1. Introduction
There is accumulating evidence indicating that
disorders of lipid metabolism, such as atherosclerosis (AS) and
disorders of bone metabolism, such as osteoporosis, which are
multifactorial and degenerative diseases, are significant worldwide
public health problems and frequently coexist, in particular in
elderly postmenopausal females (1,2).
Observational studies have demonstrated an alteration in lipid
levels and increased adipocytes in association with decreased bone
mineral density (BMD) and bone mass loss in patients with OP.
Furthermore, in a mouse model of glucocorticoid (GC)-induced OP, a
decline in bone mass was observed concomitantly with an
accumulation of adipocytes (3). It
is notable that stains, which are used to treat hyperlipidemia, are
associated with increased BMD and reduced fracture risk.
Conversely, estrogen therapy for postmenopausal OP has been shown
to reduce levels of total cholesterol (TC) and low density
lipoprotein cholesterol (LDL-C) (4,5).
Furthermore, fat and bone share a common progenitor: Multipotent
mesenchymal stem cells (MSCs) in bone marrow (BM), which are able
to differentiate into osteoblasts, adipocytes and chondrocytes, in
addition to other cell phenotypes. Therefore, it is postulated in
the present review that lipid and bone metabolism are interrelated
and mutually constrained.
2. Co-existence of disordered lipid
metabolism and bone dysfunction/osteoporosis (OP)
Lipid metabolism disorder
The definition of disorders to plasma lipid
metabolism is extensive and complex, and includes the quantity and
quality of lipids and their metabolites in blood, and abnormalities
in other tissues as a result of congenital and acquired factors.
The commonly observed parameters used in clinical practice in order
to measure plasma lipid metabolism are triglycerides (TG), TC,
LDL-C and high density lipoprotein cholesterol (HDL-C). Levels of
phospholipids (PL), free fatty acid (FFA), apolipoproteins (apos),
lipoprotein (Lp) a and certain lipoprotein subclasses, along with
the products of lipid oxidation may also be included. The
atherogenic lipid profile is characterized by increased levels of
plasma TG and LDL-C, together with decreased levels of HDL-C.
Bone dysfunction and OP
Bone is an organ which constantly undergoes
processes of destruction and regeneration, termed bone remodeling.
In order to perform these dual functions, bone contains two
antagonistic cell populations. Osteoblasts are bone-forming cells,
which deposit new matrix that eventually becomes mineralized,
whereas osteoclasts are bone-resorbing cells, which resorb
mineralized extracellular matrix. This process of destroying and
forming bone occurs constantly and maintains the balance of bone
metabolism. When this balance is disturbed, for example by aging,
the menopause, or hormonal or dietary changes, there may be a loss
of bone mass and in minerals conducive to the density and strength
of bone.
OP is a systemic skeletal disease, characterized by
decreased bone mass and deterioration of the microarchitecture of
bone tissue, which results in increased bone fragility and
susceptibility to fractures (6).
OP is diagnosed when the BMD is >2.5 standard deviations (SD)
below peak bone mass, while osteopenia is diagnosed when the BMD is
1–2.5 SD below peak bone mass, in accordance with the criteria of
the World Health Organization (WHO) (7). There are two subtypes of OP. The
first is termed primary, or age-associated, OP. This refers to the
development of this disease without any apparent cause. It is more
common in females but is also recognized in males, in particular in
groups of higher age. In addition, in certain females who are
undergoing menopause, the ratio of bone loss to production is very
high and fractures may occur at a comparatively early age in these
individuals. The second type, termed secondary OP, refers to bone
loss due to a separate disease process and affects males as well as
females. Such diseases include a number of metabolic disorders,
including rheumatoid arthritis, hyperparathyroidism, Cushing’s
disease and chronic kidney disease; furthermore, the side effects
of particular drugs, including anti-epileptics, glucocorticoids and
lithium as well as eating disorders, cancer and organ
transplantation can cause secondary OP.
Effect of disorders of lipid metabolism
on bone status
OP and AS are often present in the same individual
and a number of clinical studies support a role for lipid
metabolism in the development of OP (8), although this is not a universal
finding. The data on the impact of lipid metabolism disorders on
bone status were obtained from clinical reports (5,9–18) as
well as from animal studies (19–22).
Clinical investigations
A summary of the results of clinical studies is
presented in Table I and
demonstrates a number of inconsistencies. For instance, one study
showed that in OP occurring in postmenopausal females, there was a
negative association between bone mass and TC levels (5). Furthermore, Yamaguchi et al
(9) investigated the correlation
between plasma lipid levels and BMD, in addition to the presence of
vertebral fractures in 214 Japanese postmenopausal females. The
authors found that plasma LDL-C levels were inversely correlated
with the BMD. By contrast, low levels of HDL and TG were associated
with an increased risk of vertebral fracture. A study by Orozco
(10) recruited 52 overweight
early postmenopausal females. The results demonstrated that females
with an atherogenic lipid profile (TC≥240 mg/dl, LDL-C≥160 mg/dl
and Lpa≥25 mg/dl) had a lower BMD in the lumbar spine and femur, in
addition to a higher risk of osteopenia, compared with individuals
with a normal lipid profile. This indicated that hyperlipidemia may
be associated with the development of OP. A British study,
conducted by Dennison et al (11), demonstrated an association between
lipid profile and BMD that was markedly attenuated by controlling
for total body fat. In a study by Broulik et al (12) assessing 241 Czech females with
osteoporosis and 98 age-matched controls, it was observed that
osteoporotic females with vertebral fractures had markedly higher
cholesterol levels. OP in males is increasingly recognized as an
important health problem. In a cross-sectional study, Tang et
al (13) assessed calcaneal
bone mass alongside metabolic parameters in 368 older males (mean
age, 78.8 years). Among these subjects, 36.4% were found to be
osteopenic and 16.3% were osteoporotic, and calcaneal bone mass was
found to be correlated with higher plasma TG levels.
 | Table IAssociation between lipid levels and
bone metabolism (represented as BMD, bone mass and risk of
fracture) in clinical trials. |
Table I
Association between lipid levels and
bone metabolism (represented as BMD, bone mass and risk of
fracture) in clinical trials.
| Study cohort | Lipid levels vs.
BMD, bone mass and risk of fracture | Ref. |
|---|
| 45 asymptomatic
post-menopausal females | Negative
association between TC levels and bone mass | (5) |
| 214 post-menopausal
Japanese females (aged 47–86 years) | Levels of LDL-C
negatively, but those of HDL-C positively, related to lumbar spine
radius and BMD
There is an inverse association between TG levels and the risk of
vertebral fractures | (9) |
| 52 overweight early
postmenopausal females from Spain | The levels of TC,
LDL-C and Lp(a) were negatively associated with BMD of the lumbar
spine and femoral neck | (10) |
| 465 males and 448
females from the UK | Positive
association between TG levels and BMD of the lumbar spine and total
femoral region
Negative association between HDL-C levels and BMD of the lumbar
spine and total femoral region | (11) |
| 241 osteoporotic
Czech females and 98 age-matched controls | Negative
association between cholesterol levels and bone mass | (12) |
| 368 older males
(age, 78.8 years), half of them with osteopenia | The TG levels were
negatively correlated with calcaneal bone mass | (13) |
| 107 post-menopausal
Turkish females (aged 45–79 years) | An increase in TC
levels by 1 mg/dl reduced the risk of vertebral fracture by
2.2%
Weak inverse correlation between TC and LDL-C levels, and BMD at
the forearm UD region, after controlling for confounders | (14) |
| 762 older males
followed up for 10 years | Negative
association between TG levels and incidence of fractures | (15) |
| 712 females and 450
males enrolled in the Framingham osteoporosis study (aged 32–61
years) | No association
between TC levels and BMD was found for any of the bone sites | (16) |
| 340 post-menopausal
females from Denmark (aged 50–75 years) | Negative
association between TC levels and BMD of the lumbar spine and
distal forearm
Negative association between TC levels and BMD of the lumbar spine
after adjustment for age and BMI. No association between TC levels
and BMD of the spine | (17) |
| 7137 men, 4585
premenopausal females, and 2248 postmenopausal females from
China | Negative
association between TC, TG, LDL-C and LDL-C/HDL-C ratio and
whole-body bone mineral content | (18) |
Whilst the results of the studies by Broulik and
Tang (12,13) suggested that disorders of lipid
metabolism, specifically hyperlipidemia, negatively affect the bone
status, Pharhami et al (23) demonstrated that a baseline level of
cholesterol synthesis is required for the osteoblastic
differentiation of MSCs.
Furthermore, Sivas et al (14) used multivariate binary logistic
regression analysis in order to identify possible risk factors for
vertebrae fracture. This study suggested that TC levels were the
strongest factor affecting the risk of sustaining vertebral
fractures, and that an increase by 1 mg/dl in levels of TC reduced
the risk of vertebrae fracture by 2.2% (P=0.009). Likewise, Szulc
et al (15) investigated
the association between BMD, bone fragility and metabolic syndrome
(MetS) in 762 elderly males who were followed up for 10 years. In
contrast to the results of other studies, the study showed that the
participants with MetS had a higher BMD and a lower fracture risk.
In addition, higher TG levels were significantly correlated with a
lower incidence of fractures. These findings indicate that MetS has
a protective impact on bone, which may be associated with higher TG
levels, and the other components of MetS, including insulin
resistance, dysglycemia, central obesity, low high density
lipoprotein cholesterol and hypertension, and fracture risk are
scanty (15). An experimental
study demonstrated that TGs usually form a layer between collagen
fibers and mineral crystals, and that they also regulate the
attachment of protein matrix and bone mineral that leads to
enhancement of the qualitative properties of bone (24). A study by Dennison et al
(11), investigating the
correlation between BMD and lipid profiles, also observed that
fasting TG levels were significantly correlated with the BMD in the
lumbar spine and femurs in males and females. Furthermore, fasting
HDL-C levels were correlated with the BMD of the lumbar spine in
females. The ratio of HDL-C/LDL-C was shown to be negatively
correlated with total femoral BMD in males and females. In
addition, total spine BMD was inversely correlated with levels of
apoA but positively associated with levels of apoB in males and
females (11). These results led
to the conclusion that increased plasma TG, LDL-C and apoB levels,
along with decreased levels of HDL-C and apoA, had a beneficial
impact on bone metabolism, which, however, contradicted the
conclusions of other studies (16,17).
The Framingham Osteoporosis Study, conducted in 712
females and 450 males, demonstrated no significant association
between cholesterol levels and subsequent development of OP in
either gender (16). Similarly,
Tanko et al (17), in a
study on 340 postmenopausal females aged <76 years, found no
correlation between cholesterol and the mean BMD eight years
later.
A number of factors may be responsible for these
discrepancies. Firstly, the differences in non-modifiable
characteristics of the subjects, including age, duration of the
menopause and drug history [(for example use of statins or hormone
replacement therapy (HRT)] may have introduced bias. Secondly, the
differences in modifiable characteristics, including cigarette and
alcohol consumption, or physical activity among the study subjects
may also have led to bias. Apart from these reasons, the use of
different research methodologies may have affected the results.
Previous studies into the association between lipid
levels and bone metabolism have primarily focused on Western
populations. By contrast, there is limited literature on this
subject in the Chinese population. Clearly, environmental, genetic
and dietary differences between Western and Chinese populations may
have produced conflicting results. Wang et al (25) investigated the prevalence of OP in
mainland China, Hong Kong and Taiwan by reviewing relevant studies.
Compared with caucasian populations, the OP prevalence was lower in
the Chinese population. The prevalence of OP in mainland China
ranged from 6.6 to 19.3% (average, 13.0%). In Hong Kong, in females
≥50 years of age the prevalence of OP ranged from 34.1 to 37.0%,
although it was only 7% in males. In Taiwan, the mean prevalence of
OP in females was 11.4% and in males it was 1.6%. According to this
study, the prevalence differed predominantly as a result of the
differences in region, gender and bone sites (25). Hsu et al (18) conducted a study that aimed to
analyze the association between plasma lipid profile and BMD, bone
mineral content (BMC) and osteoporotic fractures in 7137 Chinese
males, and 4585 premenopausal and 2248 postmenopausal females. A
significant inverse correlation was found between levels of
cholesterol, TG and LDL, the LDL-to-HDL ratio and the whole-body
BMC. No significant correlation between whole-body BMC and levels
of HDL was detected (18).
Due to China’s aging population there has been a
marked increase in levels of obesity and diabetes mellitus (DM) in
this population, which has significantly affected the prevalence of
OP and brought attention to this association. Thus, further
investigation of this correlation in different populations is
required, particularly in the Chinese population.
Experimental animals studies
In experimental animal models, TC and LDL-C levels
were significantly increased, while the BMC and BMD of femurs were
significantly decreased in ovariectomized (OVX) rats (19). Liu et al (20) investigated the prophylactic and
therapeutic action of the Gengnianchun medicinal preparation on
OVX-induced OP and hyperlipidemia. This study demonstrated a marked
reduction in BMD and biomechanical markers in the lumbar vertebrae
along with an increase in serum TC and LDL-C levels in rats
following OVX. Another study incorporated an atherogenic diet
(high-fat/high-cholesterol with sodium cholate), which resulted in
a marked reduction in femoral BMC (43%) and BMD (15%) compared with
those of control mice fed a low-fat/no-cholesterol diet (21). A previous study demonstrated that a
hypercholesterolemic non-atherogenic diet contributes to the
development of an osteoporotic phenotype in mice, including an
increase in osteoclasts, the loss of trabeculae, thinning of the
trabeculae and cortex, and a reduction in failure load and energy
to failure (22).
These findings suggest that elderly patients with
hyperlipidemia are at risk of developing OP. Therefore, it is
recommended in the present review that in clinical practice, a diet
low in fat, cholesterol, carbohydrate and sodium, and high in
protein should be followed by this population due to the beneficial
effects of an improved lipid profile on bone status and fracture
risk.
In conclusion, the results of clinical human and
experimental animals studies indicate that dysplipidemia is
involved in bone metabolism. However, further investigation is
required to confirm these findings and to clarify the
inconsistencies identified.
3. Trans-differentiation between adipocytes
and osteoblasts
As shown in Fig. 1,
adipocytes and osteoblasts are derived from a common bone marrow
stromal cell (BMSC) pool. The balance between adipocyte and
osteoblast differentiation is regulated by a signaling
communication pathway that requires extracellular stimuli, the
coordination of receptors and a series of cascade events and
transcription factors in the nucleus, including the peroxisome
proliferator-activated receptor γ (PPARγ), receptor activator of
nuclear factor κB ligand (RANKL)/receptor activator of nuclear
factor κB (RANK)/osteoprotegerin (OPG) and Wnt/β-catenin signaling
pathways. Disorders of lipid metabolism may affect osteoblast
differentiation by interfering with key signaling pathways.
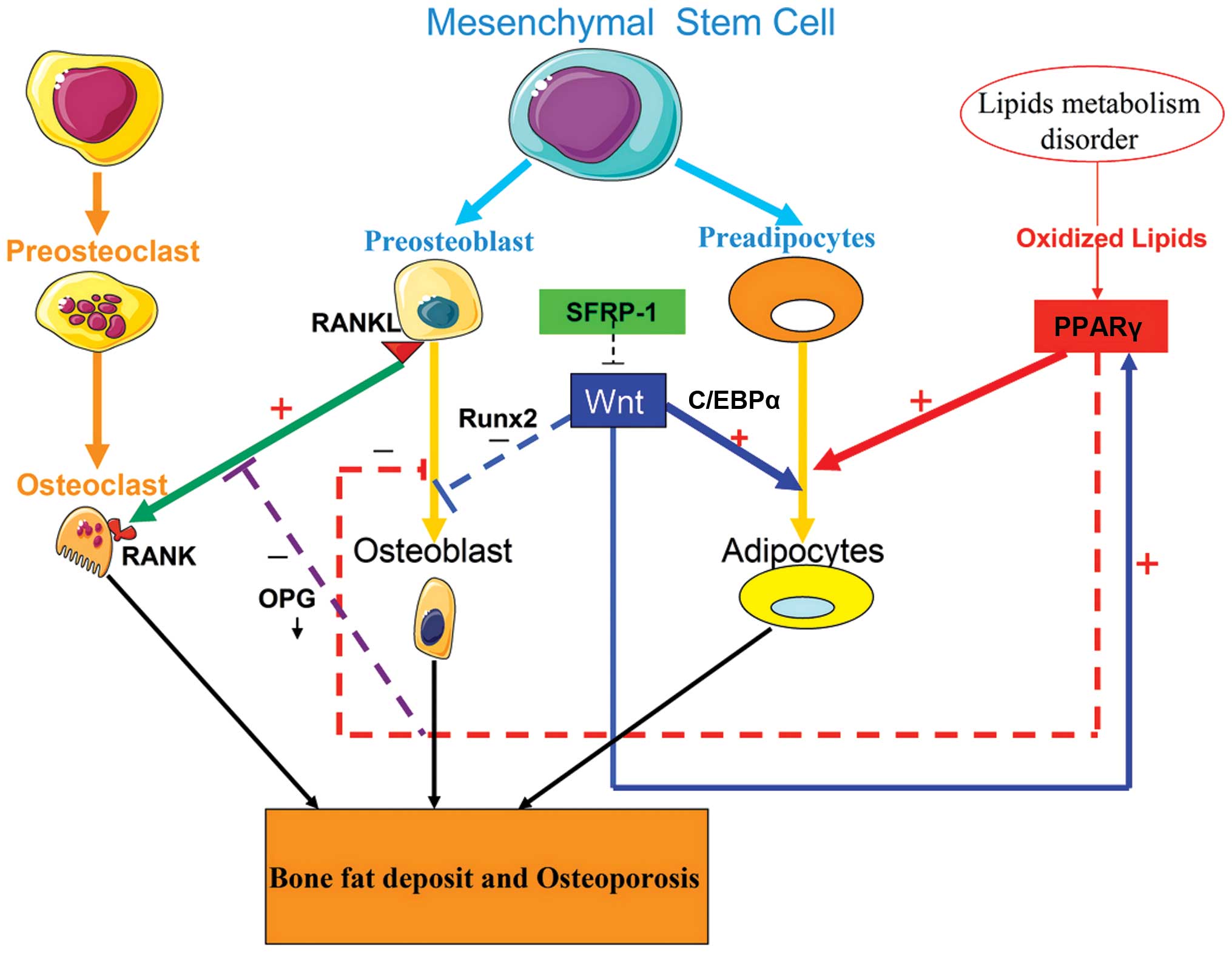 | Figure 1Signaling pathway regulation of the
differentiations of adipocytes and osteoblasts. Adipocytes and
osteoblasts are derived from a common MSC pool. The balance of
adipocyte and osteoblast differentiation requires communication
between extracellular stimuli, as well as a coordinated network of
receptors and transcription factors in the nucleus, including the
RANKL/RANK/OPG and Wnt/β-catenin signaling pathway, along with
PPARγ. Disordered lipid metabolism disorder may result in an
increase in oxidized lipids. Oxidized lipids promote the
differentiation of adipocytes and inhibit that of osteoblasts, by
activating PPARγ. The sFRP-1 binds to the Wnt receptor and blocks
the Wnt signaling pathway, thereby inhibiting osteoblast
differentiation and promoting adipocyte differentiation. The
low-activity Wnt signaling pathway in BMSCs enhances adipocyte
differentiation by upregulating C/EBPα and PPARγ, whereas it
suppresses osteoblast differentiation by downregulating Runx2
expression. Furthermore, low activity of the Wnt signaling pathway
in osteoblasts down-regulates the expression of OPG, which leads to
the enhancement of bone resorption by enforcing RANKL-induced
osteoclastic differentiation based on the RANKL/RANK/OPG pathway.
These changes ultimately increase bone fat deposition and promote
the development of osteoporosis. MSC, mesenchymal stem cell; RANKL,
receptor activator for nuclear factor κB ligand; OPG,
osteoprotegerin, BMSC, bone marrow stromal cell; C/EBPα,
CCAAT-enhancer binding proteins α; PPARγ, peroxisome
proliferator-activated receptor γ; sFRP-1, secreted
frizzled-related protein 1; Runx2, runt-related protein 2. |
Role of PPARγ2 in fat and bone
PPARγ2 is a member of the nuclear hormone receptor
subfamily of transcription factors, which is involved in promoting
and mediating the differentiation and proliferation of adipocytes.
It is expressed predominantly and specifically in adipocytes
(26). The PPARγ promoter contains
transcription factor CCAAT-enhancer binding protein (C/EBP) binding
sites, and C/EBPβ, and C/EBPδ may directly activate the PPARγ. In
turn, the PPARγ may upregulate the expression of C/EBPα. Thus,
PPARγ and C/EBPα exert a coordinated effect on the regulation of
the expression of a series of genes that are required for adipocyte
differentiation and lipid metabolism (26). The long chains and oxidized
derivatives of fatty acids have been shown to bind to and activate
PPARγ. Furthermore, thiazolidinedione, an anti-diabetic drug, has
been demonstrated to activate PPARγ. Activation of PPARγ2 induces
terminal adipocyte differentiation and cell cycle arrest in various
mesenchymal cell lines. In the BM, activation of the PPARγ2
receptor suppresses osteoblast and bone formation, and promotes
adipocyte differentiation (27).
Fatty acids have been shown to activate PPARγ2 expression in BM
cells, which results in fat accumulation in the BM (28). It has been established that the
PPARγ2 pathway is not only involved in fat redistribution, but also
in bone loss with aging. With advanced age, fat deposition is
increased, not only in subcutaneous and visceral, but also in the
BM of menopausal females (29).
RANKL/RANK//OPG pathway and bone
turnover
OPG, a member of the tumor necrosis factor (TNF)
receptor family, is secreted by osteoblasts (30). OPG binds to RANK, a surface
molecule of osteoclasts, thus competing with RANKL. RANKL is
induced and mediates the differentiation and activation of
osteoclasts (31). The
RANKL/RANK/OPG pathway is required for bone remodeling.
OPG-knockout mice develop OP with multiple fractures, and these
abnormalities were reversed following transgenic OPG restoration.
Similarly, a study by Jabbar et al (32) indicated that higher levels of OPG
in postmenopausal females with OP are positively associated with
increased bone turnover and lower BMD, which may be due to
increased bone resorption. In humans, Bekker et al (33) suggested that subcutaneous injection
of a single dose of OPG markedly reduces bone turnover and bone
resorption in menopausal females after six weeks. In postmenopausal
females, OPG levels were significantly and independently positively
correlated with bone mass and prevalent vertebral fractures
(34). Indridason et al
(35) concluded that serum OPG
levels were beneficial for bone formation and protected against
age-associated bone loss. Based on the known effects of the
RANKL/RANK/OPG system on bone metabolism, a human monoclonal
antibody against RANKL, denosumab, was developed. Subcutaneous
administration of denosumab produced a positive effect on the
incidence of fractures in postmenopausal females with OP (36).
Wnt signaling pathway switches between
adipocyte and osteoblast differentiation
Studies have indicated that fat content and bone
mass are closely correlated and mutually constrained processes.
Futhermore, in vivo and in vitro studies have
indicated that T2531 mutation of the LDL receptor-related protein
(LRP)-5 promotes osteogenesis and suppresses adipogenesis. However,
the inactivating mutation, T244 M, of LRP 5 produces opposite
effects (37). The canonical
Wnt-β-catenin signaling pathways have been shown to regulate the
differentiation of BMSCs into osteoblasts and adipocytes (38). The Wnts are a family of 19 secreted
signaling glycoproteins, which regulate the differentiation and
proliferation of cells. The canonical Wnt pathway is triggered via
binding of Wnt ligands to membrane receptors, including LRP-5 and
LRP-6, as well as frizzled proteins (39). This drives an intracellular cascade
of reactions resulting in the stabilization of β-catenin, which
translocates into the nucleus. There, it binds to the transcription
factors T-cell factor and lymphoid enhancing factor, thereby
regulating gene expression (40).
A previous study also showed a significantly higher activity of Wnt
signaling in bone murine MSCs compared with that in adipocyte MSCs
as a result of the upregulation of Wnt-associated genes and
factors, along with increased Wnt activity, in bone mMSCs (41). A low activity of Wnt signaling
leads to differentiation into adipocytes. Canonical Wnt-β catenin
signaling in BMSCs has been shown to enhance osteoclast
differentiation by upregulating the expression of Runx2 expression.
However, it suppresses adipocyte differentiation by downregulating
the expression of C/EBPα and PPARγ, whilst increasing that of
osterix and Runx2 (42). The
secreted frizzled-related protein 1 (sFRP-1) acts as a canonical
Wnt pathway antagonist by binding to Wnt ligands (43). Mice with a deficiency in sFRP-1
exhibited a higher bone mass profile as a result of the promotion
of bone formation and reduced bone loss, in addition to the
accelerated maturation of hypertrophic chondrocytes (44). However, overexpression of sFRP-1
inhibited osteoblast differentiation in vitro and bone
formation in vivo (45).
Furthermore, sFRP-1 promotion of adipocyte differentiation was
associated with physiological conditions, such as obesity (46). A number of clinical conditions lead
to bone loss, including aging, OP and GC therapy, and which are
also associated with fat accumulation in the BM. Based on this
context and the results of the study investigating the association
between obesity and the sFRP-1 promotion of adipocyte
differentiation, the researchers concluded that the association
between osteogenesis and adipogenesis is a result of the selective
differentiation of BMSC into either osteoblasts or adipocytes, at
the expense of the alternative lineage, and that the Wnt signaling
pathway is important in regulating the balance between
osteoblastogenesis and adipogenesis (41).
4. Regulatory action of adipocyte-secreted
cytokines-adipokines on bone status
Leptin exerts its effects on bone via
central and peripheral pathways
Previous studies have indicated that adipose tissue
is not only an energy-storing organ, but also secretes various
cytokines. Leptin is an adipocytokine that is predominantly
produced in white adipose tissue. Circulating leptin levels are
proportional to the quantity of body fat (47). The adipokine leptin has emerged as
a significant factor involved in bone metabolism. Leptin acts on
the peripheral and central nervous systems in order to regulate
bone metabolism (48). To date,
experimental animal studies investigating the impact of leptin on
the skeleton have produced conflicting results. Turner et al
(49) investigated the effects of
leptin deficiency on bone metabolism. The results demonstrated that
in comparison with wild-type (WT) mice, leptin-deficient (ob/ob)
mice and leptin receptor-deficient (db/db) mice had a lower rate of
bone formation and a reduced osteoblast-lined perimeter. However,
subcutaneous replacement of leptin in ob/ob mice resulted in an
increase in bone formation, along with an increased
osteoblast-lined perimeter (49).
In addition, the authors utilized gene therapy in order to
selectively increase leptin levels in the hypothalami of ob/ob
mice. This resulted in normalization of the mouse bone mass, in
accordance with the results obtained using subcutaneous
administration of the hormone (49). The leptin replacement study
demonstrated no difference between the indirect central and direct
peripheral actions of leptin on bone metabolism, as peripherally
administered leptin is able to cross the blood-brain barrier. The
peripheral and central pathways involving leptin exert a positive
effect on bone metabolism (49).
In accordance with these results, a study by Cornish
et al (50) demonstrated
that leptin-deficient ob/ob mice have a significantly lower total
BMC and BMD compared with those of WT mice. Furthermore, the bone
mass and BMD were reduced in the femurs of leptin-deficient ob/ob
mice, compared with those in WT mice. This is due to a reduction in
cortical thickness and trabecular density (BV/TV) (51). With administration of leptin gene
treatment to the hypothalamus, there was an increase in bone mass
and bone length in ob/ob mice (52). Furthermore, serum levels of
osteocalcin, a biochemical marker of bone formation, were also
significantly increased in ob/ob mice that received hypothalamic
leptin gene therapy (53) or
direct administration of leptin into the hypothalamus (54).
Other studies have produced findings inconsistent
with those discussed thus far. A study on leptin-deficient ob/ob
mice found that these animals initially exhibited a high bone mass,
whereas following administration of leptin into the hypothalamus
there was a reduction in bone mass, which may have been a result of
the promotion of bone resorption and suppression of bone formation
(55). Blocking of the sympathetic
nervous system (SNS) has been shown to abrogate these effects, a
response which appears to be mediated by the influence of
β-adrenoreceptors (β-AR) on osteoblasts; furthermore, leptin
stimulates bone resorption in part through increased RANKL
expression (Fig. 2). It is thus
thought that the indirect central action of leptin on bone
metabolism is to exert an anti-osteogenic effect.
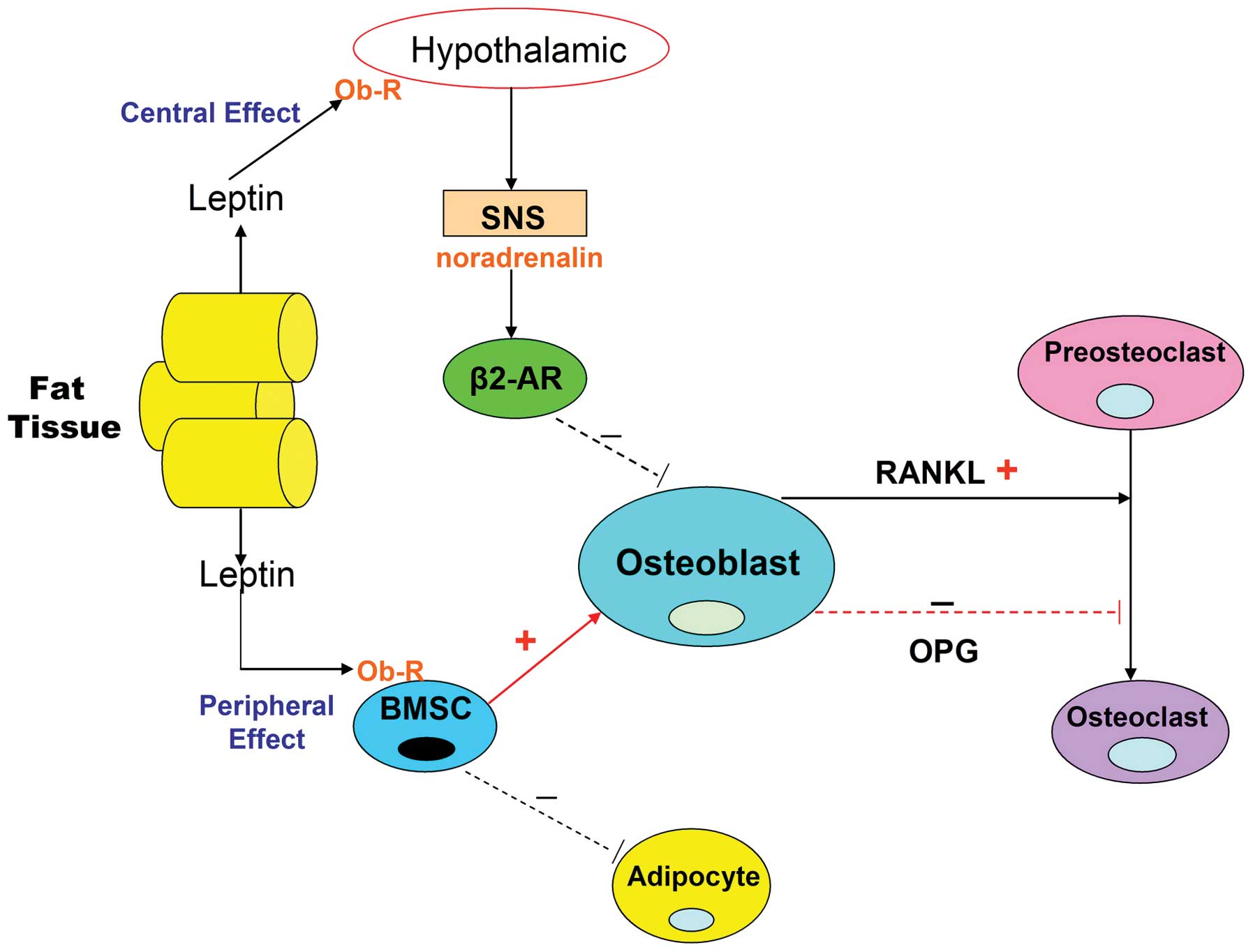 | Figure 2Leptin action on bone metabolism via
the peripheral and central nervous systems. Leptin is an
adipocytokine, which is primarily produced in white adipose tissue.
Osteoblasts, osteoclasts and BMSCs express the leptin receptor. The
direct peripheral action of leptin on bone occurs via binding to
the Ob-R on BMSCs. Leptin suppresses adipogenic differentiation of
BMSCs and promotes osteoblastic differentiation. In addition,
leptin increases OPG production in association with reducing that
of RANKL, thereby inhibiting osteoclastic differentiation. Leptin
also exhibits a central indirect effect on bone by binding to its
receptor in the hypothalamus and activating the SNS, enabling
binding of noradrenalin to β2-AR on osteoblasts and thus inhibiting
bone formation. It also increases RANKL expression and promotes the
differentiation of osteoclasts. BMSC, bone marrow stromal cell;
Ob-R, leptin receptor; OPG, osteoprotegerin; RANKL, receptor
activator for nuclear factor κB ligand; SNS, sympathetic nervous
system; β2-AR, β2-adrenergic receptor. |
Osteoblasts, osteoclasts and BMSCs express the
leptin receptor (Ob-R). Leptin treatment increases OPG production,
in association with a reduction in the expression of RANKL, thereby
inhibiting osteoclastic differentiation (Fig. 2) (56). In addition, leptin suppresses the
adipogenic differentiation of BMSCs, thus promoting osteogenic
differentiation (Fig. 2) (57). The peripheral action of leptin
produces an anabolic effect on bone metabolism. However, higher
levels of leptin may contribute to BMSC apoptosis (58), as well as increased bone resorption
and reduced bone formation. In accordance with these findings,
Hamrick et al (59)
postulated that increased adipogenesis in bone may increase the
leptin levels in the BM microenvironment, leading to bone loss.
Central infusions of leptin have been shown to significantly reduce
adipocytes in the BM (60).
Idelevich et al (61) questioned whether the effects that
leptin exert on bone metabolism via peripheral and central pathways
are the same or opposite. This question has not been fully
addressed by the current body of evidence, and further
investigation is therefore required.
Studies into the effects of leptin therapy in humans
are limited. Farooqi et al (62) reported a case study of a 9 year-old
girl, diagnosed with leptin deficiency, who was treated with leptin
replacement. The authors found that weight and bone mass increased.
Similarly, Paz-Filho et al (63) reported the case of a 27 year-old
adult male, who had been identified as leptin-deficient and
received r-metHuLeptin treatment for six years. The authors found
that weight and plasma lipid levels tended towards normal values,
and the BMD of the lumbar spine increased by 11% (64).
Complexity mechanisms of adiponectin on
bone metabolism
Adiponectin (also termed Acrp30, AdipoQ, apM1 and
GBP28) is a 28-kDa protein secreted from adipocytes. Plasma
concentrations of adiponectin are negatively correlated with fat
mass and body mass index (BMI) (64). Adiponectin is involved in the
regulation of glucose, lipid metabolism, energy homeostasis and the
inflammatory response (65,66).
The receptors for adiponectin, AdipoR1 and AdipoR2, have been
identified in osteoblasts and osteoclasts, and the proliferation
and differentiation of osteoblasts is enhanced by adiponectin,
while the differentiation of osteoclasts is inhibited by this
hormone in vitro (67,68).
This indicates that adiponectin is also involved in bone
metabolism.
Kajimura et al (69) showed that adiponectin has the
unusual ability to modulate the same function in opposite
directions, according to where it is acting and what it
antagonizes. During direct adiponectin signaling in osteoblasts,
inhibition of the proliferation of osteoblasts is observed, whereas
during central signaling, the proliferation of osteoblasts was
promoted. Similarly, adiponectin has been shown to upregulate RANKL
expression in osteoblasts. However, downregulation of RANKL
expression occurs in reponse to adiponectin signaling in the brain
(69). The findings of a study by
Luo et al (70) suggested
that adiponectin has no direct effect on the differentiation of
osteoclast precursors, but that it indirectly enhances the
formation of osteoclasts by stimulating the expression of RANKL and
inhibiting that of OPG in human osteoblasts. By contrast, other
studies have proposed that adiponectin inhibits RANKL-induced
osteoclastogenesis in RAW264.7 cells by downregulating the
expression of RANKL-stimulated osteoclast regulators and markers,
including nuclear factor of activated T-cells 2 (NFAT2), TNF
receptor-associated factor 6, cathepsin K and triiodothyronine
receptor auxiliary protein (71).
It has also been shown to suppress the proliferation and survival
of osteoclast precursor cells, in addition to increasing
osteoclastic apoptosis (71).
Further investigations have indicated that the inhibitory actions
of adiponectin on osteoclasts are mediated by β amyloid precursor
protein-like 1 downregulation of protein kinase B activity
(71). A separate study suggested
that adiponectin may suppress RANKL-induced osteoclast
differentiation, and that adiponectin functions as a negative
regulator of RANKL-induced osteoclastogenesis via downregulation of
NFATc1 expression, which is associated with the AMP-activated
protein kinase signaling pathway (72). Luo et al (73) also demonstrated that adiponectin
acts directly on osteoblasts and stimulates the differentiation and
proliferation of human osteoblast cells. The process of
differentiation is regulated through AdipoR/p38 mitogen-activated
protein kinase pathway, and that of proliferation is regulated via
the AdipoR/c-Jun N-terminal kinase signaling pathway (73). A study by Lee et al
(74) suggested that adiponectin
promotes osteoblastic differentiation in mesenchymal progenitor
cells via an increase in the expression of
prostaglandin-endoperoxide synthase 2, which is mediated by the
AdipoR1/p38 MAPK/c-Jun pathway (74). Overall, these results indicate that
the action of adiponectin in bone metabolism is complex, and that
the association between adiponectin and bone metabolism requires
further investigation.
Overexpression and knockout models of adiponectin
have been used to explore the role of adiponectin in the skeleton.
Mitsui et al (75) used 12
week-old transgenic (AdTg) mice that overexpressed human
full-length adiponectin. They showed that the bone mass of these
mice was significantly higher and that bone formation was markedly
increased compared with those in the WT littermates (75). The overexpression of adiponection
mice model had conflicting effect on bone metabolism; and Ealey
et al is contrary to the other study mentioned in the
previous sentence, and the subsequent paragraph we mainly discuss
which factor contribute to these differences in results. Ealey
et al (76) demonstrated
that AdTg mice had a significantly lower femoral BMC and the peak
load of the femur neck peak was significantly lower compared with
that in the control group. Adiponectin is known to increase insulin
sensitivity and decrease insulin resistance (77). A number of studies have reported
that in transgenic mice with hyperadiponectinemia, fat accumulation
was inhibited, and fat mass and adipocyte size decreased within the
adipose tissue. In addition, premature death induced by a high-fat
diet was prevented in these mice (78,79),
which resulted in enhanced insulin sensitivity. Thus, it is
possible that the increased osteoblastogenesis may be due to the
anabolic effects of insulin on bone (75).
A separate study demonstrated an increase in
trabecular bone volume by 30% and an increase in the number of
trabecualae by ~38% at 14 weeks of age observed in Ad knockout
(ADKO) male mice (80). The
majority of studies have found that AdKO mice exhibit either
spontaneous or diet-induced insulin resistance and
hyperinsulinemia, which leads to the increased bone mass observed
in AdKO mice (81–84). Due to the complex and contradictory
results obtained in these studies, Kanazawa (85) suggested that the bone-specific
effects of adiponectin over- and underexpression require further
investigation.
The results of a number of clinical studies have
indicated an association between adiponectin levels and bone
metabolism in certain individuals; however, there have been
conflicting results. Among 81 non-diabetic patients with OP, the
fasting plasma levels of adiponectin were significantly negatively
correlated with femoral neck and lumbar spine BMD (86). Wu et al (87) investigated the association between
adiponectin levels and BMD, along with bone turnover markers, in
336 postmenopausal Chinese females. The results demonstrated a
significant negative correlation between adiponectin levels and
BMD, and suggested that adiponectin regulates bone metabolism by
enhancing bone resorption in postmenopausal females (87). A separate study, including 81
post-menopausal females, 43 of which were osteopenic/osteoporotic,
demonstrated no significant association between total adiponectin
levels and BMD (88). Thus, the
effects of adiponectin on bone metabolism in humans also require
further investigation.
5. Negative regulatory effect of disordered
lipid metabolism on bone microcirculation
Studies have shown that higher marrow fat content is
correlated with lower trabecular BMD and with increased prevalence
of vertebral fracture (89).
Subjects with OP or osteopenia have significantly increased marrow
fat content compared with that of subjects with a normal BMD. A
number of potential mechanisms, whereby disordered lipid metabolism
interacts with bone microcirculation and the development of OP,
have been proposed. Dysfunction of lipid metabolism may contribute
to impaired nitric oxide and enhanced endothelin production,
resulting in endothelial cell dysfunction and an increased risk of
thrombus formation (90). In
addition, high doses of corticosteroids contribute to cholesterol
synthesis, which results in fat deposition, liver steatosis and fat
emboli (91). Disordered lipid
metabolism increases the size of adipocytes in the medullary
cavity, which results in an increase in the pressure of the marrow
cavity, which in turn compromises perfusion via triggering of the
coagulation pathway (92,93). The average diameter of adipocytes
in the BM has been shown to increase by >10 μm (94). Furthermore, increased levels of
circulating lipids lead to accumulation of lipids in the BM, with
subsequent occlusion of subchondral vessels as a result of fat
emboli (95).
Savopoulos et al (96) stated that, in their opinion, the
‘dynamic equilibrium between adipogenesis and bone formation is the
‘melting point’ in the prevention or therapy of clinical diseases
characterized by the balance disorder’. The common regulatory
factors between adipocyte and osteoblast differentiation may be a
target for novel agents for the prevention and treatment of these
metabolism-associated diseases.
6. Clinical drug treatments of OP are
associated with lipid metabolism
Pleiotropic properties of statins
Statins may produce beneficial effects on bone
metabolism. Due to the involvement of hyperlipidemia in the
pathophysiology of OP, the beneficial effects of these drugs may be
ascribed to their lipid-lowering activity. Statins act by
competitively inhibiting 3-hydroxy-3-methylglutaryl-CoA reductase,
the key rate-limiting enzyme of the endogenous cholesterol
biosynthesis pathway, which catalyses the reduction of mevalonate.
However, the effects of statins on bone also appear to be
independent of their lipid-lowering actions. Statins have been
reported to have additional pleiotropic properties; among them a
beneficial effect on BMD (97).
The promotion of bone formation in cultured mouse
and human bone cells by statins was first reported by Mundy et
al (8) in 1999. Simvastatin
and lovastatin were shown to activate the promoter of bone
morphogenetic protein 2, a gene that increases osteoblast
differentiation and contributes to bone formation. Esposito et
al (98) postulated that
statins may suppress the apoptosis of osteoblasts via the
TGFβ/Smad3 pathway, which leads to increased bone formation. The
phosphorylation type I receptor activates Smad3, which is required
for the differentiation and survival of osteoblasts (99). Furthermore, statins have been shown
to inhibit osteoclast proliferation via an increase in the
expression of the estrogen receptor and regulation of the
RANKL/RANK/OPG signaling pathway (100). Studies on animals have confirmed
the beneficial impact of statins on bone metabolism in vitro
and in vivo, as anabolic and anti-resorptive agents
(101–104). Statins exhibited dual effects by
enhancing the activity of osteoblasts and inhibiting that of
osteoclasts, leading to bone formation.
In accordance with previous studies, Uzzan et
al (105) demonstrated that
statins exert a positive effect on BMD at various sites. A
meta-analysis of 19 studies evaluated the influence of statins on
BMD and the risk of fractures. Among these studies, 12 reported
that statins increased BMD. However, one study reported a
deleterious effect, and the remaining six demonstrated no effect. A
number of reasons may account for differences between the results
obtained from clinical and experimental studies on the effects of
statins on bone metabolism. Statins are poorly distributed to bone
due to the hepatic first-pass effect. As the type of statins used
varied and the doses administered were higher in the animal model
than in humans, experimental studies were more likely to
demonstrate a positive effect. In addition, in the clinical
studies, the treatment periods were relatively short compared with
those in normal clinical practice and it is possible that the
statins were prescribed preferentially to subjects with a lower
fracture risk, thus introducing selection bias (97,105). A number of other factors may also
be involved in the process, which have yet to be determined.
Further investigation is thus required in order to
clarify the action of statins on bone in clinical practice. In
addition, the doses of statins that produce a significant effect in
the management of OP necessitate additional evaluation. In the
future, statins may be candidates for the prevention and treatment
of OP.
Bisphosphonates (BPs) have
anti-resorptive effects in the treatment of OP
The interference with mevalonate generation by
statins may also be an important regulatory mechanism underlying
the action of BPs. BPs are widely used in the treatment of OP due
to their suppression of osteoclastic activity (106). Considering that BPs and statins
affect the same metabolic pathway, it has remained elusive why BPs
exhibit only anti-resorptive effects, whereas statins inhibit bone
loss but also stimulate bone formation. The difference in
sensitivity of osteoblasts and osteoclasts to statins compared with
that to BPs may be one explanation for this discrepancy (105). BPs are potent inhibitors of bone
resorption, among the therapeutic options available for the
treatment of OP. They are synthetic compounds with a high affinity
for calcium-containing crystals, which selectively concentrate in
the bones by binding to hydroxyapatite crystals. BPs are absorbed
onto the bone mineral surfaces and are released during phases of
bone remodeling. They interfere with the action of bone-resorbing
osteoclasts, induce the apoptosis of these cells and reduce bone
turnover, thereby reducing the risk of fractures. BPs are
considered to be the current gold standard for treatment of
GC-induced OP (107). A number of
large-scale trials have shown that BPs increase BMD and decrease
the incidence of vertebral fractures in patients with GC-induced OP
(108–110).
Estrogen therapy for OP in postmenopausal
females and aging men
Bone cells have two classes of intracellular steroid
receptors for estrogen, ERα and ERβ. When estrogen binds to the
receptors, the relevant genes are activated (111). The principal action of estrogen
on bone is to decrease bone turnover and maintain a balance between
bone formation and bone resorption. Estrogen deficiency is the
primary factor underlying the development of OP in postmenopausal
females and may also result in bone loss in aging men (111). For postmenopausal females, the
adipose tissue is the leading source of estrogen, which is created
in these cells through the aromatization of androgens. Osteoclast
apoptosis is regulated by estrogens (112,113). Estrogen deficiency has an
indirect effect on bone, besides its direct effects on bone loss.
Estrogen levels are negatively associated with the serum
parathyroid hormone (PTH) levels (114). Increased PTH secretion leads to
accelerated bone loss. In addition, recent data have demonstrated
that estrogen deficiency stimulates osteoclastogenesis through
enhancing the production of TNF-α and RANKL by monocytes and T
cells. T-cell activity was shown to be significantly higher in
postmenopausal females with OP than that in healthy postmenopausal
subjects (115). The results of
this study suggested that T cells are involved in bone loss due to
estrogen deficiency. A meta-analysis of 57 studies, which
randomized postmenopausal females to HRT or a control and had a
duration of >1 year, revealed that HRT had a persistent and
significant positive effect on BMD at all sites. However, although
the evidence demonstrated a decrease in the risk of vertebral and
non-vertebral fractures, this reduction was not significant in
postmenopausal females (116).
Action of β-blockers on bone through the
SNS
Studies have illustrated that β-blockers have a
marked effect on bone metabolism and fracture healing. β-blockers
appear to promote bone formation and/or suppress bone resorption in
animals, and to reduce fracture risk in humans (106,117). Observational studies and
experimental animal studies have demonstrated that the SNS has a
catabolic act on bone, reduces BMD and leads to a deterioration of
trabecular microarchitecture as well as changes in cortical width
(117,118). The β-blocker propranolol was
shown to increase bone formation in OVX female rats (119). In a population-based study, Pasco
et al (120) investigated
this association between β-blockers and BMD and fracture risk in
569 females. The authors found that β-blocker administration was
associated with an increased BMD and a reduced fracture risk
(120). Similarly, a study by
Schlienger et al (121)
also indicated that use β-blockers significantly decreased the risk
of fractures. In the Framingham study, Ferrari et al
(122) reported an increase in
BMD in association with treatment with β-blockers. By contrast, no
difference in BMD was detected between β-blocker users and nonusers
in the study by Rejnmark et al (123). These authors also found that the
risk of fracture is higher in people using β-blockers than in those
not receiving this medication. However, this study had a small
sample size, meaning that it may have had insufficient statistical
power to detect such difference.
Nevertheless, the results of human studies are
conflicting and further observational studies and randomized
controlled trials are required in order to confirm the potential
beneficial effects of β-blockers on bone metabolism.
Currently, drugs not only treat OP, but also
modulate lipid metabolism, which indicates that the metabolism of
bone and lipids is correlated. Furthermore, it is proposed in the
present review that in clinical practice, physicians may adopt
combinations of therapy in accordance with an individual patient’s
characteristics, in order to provide maximal benefit.
7. Conclusion
In conclusion, there is accumulating evidence in
support of the hypothesis that the metabolism of lipids and that of
bone are closely associated and mutually regulated. However, some
of the studies on this topic have produced conflicting results.
Therefore, further investigations in human subjects are required,
in particular in the Chinese population, due to the ageing
population of China and the concomitant increase in obesity and DM,
which significantly affect this association. Understanding the
correlation between lipid metabolism and bone dysfunction may aid
in comprehending the clinical association between AS and OP, and in
the safe and rational choice of pharmacological therapies in the
prevention and treatment of these prevalent diseases.
Acknowledgements
The authors would like to thank Professor Yingqun
Wang for the suggestions for this manuscript. This study was
supported by the National Natural Science Foundation of China
(grant nos. 81072190 and 81370969 to X Yu, and grant no. 31300648
to L Tian).
References
|
1
|
Bagger YZ, Tankó LB, Alexandersen P, Qin G
and Christiansen C: Prospective epidemiological risk factors study
group: radiographic measure of aorta calcification is a
site-specific predictor of bone loss and fracture risk at the hip.
J Intern Med. 259:598–605. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sinnott B, Syed I, Sevrukov A and
Barengolts E: Coronary calcification and osteoporosis in men and
postmenopausal females are independent processes associated with
aging. Calcif Tissue Int. 78:195–202. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yao W, Cheng Z, Busse C, Pham A, Nakamura
MC and Lane NE: Glucocorticoid excess in mice results in early
activation of osteoclastogenesis and adipogenesis and prolonged
suppression of osteogenesis: a longitudinal study of gene
expression in bone tissue from glucocorticoid-treated mice.
Arthritis and Rheum. 58:1674–1686. 2008. View Article : Google Scholar
|
|
4
|
Edwards CJ, Hart DJ and Spector TD: Oral
statins and increased bone-mineral density in postmenopausal
females. Lancet. 355:2218–2219. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Barengolts EI, Berman M, Kukreja SC,
Kouznetsova T, Lin C and Chomka EV: Osteoporosis and coronary
atherosclerosis in asymptomatic postmenopausal females. Calcif
Tissue Int. 62:209–213. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Consensus development conference:
diagnosis, prophylaxis and, treatment of osteoporosis. Am J Med.
94:646–650. 1993. View Article : Google Scholar
|
|
7
|
Bonjour JP, Ammann P and Rizzoli R:
Importance of preclinical studies in the development of drugs for
treatment of osteoporosis: a review related to the 1998. WHO
guielelines. 9:379–393. 1999.
|
|
8
|
Mundy G, Garrett R, Harris S, et al:
Stimulation of bone formation in vitro and in rodents by statins.
Science. 286:1946–1949. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yamaguchi T, Sugimoto T, Yano S, Yamauchi
M, Sowa H, Chen Q and Chihara K: Plasma lipids and osteoporosis in
postmenopausal females. Endocr J. 49:211–217. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Orozco P: Atherogenic lipid profile and
elevated lipoprotein (a) are associated with lower bone mineral
density in early postmenopausal overweight females. Eur J
Epidemiol. 19:1105–1112. 2004. View Article : Google Scholar
|
|
11
|
Dennison EM, Syddall HE, Aihie Sayer A,
Martin HJ and Cooper C: Lipid profile, obesity and bone mineral
density: the Hertfordshire Cohort Study. QJM. 100:297–303. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Broulik PD and Kapitola J: Interrelations
between body weight, cigarette smoking and spine bone mineral
density in osteoporotic Czech females. Endocr Regul. 27:57–60.
1993.PubMed/NCBI
|
|
13
|
Tang YJ, Sheu WH, Liu PH, Lee WJ and Chen
YT: Positive associations of bone mineral density with body mass
index, physical activity and blood triglyceride level in men over
70 years old: a TCVGHAGE study. J Bone Miner Metab. 25:54–59. 2007.
View Article : Google Scholar
|
|
14
|
Sivas F, Alemdaroǧlu E, Elverici E, Kuluǧ
and Ozoran K: Serum lipid profile: its relationship with
osteoporotic vertebrae fractures and bone mineral density in
Turkish postmenopausal females. Rheumatol Int. 29:885–890. 2009.
View Article : Google Scholar
|
|
15
|
Szulc P, Varennes A, Delmas PD, Goudable J
and Chapurlat R: Men with metabolic syndrome have lower bone
mineral density but lower fracture risk-the MINOS study. J Bone
Miner Res. 25:1446–1454. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Samelson EJ, Cupples LA, Hannan MT, et al:
Long-term effects of serum cholesterol on bone mineral density in
females and men: the Framingham Osteoporosis Study. Bone.
34:557–561. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tankó LB, Bagger YZ, Nielsen SB and
Christiansen C: Does serum cholesterol contribute to vertebral bone
loss in postmenopausal females? Bone. 32:8–14. 2003. View Article : Google Scholar
|
|
18
|
Hsu YH, Venners SA, Terwedow HA, et al:
Relation of body composition, fat mass and serum lipids to
osteoporotic fractures and bone mineral density in Chinese men and
females. Am J Clin Nutr. 83:146–154. 2006.PubMed/NCBI
|
|
19
|
Czerny B, Pawlik A, Juzyszyn Z and
Myśliwiec Z: Effect of tamoxifen on bone mineral density and blood
lipids in ovariectomized rats. Pol J pharmacol. 55:1137–1142.
2003.
|
|
20
|
Liu KJ, Wang WJ, Li DJ, Jin HF and Zhou
WJ: Effect of Gengnianchun Recipe on bone mineral density, bone
biomechanical parameters and serum lipid level in ovariectomized
rats. Chin J Integr Med. 12:132–136. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Parhami F, Tintut Y, Beamer WG, Gharavi N,
Goodman W and Demer LL: Atherogenic high-fat diet reduces bone
mineralization in mice. J Bone Miner Res. 16:182–188. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pelton K, Krieder J, Joiner D, Freeman MR,
Goldstein SA and Solomon KR: Hypercholesterolemia promotes an
osteoporotic phenotype. Am J Pathol. 181:928–936. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Parhami F, Mody N, Gharavi N, Ballard AJ,
Tintut Y and Demer LL: Role of cholesterol biosynthetic pathway in
osteoblastic differentiation of marrow stromal cells. J Miner Res.
17:1997–2003. 2002. View Article : Google Scholar
|
|
24
|
Xu S and Yu JJ: Beneath the minerals, a
layer of round lipid particles was identified to mediate collagen
calcification in compact bone formation. Biophys J. 91:4221–4229.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang Y, Tao Y, Hyman ME, Li J and Chen Y:
Osteoporosis in China. Osteoporosis Int. 20:1651–1662. 2009.
View Article : Google Scholar
|
|
26
|
Gimble JM, Robinson CE, Wu X, et al:
Peroxisome proliferator-activated receptorgamma activation by
thiazolidinediones induces adipogenesis in bone marrow stromal
cells. Mol Pharmacol. 50:1087–1094. 1996.PubMed/NCBI
|
|
27
|
Lecka-Czernik B, Moerman EJ, Grant DF,
Lehman JM, Manolagas SC and Jilka RL: Divergent effects of
selective peroxisome proliferators-activated receptor-gamma2
ligands on adipocyte versus osteoblast differentiation.
Endocrinology. 143:2376–2384. 2002.PubMed/NCBI
|
|
28
|
Huang JT, Welch JS, Ricote M, et al:
Interleukin-4-dependent production of PPAR gamma ligands in
macrophages by 12/15-lipoxygenase. Nature. 400:378–382. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kirkland JL, Tchkonia T, Pirtskhalava T,
Han J and Karagiannides I: Adipogenesis and aging: does aging make
fat go MAD? Exp Gerontol. 37:757–767. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Simonet WS, Lacey DL, Dunstan CR, et al:
Osteoprotegerin: a novel secreted protein involved in the
regulation of bone density. Cell. 89:309–319. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schoppet M, Preissner KT and Hofbauer LC:
RANK ligand and osteoprotegerin: paracrine regulators of bone
metabolism and vascularfunction. Arterioscler Thromb Vasc Biol.
22:549–553. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jabbar S, Drury J, Fordham JN, Datta HK,
Francis RM and Tuck SP: Osteoprotegerin, RANKL and bone turnover in
postmenopausal osteoporosis. J Clin Pathol. 64:354–357. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bekker PJ, Holloway D, Nakanishi A,
Arrighi M, Leese PT and Dunstan CR: The effect of a single dose of
osteoprotegerin in postmenopausal females. J Bone Miner Res.
16:348–360. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mezquita-Raya P, de la Hiquera M, García
DF, et al: The contribution of serum osteoprotegerin to bone mass
and vertebral fractures in postmenopausal females. Osteoporos Int.
16:1368–1374. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Indridason OS, Franzson L and Sigurdsson
G: Serum osteoprotegerin and its relationship with bone mineral
density and markers of bone turnover. Osteoporos Int. 16:417–423.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cummings SR, Martin JS, McClung MR, et al:
Denosumab for prevention of fractures in postmenopausal females
with osteoporosis. N Engl J Med. 361:756–765. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Qiu W, Andersen TE, Bollerslev J, Mandrup
S, Abdallah BM and Kassem M: Patients with high bone mass phenotype
exhibit enhanced osteoblast differentiation and inhibition of
adipogenesis of human mesenchymal stem cells. J Bone and Mineral
Res. 22:1720–1731. 2007. View Article : Google Scholar
|
|
38
|
Abdallah BM and Kassem M: New factors
controlling the balance between osteoblastogenesis and
adipogenesis. Bone. 50:540–545. 2012. View Article : Google Scholar
|
|
39
|
Tamai K, Semenov M, Kato Y, et al:
LDL-receptor related proteins in Wnt signal transduction. Nature.
407:530–535. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bienz M: TCF: transcriptional activator or
repressor? Curr Opin Cell Biol. 10:366–372. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Taipaleenmäki H, Abdallah BM, AlDahmash A,
Säämänen AM and Kassem M: Wnt signaling mediates the cross-talk
between bone marrow derived pre-adipocytic and pre-osteoblastic
cell populations. Exp Cell Res. 317:745–756. 2011. View Article : Google Scholar
|
|
42
|
Kang S, Bennett CN, Gerin I, Rapp LA,
Hankenson KD and Macdougald OA: Wnt signaling stimulates
osteolastogenesis of mesenchymal precursors by suppressing
CCAAT/enhancer-binding protein alpha and peroxisome
proliferators-activated receptor gamma. J Biol Chem.
282:14515–14524. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jones SE and Jomary C: Secreted
frizzled-related proteins: searching for relationships and
patterns. Bioessays. 24:811–820. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gaur T, Rich L, Lengner CJ, et al:
Secreted frizzled related protein 1 regulates Wnt signaling for
BMP2 induced chondrocyte differentiation. J Cell Physiol.
208:87–96. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yao W, Cheng Z, Shahnazari M, Dai W,
Johnson ML and Lane NE: Overexpression of secreted frizzled-related
protein 1 inhibits bone formation and attenuates parathyroid
hormone bone anabolic effects. J Bone Miner Res. 25:190–199. 2010.
View Article : Google Scholar
|
|
46
|
Lagathu C, Christodoulides C, Tan CY, et
al: Secreted frizzled-related protein 1 regulates adipose tissue
expansion and is dysregulated in severe obesity. Int J Obes.
34:1695–1705. 2010. View Article : Google Scholar
|
|
47
|
Denver RJ, Bonett RM and Boorse GC:
Evolution of leptin structure and function. Neuroendocrinology.
94:21–38. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Thomas T: The complex effects of leptin on
bone metabolism through multiple pathways. Curr Opin Pharmacol.
4:295–300. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Turner RT, Kalra SP, Wong CP, Philbrick
KA, Lindenmaier LB, Boghossian S and Iwaniec UT: Peripheral leptin
regulates bone formation. J Bone Mineral Res. 28:22–34. 2013.
View Article : Google Scholar
|
|
50
|
Cornish J, Callon KE, Bava U, et al:
Leptin directly regulates bone cell function in vitro and reduces
bone fragility in vivo. J Endocrinol. 175:405–415. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hamrick MW, Pennington C, Newton D, Xie D
and Isales C: Leptin deficiency produces contrasting phenotypes in
bones of the limb and spine. Bone. 34:376–383. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Iwaniec UT, Boghossian S, Lapke PD, Turner
RT and Kalra SP: Central leptin gene therapy corrects skeletal
abnormalities in leptin-deficient ob/ob mice. Peptides.
28:1012–1019. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kalra SP, Dube MG and Iwaniec UT: Leptin
increases osteoblast-specific osteocalcin release through a
hypothalamic relay. Peptides. 30:967–973. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Bartell SM, Rayalam S, Ambati S, et al:
Central (ICV) leptin injection increases bone formation, bone
mineral density, muscle mass, serum IGF-1, and the expression of
osteogenic genes in leptin-deficient ob/ob mice. J Bone Miner Res.
26:1710–1720. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ducy P, Amling M, Takeda S, et al: Leptin
inhibits bone formation through a hypothalamic relay: a central
control of bone mass. Cell. 100:197–207. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Holloway WR, Collier FM, Aitken CJ, et al:
Leptin inhibits osteoclast generation. J Bone Miner Res.
17:200–209. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Thomas T, Gori F, Khosla S, Jensen MD,
Burguera B and Riggs BL: Leptin acts on human marrow stromal cells
to enhance differentiation to osteoblasts and to inhibit
differentiation to adipocytes. Endocrinology. 140:1630–1638.
1999.PubMed/NCBI
|
|
58
|
Kim GS, Hong JS, Kim SW, Koh JM, An CS,
Choi JY and Cheng SL: Leptin induces apoptosis via
ERK/cPLA2/cytochrome pathway in human bone marrow stromal cells. J
Biol Chem. 278:21920–21929. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Hamrick MW and Ferrari SL: Leptin and the
sympathetic connection of fat to bone. Osteoporos Int. 19:905–912.
2008. View Article : Google Scholar
|
|
60
|
Hamrick MW, Della-Fera MA, Hartzell D,
Pennington C and Baile CA: Intrahypothalamic injections of leptin
increase adipocyte apoptosis in peripheral fat pad and in bone
marrow. Cell Tissue Res. 327:133–141. 2007. View Article : Google Scholar
|
|
61
|
Idelevich A, Sato K and Baron R: What are
the effects of leptin on bone and where are they exerted? J Bone
Miner Res. 28:18–21. 2013. View Article : Google Scholar
|
|
62
|
Farooqi IS, Jebb SA, Langmack G, et al:
Effects of recombinant leptin therapy in a child with congenital
leptin deficiency. N Engl J Med. 341:879–884. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Paz-Filho G, Mastronardi C, Delibasi T,
Wong ML and Licinio J: Congenital leptin deficiency: diagnosis and
effects of leption replacement therapy. Arq Bras Endocrinol Metab.
54:690–697. 2010. View Article : Google Scholar
|
|
64
|
Arita Y, Kihara S, Ouchi N, et al:
Paradoxical decrease of an adipose-specific protein, adiponectin,
in obesity. Biochem Biophys Res Commun. 257:79–83. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kumada M, Kihara S, Dumitsuji S, et al:
Association of hypoadiponectinemia with coronary artery disease in
men. Arterioscler Thromb Vasc Biol. 23:85–89. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Hara K, Horikoshi M, Yamauchi T, et al:
Measurement of the high-molecular weight form of adiponectin in
plasma is useful for the prediction of insulin resistance and
metabolic syndrome. Diabetes Care. 29:1357–1362. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Oshima K, Nampei A, Matsuda M, et al:
Adiponectin increases bone mass by suppressing osteoclast and
activating osteoblast. Biochem Biophys Res Commun. 331:520–526.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Yamaguchi N, Kukita T, Li YJ, Marinez
Argueta JG, Saito T, Hanazawa S and Yamashita Y: Adiponectin
inhibits osteoclast formation stimulated by lipoplysaccharide from
Actinobacillus actinomycetemcomitans. FEMS Immunol Med Microbiol.
49:28–34. 2007. View Article : Google Scholar
|
|
69
|
Kajimura D, Lee WH, Riley JK, et al:
Adiponectin regulates bone mass via opposite central and peripheral
mechanisms through FoxO1. Cell Metab. 17:901–915. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Luo XH, Guo LJ, Xie H, Yuan LQ, Wu XP,
Zhou HD and Liao EY: Adiponectin stimulates RANKL and inhibits OPG
expression in human osteoblasts through the MAPK signaling pathway.
J Bone Miner Res. 21:1648–1656. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Tu QS, Zhang J, Dong LQ, Saunders E, Luo
E, Tang J and Chen J: Adiponectin inhibits osteoclastogenesis and
bone resorption via APPL1-mediated suppression of Akt1. J Bio Chem.
286:12542–12553. 2011. View Article : Google Scholar
|
|
72
|
Yamaguchi N, Kukita T, Li YJ, et al:
Adiponectin inhibits induction of TNF-alpha/RANKL-stimulated NFATc1
via the AMPK signaling. FEBS Letters. 582:451–456. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Luo XH, Guo LJ, Yuan LQ, Xie H, Zhou HD,
Wu XP and Liao EY: Adiponectin stimulates human osteobalsts
proliferation and differentiation via the MAPK signaling pathway.
Exp Cell Res. 309:99–109. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Lee HW, Kim SY, Kim AY, Lee EJ, Choi JY
and Kim JB: Adiponection stimulates osteoblast differentiation
through induction of COX2 in mesenchymal progenitor cells. Stem
Cells. 27:2254–2262. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Mitsui Y, Gotoh M, Fukushima N, et al:
Hyperadiponectinemia enhances bone formation in mice. BMC
Musculoskelet Disord. 12:182011. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Ealey KN, Kaludjerovic J, Archer M and
Ward WE: Adiponectin is a negative regulator of bone mineral and
bone strength in growing mice. Exp Biol Med (Maywood).
233:1546–1553. 2008. View Article : Google Scholar
|
|
77
|
Hotta K, Funahashi T, Bodkin NL, et al:
Circulating concentrations of the adipocyte protein adiponetin are
decreased in parallel with reduced insulin sensitivity during the
progression to type 2 diabetes in rhesus monkeys. Diabetes.
50:1126–1133. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Bauche IB, E1 Mkadem SA, Pottier AM, et
al: Overexpression of adiponectin targeted to adipose tissue in
transgenic mice: impaired adipocyte differentiation. Endocrinology.
148:1539–1549. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Otabe S, Yuan X, Fukutani T, et al:
Overexpression of human adiponectin in transgenic mice results in
suppression of fat accumulation and prevention of premature death
by high-calorie diet. Am J Physiol Endocrinol Metab. 293:E210–E218.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Williams GA, Wang Y, Callon KE, et al: In
vitro and in vivo effects of adiponectin on bone. Endocrinology.
150:3603–3610. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Yano W, Kubota N, Itoh S, et al: Molecular
mechanism of moderate insulin resistance in adiponectin-knockout
mice. Endocr J. 55:515–522. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Nawrocki AR, Rajala MW, Tomas E, et al:
Mice lacking adiponectin show decreased hepatic insulin sensitivity
and reduced responsiveness to peroxisome proliferator-activated
receptor gamma agonists. J Biol Chem. 281:2654–2660. 2006.
View Article : Google Scholar
|
|
83
|
Kubota N, Terauchi Y, Yamauchi T, et al:
Disruption of adiponectin causes insulin resistance and neointimal
formation. J Biol Chem. 277:25863–25866. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Maeda N, Shimomura I, Kishida K, et al:
Diet-induced insulin resistance in mice lacking adiponectin/ACRP30.
Nat Med. 8:731–737. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
85
|
Kanazawa I: Does adiponectin have adverse
effects on bone mass and fracture. Internal Med. View Article : Google Scholar : 2011.
|
|
86
|
Mohiti-Ardekani J, Soleymani-Salehabadi H,
Owlia MB and Mohiti A: Relationships between serum adipocyte
hormones (adiponectin, leptin, resistin), bone mineral density and
bone metabolic markers in osteoporosis patients. J Bone Miner
Metab. 32:400–404. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Wu N, Wang QP, Li H, Wu XP, Sun ZQ and Luo
XH: Relationships between serum adiponectin, leptin concentrations
and bone mineral density and bone biochemical markers in Chinese
females. Clin Chim Acta. 411:771–775. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Tenta R, Kontogianni MD and Yiannakouris
N: Association between circulating levels of adiponectin and
indices of bone mass and bone metabolism in middle-aged
post-menopausal females. J Endocrinol Invest. 35:306–311. 2012.
|
|
89
|
Schwartz AV, Sigurdsson S, Hue TF, et al:
Vertebral bone marrow fat associated with lower trabecular BMD and
prevalent vertebral fracture in older adults. J Clin Endocrinol
Metab. 98:2294–2300. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Widlansky ME, Gokce N, Keaney JF and Vita
JA: The clinical implications of endothelial dysfunction. J Am Coll
Cardiol. 42:1149–1160. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Wang GJ, Maga DB, Richemer WG, Sweet DE,
Reger SI and Thompson RC: Cortisone induced bone changes and its
response to lipid clearing agents. Clin Orthop. 130:81–85.
1978.PubMed/NCBI
|
|
92
|
Miyanishi K, Yamamoto T, Irisa T,
Yamashita A, Jingushi S, Noguchi Y and Iwamoto Y: Bone marrow fat
cell enlargement and a rise in intraosseous pressure in
steroid-treated rabbits with osteonecrosis. Bone. 30:185–190. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Zhou Q, Li Q, Yang L and Liu F: Changes of
blood vessels in glucocorticoid-induced avascular necrosis of
femoral head in rabbits. Zhonghua Wai Ke Za Zhi. 38:212–215.
2000.In Chinese.
|
|
94
|
Kitajima M, Shigematsu I, Ogawa K,
Sugihara H and Hotokebuchi T: Effects of glucocorticoid on
adipocyte size in human bone marrow. Med Mol Morphol. 40:150–156.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Kerachian MA, Séguin C and Harvey EJ:
Glucocorticoids in osteonecrosis of the femoral head: a new
understanding of the mechanisms of action. J Steroid Biochem Mol
Biol. 114:121–128. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Savopoulos CH, Dokos CH, Kaiafa G and
Hatzitolios A: Adipogenesis and osteoblastogenesis:
trans-differentiation in the pathophysiology of bone disorders.
Hippokratia. 15:18–21. 2011.PubMed/NCBI
|
|
97
|
Tsartsalis AN, Dokos C, Kaiafa GD,
Tsartsalis DN, Kattamis A, Hatzitolios AI and Savopoulos CG:
Statins, bone formation and osteoporosis: hope or hype? Hormones
(Athens). 11:126–139. 2012. View Article : Google Scholar
|
|
98
|
Esposito K, Capuano A, Sportiello L,
Giustina A and Giugliano D: Should we abandon statins in the
prevention of bone fractures? Endocrine. 44:326–333. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Borton AJ, Frederick JP, Datto MB, Wang XF
and Weinstein RS: The loss of Smad3 results in a lower rate of bone
formation and osteopenia through dysregulation of osteoblast
differentiation and apoptosis. J Bone Miner Res. 16:1754–1764.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Kaji H, Kanatani M, Sugimoto T and Chihara
K: Stains modulate the levels of osteoprotegerin/receptor activator
of NF kappaB ligand mRNA in mouse bone-cell cultures. Horm Metab
Res. 37:589–592. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Garrett IR and Mundy GR: The role of
statins as potential targets for bone formation. Arthritis Res.
4:237–240. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
102
|
Sugiyama M, Kodama T, Konishi K, Abe K,
Asami S and Oikawa S: Compactin and simvastatin, but not
pravastatin, induce bone morphogenetic protein-2 in human
osteosarcoma cells. Biochem Biophys Res Commun. 271:688–692. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Maeda T, Matsunuma A, Kawane T and
Horiuchi N: Simvastatin promotes osteoblast differentiation and
mineralization in MC3T3-E1 cells. Biochem Biophys Res Commun.
280:874–877. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Oxlund H, Dalstra M and Andreassen TT:
Statin given perorally to adult rats increases cancellous bone mass
and compressive strength. Calcif Tissue Int. 69:299–304. 2001.
View Article : Google Scholar
|
|
105
|
Uzzan B, Cohen R, Nicolas P, Cucherat M
and Perret GY: Effects of statins on bone mineral density: a
meta-analysis of clinical studies. Bone. 40:1581–1587. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Anagnostis P, Karagiannis A, Kakafika AI,
Tziomalos K, Athyros VG and Mikhailidis DP: Atherosclerosis and
osteoporosis: age-dependent degenerative processes or related
entities? Osteoporos Int. 20:197–207. 2009. View Article : Google Scholar
|
|
107
|
de Nijs RN, Jacobs JW, Lems WF, et al:
Alendronate or alfacalcidol in glucocorticoid-induced osteoporosis.
N Engl J Med. 355:675–684. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Reid DM, Devogelaer JP, Saag K, et al:
Zoledronic acid and risedronate in the prevention and treatment of
glucocorticoid-induced osteoporosis (HORIZON): a multicentre,
double-blind, double-dummy, randomized controlled trial. Lancet.
373:1253–1263. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Saag KG, Shane E, Boonen S, et al:
Teriparatide or alendronate in glucocorticoid-induced osteoporosis.
N Engl J Med. 357:2028–2039. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Wallach S, Cohen S, Reid DM, et al:
Effects of risedronate treatment on bone density and vertebral
fracture in patients on corticosteroid therapy. Calcif Tissue Int.
67:277–285. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Riggs BL, Khosla S and Melton LJ III: A
unitary model for involutional osteoporosis: Estrogen deficiency
causes both type I and type II osteoporosis in postmenopausal
females and contributes to bone loss in aging men. J Bone Miner
Res. 13:763–773. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Heaney RP, Recker RR and Saville PD:
Menopausal changes in bone remodeling. J Lab Clin Med. 92:964–970.
1978.PubMed/NCBI
|
|
113
|
Manolagas SC and Jilka RL: Bone marrow,
cytokines, and bone remodeling. Emerging insights into the
pathophysiology of osteoporosis. N Engl J Med. 332:305–311. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Khosla S, Atkinson EJ, Melton LJ III and
Riggs BL: Effects of age and estrogen status on serum parathyroid
hormone levels and biochemical markers of bone turnover in females:
a population based study. J Clin Endocrinol Metab. 82:1522–1527.
1997.PubMed/NCBI
|
|
115
|
D’Amelio P, Grimaldi A, Di Bella S, et al:
Estrogen deficiency increases osteoclastogenesis up-regulating T
cells activity: a key mechanism in osteoporosis. Bone. 43:92–100.
2008. View Article : Google Scholar
|
|
116
|
Wells G, Tugwell P, Shea B, et al:
Meta-analysis of the efficacy of hormone replacement therapy in
treating and preventing osteoporosis in postmenopausal females.
Endocr Rev. 23:529–539. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Bonnet N, Gadois C, McCloskey E, Lemineur
G, Lespessailles E, Courteix D and Benhamou CL: Protective effect
of beta blockers in postmenopausal females: influence on fractures,
bone density, micro and macroarchitecture. Bone. 40:1209–1216.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Bonnet N, Benhamou CL, Brunet-lmbault B,
et al: Severe bone alterations under beta2 agonist treatment: bone
mass, microarchitecture and strength analyses in female rats. Bone.
37:622–633. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Bonnet N, Benhamou CL, Malaval L, et al:
Low dose beta-blocker prevents ovariectomy-induced bone loss in
rats without affecting heart functions. J Cell Physiol.
217:819–827. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Pasco JA, Henry MJ, Sanders KM, Kotowicz
MA, Seeman E and Nicholson GC: beta-adrenergic blockers reduce the
risk of fracture partly by increasing bone mineral density: Geelong
Osteoporosis Study. J Bone Miner Res. 19:19–24. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Schlienger RG, Kraenzlin ME, Jick SS and
Meier CR: Use of beta-blockers and risk of fractures. JAMA.
292:1326–1332. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Ferrari SL, Demissie S, Karasik D, Cupples
LA, Imamovic A, Dupuis J and Kiel DP: Beta 2 adrenergic receptor,
beta-blockers and their influence on bone mass in humans: the
Framingham osteoporosis study. J Bone Miner Res. 20(Suppl 1):
11–12. 2005.
|
|
123
|
Rejnmark L, Vestergaard P, Kassem M,
Christoffersen BR, Kolthoff N, Brixen K and Mosekilde L: Fracture
risk in perimenopausal females treated with beta-blockers. Calcif
Tissue Int. 75:365–372. 2004. View Article : Google Scholar : PubMed/NCBI
|














