Introduction
Langerhans cell histiocytosis (LCH) is a rare
hematological disorder caused by the clonal proliferation and
accumulation of LCH cells, which have similar phenotypic features
and are thought to be a myeloid-derived precursor of epidermal
Langerhans cells (1). Its exact
aetiology remains unknown but recent data show that in a
significant percentage of LCH patients mutations that activate the
RAS-RAF-MEK-ERK pathway are identified [BRAF oncogene (V600E):
50–60%, MAP2KI genes: 10–20%, other genes (ERBB3 and ARAF)]
(2). The disease has an extreme
variety of possible clinical presentations ranging from a single
focal bone lesion to a life-threatening multisystem disease
requiring emergency and aggressive chemotherapy (3). LCH is a rare condition affecting <1
in 200,000 children younger than 15 years of age. Its incidence
ranges from 2 to 9 cases per million per year, with a male to
female ratio close to one. It can present at any age but among
children its peak age at presentation is 3–4 years. There seems to
be a seasonal variation with spikes in fall and winter (4). Whether this rare disorder is a
neoplasia or a cancer-like condition of the immune system is still
hotly debated and international collaborative trials aiming to
identify the optimal treatment duration for the prevention of
recurrent and the treatment of resistant disease are ongoing.
Herein, we report a striking series of four patients
diagnosed with LCH in a single Pediatric Department with a
catchment area of approximately 270,000 inhabitants during an
eight-month period. We aim to highlight the heterogeneity of
disease's presentation, to present the management and treatment
options and to raise the index of suspicion for health care
practitioners.
Case report
Four patients that were diagnosed with LCH in the
Pediatric Department of a University Affiliated Hospital between
September 2014 and April 2015 are presented after retrospective
review of all patients' notes. Informed consent was obtained by all
the parents of our patients. The present study was approved by the
Papageorgiou Hospital Ethic Committee. Their demographic
characteristics and initial clinical signs and symptoms are
summarized in Table I.
 | Table I.Demographic characteristics, clinical
and radiological findings as well as treatment stratification group
of all patients. |
Table I.
Demographic characteristics, clinical
and radiological findings as well as treatment stratification group
of all patients.
| Patient | No. 1 | No. 2 | No. 3 | No. 4 |
|---|
| Age | 5 months | 7 years | 4 months | 4 ½ years |
| Gender | Female | Male | Female | Male |
| Location | Left frontal bone,
Skin | Left iliac bone | Perianal area | Brain, Left
femur |
| Clinical
manifestation | Oedema and redness,
refractory eczema, early teething | Persistent pain after
trauma | Refractory
eczema | Diabetes insipidus
Limp |
| Symptom's
duration | ½ month | 1 month | 3.5 months | 6 months |
| Imaging findings | Lytic lesion (Brain
MRI) | Lytic lesion (Pelvic
and lower limb CT) | (−) | Ectopic
neurohypophysis, thickened pituitary stalk, loss of ‘bright spot’
(Brain MRI) Lytic lesion (Lower limb CT) |
| Diagnosis (LCH
III) | Multisystem, Group
2-low risk group/Group 3-localized special site involvement | Single
system/Unifocal, Group 2-low risk group | Unifocal, Group 2-low
risk group | Multisystem, Group
3-localized special site involvement |
Clinical presentation
Patient no. 1
A 5-month old female infant was admitted for
erythema and edema of the left orbit, after a reported insect bite
that had been treated with oral amoxicillin for seven days without
improvement. Her family and medical histories were unremarkable,
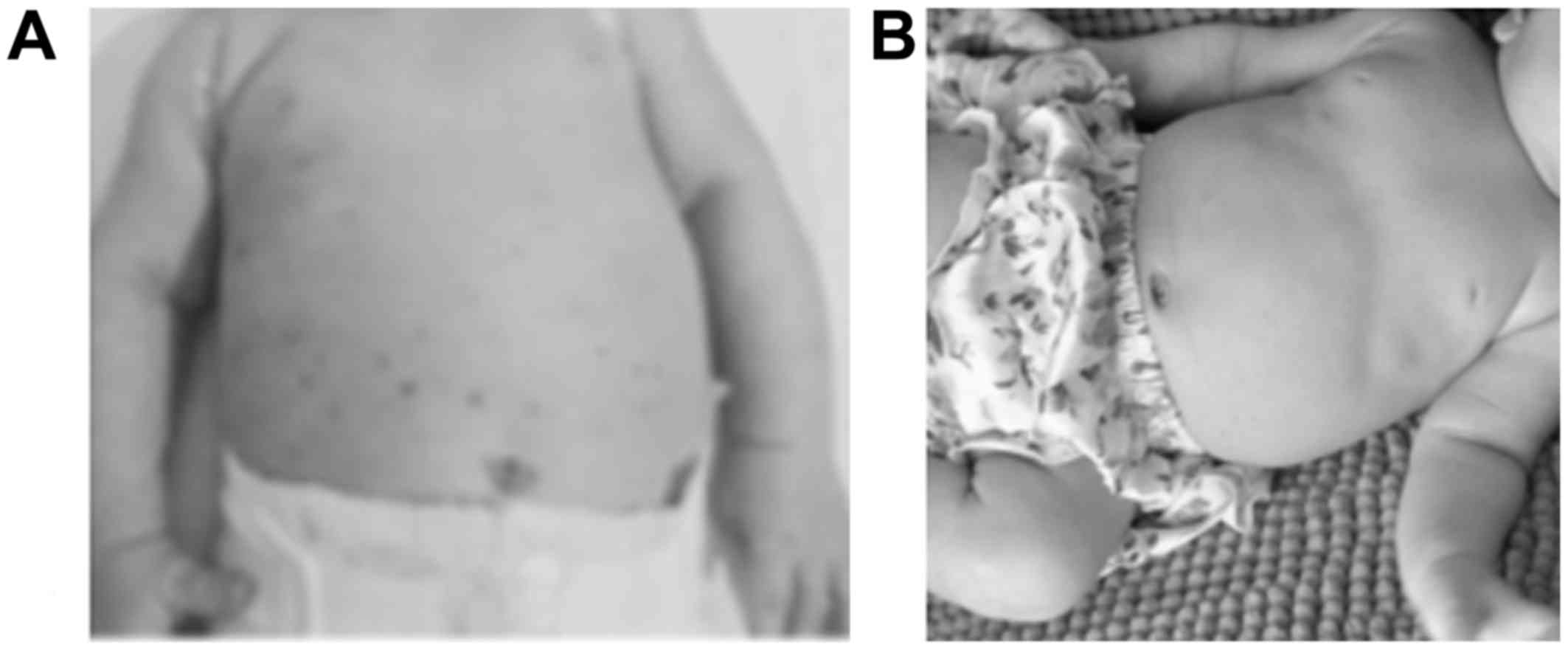
except for a persistent rash on the trunk since birth (Fig. 1A). She was treated with intravenous
ampicillin/sulbactam with prompt improvement. A skin biopsy of the
trunkal rash was scheduled and the patient was discharged but
returned three days later with recurrence of the inflammation in
the left orbit. A plain film and MRI of the head revealed a lytic
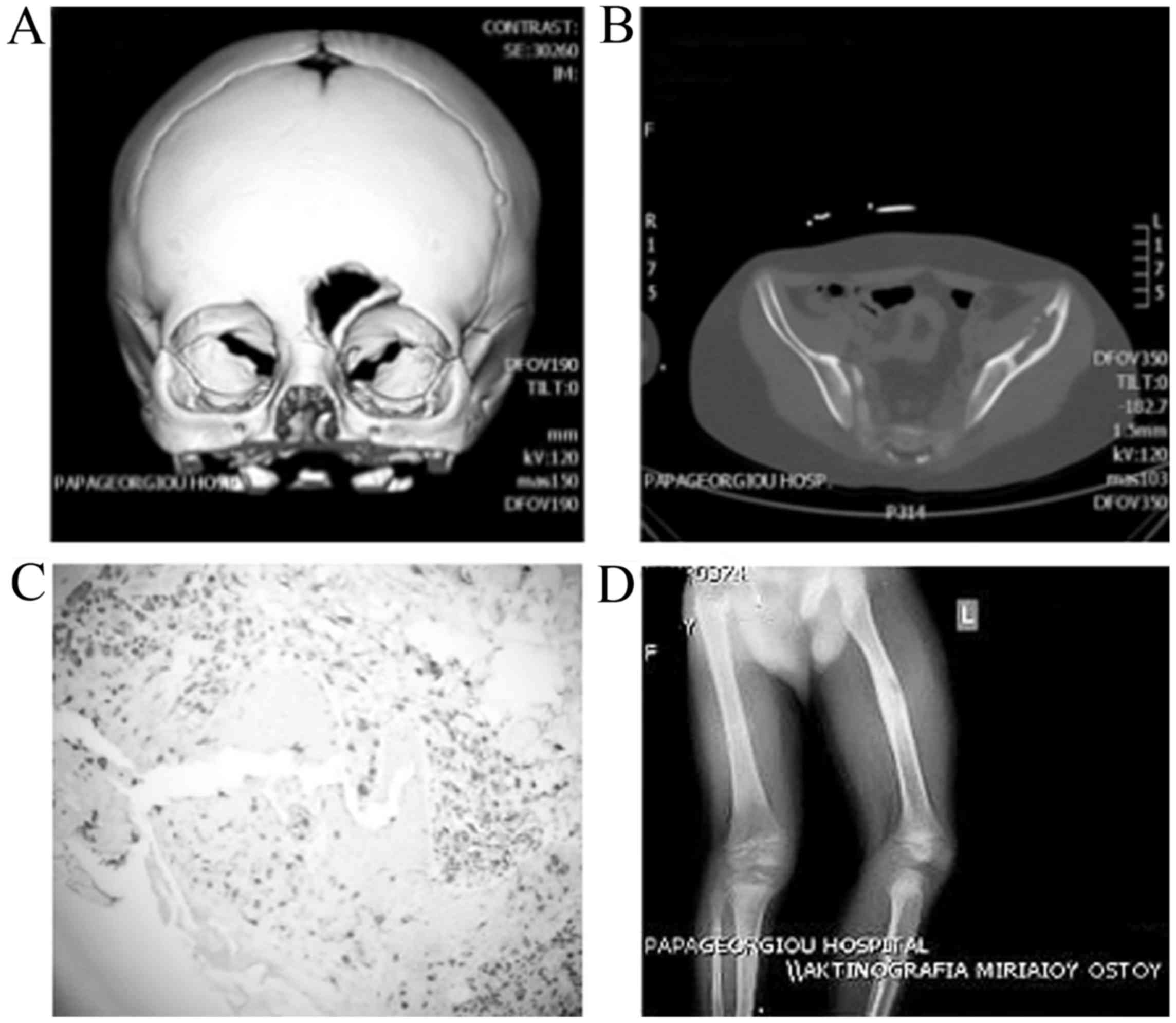
lesion in the frontal bone (Fig.
2A).
Patient no. 2
A 7-year-old boy with a history of
Wolf-Parkinson-White syndrome presented with localized pain on the
left hip with concomitant limp during the last month. The pain
started after an injury while playing. A plain film of the hip at
the time of the injury was normal. However, due to symptom
persistence, imaging studies were performed (pelvic MRI and CT
scan) that demonstrated a lytic lesion of the left iliac bone
(Fig. 2B).
Patient no. 3
A 4-month old, otherwise healthy female infant was
referred for a persistent circular, eczematous, perianal skin
lesion approximately 5 cm in diameter. The lesion was initially
treated with a local corticosteroid and antibiotic cream for a
month without improvement. Because of the persistence of the lesion
and of the non-typical location a biopsy was performed (Fig. 2C).
Patient no. 4
A 4.5 year-old boy with a known history of diabetes
insipidus (DI) under hormone replacement treatment for the past two
years was admitted for investigation of a limp and left lower limb
pain. The pain had progressed over the past few weeks and was worse
at night. At the time of DI diagnosis a brain MRI had showed an
ectopic posterior pituitary gland. A plain film of the left lower
limb revealed a 6 mm lytic lesion of the femur (Fig. 2D). A repeat brain MRI showed
thickening of the pituitary stalk and loss of the ‘bright spot’
findings that are pathognomonic for pituitary gland LCH, that
preceded the bone involvement.
Management and outcome
LCH was confirmed in all patients with biopsy of the
affected sites. All pathology specimens stained positive for CD1a,
protein S-100 and CD207 (Langerin) confirming the diagnosis
(Fig. 2C) (5). All the patients underwent a thorough
hematological evaluation and further imaging (skeletal survey,
abdominal ultrasound and CT or MRI scan) to exclude multifocal bone
disease and risk organ involvement.
All patients were referred to a Pediatric
Hematology-Oncology unit where they were managed according to the
LCH III protocol after risk stratification (Table I) (6).
Patient 4 had the femur lesion surgically resected before
chemotherapy. All patients received induction treatment with IV
vinblastine weekly and oral prednisolone daily for 6 weeks,
followed by maintenance treatment for 6 months (IV vinblastine and
5 day pulses of oral prednisolone every 3 weeks). They all
demonstrated a good response except patient 1 who had an immediate
skin disease response (Fig. 1B), but
quick recurrence of the lytic lesion at the same site while on
treatment. A surgical excision of the patient's left orbital mass
was performed and maintenance treatment was further continued for
12 months in total. Two months after the end of treatment, a
routine imaging unveiled multiple new lytic lesions of the skull. A
water deprivation test performed because of polyuria-polydipsia
confirmed DI as a result of pituitary gland involvement. The
patient received 3 cycles of cytarabine and cladribine (2-CDA) as
second line therapy for persistent/relapsed disease and maintenance
treatment (with mercaptopurine and methotrexate) for 12 months and
is now in remission.
Discussion
The clinical presentation of LCH is heterogeneous as
practically every organ or system may be affected. The disease is
divided into single-system (uni- or multi-focal) or multi-system.
Depending on the organs involved patients are divided as high-risk
(lungs, liver, spleen, bone marrow) or as low-risk (skin, bones,
lymph nodes, gastrointestinal tract) that can be either unifocal or
multifocal. There is significant difference in mortality rates in
the two risk groups. In agreement with the literature in our case
series the organs most frequently affected were the skeleton (80%),
the skin (33%), and the pituitary gland (25%) (3).
Bone involvement may present as a painful bone
lesion (patients nos. 2 and 3) or as a painless skull lytic lesion
(patient no. 1). The bones most frequently affected are the skull,
femur, ribs, humerus and the vertebrae. A skeletal survey is
necessary to reveal asymptomatic sites (3). Lesions of the orbital, temporal,
mastoid, sphenoid, zygomatic and ethmoid bones, as well as of the
maxilla, sinuses and the anterior or middle cranial fossa are
considered ‘CNS-risk’ lesions (patient no. 1) (3).
Diabetes insipidus (DI) is the most frequent type of
CNS involvement and may precede or follow other manifestations
(patient nos. 4 and 1, respectively). LCH is the most common cause
of isolated DI and of a thickened pituitary stalk in brain MRI
(7). Treatment should be initiated
even without biopsy of the lesion, provided a germ cell tumor and a
lymphoma have been excluded (7).
Lacking specific imaging criteria, other MRI lesions resembling LCH
that involve the skull need to be confirmed with biopsy in order to
differentiate from other malignancies as meningioma,
rhabdomyosarcoma and Ewing's sarcoma (8).
The incidence of the disease during early infancy is
estimated to be 1–2/1.000.000, and the predominant initial symptom
is a skin rash on the scalp or body, ranging from red or necrotic
papules to hypopigmented macules or persistent ‘cradle cap’.
Underdiagnosis is usual when LCH manifests as a skin rash during
early infancy (patients nos. 1 and 3, Fig. 1) because of the difficulty to
differentiate it from benign skin conditions like eczema or
bacterial infections (9). Although
isolated cutaneous LCH may be self-limited it is essential for
primary health care providers to be aware of the disorder and
closely follow-up their patients because it may progress to
multisystem disease (40% of cases) (10), like patient no. 1. Among infants LCH
is not only rarer but also carries a worse prognosis in comparison
to older children, with a mortality rate reaching 50% (9,10).
The overall survival of low-risk LCH in children is
estimated to be 99%, but for high-risk LCH is around 80% with high
possibility of relapse (11). The
LCH-IV is an international collaborative treatment protocol that is
expected to be completed by 2018. Its main objectives are to reduce
mortality and reactivation rates by intensification and
prolongation of treatment especially in multisystem LCH and to
devise strategies to avoid permanent consequences in treated
patients (12).
BRAF is a kinase in the MAPK pathway playing a
central role in cell growth. The BRAF-V600E mutation induces
activation of the RAS/RAF/MAPK/ERK pathway. The discovery of
recurrent BRAF-V600E and MAP2K1 mutations in LCH patients has
revolutionized our understanding of the biology of the disease and
has offered significant alternatives for its management. Therefore,
despite ongoing considerations regarding safety and toxicity
especially in childhood, agents such as vemurafenib, dabrafenib and
trametinib have been recently introduced in the management of
persistent and refractory LCH cases that harbor the mutations and
have induced prolonged remissions (7).
Despite the advancements in our understanding of the
disease's etiology and natural history and the novel therapeutic
approaches for LCH, there are pitfalls that may delay the diagnosis
from three months to several years (11,13),
especially when the initial presentation is a common and
non-specific symptom (patients nos. 2 and 3). Pediatricians,
pediatric oncologists and other specialists treating children
(family physicians, dermatologists, orthopedic surgeons,
otorhinolaryngologists and dentists) should be familiar with the
multiple faces of LCH in order to achieve timely diagnosis and to
ensure appropriate treatment and optimal outcome.
References
|
1
|
Badalian-Very G, Vergilio JA, Fleming M
and Rollins BJ: Pathogenesis of Langerhans cell histiocytosis. Annu
Rev Pathol. 8:1–20. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chakraborty R, Hampton OA, Shen X, Simko
SJ, Shih A, Abhyankar H, Lim KP, Covington KR, Trevino L, Dewal N,
et al: Mutually exclusive recurrent somatic mutations in MAP2K1 and
BRAF support a central role for ERK activation in LCH pathogenesis.
Blood. 124:3007–3015. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Haupt R, Minkov M, Astigarraga I, Schäfer
E, Nanduri V, Jubran R, Egeler RM, Janka G, Micic D,
Rodriguez-Galindo C, et al: Langerhans cell histiocytosis (LCH):
Guidelines for diagnosis, clinical work-up and treatment for
patients till the age of 18 years. Pediatr Blood Cancer.
60:175–184. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stålemark H, Laurencikas E, Karis J,
Gavhed D, Fadeel B and Henter JI: Incidence of Langerhans cell
histiocytosis in children: A population-based study. Pediatr Blood
Cancer. 51:76–81. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Drutz JE: Histiocytosis. Pediatr Rev.
32:218–219. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gadner H, Minkov M, Grois N, Pötschger U,
Thiem E, Aricò M, Astigarraga I, Braier J, Donadieu J, Henter JI,
et al: Therapy prolongation improves outcome in multisystem
Langerhans cell histiocytosis. Blood. 121:5006–5014. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Allen CE, Ladisch S and McClain KL: How I
treat Langerhans cell histiocytosis. Blood. 126:26–35. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liang C, Liang Q, Du C, Zhang X and Guo S:
Langerhans' cell histiocytosis of the temporal fossa: A case
report. Oncol Lett. 11:2625–2628. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Minkov M, Prosch H, Steiner M, Grois N,
Pötschger U, Kaatsch P, Janka-Schaub G and Gadner H: Langerhans
cell histiocytosis in neonates. Pediatr Blood Cancer. 45:802–807.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lau L, Krafchik B, Trebo MM and Weitzman
S: Cutaneous Langerhans cell histiocytosis in children under one
year. Pediatr Blood Cancer. 46:66–71. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Grana N: Langerhans cell histiocytosis.
Cancer Contr. 21:328–334. 2014. View Article : Google Scholar
|
|
12
|
The Histiocyte society, . LCH-IV
International Collaborative Treatment Protocol for Langerhans Cell
Histiocytosis. http://www.bspho.be/wp-content/uploads/LCH-IV-protocol-amended-version-Nov.-2015v1.3.pdfJanuary
8–2017
|
|
13
|
Abla O, Egeler RM and Weitzman S:
Langerhans cell histiocytosis: Current concepts and treatments.
Cancer Treat Rev. 36:354–359. 2010. View Article : Google Scholar : PubMed/NCBI
|