Introduction
Mesenchymal stem cells (MSCs) are multipotent cells
that are able to differentiate into cardiomyocytes and vascular
endothelial cells (1). Therefore
stem-cell therapy offers the prospect of a novel and effective
treatment with which to repair ischemic heart tissue following
acute myocardial infarction (AMI) (2). Injection of allogeneic MSCs into
regions of damaged myocardium, 3 days after AMI has been shown to
stimulate cardiac regeneration and to markedly decrease myocardial
infarct size (3). The results from
clinical trials have revealed that MSC engraftment via
intramyocardial injection or intracoronary infusion is able to
induce a moderate, but significant improvement in myocardial
infarct size and left ventricular function (4). However, graft cell death is an
important factor, which should be addressed in order to enable the
development of cell therapy for cardiac repair. Ischemia and a
hypoxic microenvironment may make the largest contribution to poor
graft survival rate (5).
Hydrogen sulfide (H2S) is a colorless,
water soluble, flammable gas, which has a characteristic smell of
rotten eggs. As with other members of the gasotransmitter family
(nitric oxide and carbon monoxide), H2S has been shown
to possess extensive biological functions and may therefore be
viewed as an important signaling molecule, involved in multiple
signaling mechanisms under normal physiological conditions
(6). Accumulating evidence
suggests that exogenously applied H2S and endogenously
altered H2S production are cytoprotective and regulate
cell apoptosis in various models of cellular injury, including
hypoxia (7), ischemia and
reperfusion injury (8), oxidative
stress (9) and inflammation
(10). A previous study by this
group demonstrated that hypoxia and serum deprivation (H/SD) is
able to reduce endogenous H2S production by inhibiting
the expression and activity of cystathionine γ-lyase (CSE), a key
enzyme involved in H2S synthesis in MSCs (11). Upregulation of the
CSE/H2S system prevents the H/SD-induced decrease in
endogenous H2S generation and protects MSCs from
apoptosis (11). However, the
mechanism underlying the ability of endogenous H2S to
protect MSCs from apoptosis under H/SD cultivation remains to be
elucidated.
In the present study, a model of MSC apoptosis
induced by H/SD was developed, and the overexpression of CSE in
MSCs was achieved using lentivirus delivery, in order to examine
the mechanisms underlying the antiapoptotic effect mediated by
endogenous H2S.
Materials and methods
Materials
Low-glucose Dulbecco’s modified Eagle’s medium
(L-DMEM) and fetal bovine serum (FBS) were obtained from Hyclone
(Logan, UT, USA). Propidium iodide (PI), RNase and
DL-propargylglycine (PPG) were obtained from Sigma-Aldrich (St.
Louis, MO, USA). A cell mitochondria isolation kit and trypsin-EDTA
Solution were obtained from Beyotime Institute of Biotechnology
(Haimen, China). Polyclonal rabbit CSE (sc-135203) and monoclonal
mouse cytochrome c (sc-13561) antibodies were obtained from
Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Rabbit
poly-clonal Bax (BS6420) and rabbit polyclonal Bcl-2 (BS6421)
antibodies were obtained from Bioworld Technology, Inc. (St. Louis
Park, MN, USA). Rabbit polyclonal Akt (#9272), and rabbit
polyclonal phospho-Akt (Ser473; #9271), rabbit polyclonal binding
immunoglobulin protein (BiP; #3183) and rabbit polyclonal C/EBP
homologous protein (CHOP; #2895) antibodies were obtained from Cell
Signaling Technology, Inc. (Danvers, MA, USA). The enhanced
chemiluminescence western blotting system was purchased from EMD
Millipore (Billerica, MA, USA). Lipofectamine 2000® was
purchased from Invitrogen Life Technologies (Paisley, UK).
Polybrene was obtained from Chemicon (Temecula, CA, USA).
Cell culture and model of H/SD
MSCs were isolated from Sprague-Dawley rats (30 male
rats; 4 weeks old; ~80g; Shanghai Laboratory Animals Center,
Shanghai, China) as previously described (11). Briefly, bone marrow was harvested
from the tibia and femur of male rats, plated in L-DMEM
supplemented with 20% inactivated FBS and 100 units/ml
penicillin/streptomycin, and incubated at 37°C in a humidified
tissue culture incubator containing 5% CO2. The medium
was replaced after 24 h to discard non-adherent hematopoietic
cells. The adherent spindle-shaped MSCs were expanded and cultured
for no more than two or three passages. Subsequently, cells were
analyzed for the expression of surface markers [CD44 and CD90
(positive), CD34 and CD45 (negative)], using flow cytometry (BD
FACSCalibur; BD Biosciences, Baltimore, MD, USA) as described
previously (12). All procedures
performed on animals were approved by the University of Anhui
Animal Care Committee (Hefei, China) and were conducted in
accordance with national guidelines. Cell apoptosis was induced by
H/SD, Briefly, MSCs were washed with serum-free L-DMEM, placed in
serum-free medium and then incubated in a sealed hypoxic GENbox jar
for 12 h fitted with a catalyst (GENbox anaer, Biomérieux, Marcy
l’Etoile, France) in order to sequester free oxygen. The
O2 concentration was <0.1% after 2.5 h of using the
GENbox anaer.
Plasmid construction, lentivirus
production and transduction
Polymerase chain reaction was used to amplify the
CSE gene (GenBank accession number, AY032875) from rat liver
tissues using the following primer sequences: Forward,
5′-GTATGGAGGCACCAACAGGT-3′ and reverse, 5′-GTTGGGTTTGTGGGTGTTTC-3′
(Sangon Biotech, Co., Ltd., Shanghai, China). The cycling
conditions were as follows: Initial denaturation at 95°C for 5 min,
5 cycles of 95°C for 30 sec, 63°C for 30 sec (−1°C/cycle) and 72°C
for 20 sec, 15 cycles of 95°C for 30 sec, 58°C for 30 sec
(−0.5°C/cycle) and 72°C for 20 sec, then 19 cycles of 95°C for 30
sec, 51°C for 30 sec (−1°C/cycle) and 72°C for 20 sec. The
amplified CSE gene was subcloned into the pLVX-IRES-ZsGreen vector
using in vitro recombination methods. The pseudo-lentivirus
was produced via transient transfection of 293FT packaging cells.
On the day prior to transfection, 1.6×106 293FT cells
were plated in 6-cm dishes. Subsequently, cells were cotransfected
with either 1.7 µg pLVX-IRES-ZsGreen vector or
pLVX-IRES-ZsGreen-CSE with all cells receiving 1.13 µg pCMV
Δ8.91 and 0.57 µg pMD.G, using Lipofectamine 2000. The
culture supernatants were harvested at 72 h following transfection
and filtered through a 0.45-µm low protein binding
polysulfonic filter (Millipore, Bedford, MA, USA). For
transduction, 2×106 MSC cells were seeded into a 10-cm
dish and incubated with lentiviruses and 8 µg/ml polybrene
in the incubator for 48 h.
Flow cytometry assay
Treated MSCs were digested with trypsin (2.5 g/l)
and centrifuged at 250 × g for 5 min. The supernatant was then
removed. Cells were washed twice with phosphate-buffered saline
(PBS) and fixed with 70% ethanol at −20°C overnight. Cells were
then centrifuged at 250 × g for 5 min, washed twice with PBS and
adjusted to a concentration of 1×106 cells/ml. A
quantity of 0.5 ml RNase (1 mg/ml in PBS; Sigma-Aldrich) was added
into the 0.5 ml cell sample and incubated at 37°C for 30 min.
Following addition of the PI, to a final concentration of 50 mg/l,
cells were filtered and incubated in darkness at 4°C for 30 min,
prior to flow cytometric analysis (Beckman-Coulter, Miami, FL,
USA). In the DNA histogram, the amplitude of the sub-G1 DNA peak
represents the quantity of apoptotic cells.
Cell mitochondria isolation
The mitochondria were isolated using a cell
mitochondria isolation kit (Beyotime Institute of Biotechnology)
according to the manufacturer’s instructions. Briefly,
5×107 cells were harvested and washed with ice-cold PBS.
Cells were incubated with 1.0 ml mitochondria extraction mixed
buffer provided in the kit for 15 min and then homogenized using an
ice-cold dounce tissue grinder (Hede Biotechnology, Beijing,
China). The homogenates were centrifuged at 600 × g for 10 min and
then the supernatants were further centrifuged at 11,000 × g for 10
min at 4°C. The supernatants were collected and the precipitate
consisted of the cell mitochondria. The cytosolic proteins were
isolated from the supernatant following further centrifugation at
12,000 × g for 10 min, at 4°C. The samples containing the cell
mitochondria were then separated using the mitochondria lysis mixed
buffer for analysis of mitochondrial proteins.
Western blotting
Cultured cells were harvested and lysed. Equal
quantities of proteins were boiled and separated by SDS-PAGE, then
electrophoretically transferred onto a nitrocellulose membrane (EMD
Millipore). The membranes were blocked with Tris-buffered saline
with Tween 20 (TBST) containing 5% bovine serum albumin
(Sigma-Aldrich) for 2 h. The primary antibody dilutions were 1:500
for CSE, Bax, Bcl-2 and cytochrome c, and 1:1,000 for CHOP,
BiP, Akt and p-Akt. The membranes were then incubated with primary
antibodies at 4°C overnight. Following washing with TBST, the
membranes were incubated with goat anti-rabbit (#7074) or horse
anti-mouse (#7076) horseradish peroxidase-conjugated IgG antibodies
(Cell Signaling Technology, Inc., Beverly, MA, USA) diluted to
1:1,000 at room temperature for 2 h. The membranes were washed
again and developed with an enhanced chemiluminescence system
followed by apposition of the membranes with autoradiographic films
(Eastman Kodak Company, Shanghai, China). The optical density of
the protein band on western blots was calculated using Quantity One
1-D software, version 4.6.6 (Bio-Rad, Hercules, CA, USA).
Statistical analysis
Data are expressed as the mean ± standard error of
the mean. Differences among groups were assessed using a one-way
analysis of variance. Comparisons between the two groups were
evaluated using post hoc tests. P<0.05 was considered to
indicate a statistically significant difference.
Results
Characteristics of cultured MSCs
MSCs were isolated and cultured from the bone marrow
of male Sprague-Dawley rats. At 5 days following isolation,
fusiform and fibroblast-like adherent cells were apparent, and
formed cell colonies (Fig. 1). The
determination of the surface markers of MSCs at passage 3 using
flow cytometry, was the same as in a previous study by this group
(12). The MSCs exhibited a
positive expression of cluster of differentiation (CD)44, CD54 and
CD90, while the expression of CD31, CD34 and CD45 was not
observed.
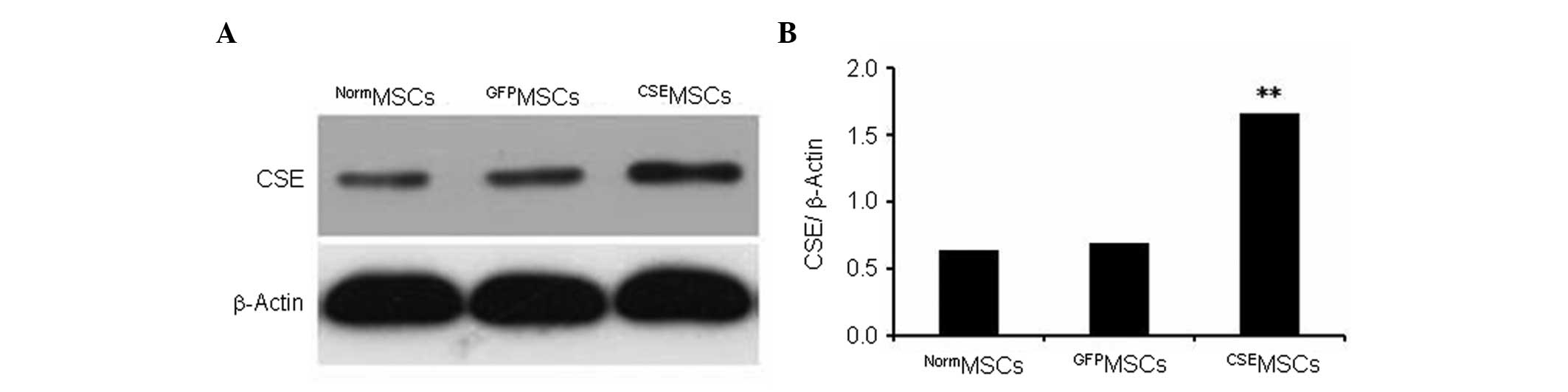
Overexpression of CSE in genetically
modified MSCs
CSE overexpression was mediated by lentiviral
transduction in MSCs. The present data showed that CSE expression
in MSCs infected with the pLV-ZsGreen-CSE lentivirus
(CSEMSCs) was upregulated by >2.5-fold compared with
MSCs infected with the pLV-ZsGreen lentivirus (GFPMSCs)
or with untransduced MSCs (NormMSCs; Fig. 2).
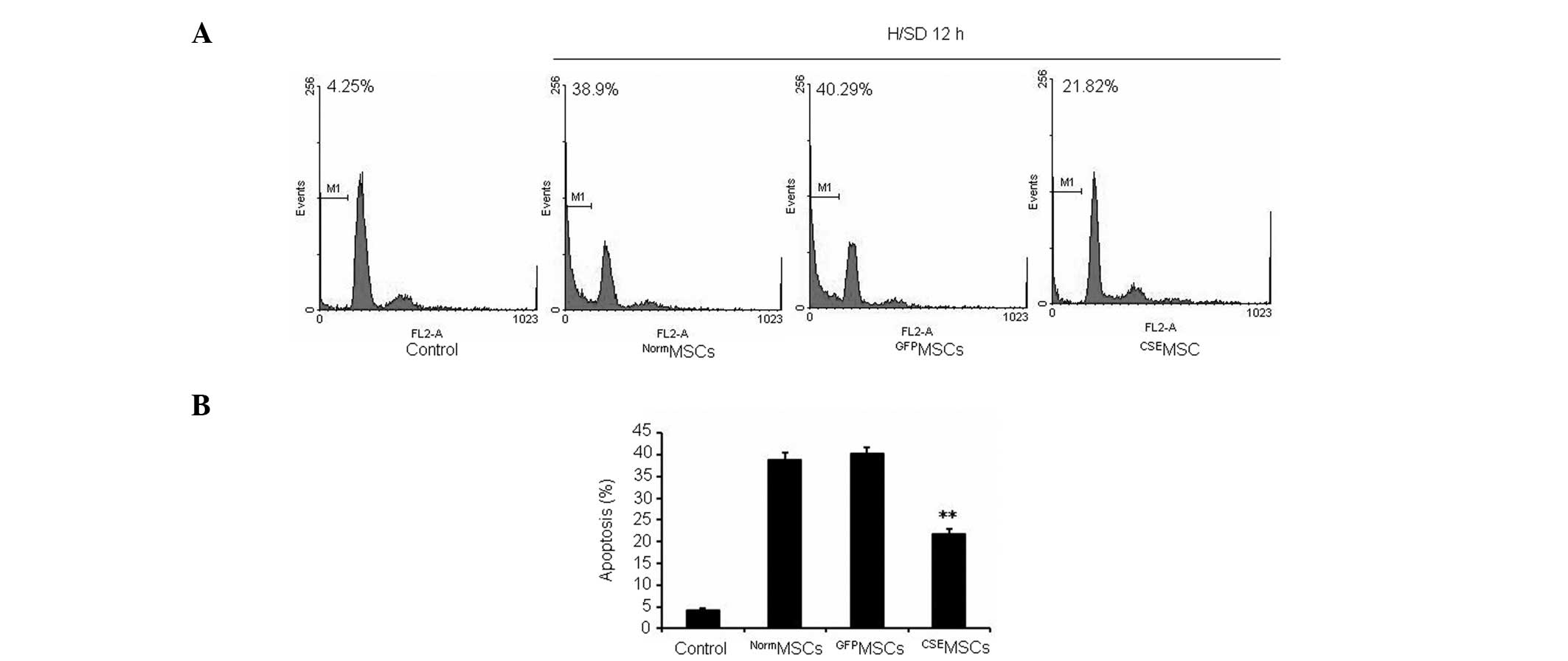
Overexpression of CSE protects MSCs from
H/SD-induced apoptosis in vitro
In order to further examine the regulatory role of
CSE overexpression in H/SD-induced apoptosis in MSCs, the modified
and normal MSCs were exposed to H/SD for 12 h. As shown in Fig. 3, it was observed that
CSEMSCs had a significantly lower level of apoptosis
compared with NormMSCs or GFPMSCs. Therefore,
the present data indicated that upregulation of the
CSE/H2S system protects MSCs from H/SD-induced
apoptosis.
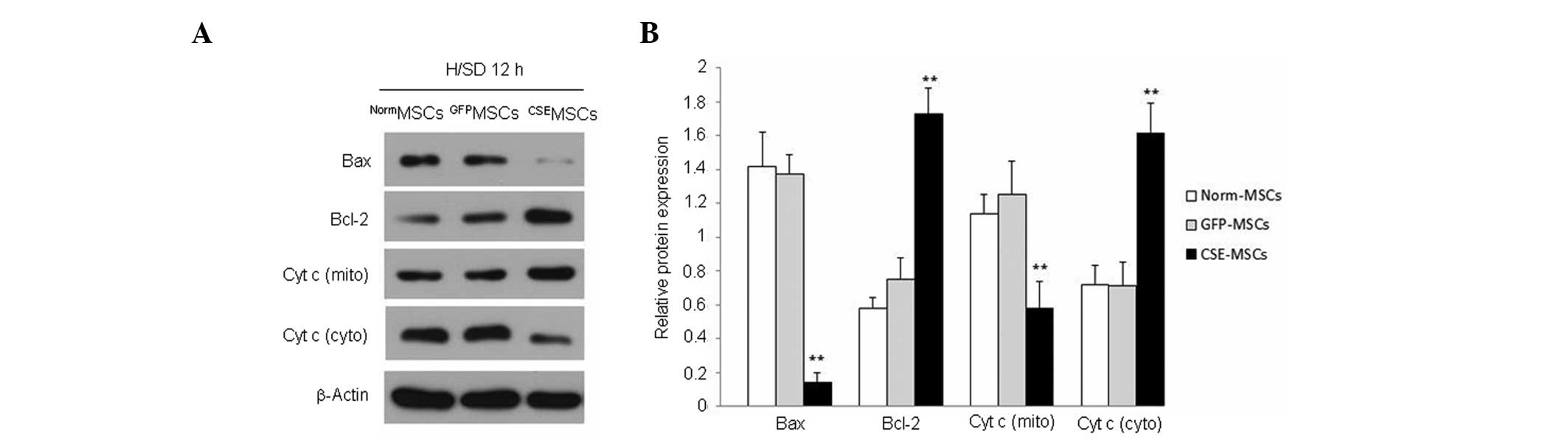
CSE protects MSCs from H/SD-induced
apoptosis via inhibition of the mitochondrial injury pathway
Based on these initial results, the mechanism
underlying the protection of MSCs from H/SD-induced apoptosis by
CSE was further investigated. It has been reported that
H/SD-induced apoptosis of MSCs is mediated by changes in the
mitochondrial integrity and function, but may be independent of the
death receptor pathway (13).
Therefore, the role of the mitochondrial injury pathway was
examined, primarily with regards to the protective effect of CSE
overexpression against H/SD-induced apoptosis in MSCs. As shown in
Fig. 4, following 12 h H/SD
cultivation, the expression of Bax protein was reduced. However,
Bcl-2 protein expression was increased in CSEMSCs
compared with NormMSCs and GFPMSCs. Changes
in the level of cytochrome c in the cytosolic and
mitochondrial fractions were also measured using western blotting.
It was observed that cytochrome c was significantly
increased in the cytosol but decreased in the mitochondria in
NormMSCs and GFPMSCs, compared with levels in
CSEMSCs (Fig. 4). The
present data demonstrated that H/SD cultivation promotes cytochrome
c release from the mitochondria into the cytosol in MSCs.
However, CSE overexpression may contain the cytochrome c
within the mitochondria and inhibit the release of cytochrome
c into the cytosol. The present data indicated that
upregulation of the CSE/H2S system inhibits
mitochondrial injury and protects MSCs against H/SD-induced
apoptosis.
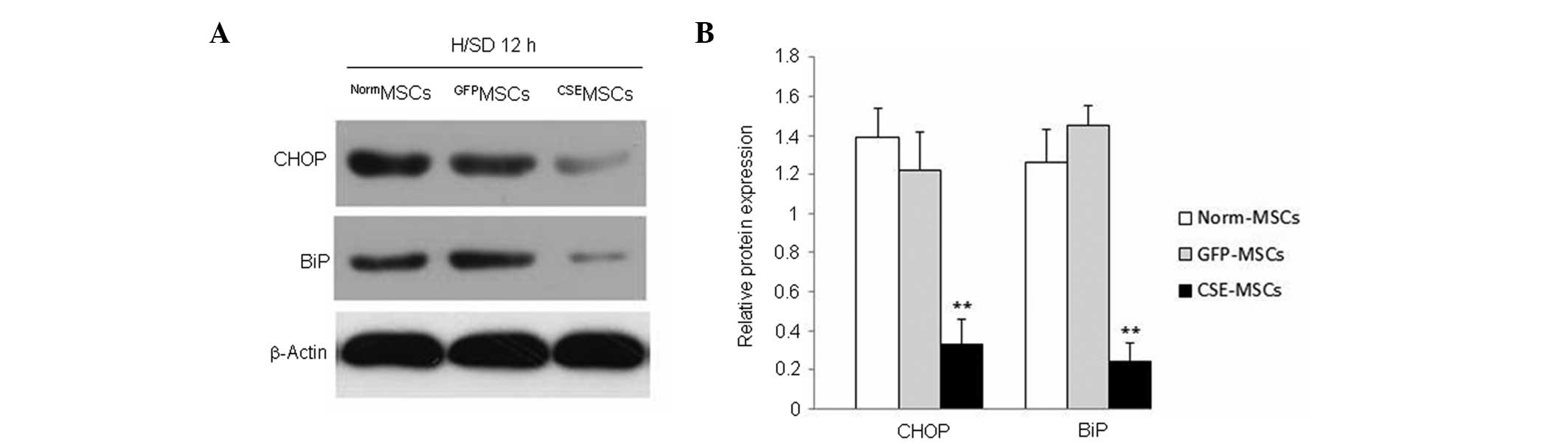
CSE overexpression inhibits the
H/SD-induced increase in CHOP and BiP expression in MSCs
Previous evidence has demonstrated that ERS is vital
for cell apoptosis (14). The role
of ERS has been investigated in multiple models of cell damage and
apoptosis (15). It has been
demonstrated that ERS is involved in H/SD-induced and
H2O2-induced apoptosis of MSCs (16,17).
In order to elucidate whether ERS was associated with the
protective effect of CSE overexpression against H/SD-induced
apoptosis in MSCs, the expression of two indicators of ERS
(18), CHOP and BiP, was observed
in genetically modified and normal MSCs. The result revealed that
the expression of CHOP and BiP was downregulated in
CSEMSCs subjected to H/SD for 12 h, compared with that
in NormMSCs or GFPMSCs (Fig. 5). These results indicated that the
ERS response is inhibited by CSE overexpression and may be involved
in the protective effect of endogenous H2S on MSCs
subjected to H/SD.
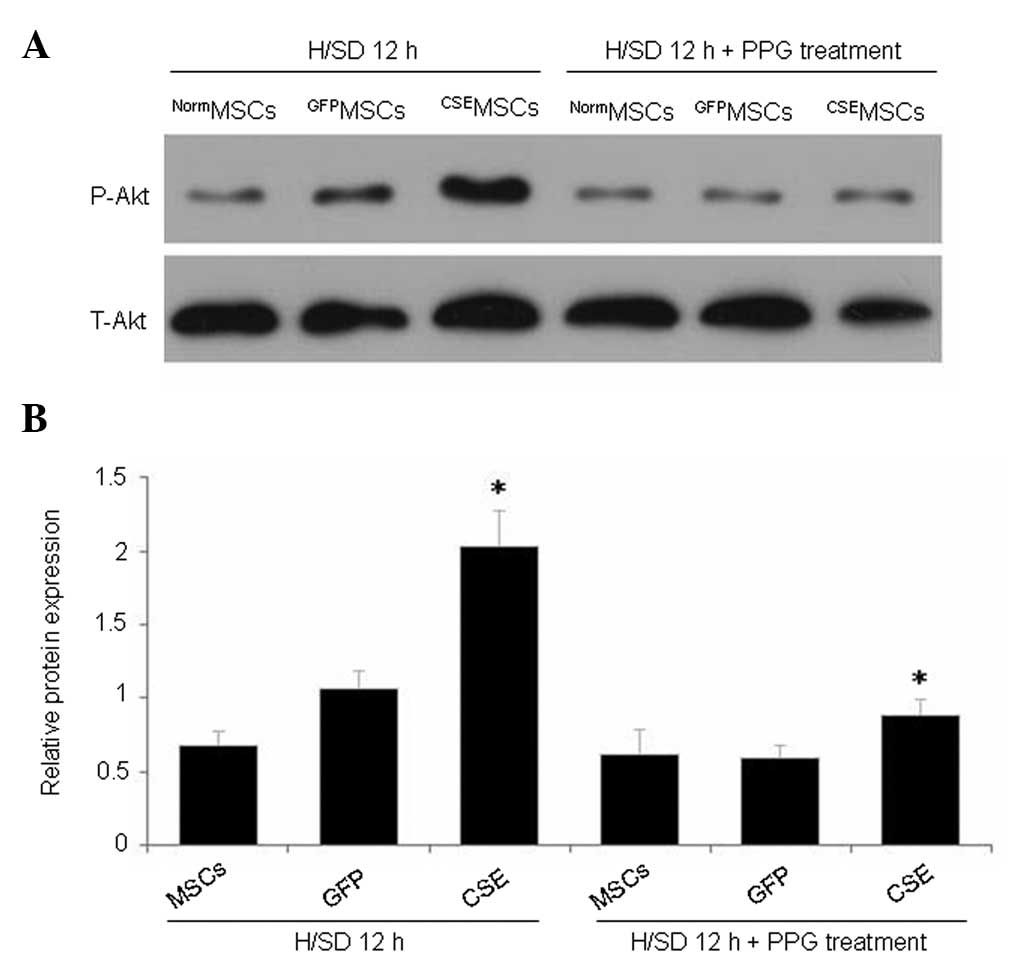
CSE overexpression activates the PI3K/Akt
signaling pathway in MSCs
Overexpression of CSE promotes the phosphorylation
of Akt in MSCs, and PI3K/Akt is an important cell survival pathway
in various types of cells (19–21).
Therefore, levels of total and phosphorylated Akt were measured
using western blotting, in order to detect whether the
overexpression of CSE activates the PI3K/Akt pathway. The results
demonstrated that overexpression of CSE significantly increased the
phosphorylation of Akt in CSEMSCs compared with that in
NormMSCs or GFPMSCs (Fig. 6). In addition, H2S
production was inhibited following treatment of MSCs with the CSE
inhibitor, PPG (5 mmol/l), in the presence of H/SD for 12 h. As
shown in Fig. 6, PPG significantly
inhibited the phosphorylation of Akt in CSEMSCs. These
findings demonstrated that CSE overexpression activates the
phosphorylation of Akt in MSCs subjected to H/SD. In addition, the
CSE inhibitor, PPG, inhibits the phosphorylation of Akt in
CSEMSCs following 12 h H/SD cultivation. Therefore, the
present data indicated that endogenous H2S protects MSCs
from H/SD-induced apoptosis through the activation of the PI3K/Akt
signaling pathway.
Discussion
The technique of in vitro cell culture has
been widely utilized to imitate the ischemic microenvironment. The
survival rate of MSCs in an ischemic microenvironment is critical
to the success of cell-based transplantation therapy for ischemic
heart disease. Therefore, it is important to elucidate the
molecular mechanism responsible for the survival rate of
transplanted MSCs.
It has previously been demonstrated that
lysophosphatidic acid rescues ERS-associated apoptosis in
H/SD-stimulated MSCs (16,22). Dong et al (23) observed that atorvastatin protects
MSCs from H/SD injury via activation of an amp-activated protein
kinase. Nie et al (24)
found that the upregulation of microRNA (miR)-21, miR-23a and
miR-210, induced by H/SD, may be involved in protecting MSCs from
apoptosis (24). It has also been
shown that heat shock protein 90 protects rat MSCs against
H/SD-induced apoptosis via the PI3K/Akt and the
extracellular-signal-regulated kinase (ERK)1/2 pathways (25).
After NO and CO, H2S is considered to be
the third identified endogenous ‘gaseous transmitter’ (26). Accumulating evidence has suggested
that H2S exerts protective effects against various
ischemic and hypoxic injuries, in numerous tissues and cell models.
It has been reported that H2S protects HaCaT cells from
cobalt chloride-induced cytotoxicity and inflammation, by
negatively regulating the reactive oxygen species (ROS)/nuclear
factor-κB/COX-2 pathway (27). Yao
et al (28) demonstrated
that H2S protects cardiomyocytes from
hypoxia/reoxygenation-induced apoptosis by preventing glycogen
synthase kinase (GSK)-3β-dependent opening of mitochondrial
permeability transition pores (28). The KATP/PKC/ERK1/2 and
PI3K/Akt pathways have been shown to contribute to H2S
preconditioning-induced cardioprotection (29). Elrod et al (30) observed that H2S
attenuates myocardial ischemia-reperfusion injury by preserving
mitochondrial function. H2S also protects endothelial
cells from high glucose-induced apoptosis by inhibiting oxidative
stress injury, leading to a decrease in intracellular ROS
generation and malondialdehyde levels, and an increase in
superoxide dismutase activity (31). Xie et al (32) revealed that exogenous
H2S preconditioning protects MSCs from hypoxia-induced
cell death, an effect which was accompanied by a significantly
increased level of phosphorylation of Akt, ERK1/2 and GSK-3β.
The signaling cascades that control
caspase-dependent apoptosis may be classified into the
mitochondrial injury and the death receptor pathways (33). The mitochondrial injury pathway may
be induced by a wide variety of signals, including Bax and Bcl-2,
amongst others, which result in the release of cytochrome c
from the mitochondria into the cytoplasm. Bcl-2, as an
antiapoptotic factor, is able to detain cytochrome c in the
mitochondria, but Bax, as a proapoptotic factor is able to promote
the release of cytochrome c into the cytoplasm. The
endoplasmic reticulum (ER) is a multifunctional signaling organelle
with sophisticated stress-signaling pathways that control the entry
and release of Ca2+, sterol biosynthesis, membrane
protein translocation and apoptosis (34). GRP78, also known as BiP, is a
critical ER chaperone protein, which performs multiple functions
and is upregulated under conditions of stress to restore the
function of the ER. CHOP exhibits low expression under
physiological conditions, but is markedly induced in response to
ERS. BiP and CHOP are accepted as markers of ERS (35). The PI3K/Akt pathway has been
observed to respond to a variety of stimuli, including serum
withdrawal, cell cycle disturbances, loss of cell adhesion and DNA
damage, in a variety of cell types. In addition, the PI3K/Akt
signaling pathway is important in mediating survival signaling in
MSCs (36).
Using in vitro experiments, it was shown that
38.9% MSCs underwent apoptosis following H/SD for 12 h. By
contrast, overexpression of CSE reduces the levels of apoptosis of
MSCs by 21.82%. Compared with the control, the expression of Bax,
CHOP and BiP was reduced. However, that of Bcl-2 increased.
Additionally, cytochrome c remained in the mitochondria.
Furthermore, it was found that overexpression of CSE promotes the
phosphorylation of Akt, an effect which was eliminated following
administration of the CSE inhibitor, PPG.
In conclusion, the present study demonstrated the
harmful effect of H/SD on MSCs. The raised level of endogenous
H2S produced by CSE overexpression was shown to protect
MSCs from H/SD-induced apoptosis, via negative regulation of the
mitochondrial injury pathway, inhibition of ERS and activation of
the PI3K/Akt signaling pathway. Therefore, the antiapoptotic
effects of CSE and H2S may be an effective approach to
improve the cellular survival rate following cell-based therapy in
transplantation.
References
|
1
|
Miyahara Y, Nagaya N, Kataoka M, et al:
Monolayered mesenchymal stem cells repair scarred myocardium after
myocardial infarction. Nat Med. 12:459–465. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mathur A and Martin JF: Stem cells and
repair of the heart. Lancet. 364:183–192. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Amado LC, Saliaris AP, Schuleri KH, et al:
Cardiac repair with intramyocardial injection of allogeneic
mesenchymal stem cells after myocardial infarction. Proc Natl Acad
Sci USA. 102:11474–11479. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gyongyosi M, Lang I, Dettke M, et al:
Combined delivery approach of bone marrow mononuclear stem cells
early and late after myocardial infarction: the MYSTAR prospective,
randomized study. Nat Clin Pract Cardiovasc Med. 6:70–81. 2009.
View Article : Google Scholar
|
|
5
|
Zhang M, Methot D, Poppa V, Fujio Y, Walsh
K and Murry CE: Cardiomyocyte grafting for cardiac repair: graft
cell death and anti-death strategies. J Mol Cell Cardiol.
33:907–921. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Calvert JW, Coetzee WA and Lefer DJ: Novel
insights into hydrogen sulfide-mediated cytoprotection. Antioxid
Redox Signal. 12:1203–1217
|
|
7
|
Dong XB, Yang CT, Zheng DD, et al:
Inhibition of ROS-activated ERK1/2 pathway contributes to the
protection of H2S against chemical hypoxia-induced
injury in H9c2 cells. Mol Cell Biochem. 362:149–157. 2012.
View Article : Google Scholar
|
|
8
|
Biermann J, Lagrèze WA, Schallner N,
Schwer CI and Goebel U: Inhalative preconditioning with hydrogen
sulfide attenuated apoptosis after retinal ischemia/reperfusion
injury. Mol Vis. 17:1275–1286. 2011.PubMed/NCBI
|
|
9
|
Taniguchi S, Kang L, Kimura T and Niki I:
Hydrogen sulphide protects mouse pancreatic β-cells from cell death
induced by oxidative stress, but not by endoplasmic reticulum
stress. Br J Pharmacol. 162:1171–1178. 2011. View Article : Google Scholar :
|
|
10
|
Sivarajah A, Collino M, Yasin M, et al:
Anti-apoptotic and anti-inflammatory effects of hydrogen sulfide in
a rat model of regional myocardial I/R. Shock. 31:267–274. 2009.
View Article : Google Scholar
|
|
11
|
Li C, Guo Z and Guo B: Inhibition of
endogenous CSE/H2S system contributes to hypoxia and
serum deprivation-induced apoptosis in mesenchymal stem cells. Mol
Med Rep. 9:2467–2472. 2014.PubMed/NCBI
|
|
12
|
Wang A, Shen F, Liang Y and Wang J:
Marrow-derived MSCs and atorvastatin improve cardiac function in
rat model of AMI. Int J Cardiol. 150:28–32. 2011. View Article : Google Scholar
|
|
13
|
Zhu W, Chen J, Cong X, Hu S and Chen X:
Hypoxia and serum deprivation-induced apoptosis in mesenchymal stem
cells. Stem Cells. 24:416–425. 2006. View Article : Google Scholar
|
|
14
|
Ferri KF and Kroemer G: Organelle-specific
initiation of cell death pathways. Nat Cell Biol. 3:E255–E263.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar
|
|
16
|
Li Z, Wei H, Liu X, Hu S, Cong X and Chen
X: LPA rescues ER stress-associated apoptosis in hypoxia and serum
deprivation-stimulated mesenchymal stem cells. J Cell Biochem.
111:811–820. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wei H, Li Z, Hu S, Chen X and Cong X:
Apoptosis of mesenchymal stem cells induced by hydrogen peroxide
concerns both endoplasmic reticulum stress and mitochondrial death
pathway through regulation of caspases, p38 and JNK. J Cell
Biochem. 111:967–978. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Harding HP and Ron D: Endoplasmic
reticulum stress and the development of diabetes: a review.
Diabetes. 51(Suppl 3): 455–461. 2002. View Article : Google Scholar
|
|
19
|
Franke TF, Kaplan DR and Cantley LC: PI3K:
Downstream AKTion blocks apoptosis. Cell. 88:435–437. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Datta SR, Brunet A and Greenberg ME:
Cellular survival: A play in three Akts. Genes Dev. 13:2905–2927.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liao Y and Hung MC: Regulation of the
activity of p38 mitogen-activated protein kinase by Akt in cancer
and adenoviral protein E1A-mediated sensitization to apoptosis. Mol
Cell Biol. 23:6836–6848. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen J, Baydoun AR, Xu R, et al:
Lysophosphatidic acid protects mesenchymal stem cells against
hypoxia and serum deprivation-induced apoptosis. Stem Cells.
26:135–145. 2008. View Article : Google Scholar
|
|
23
|
Dong Q, Yang Y, Song L, et al:
Atorvastatin prevents mesen-chymal stem cells from hypoxia and
serum-free injury through activating AMP-activated protein kinase.
Int J Cardiol. 153:311–316. 2011. View Article : Google Scholar
|
|
24
|
Nie Y, Han BM, Liu XB, et al:
Identification of MicroRNAs involved in hypoxia- and serum
deprivation-induced apoptosis in mesenchymal stem cells. Int J Biol
Sci. 7:762–768. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gao F, Hu XY, Xie XJ, et al: Heat shock
protein 90 protects rat mesenchymal stem cells against hypoxia and
serum deprivation-induced apoptosis via the PI3K/Akt and ERK1/2
pathways. J Zhejiang Univ Sci B. 11:608–617. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang R: Two’s company, three’s a crowd:
Can H2S be the third endogenous gaseous transmitter. FASEB J.
6:1792–1798. 2002. View Article : Google Scholar
|
|
27
|
Yang C, Yang Z, Zhang M, et al: Hydrogen
sulfide protects against chemical hypoxia-induced cytotoxicity and
inflammation in HaCaT cells through inhibition of ROS/NF-κB/COX-2
pathway. PLoS One. 6:e219712011. View Article : Google Scholar
|
|
28
|
Yao LL, Huang XW, Wang YG, Cao YX, Zhang
CC and Zhu YC: Hydrogen sulfide protects cardiomyocytes from
hypoxia/reoxygenation-induced apoptosis by preventing
GSK-3beta-dependent opening of mPTP. Am J Physiol Heart Circ
Physiol. 298:H1310–H1319. PubMed/NCBI
|
|
29
|
Hu Y, Chen X, Pan TT, et al:
Cardioprotection induced by hydrogen sulfide preconditioning
involves activation of ERK and PI3K/Akt pathways. Pflugers Arch.
455:607–616. 2008. View Article : Google Scholar
|
|
30
|
Elrod JW, Calvert JW, Morrison J, et al:
Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury
by preservation of mitochondrial function. Proc Natl Acad Sci USA.
104:15560–15565. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guan Q, Zhang Y, Yu C, Liu Y, Gao L and
Zhao J: Hydrogen sulfide protects against high-glucose-induced
apoptosis in endo-thelial cells. J Cardiovasc Pharmacol.
59:188–193. 2012. View Article : Google Scholar
|
|
32
|
Xie X, Sun A, Zhu W, et al:
Transplantation of mesenchymal stem cells preconditioned with
hydrogen sulfide enhances repair of myocardial infarction in rats.
Tohoku J Exp Med. 226:29–36. 2012. View Article : Google Scholar
|
|
33
|
Kumar S: Caspase function in programmed
cell death. Cell Death Differ. 14:32–43. 2007. View Article : Google Scholar
|
|
34
|
Berridge MJ: The endoplasmic reticulum: a
multifunctional signaling organelle. Cell Calcium. 32:235–249.
2002. View Article : Google Scholar
|
|
35
|
Wang XY, Yang CT, Zheng DD, et al:
Hydrogen sulfide protects H9c2 cells against doxorubicin-induced
cardiotoxicity through inhibition of endoplasmic reticulum stress.
Mol Cell Biochem. 363:419–426. 2012. View Article : Google Scholar
|
|
36
|
Mangi AA, Noiseux N, Kong D, et al:
Mesenchymal stem cells modified with Akt prevent remodeling and
restore performance of infarcted hearts. Nat Med. 9:1195–1201.
2003. View Article : Google Scholar : PubMed/NCBI
|