was
synthesized using a ABScript II cDNA First Strand Synthesis kit
(ABclonal, Wuhan, China). The reaction conditions were set at 85°C
for 15 min. Subsequently, cDNA was amplified using the TeloPrime
Full-Length cDNA Amplification kit (Lexogen, Beijing, China). qPCR
experiments were performed using a SYBR Premix Ex Taq™ Real-Time
PCR Kit (Takara Bio, Inc., Otsu, Japan). The reaction conditions
were set at 90°C for 20 sec, (at 90°C for 10 sec and at 58°C for 40
sec) for 35 cycles, at 72°C for 3 min. The sequences of the primers
used are listed in Table I. U6 and
GAPDH were used as internal controls. Gene expression was
quantified using the 2-ΔΔCq method (33).
 | Table IThe sequences of the primers used for
RT-qPCR. |
Table I
The sequences of the primers used for
RT-qPCR.
| Primer name | Sequence
(5′-3′) | Product size
(bp) |
|---|
| miR-9 | F:
GTGCAGGGTCCGAGGT | |
| R:
GCGCTCTTTGGTTATCTAGC | 206 |
| p16 | F:
CAGGTCATGATGATGGGCAG | |
| R:
GATGGCCCAGCTCCTCAG | 223 |
| Cyclin A | F:
TAACCAGGTGATCGTGCAGT | |
| R:
TGGAGTCTGTGCCAAATCCT | 217 |
| Cyclin D1 | F:
CCCTCGGTGTCCTACTTCAA | |
| R:
CTTAGAGGCCACGAACATGC | 219 |
| c-Myc | F:
GGACGACGAGACCTTCATCA | |
| R:
CGTTGAGAGGGTAGGGGAAG | 245 |
| U6 | F:
ACACCAAGCAGTCCGAAGAG | |
| R:
ACAAAATTTCTCACGCCGGT | 220 |
| GAPDH | F:
CCATCTTCCAGGAGCGAGAT | |
| R:
TGCTGATGATCTTGAGGCTG | 222 |
Western blot analysis
Total protein was isolated from the cells and
tissues were using RIPA buffer (Solarbio). The BCA protein assay
(Thermo Fisher Scientific) was carried out to measure the contents
of the proteins. Each protein (10 μg) was separated by 12%
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) and blotted onto a PVDF membrane (Reno, Hangzhou,
China). Subsequently, the PVDF membrane was soaked in TBST-soluble
5% dried skimmed milk at room temperature for 2 h. The membrane was
respectively bound to the corresponding primary antibodies
[anti-p16, ab118459, 1:800; anti-ERK, ab224313, 1:700; anti-p-ERK,
ab214036, 1:700; anti-JNK, ab208035, 1:1,000; anti-p-JNK, ab124956,
1:1,000; anti-GAPDH, ab181602, 1:800; all from Abcam (Cambridge,
MA, USA); anti-cyclin A, sc-271682, 1:800; anti-cyclin D1 sc-246,
1:1,000; anti-c-Myc, sc-373712, 1:600; anti-p-38, sc-7972, 1:800;
anti-p-p38 sc-7975-R, 1:800; all from Santa Cruz Biotechnology
(Santa Cruz, CA, USA)] at 4°C for 24 h. After binding, the PVDF
membrane was bound to the corresponding secondary antibodies (goat
anti-rabbit IgG H&L, ab150077, 1:6,000; rabbit anti-mouse IgG
H&L, ab6726, 1:6,000; Abcam) at 37°C for 1 min. The blots were
assessed using ECL detection reagent (Yeasen, Shanghai, China), and
densitometry was performed using the Bio-Rad ChemiDoc system with
Image Lab software version 6.0 (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
Cell counting kit-8 (CCK-8) assay
The Saos-2 cells were seed in a 96-well plate
(1.5×103 cell/well) in an incubator at 37°C for 24 h.
The cells were subjected to transfection with the corresponding
plasmids for 0, 12 and 24 and 48 h. Subsequently, the cells were
treated with CCK-8 reagent at 37°C for 4 h. The value of OD at 450
nm was read using a SpectraMax iD5 microplate reader (Molecular
Devices, Shanghai, China).
Colony formation assay
The Saos-2 cells were seeded in a 60-mm culture dish
(2.5×104 cell/ml) in an incubator for 24 h at 37°C. The
cells were then exposed to miRNA negative control, hsa-miR-9 mimics
and hsa-miR-9 inhibitors. After the cells have been transfected for
24 h, the cells were fixed using polyoxymethylene at room
temperature for 20 min. The cells were then stained by 0.1% crystal
violet (Solarbio) at room temperature for 30 min.
Cell cycle assay
The Saos-2 cells were seeded in a 6-well plate
(3×104 cell/well) in an incubator at 37°C for 24 h. The
cells were then exposed to the corresponding plasmids for 48 h. The
cells were then fixed by paraformaldehyde for 25 min at 4°C. The
cells were subsequently stained with propidium iodide (PI; Leagene,
Beijing, China) for 30 min at room temperature. At least
4×103 cells were collected for assay using a FACScan
flow cytometer (BD Biosciences, San Jose, CA, USA).
Establishment of mouse model of OS
All animal experiments were performed following the
approval of the Henan Provincial People’s Hospital Animal Ethics
Committee and according to the Guidelines for the Care and Use of
Laboratory Animals. A total of 6 Balb/c nude mice per group (6-8
weeks old; weighing 20±2 g) were provided by Rochen (Shanghai,
China). Following transfection, the Saos-2 cells suspension
(106 cells per mouse) was injected into the subcutaneous
tissue of the nude mice to produce OS tumor xenografts.
Tumor volume and weight assay
After 15, 20, 25, 30, 35, 40 and 45 days of culture,
the length and width of the tumors in each group of animals (6 in
each group; 3 groups) were counted using the caliper, tumor volume
= (width2 × length)/2, as previously described (34). The animals were sacrificed after 45
days. Tumor mass was calculated. The tumor weights were detected
using an electronic balance (SECURA213-1CN; Sartorius, Goettingen,
Germany).
Immunohistochemistry (IHC)
The animals were sacrificed and tumors were
collected. The samples were dehydrated in a graded ethanol followed
by routine paraffin embedding and sectioning. The paraffin-embedded
tissue slices were then washed with distilled water 3 times. The
slices were soaked in a citrate buffer (pH 6.0) at room temperature
for 30 min. The hydrogen peroxide reagent was then dripped into the
slices at room temperature for 10 min. Subsequently, the slices
were sealed using goat serum at room temperature for 20 min. The
anti-p16 antibody (ab151303, 1:800) was added to the slices at 4°C
for 24 h. The following day, the secondary antibodies (goat
anti-rabbit IgG-HRP; ab205718, 1:2,000) (both from Abcam) were
dripped onto the slices at room temperature for 20 min. The slices
were then stained by DAB staining solution (Leica, Shanghai, China)
at room temperature for 15 min. The slices were then cleaned with
distilled water for 3 times. Subsequently, the slices were dyed
using hematoxylin (Solarbio) for 3 min at room temperature. The
slices were observed under a fluorescence microscope (MF43; Mshot,
Guangzhou, China).
Statistical analysis
The data are presented as the means ± standard
deviation (SD) and were analyzed using IBM SPSS 20 statistical
software. The Student’s t-test was used to analyze differences
between 2 groups and differences between the paired patient
tissues. The differences among groups were analyzed by one-way
analysis of variance (ANOVA) followed by Dunnett’s t-test. The
Chi-square test was carried out to examine the association between
p16 expression and the clinicopathological characteristics of the
patients with OS. Pearson’s correlation test (2-tailed) was applied
to analyze the correlation between p16 and miR-9 expression. A
value of P<0.05 was considered to indicate a statistically
significant difference.
Results
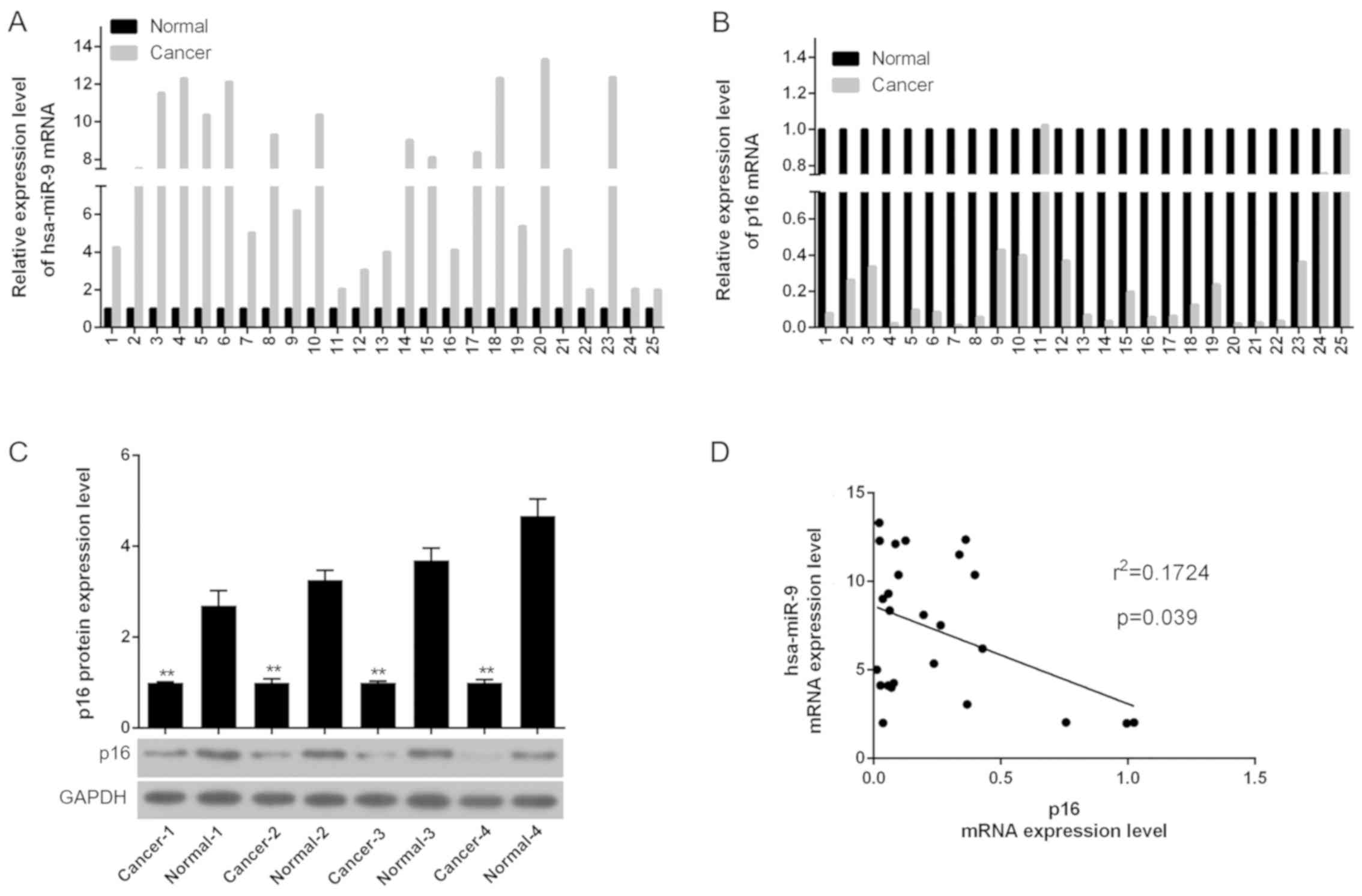
miR-9 is expressed at high levels and p16
is expressed at low levels in patients with OS
In order to examine the expression of miR-9 and p16
in normal tissue and OS tissue, western blot analysis and RT-qPCR
were performed. As shown by the RT-qPCR data, the mRNA level of
miR-9 in the OS tissue was higher than that in the normal tissue;
however, the mRNA level of p16 in the OS tissue was lower than that
in the normal tissue (Fig. 1A and
B). In addition, the western blot analysis results revealed
that compared to the normal tissue, the p16 protein level was
decreased in the OS tissue (Fig.
1C). A negative correlation between the expression of miR-9 and
p16 in 4 randomly selected pairs of OS patient tissue (Fig. 1D) was also observed. The
clinicopathological data demonstrated that rather than age or sex,
a low expression of p16 in patients with OS was associated with
tumor size, TNM stage and lung metastasis (Table II).
| Table IIAssociation between p16 expression
and the clini-copathological characteristics of patients with
osteosarcoma. |
Table II
Association between p16 expression
and the clini-copathological characteristics of patients with
osteosarcoma.
| Groups | No. of
patients | Low p16
expression | High
p16expression | P-value |
|---|
| Age (years) | | | | |
| <60 | 15 | 8 | 7 | 0.513 |
| ≥60 | 10 | 4 | 6 | |
| Sex | | | | 0.821 |
| Male | 12 | 5 | 7 | |
| Female | 13 | 6 | 7 | |
| Tumor size
(cm) | | | | 0.011a |
| <3 | 9 | 2 | 7 | |
| ≥3 | 16 | 12 | 4 | |
| TNM stage | | | | 0.025a |
| I and II | 8 | 3 | 5 | |
| III and IV | 17 | 14 | 3 | |
| Lung
metastasis | | | | 0.008b |
| No | 14 | 4 | 10 | |
| Yes | 11 | 9 | 2 | |
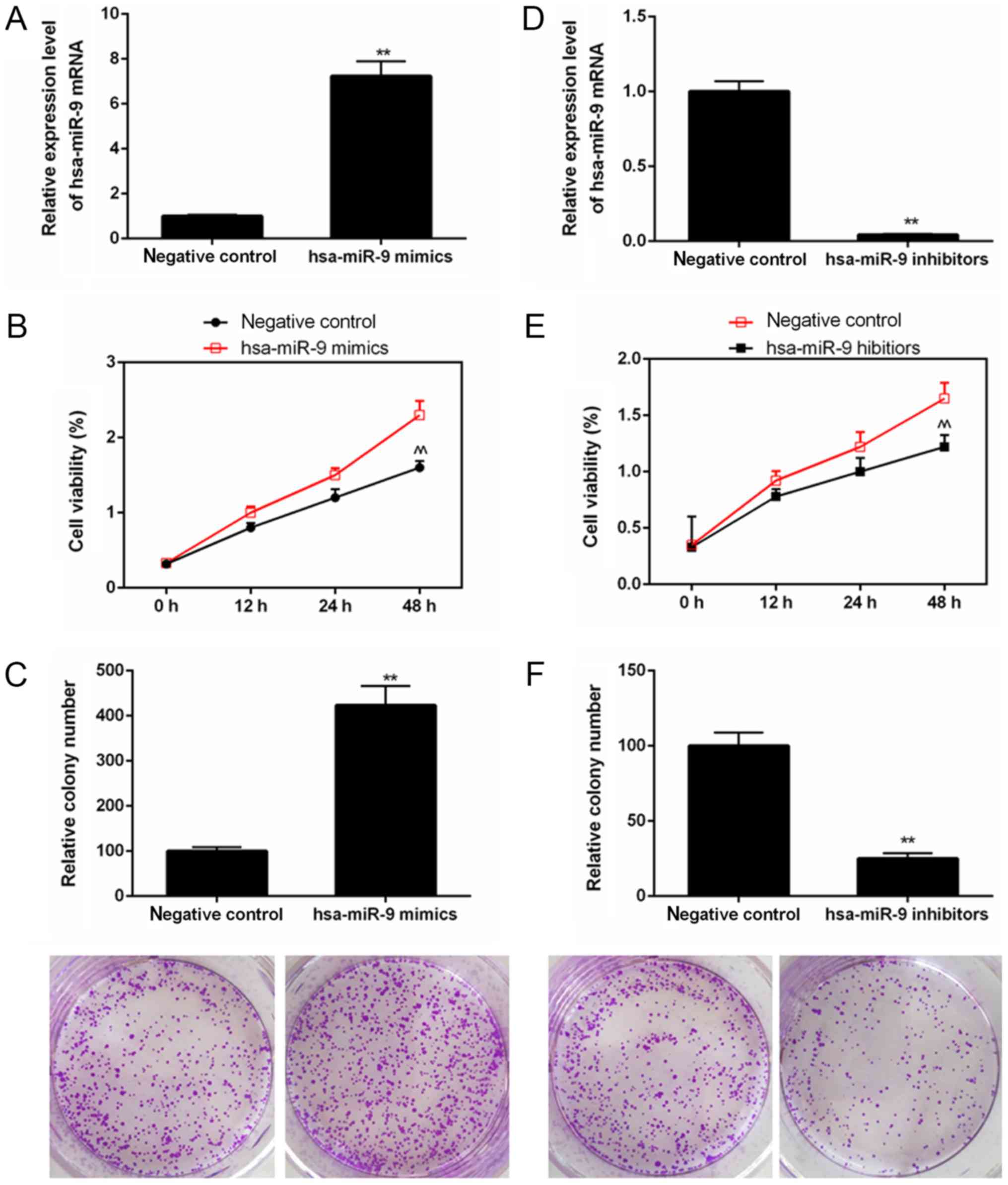
miR-9 depletion suppresses the
proliferation of Saos-2 cells
The results of RT-qPCR revealed that the miR-9 mRNA
level was increased in the cells exposed to hsa-miR-9 mimics, while
the mRNA levels of miR-9 were decreased in the cells transfected
with hsa-miR-9 inhibitors (Fig. 2A and
D). As shown by the results of CCK-8 assay, the value of OD at
450 nm was enhanced in the cells transfected with hsa-miR-9 mimics
(Fig. 2B); however, it was
decreased in the cells transfected with hsa-miR-9 inhibitors
(Fig. 2E). The colony formation
assay results indicated that the relative colony number was
increased in the cells transfected with hsa-miR-9 mimics (Fig. 2C), whereas it was decreased in the
cells transfected with hsa-miR-9 inhibitors (Fig. 2F).
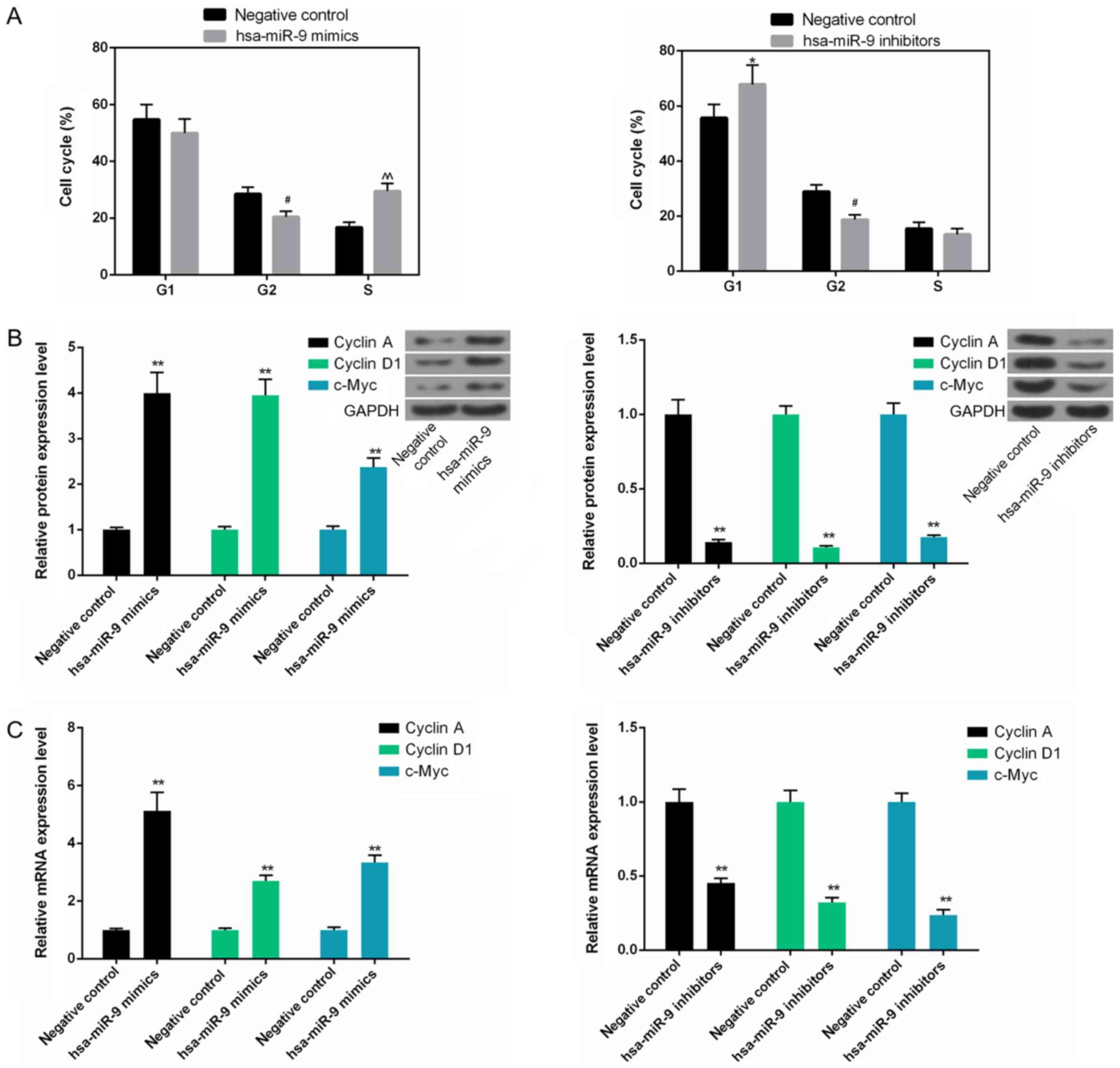
miR-9 depletion arrests the cell cycle at
the G1 phase by downregulating cyclin A, cyclin D1 and c-Myc
expression in Saos-2 cells
The cell cycle analysis data revealed that the
number of S phase cells was high in the hsa-miR-9 mimics group,
whereas the number of cells in the G1 phase was elevated in the
hsa-miR-9 inhibitors group. However, the number of cells in the G2
phase in the hsa-miR-9 mimics and hsa-miR-9 inhibitors group
decreased (Fig. 3A). In addition,
the protein and mRNA levels of cyclin A, cyclin D1 and c-Myc were
upregulated in the hsa-miR-9 mimics, but downregulated in the
hsa-miR-9 inhibitors group (Fig. 3B
and C).
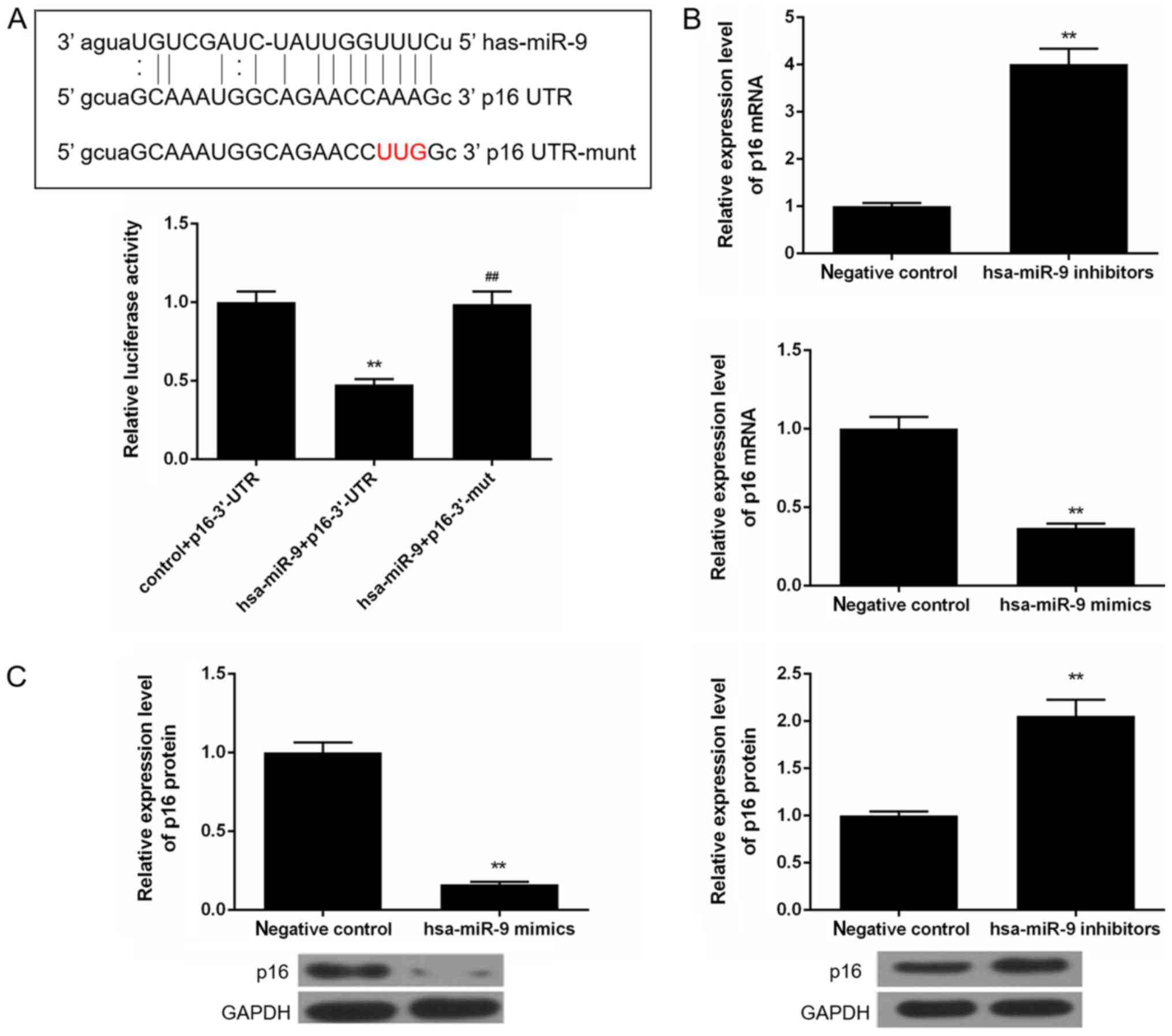
miR-9 depletion increases p16 expression
in Saos-2 cells
According to the microRNA.org
website, p16 was a target gene for miR-9. By performing luciferase
assay, we then found that the luciferase activity was suppressed in
the cells transfected with hsa-miR-9 mimics and p16-3′UTR; however,
it remained stable in the hsa-miR-9 + p16-3′UTR mut group cells
(Fig. 4A). The mRNA and protein
expression levels of p16 were also found to be elevated when the
cells were transfected with hsa-miR-9 inhibitors. However, when the
cells were transfected with hsa-miR-9 mimics, the mRNA and protein
levels of p16 were suppressed (Fig. 4B
and C).
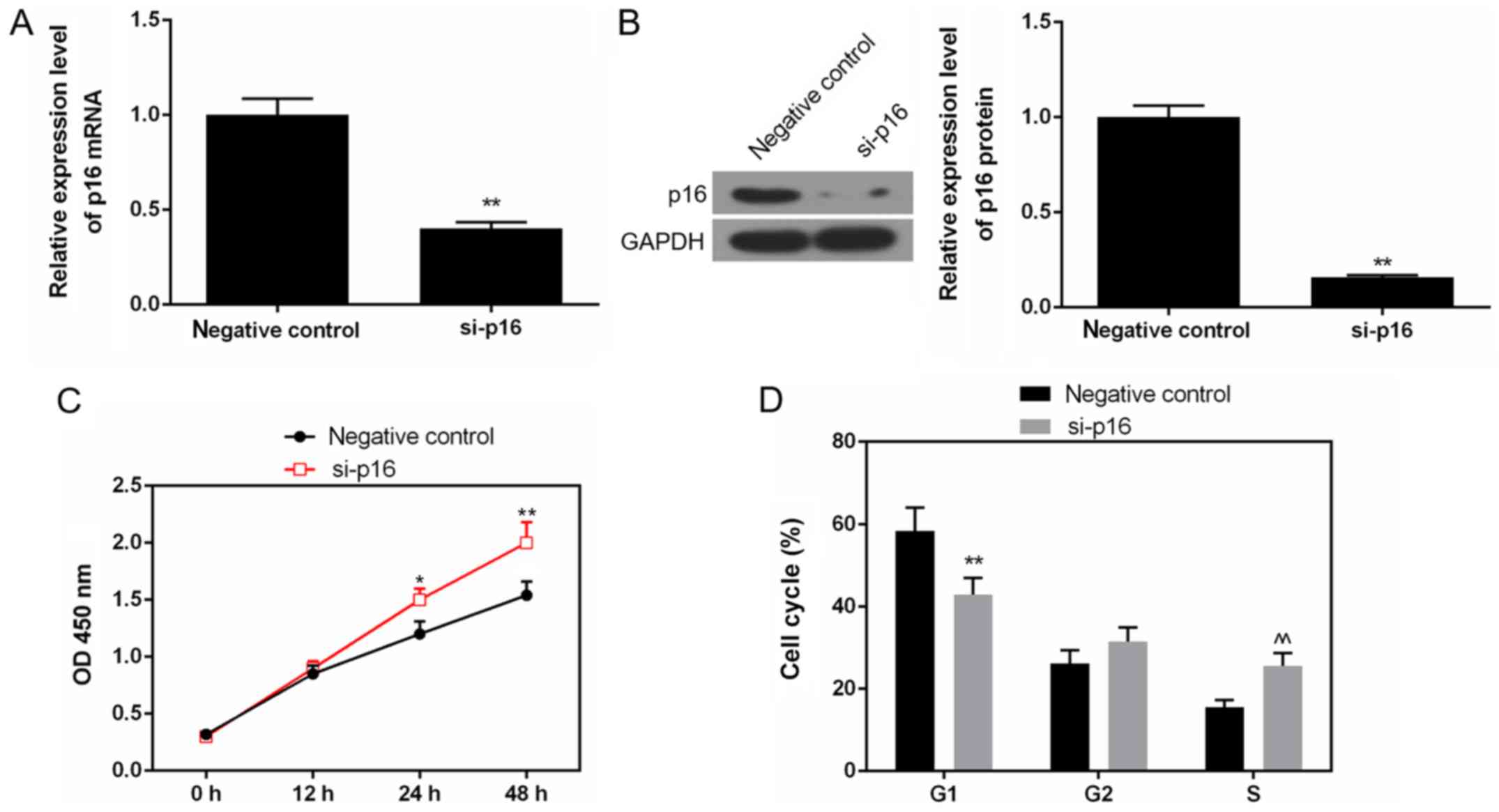
Silencing of p16 enhances the
proliferation of Saos-2 cells by promoting cell cycle G1 phase to S
phase transformation
As shown by the results of RT-qPCR and western blot
analysis, the mRNA and protein levels of p16 were decreased in the
cells transfected with siRNA p16, compared to the negative control
(Fig. 5A and B). The CCK-8 data
also revealed that the OD value was elevated in the cells
transfected with siRNA p16 (Fig.
5C). The depletion of p16 significantly increased the number of
cells in the S phase, while it reduced the number of cells in the
G1 phase (Fig. 5D).
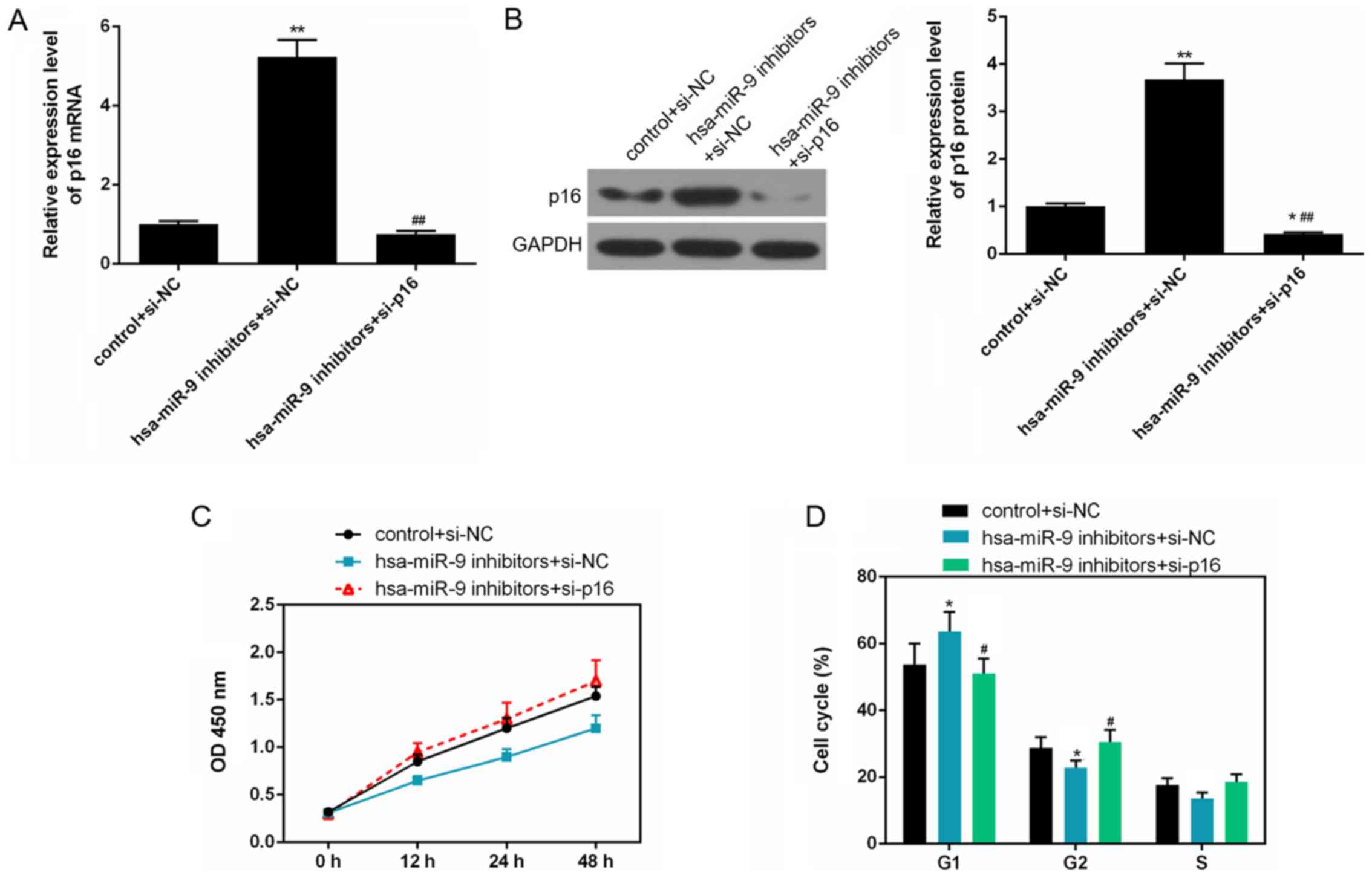
Silencing of p16 reverses the inhibitory
effects of miR-9 inhibitor on Saos-2 cell proliferation by
promoting cell cycle G1 phase to S phase transformation
The results of qRT-qPCR and western blot analysis
revealed that the mRNA and protein levels of p16 were markedly
decreased in the hsa-miR-9 inhibitors + si-p16, compared to the
hsa-miR-9 inhibitors + si-NC group (Fig. 6A and B). When the cells were
transfected with has-miR-9 inhibitors and siRNA p16, the OD value
was elevated (Fig. 6C), and the
number of cells in the G1 phase decreased, and the number of cells
in the G2 phase increased (P<0.05; Fig. 6D).
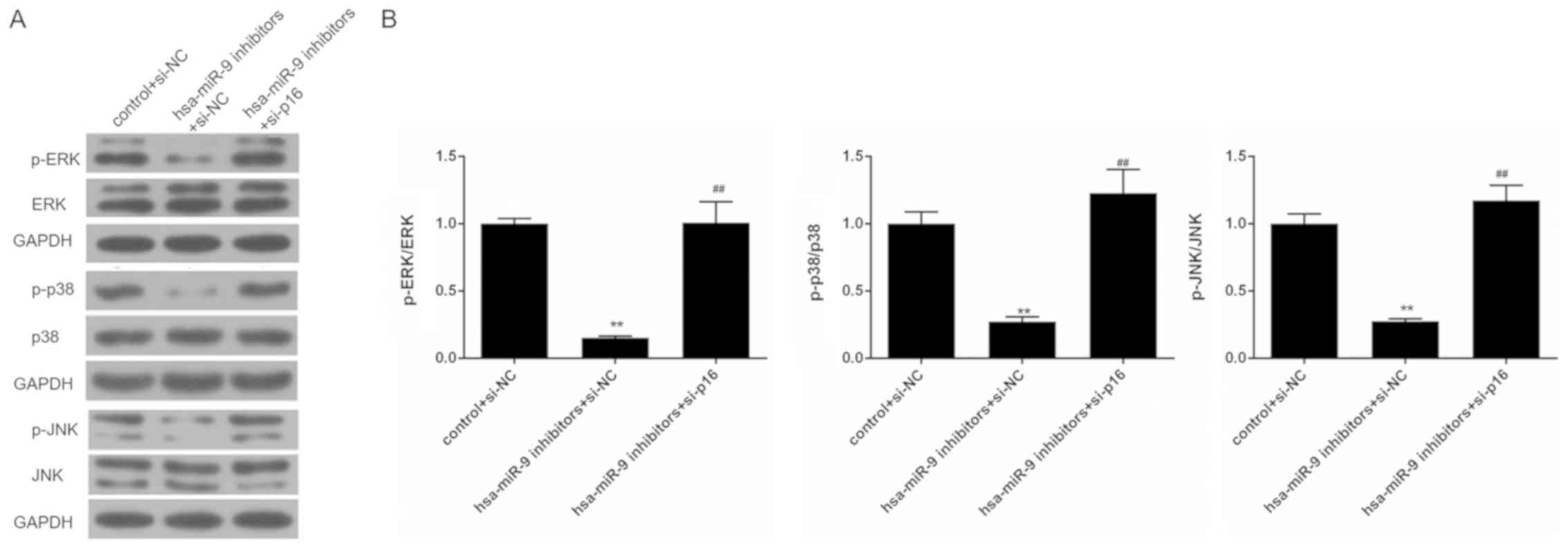
Silencing of p16 reverses the inhibitory
effects of miR-9 inhibitor on the activation of the ERK/p38/JNK
pathway in Saos-2 cells
To examine the effects of miR-9 inhibitor and siRNA
p16 on the ERK/p38/JNK pathway in Saos-2 cells, western blot
analysis was performed. The results revealed that miR-9 inhibitor
suppressed the phosphorylation of ERK, p38 and JNK. Nevertheless,
the expression levels of total ERK, p38 and JNK remained stable.
The proportions of p-ERK/ERK, p-p38/p38 and p-JNK/JNK were markedly
reduced in the hsa-miR-9 inhibitors + si-NC group. However, in
comparison with the hsa-miR-9 inhibitors + si-NC group, the
phosphorylation levels of ERK, p38 and JNK were elevated in the
hsa-miR-9 inhibitors + si-p16 group. The proportions of p-ERK/ERK,
p-p38/p38 and p-JNK/JNK were also increased in the hsa-miR-9
inhibitors + si-p16 group (Fig.
7).
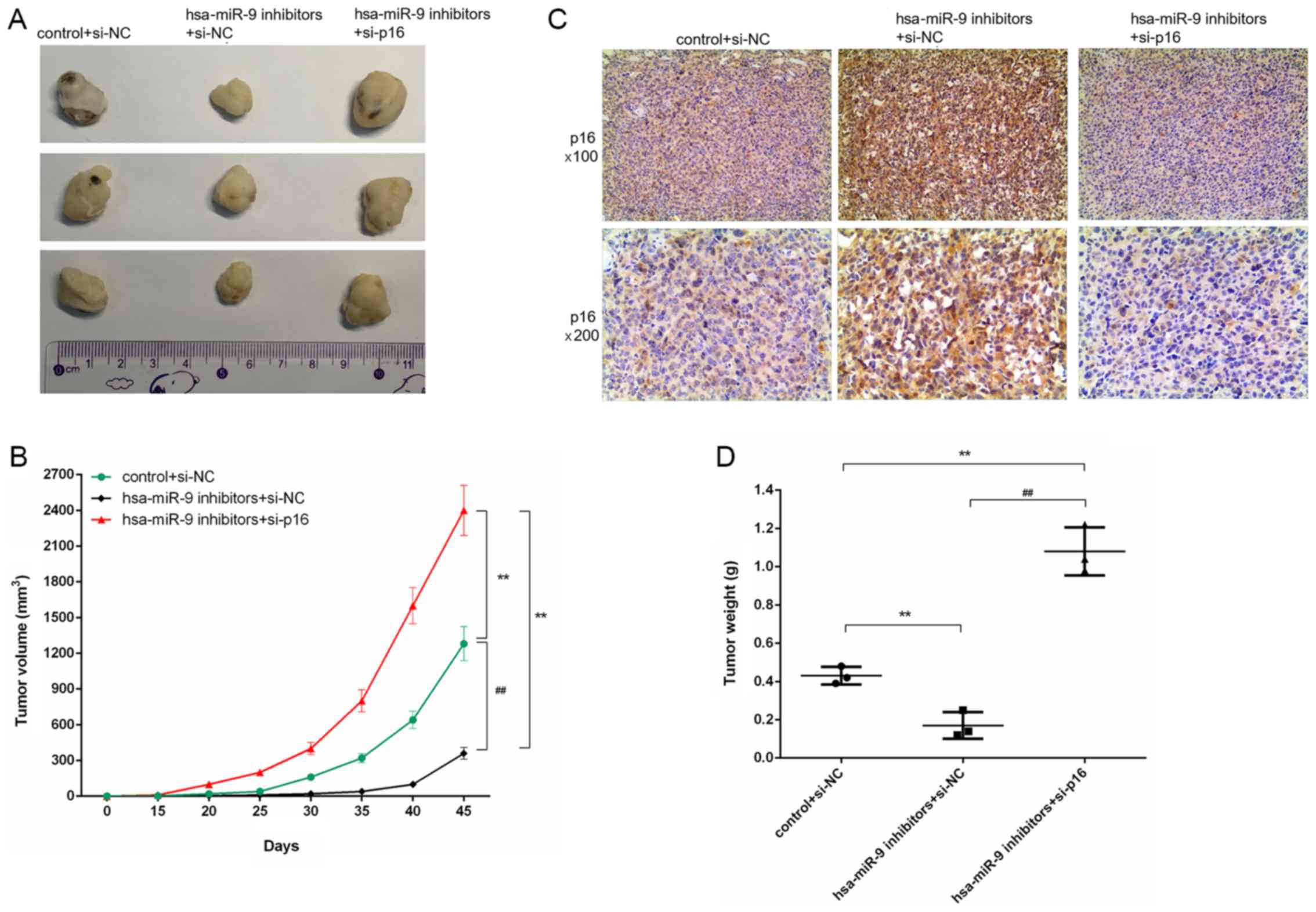
Silencing of p16 reverses the inhibitory
effects of miR-9 inhibitor on tumor growth in mice with OS
As shown by the images in Fig. 8A, the tumors in the hsa-miR-9
inhibitors + si-NC group were smaller than those in the hsa-miR-9
inhibitors + si-p16 group. In addition, according to the results of
our calculations, as time progressed, the volume of the tumors
increased. The tumor volume in the hsa-miR-9 inhibitors + si-p16
group was larger than that in the hsa-miR-9 inhibitors + si-NC
group (Fig. 8B). The IHC results
also revealed a large number of evident brown particles in the
hsa-miR-9 inhibitors + si-NC group (Fig. 8C). Additionally, the tumor weight
in the hsa-miR-9 inhibitors + si-p16 group was greater than that in
the hsa-miR-9 inhibitors + si-NC group (Fig. 8D).
Discussion
miRNAs, which are the key regulators of cancer and
can regulate the proliferation, migration and invasion of tumor
cells, can function both as oncogenes and tumor suppressors. miR-9
has been shown to be highly expressed in lung cancer,
hepatocellular carcinoma and OS (35-37)
and to be expressed at low levels in ovarian cancer and
nasopharyngeal carcinoma (38,39).
In this study, we found that the expression of miR-9 in OS tissue
was higher than that in adjacent normal tissue. Furthermore, Zhu
et al have proven that miR-9 facilitates the growth of OS
cells (37). Similar to the
findings of this previous study, in this study, we observed that
miR-9 mimics increased the proliferation and colony numbers of
Saos-2 cells, while miR-9 inhibitors reduced the proliferation and
colony numbers of the cells. This phenomenon suggest that miR-9
exerts oncogenic effects on OS.
An uncontrolled cell cycle is the cause of tumor
growth and malignant cell proliferation (40,41).
Hence, in this study, we examined the role of miR-9 in the cell
cycle of Saos-2 cells. Liu et al demonstrated that miR-9
depletion blocked N2a cells cells at the G1 phase (42). The data from this study revealed
that miR-9 mimics arrested the cells in the S phase, and miR-9
inhibitors arrested the cells in the G1 phase. Cyclin A, cyclin D1
and c-Myc expression levels were detected to examine the molecular
mechanisms of the cell cycle. The main function of cyclin D1 is to
promote cell proliferation. Cyclin D1 combines and activates the
cyclin-dependent kinase 4 (CDK4) specific to the G1 phase, the
inhibitor protein (Rb) of which is therefore phosphorylated. The
phosphorylated Rb protein dissociates from its E2F transcription
factor, and the E2F transcriptional factor begins to transcribe the
gene of the living cell cycle, thereby promoting the cell cycle
into the S phase from the G1 phase (43). Cyclin A mainly activates CDK2 and
CDK1 to regulate chromosome replication and mitosis (44,45).
The c-Myc gene is an important member of the myc gene family and
promotes cell division (46).
According to a previous study, miR-9 promotes the growth and cell
cycle progression of bladder cancer cells by increasing cyclin D1
expression (47). Zhou et
al testified that miR-632 accelerated cell proliferation in
laryngeal cancer by upregulating cyclin D1 and c-Myc expression
(48). The findings of this study
demonstrated that miR-9 mimics promoted cyclin A, cyclin D1 and
c-Myc expression levels, while miR-9 inhibitors suppressed cyclin
A, cyclin D1 and c-Myc expression levels. These results suggest
that miR-9 facilitates the proliferation of Saos-2 cells by
arresting the cells in the S phase, and upregulating cyclin A,
cyclin D1 and c-Myc expression levels.
p16 is one of the most common suppressor genes of
human tumor cells, encoding a 16 kDa protein, namely the p16
protein (49,50). In 1994, Nobori et al
suggested that the role of p16 in cancer suppression was far
greater than the role of p53, and its deletion or mutation was
associated with the occurrence of a variety of tumors (51). In this study, p16 was found to be
expressed at low levels in OS tissue compared to adjacent normal
tissue. In addition, p16 is a cyclin dependent inhibitor (CDI),
which can negatively regulate the cell cycle by inhibiting CDK
(50,52,53).
The inactivation of the p16 gene will lead to excessive cell
proliferation (54). Thus, we
hypothesized that p16 may affect cell proliferation in OS. As
expected, we observed that the silencing of p16 significantly
promoted the proliferation of Saos-2 cells by arresting the cells
in the S phase.
miRNAs are involved in the progression of various
types of cells by regulating their target genes (55-57).
O’Loghlen et al demonstrated that the miR-9 family control
senescence by mediating p16 gene in cancer (58). Hence, we hypothesized that miR-9
and p16 might have a certain connection in OS. Notably, in this
study, according to the microRNA.org
website prediction, the 3′-UTR of p16 mRNA had the complementary
sites for hsa-miR-9. The luciferase activity was attenuated in the
hsa-miR-9 + p16-3′UTR group; however, no change was observed in the
hsa-miR-9 + p16-3′UTR mut group. Moreover, miR-9 mimics decreased
the expression of p16; however, miR-9 inhibitors increased p16
expression. These data suggest a negative correlation between miR-9
and p16 in OS. Importantly, p16 was found to be a target gene of
miR-9 in OS.
To further verify that p16 is a target gene of miR-9
in OS, the growth of Saos-2 cells was measured following
transfection with hsa-miR-9 inhibitors and siRNA p16. Furthermore,
the effects of miR-9 inhibitor and si-p16 on tumor growth in mice
with OS were investigated. The results revealed that the silencing
p16 reversed the repression impact of miR-9 on cell growth by
arresting G1 phase. In vivo, the data also revealed that p16
depletion reversed the suppressive effects of miR-9 inhibitor on
tumor growth. This phenomenon indicated that the carcinogenic
effects of miR-9 in OS involve the direct targeting of p16.
Researchers have demonstrated that ERK/p38/JNK
pathway involves the regulation of the proliferation,
differentiation, apoptosis and metastasis of various cancer cells
(23,24,28,29).
Wang et al confirmed that miR-155 increases OS cell growth
by regulating MAPK signaling (59). Gui et al indicated that
miR-497 inhibited the proliferation of OS cells by targeting the
MAPK/ERK pathway (60). Zhang
et al demonstrated that miR-9 inhibited the growth of
hepatocellular carcinoma cells by mediating ERK signaling (61). Ben-Hamo and Efroni proved that
miR-9 regulated the p38 pathway in glioblastoma multiforme
(62). Herein, we hypothesized
that miR-9 may regulate the ERK-p38-JNK pathway in OS. Fortunately,
our data revealed that miR-9 inhibitors downregulated the
phosphorylation levels of ERK, p38 and JNK. Nevertheless, the
silencing of p16 reversed the suppressive effects of miR-9
inhibitor on the ERK/p38/JNK pathway. These results demonstrated
that miR-9 depletion downregulates the ERK/p38/JNK pathway by
targeting p16.
This study also had some limitations, for example,
the number of patients with OS enrolled was not sufficient and the
trial period was too short. We thus aim to carry out further
studies in the future with a larger sample size in order to
validate the results of this study.
In conclusion, the findings of this study
demonstrated that miR-9 depletion suppresses cell proliferation by
targeting p16 and mediating the activation of the ERK/p38/JNK
pathway in OS. These results provide the foundation for future
studies on the pathogenesis of OS.
Funding
This work study supported by the Medical Science and
Technology Research Project of Henan Provincial Health Department
(grant no. 201203106).
Availability of data and materials
The analyzed datasets generated during this study
are available from the corresponding author on reasonable
request.
Authors’ contributions
SG made substantial contributions to the conception
and design of the study. JW, ST and JL contributed to data
acquisition, and data analysis and interpretation. SG, ST and JL
contributed to the drafting of the article or critically revising
it for important intellectual content. All authors agree to be
accountable for all aspects of the work in ensuring that questions
related to the accuracy or integrity of the work are appropriately
investigated and resolved. All authors have read and approved the
final manuscript.
Ethics approval and consent to
participate
All procedures performed involving human
participants were in accordance with the ethical standards of the
institutional and/or national research committee and with the 1964
Helsinki declaration and its later amendments or comparable ethical
standards. All patients signed informed consent forms, allowing
their tissues to be used in this study. The Ethics Committee of
Henan Provincial People’s Hospital authorized this research. All
animal experiments were performed following the approval of the
Henan Provincial People’s Hospital Animal Ethics Committee and
according to the Guidelines for the Care and Use of Laboratory
Animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare no conflicts of interest.
Acknowledgments
Not applicable.
References
|
1
|
Garcia-Moure M, Martinez-Vélez N,
Patiño-García A and Alonso MM: Oncolytic adenoviruses as a
therapeutic approach for osteosarcoma: A new hope. J Bone Oncol.
9:41–47. 2016. View Article : Google Scholar
|
|
2
|
Misaghi A, Goldin A, Awad M and Kulidjian
AA: Osteosarcoma: A comprehensive review. SICOT J. 4:122018.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Guenther LM, Rowe RG, Acharya PT, Swenson
DW, Meyer SC, Clinton CM, Guo D, Sridharan M, London WB, Grier HE,
et al: Response Evaluation Criteria in Solid Tumors (RECIST)
following neoadjuvant chemotherapy in osteosarcoma. Pediatr Blood
Cancer. 65:652018. View Article : Google Scholar
|
|
4
|
Huang Z and Lou C: Application of the
alteration uptake ratio of 99mTc-MIBI scintigraphy for evaluating
the efficacy of neoadjuvant chemotherapy in osteosarcoma patients.
Hell J Nucl Med. 21:55–59. 2018.PubMed/NCBI
|
|
5
|
Cavit A, Özcanli H, Sançmiş M, Ocak GA and
Gürer EI: Tumorous conditions of the hand: A retrospective review
of 402 cases. Turk Patoloji Derg. 34:66–72. 2018.
|
|
6
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee J, Park H, Eom J and Kang SG:
MicroRNA-mediated regulation of the development and functions of
follicular helper T cells. Immune Netw. 18:e72018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee RC, Feinbaum RL and Ambros V: The C.
elegans heterochronic gene lin-4 encodes small RNAs with antisense
complementarity to lin-14. Cell. 75:843–854. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wightman B, Ha I and Ruvkun G:
Posttranscriptional regulation of the heterochronic gene lin-14 by
lin-4 mediates temporal pattern formation iC elegans. Cell.
75:855–862. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen Z, Han Y, Song C, Wei H, Chen Y,
Huang K, Li S, Ma D, Wang S, Wang J, et al: Systematic review and
meta-analysis of the prognostic significance of microRNAs in
cervical cancer. Oncotarget. 9:17141–17148. 2017.
|
|
11
|
Hershkovitz-Rokah O, Geva P, Salmon-Divon
M, Shpilberg O and Liberman-Aronov S: Network analysis of
microRNAs, genes and their regulation in diffuse and follicular
B-cell lymphomas. Oncotarget. 9:7928–7941. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ichii O and Horino T: MicroRNAs associated
with the development of kidney diseases in humans and animals. J
Toxicol Pathol. 31:23–34. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Iguchi T, Sakurai K, Tamai S and Mori K:
Circulating liver-specific microRNAs in cynomolgus monkeys. J
Toxicol Pathol. 31:3–13. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang H, Peng R, Wang J, Qin Z and Xue L:
Circulating microRNAs as potential cancer biomarkers: The advantage
and disadvantage. Clin Epigenetics. 10:592018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen M, Liu YY, Zheng MQ, Wang XL, Gao XH,
Chen L and Zhang GM: microRNA-544 promoted human osteosarcoma cell
proliferation by downregulating AXIN2 expression. Oncol Lett.
15:7076–7082. 2018.PubMed/NCBI
|
|
16
|
Ding J, Sha L, Shen P, Huang M, Cai Q and
Li J: MicroRNA-18a inhibits cell growth and induces apoptosis in
osteosarcoma by targeting MED27. Int J Oncol. 53:329–338.
2018.PubMed/NCBI
|
|
17
|
Tang W, Wang W, Zhao Y and Zhao Z:
MicroRNA-874 inhibits cell proliferation and invasion by targeting
cyclin-dependent kinase 9 in osteosarcoma. Oncol Lett.
15:7649–7654. 2018.PubMed/NCBI
|
|
18
|
Fenger JM, Roberts RD, Iwenofu OH, Bear
MD, Zhang X, Couto JI, Modiano JF, Kisseberth WC and London CA:
MiR-9 is overexpressed in spontaneous canine osteosarcoma and
promotes a metastatic phenotype including invasion and migration in
osteoblasts and osteosarcoma cell lines. BMC Cancer. 16:7842016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jang MH, Kim HJ, Gwak JM, Chung YR and
Park SY: Prognostic value of microRNA-9 and microRNA-155 expression
in triple-negative breast cancer. Hum Pathol. 68:69–78. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Snezhkina AV, Krasnov GS, Zhikrivetskaya
SO, Karpova IY, Fedorova MS, Nyushko KM, Belyakov MM, Gnuchev NV,
Sidorov DV, Alekseev BY, et al: Overexpression of microRNAs miR-9,
-98, and -199 correlates with the downregulation of HK2 expression
in colorectal cancer. Mol Biol (Mosk). 52:220–230. 2018.In Russian.
View Article : Google Scholar
|
|
21
|
Wang H, Wu Q, Zhang Y, Zhang HN, Wang YB
and Wang W: TGF-β1-induced epithelial-mesenchymal transition in
lung cancer cells involves upregulation of miR-9 and downregulation
of its target, E-cadherin. Cell Mol Biol Lett. 22:222017.
View Article : Google Scholar
|
|
22
|
Zheng L, Qi T, Yang D, Qi M, Li D, Xiang
X, Huang K and Tong Q: microRNA-9 suppresses the proliferation,
invasion and metastasis of gastric cancer cells through targeting
cyclin D1 and Ets1. PLoS One. 8:e557192013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
She H, He Y, Zhao Y and Mao Z: Autophagy
in inflammation: The p38α MAPK-ULK1 axis. Macrophage (Houst).
5:e16292018.
|
|
24
|
Tang Q, Tong M, Zheng G, Shen L, Shang P
and Liu H: Masquelet’s induced membrane promotes the osteogenic
differentiation of bone marrow mesenchymal stem cells by activating
the Smad and MAPK pathways. Am J Transl Res. 10:1211–1219.
2018.
|
|
25
|
Li S, Ma YM, Zheng PS and Zhang P: GDF15
promotes the proliferation of cervical cancer cells by
phosphorylating AKT1 and Erk1/2 through the receptor ErbB2. J Exp
Clin Cancer Res. 37:802018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pinal N, Martín M, Medina I and Morata G:
Short-term activation of the Jun N-terminal kinase pathway in
apoptosis-deficient cells of Drosophila induces tumorigenesis. Nat
Commun. 9:15412018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang S, Guo L, Su Y, Wen J, Du J, Li X,
Liu Y, Feng J, Xie Y, Bai Y, et al: Nitric oxide balances
osteoblast and adipocyte lineage differentiation via the JNK/MAPK
signaling pathway in periodontal ligament stem cells. Stem Cell Res
Ther. 9:1182018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Aroui S, Aouey B, Chtourou Y, Meunier AC,
Fetoui H and Kenani A: Naringin suppresses cell metastasis and the
expression of matrix metalloproteinases (MMP-2 and MMP-9) via the
inhibition of ERK-P38-JNK signaling pathway in human glioblastoma.
Chem Biol Interact. 244:195–203. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Speth Z, Islam T, Banerjee K and Resat H:
EGFR signaling pathways are wired differently in normal 184A1L5
human mammary epithelial and MDA-MB-231 breast cancer cells. J Cell
Commun Signal. 11:341–356. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen HJ, Lin CM, Lee CY, Shih NC, Peng SF,
Tsuzuki M, Amagaya S, Huang WW and Yang JS: Kaempferol suppresses
cell metastasis via inhibition of the ERK-p38-JNK and AP-1
signaling pathways in U-2 OS human osteosarcoma cells. Oncol Rep.
30:925–932. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Joo SS and Yoo YM: Melatonin induces
apoptotic death in LNCaP cells via p38 and JNK pathways:
Therapeutic implications for prostate cancer. J Pineal Res.
47:8–14. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhao Li T, Ge J, Yang J, Song J, Wang X,
Mao C, Zhang J, Zou Y, Liu YY, et al: Particulate matter
facilitates C6 glioma cells activation and the release of
inflammatory factors through MAPK and JAK2/STAT3 pathways.
Neurochem Res. 41:1969–1981. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
34
|
Faustino-Rocha A, Oliveira PA,
Pinho-Oliveira J, Teixeira-Guedes C, Soares-Maia R, da Costa RG,
Colaço B, Pires MJ, Colaço J, Ferreira R, et al: Estimation of rat
mammary tumor volume using caliper and ultrasonography
measurements. Lab Anim (NY). 42:217–224. 2013. View Article : Google Scholar
|
|
35
|
Drakaki A, Hatziapostolou M, Polytarchou
C, Vorvis C, Poultsides GA, Souglakos J, Georgoulias V and
Iliopoulos D: Functional microRNA high throughput screening reveals
miR-9 as a central regulator of liver oncogenesis by affecting the
PPARA-CDH1 pathway. BMC Cancer. 15:5422015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xu T, Liu X, Han L, Shen H, Liu L and Shu
Y: Up-regulation of miR-9 expression as a poor prognostic biomarker
in patients with non-small cell lung cancer. Clin Transl Oncol.
16:469–475. 2014. View Article : Google Scholar
|
|
37
|
Zhu SW, Li JP, Ma XL, Ma JX, Yang Y, Chen
Y and Liu W: miR-9 modulates osteosarcoma cell growth by targeting
the GCIP tumor suppressor. Asian Pac J Cancer Prev. 16:4509–4513.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Laios A, O’Toole S, Flavin R, Martin C,
Kelly L, Ring M, Finn SP, Barrett C, Loda M, Gleeson N, et al:
Potential role of miR-9 and miR-223 in recurrent ovarian cancer.
Mol Cancer. 7:352008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xu Lu J, Liu X, Peng X, Zhang Y, Wang B,
Luo L, Peng H, Li X, Tian GW, et al: Predictive value of miR-9 as a
potential biomarker for nasopharyngeal carcinoma metastasis. Br J
Cancer. 110:392–398. 2014. View Article : Google Scholar :
|
|
40
|
Jia GQ, Zhang MM, Wang K, Zhao GP, Pang MH
and Chen ZY: Long non-coding RNA PlncRNA-1 promotes cell
proliferation and hepatic metastasis in colorectal cancer. J Cell
Biochem. 119:7091–7104. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhou Q, You C, Zheng C, Gu Y, Gu H, Zhang
R, Wu H and Sun B: 3-Nitroacridine derivatives arrest cell cycle at
G0/G1 phase and induce apoptosis in human breast cancer cells may
act as DNA-target anticancer agents. Life Sci. 206:1–9. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu W, Liu C, Yin B and Peng XZ: Functions
of miR-9 and miR-9* during aging in SAMP8 mice and their possible
mechanisms. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 37:253–258.
2015.PubMed/NCBI
|
|
43
|
Di Sante G, Di Rocco A, Pupo C, Casimiro
MC and Pestell RG: Hormone-induced DNA damage response and repair
mediated by cyclin D1 in breast and prostate cancer. Oncotarget.
8:81803–81812. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li Y, Zhang J, Gao W, Zhang L, Pan Y,
Zhang S and Wang Y: Insights on structural characteristics and
ligand binding mechanisms of CDK2. Int J Mol Sci. 16:9314–9340.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Roskoski R Jr: Cyclin-dependent protein
kinase inhibitors including palbociclib as anticancer drugs.
Pharmacol Res. 107:249–275. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Goetzman ES and Prochownik EV: The role
for Myc in coordinating glycolysis, oxidative phosphorylation,
glutaminolysis, and fatty acid metabolism in normal and neoplastic
tissues. Front Endocrinol (Lausanne). 9:1292018. View Article : Google Scholar
|
|
47
|
Wang H, Zhang W, Zuo Y, Ding M, Ke C, Yan
R, Zhan H, Liu J and Wang J: miR-9 promotes cell proliferation and
inhibits apoptosis by targeting LASS2 in bladder cancer. Tumour
Biol. 36:9631–9640. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhou ZX, Zhang ZP, Tao ZZ and Tan TZ:
MiR-632 promotes laryngeal carcinoma cell proliferation, migration
and invasion through negative regulation of GSK3β. Oncol Res: Mar.
21:2018Epub ahead of print. View Article : Google Scholar
|
|
49
|
Lee JK, Lee KH, Kim SA, Kweon SS, Cho SH,
Shim HJ, Bae WK, Chung IJ, Chung WK, Yoon TM, et al: p16 as a
prognostic factor for the response to induction chemotherapy in
advanced hypo-pharyngeal squamous cell carcinoma. Oncol Lett.
15:6571–6577. 2018.PubMed/NCBI
|
|
50
|
Ottria L, Candotto V, Cura F, Baggi L,
Arcuri C, Nardone M, Gaudio RM, Gatto R, Spadari F and Carinci F:
HPV acting on E-cadherin, p53 and p16: Literature review. J Biol
Regul Homeost Agents. 32(Suppl 1): 73–79. 2018.PubMed/NCBI
|
|
51
|
Nobori T, Miura K, Wu DJ, Lois A,
Takabayashi K and Carson DA: Deletions of the cyclin-dependent
kinase-4 inhibitor gene in multiple human cancers. Nature.
368:753–756. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hirama T and Koeffler HP: Role of the
cyclin-dependent kinase inhibitors in the development of cancer.
Blood. 86:841–854. 1995.PubMed/NCBI
|
|
53
|
Serrano M, Hannon GJ and Beach D: A new
regulatory motif in cell-cycle control causing specific inhibition
of cyclin D/CDK4. Nature. 366:704–707. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhu Pu X, Fu L, Fan Y, Zheng Z, Zhang J,
Yang B, Guan J, Wu W, Ye HQ, et al: Companied P16 genetic and
protein status together providing useful information on the
clinical outcome of urinary bladder cancer. Medicine (Baltimore).
97:e03532018. View Article : Google Scholar :
|
|
55
|
Goody D and Pfeifer A: MicroRNAs in brown
and beige fat. Biochim Biophys Acta Mol Cell Biol Lipids.
1864:29–36. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Guo J and Cheng Y: MicroRNA-1247 inhibits
lipopolysac-charides-induced acute pneumonia in A549 cells via
targeting CC chemokine ligand 16. Biomed Pharmacother. 104:60–68.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Song Z, Cooper DKC, Cai Z and Mou L:
Expression and Regulation profile of mature MicroRNA in the pig:
Relevance to xenotransplantation. BioMed Res Int. 2018:29839082018.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
O’Loghlen A, Brookes S, Martin N,
Rapisarda V, Peters G and Gil J: CBX7 and miR-9 are part of an
autoregulatory loop controlling p16(INK) (4a). Aging Cell.
14:1113–1121. 2015. View Article : Google Scholar
|
|
59
|
Wang C, Zhang X, Zhang C, Zhai F, Li Y and
Huang Z: MicroRNA-155 targets MAP3K10 and regulates osteosarcoma
cell growth. Pathol Res Pract. 213:389–393. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Gui ZL, Wu TL, Zhao GC, Lin ZX and Xu HG:
MicroRNA-497 suppress osteosarcoma by targeting MAPK/Erk pathway.
Bratisl Lek Listy. 118:449–452. 2017.PubMed/NCBI
|
|
61
|
Zhang J, Cheng J, Zeng Z, Wang Y, Li X,
Xie Q, Jia J, Yan Y, Guo Z, Gao J, et al: Comprehensive profiling
of novel microRNA-9 targets and a tumor suppressor role of
microRNA-9 via targeting IGF2BP1 in hepatocellular carcinoma.
Oncotarget. 6:42040–42052. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Ben-Hamo R and Efroni S: Correction: Gene
expression and network-based analysis reveals a novel role for
hsa-miR-9 and drug control over the p38 network in glioblastoma
multiforme progression. Genome Med. 4:872012. View Article : Google Scholar : PubMed/NCBI
|