Introduction
Lung cancer is the main cause of cancer-associated
mortalities worldwide. Clinically, lung cancer is divided into
small cell lung cancer (SCLC) and non-SCLC (NSCLC), and NSCLC
accounts for ~85% of all lung cancers (1). To date, despite multimodality
treatments consisting of extended surgical resection, radiotherapy
and chemotherapy, NSCLC only has a five-year survival rate of
<20% (2). Hence, it is
important and urgent to develop novel effective agents and
approaches to treat NSCLC.
Flavonoids are a group of natural non-essential
compounds that are abundantly present in fruits, vegetables, tea,
seeds, nuts and red wine. Numerous flavonoid agents have been used
to treat human cancers (3).
Quercetin is a plant-derived bioflavonoid that exhibits several
biological functions in vitro and in vivo, including
anti-inflammatory, anti-oxidative and anti-cancer effects (4). A recent study has shown that an
immediate 3′-O-methylated metabolite of quercetin, named
isorhamnetin, exerts a greater anti-tumor effect as compared with
that of quercetin in human colon cancer cells (5). The anti-tumor effects of ISO been
investigated in a number of cancer types, including colorectal,
skin and gastric cancers (6–8).
However, to the best of our knowledge, the anti-cancer effect of
ISO has not been investigated in lung cancer. Thus, the present
study investigated the anti-proliferative and pro-apoptotic effects
of ISO on the growth of human NSCLC cells.
Apoptosis, or programmed cell death, is a multi-step
process that is important in controlling the cell number and
proliferation as part of normal development; however, in cancer
cells, cell cycle checkpoints and the subsequent progression of
apoptosis are frequently inactivated. Consequently, the induction
of apoptosis has been emphasized in anti-cancer strategies
(9,10). Regarding the initiation and
execution of cell death, two apoptotic pathways have been
identified: The extrinsic (Fas death receptor-mediated) and
intrinsic (mitochondrial) pathways. Either of the two pathways
involves the activation of caspases (11,12).
Autophagy is the primary mechanism to clear the cell from toxic
proteins and damaged organelles (13,14).
However, autophagy has been found to be associated with
tumorigenesis and tumor progression. Autophagy can act as a
potential tumor suppression factor by removing damaged
organelles/proteins, suppressing cell growth and avoiding genomic
instability in normal cells (15,16).
However, when tumorigenesis occurs, cancer cells can utilize
autophagy to confer stress tolerance, including an acidic
environment, hypoxia and nutrition deficiency, which serves to
promote tumor cell survival (17).
In fact, a number of existing chemotherapeutic drugs designed to
kill cancer cells by inducing apoptosis are most likely to also
induce autophagy (18).
Drug-activated autophagy in cancer cells, in turn, causes
resistance of the cells to death and decreases the curative effects
(19). In fact, inhibition of
autophagy has been shown to enhance the anti-tumor effect of
diverse chemotherapeutic drugs (20–22).
In the present study, the effects of ISO on
autophagy in A549 cells was investigated. For this, the effect of
the inhibition of autophagy on ISO-mediated cytotoxicity and
apoptosis was investigated. Furthermore, the effects of ISO on lung
cancer cells were demonstrated in vivo. To the best of our
knowledge, the present study was the first to report the effects of
ISO on human lung cancer cells.
Materials and methods
Cell lines and reagents
The NSCLC cell line A549 was obtained from the
Chinese Type Culture Collection Center (Wuhan, China), were
cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen
Life Technologies, Carlsbad, CA, USA) with 10% fetal calf serum
(FCS; Invitrogen Life Technologies). Enhanced green fluorescent
protein (EGFP)-light chain 3 (LC3) plasmid was kindly provided by
Professor Marja Jäättelä (Cell Death and Metabolism Research Unit,
Danish Cancer Society Research Center, Copenhagen, Denmark).
Transfection reagent Lipofectamine 2000 was purchased from
Invitrogen Life Technologies. The fluo-rescein isothiocyanate
(FITC) Annexin V and propidium iodide (PI) kit for apoptosis
detection was purchased from Invitrogen Life Technologies. The JC-1
kit for the mitochondrial membrane assay was from Beyotime
Institute of Biotechnology (Haimen, China). ISO, chloroquine (CQ),
3-methyladenine (3-MA) and monodansylcadaverine (MDC) were
purchased from Sigma-Aldrich (St. Louis, MO, USA). The following
antibodies were used in the present study: Rabbit
anti-human/mouse/rat caspase 3 polyclonal antibody (cat no.
GTX110543); rabbit anti-human caspase 3 (cleaved Asp175) polyclonal
antibody (cat no. GTX86909); rabbit anti-human/mouse/rat
pro-caspase 9 polyclonal antibody (cat no. GTX61008); rabbit
anti-human/mouse PARP poly-clonal antibody (cat no. GTX100573);
rabbit anti-human beta actin polyclonal antibody (cat no.
GTX109639); rabbit anti-human/rat/mouse cytochrome C polyclonal
antibody (cat no. GTX108585) (all from Genetex, Irvine, CA, USA).
LC3 antibodies (cat. no. 2775) were from Cell Signaling Technology
(Beverly, MA, USA). Beclin1 and PCNA (cat. no. sc-53407) antibodies
were from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
Secondary antibodies (anti-rabbit and anti-mouse HRP-conjugated)
were purchased from Keygen Biotech (Nanjing, China).
MTT assay
An MTT assay was used to determine the cell
viability. Briefly, 1×104 A549 cells/well in a 96-well
plate were treated with various concentrations of ISO (0-16
μM) in the presence or absence of autophagy inhibitors (10
mM 3-MA or 50 μM CQ). At the appropriate time-points, 20
μl MTT [Sangon Biotech, Shanghai, China; 5 mg/ml in
phosphate-buffered saline (PBS)] was added for 4 h at 37°C and then
all liquid was carefully removed. Optical density (OD) values were
measured with a spectrophotometer (DU-7400; Beckmann-Coulter, Brea,
CA, USA) at 490 nm following continuous agitation for 15 min with
100 μl dimethyl sulfoxide (DMSO; Sigma-Aldrich). The
inhibitory rate was calculated according to the following formula:
Inhibitory rate (%) = [1-(OD of the experimental samples/OD of the
control)] ×100%.
Colony formation assay
A549 cells were seeded in triplicate into a six-well
plate (1×104 cells/well) and cultured in DMEM with 2.5%
bovine serum albumin (Sigma-Aldrich) in the presence or absence of
ISO (2-8 μM). After 10 days, the resulting colonies were
stained with 0.05% of crystal violet (Sangon Biotech) and images
were captured.
Annexin V-fluorescein isothiocyanate/PI
apoptosis assay
A549 cells were treated with various concentrations
of ISO (0-16 μM) in the absence or presence of 10 mM 3-MA or
50 μM CQ for 24 h, and then single-cell suspensions were
prepared using EDTA-free trypsin (Sangon Biotech) digestion. Cells
were stained according to the instruction manual of the
FITC-Annexin V and PI kit and analyzed using flow cytometry (FACS
Influx SE, BD Biosciences, Franklin Lakes, NJ, USA).
DNA fragmentation assay
A549 cells were cultured in the presence or absence
of 16 μM ISO for various durations (0, 6, 12, 24, 48 or 72
h). Cells were then collected and washed with PBS. The pellet was
homogenized in 450 μl lysis buffer (20 Mm Tris-HCl, pH 8.0,
10 mM EDTA, pH 8.0, 0.2% Triton X-100; Sangon Biotech) by pipetting
through a blue pipette nozzle and incubated for 10 min on ice. The
lysates were centrifuged for 15 min at 13,000 × g and the
supernatants were incubated at 50°C overnight with proteinase K (2
mg/ml; Sangon Biotech). DNA was precipitated with two volumes of
100% ethanol and 0.1 volumes of 3 M sodium acetate for 2 h at 70°C.
DNA was pelleted at 12,000 × g for 15 min and washed twice with 70%
ethanol. DNA was dissolved in distilled water containing 1 mg/ml
RNase A (Sangon Biotech), incubated for 30 min at 37°C, and
analyzed by electrophoresis on 1.5% agarose gels (Sangon Biotech)
and staining with ethidium bromide (Sangon Biotech).
Cell cycle analysis
In order to assess whether the apoptotic effects of
ISO were dependent on caspases, A549 cells were incubated with
caspase-9 inhibitor Z-LEHD-FMK (BioVision, San Francisco, CA, USA)
or caspase-3/7 inhibitor Z-DEVD-FMK (R&D Systems, Minneapolis,
MN, USA) for 2 h followed by treatment of the cells with ISO for 48
h and subsequent flow cytometric cell cycle analysis. Following the
above treatments, cells were harvested and washed in PBS. Cells
were fixed in 1 ml cold 70% ethanol for 30 min on ice. Cells were
pelleted at by centrifugation at high speed and the supernatant was
discarded. Following two washes with PBS, the cells were incubated
with 50 μl RNAse A solution to remove any RNA. Subsequently,
400 μl PI solution per 1×106 cells was added and
mixed. Cells were incubated for 10 min at room temperature and
subjected to flow cytometric analysis (FACS Influx SE), for which
at least 10,000 events were observed.
Measurement of mitochondrial membrane
potential
The mitochondrial membrane potential was assessed
using the JC-1 assay. A549 cells were treated with 16 μM ISO
for 24 h. Subsequently, 1X JC-1 suspension was added to treat the
cells for 15 min at 37°C and images of the cells were captured
using confocal microscopy (FV100; Olympus, Tokyo, Japan). In
healthy cells with high mitochondrial membrane potential, JC-1 is
present as J-aggregate complexes with red fluorescence, while in
apoptotic or unhealthy cells with reduced mitochondrial membrane
potential, JC-1 remains in the monomeric form, which is
characterized by green fluorescence. Furthermore, the mitochondrial
membrane potential was assessed using the tetramethylrhodamine
ethyl ester (TMRE) assay. For this, A549 cells were treated with
ISO (8 μM) for 12, 24 or 48 h. The cells were then washed
with cold PBS and stained by adding 200 nM TMRE (Sangon Biotech), a
fluorescent potential-dependent indicator, followed by incubation
for 30 min at 37°C. JC-1 staining was observed by fluorescence
microscopy (Ti-E; Nikon Corporation, Tokyo, Japan) and the
mitochondrial membrane potential was detected by flow cytometry
(FACS Influx SE) at 582 nm.
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted with TRIzol (Invitrogen Life
Technologies) following the manufacturer's instructions.
First-strand cDNA was prepared by reverse transcription with
Superscript II reverse transcriptase (Invitrogen Life Technologies)
and oligo(dT) primers and stored at 20°C. Real-Time polymerase
chain reaction (Real-Time PCR) was performed using SYBR®
Premix Ex Taq™ II (Takara Bio Inc., Shiga, Japan) on an ABI 7300
QPCR system (Applied Biosystems, Thermo Fisher Scientific, Waltham,
MA, USA). The PCR cycling conditions were as follows: 95°C for 5
min, followed by 95°C for 5 sec and 60°C for 34 sec for 39 cycles.
As an internal control, levels of GAPDH were quantified in parallel
with target genes. Normalization and fold changes were calculated
using the ∆∆Ct method (23).
Primers (Beijing Genomics Institute, Beijing, China) for detection
of gene expression were as follows: GAPDH forward,
5′-TGGGGTGAGGCCGGTGCTGA-3′ and reverse, 5′-GGCATCGGCAGAAGGGGCGG-3′;
caspase-3 forward, 5′-CAGAGCTGGACTGCGGTATTGA-3′ and reverse,
5′-AGCATGGCGCAAAGTGACTG-3′; caspase-8 forward,
5′-CTGGGAAGGATCGACGATTA-3′ and reverse, 5′-CATGTCCTGCATTTTGATGG-3′;
caspase-9 forward, 5′-AGCCAGATGCTG TCCCATAC-3′ and reverse,
5′-CAGGAGACAAAACCTG GGAA-3′; B-cell lymphoma 2 (Bcl-2)-associated X
protein (Bax) forward, 5′-AGACAGGGGCCTTTTTGCTAC-3′, and reverse,
5′-AATTCGCCGGAGACACTCG-3′; Bcl-2 forward, 5′-CATGCTGGGAGCGTCACAT-3′
and reverse, 5′-CTCCACT GAACTCGTACAAACTT-3′; Bcl-2-like protein 2
(Bclw) forward, 5′-GGTGACCTACCTGGAGACACG-3′ and reverse,
5′-GTCCTCACTGATGCCCAGTTC-3′; myeloid cell leukemia 1 (Mcl-1)
forward, 5′-GAAACAGCATGAGGTGTGGTA′ and reverse,
5′-AGCCGAAGTTAAAACCTGTCC-3′; BH3 interacting-domain death agonist
(Bid) forward, 5′-GTGATG TCAGATATGGGCAGAG-3′ and reverse,
5′-ATCCCACGG ATGGATAGGTCG-3′; Bcl-2/adenovirus E1B 19 kDa
protein-interacting protein 3 (Bnip3) forward,
5′-GGTCCAGTAGACCCGAAAACA-3′ and reverse, 5′-TGT
GCTCAGTCGCTTTCCAAT-3′; Bcl-2 homologous antagonist/killer (BAK)
forward, 5′-CATCAACCGACGCTATGACTC-3′ and reverse,
5′-GTCAGGCCATGCTGGTA GAC-3′; P53 forward, 5′-ACAAGGTTGATGTGACCT
GGA-3′ and reverse, 5′-TGTAGACTCGTGAATTTCGCC-3′; P21 forward,
5′-TGCTGAATCTACGCAACCGAT-3′ and reverse,
5′-TCCAGTGGCGAATCATCTACAT-3′; and p53-upregulated modulator of
apoptosis (Puma) forward, 5′-GACCTCAACGCACAGTACGAG′ and reverse,
5′-AGGAGTCCCATGATGAGATTGT-3′.
Western blot analysis
Cells were washed twice with PBS and lysed with
lysis buffer. The concentration of the protein was determined by
ultraviolet spectrophotometry (Ruili Analysis Instruments, Beijing,
China). Equal amounts (10 μg) of protein were separated by
10% SDS-PAGE and transferred onto a polyvinylidene difluoride
membrane (Sangon Biotech). The membrane was blocked with 5% non-fat
milk in Tris-buffered saline containing Tween 20 (TBST; Sangon
Biotech) for 2 h at room temperature and incubated overnight at 4°C
with specific primary antibodies. After washing three times with
TBST, the membrane was incubated with appropriate horseradish
peroxidase-linked secondary antibody for 4 h at room temperature
and then detected with an enhanced chemiluminescence detection kit
(Sangon Biotech) in the dark.
Autophagy assay using pEGFP-LC3
A549 cells seeded in six-well plates containing
glass coverslips and transfected with pEGFP-LC3 plasmid for 24 h.
Cells were either left untreated or pre-treated with autophagy
inhibitors (10 mM 3-MA or 50 μM CQ) for 1 h. An appropriate
concentration of ISO was added for an additional 24h and the cells
were then fixed in 4% paraformaldehyde. EGFP-LC3-II punctate dots
were detected and counted using confocal microscopy as described
previously (24).
Observation of autophagic vacuoles by MDC
staining
MDC has recently been reported as another specific
marker for autophagic vacuoles (25). In the present study, MDC was
therefore used to detect autophagy. A549 cells were treated with
ISO as described above for 24 h and 0.05 mM MDC was then added to
medium for an additional 2 h at 37°C. Accumulation of MDC in
autophagy-associated vacuoles was determined using confocal
microscopy.
Cytochrome C immunoblotting
The cytosolic fraction was prepared as described
previously (26). A549 cells
treated with ISO were collected and washed with cold PBS. Cell
pellets were lysed in 40 ml lysis buffer [20 mM
4-(2-hydroxyethyl)-1-piper-azineethanesulfonic acid/NaOH, pH 7.5,
250 mM sucrose, 10 mM KCl, 2 mM MgCl2, 1 mM EDTA, 1 mM
dithiothreitol, protease inhibitor cocktail] for 20 min on ice.
They were homogenized by passing the lysate through a 22-gauge
needle 10 times. The homogenate was centrifuged at 25,000 × g for
30 min at 4°C and protein contents in the supernatant were measured
using a Bio-Rad DC Protein Assay kit II (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). The lysates containing 25 mg of protein
were analyzed by western blotting for cytochrome C (1:2,000
dilution).
A549 tumor model
The study was approved by the ethics committee of
the People's Hospital of Wuhan University (Wuhan, China). BALB/c
nu/nu mice (five weeks old) were purchased from Guangdong Medical
Laboratory Animal Center (Guangzhou, China). Mice were housed in a
specific-pathogen-free environment maintained at 25±1°C with 55%
relative humidity and given food and water ad libitum. A549
cells in sub-confluent condition were harvested and re-suspended in
sterile PBS. A549 cells (6×105 in 200 μl PBS)
were sub-cutaneously injected into the right flank of the BALB/c
nu/nu mice. Four days after tumor inoculation, mice were given a
daily intraperitoneal injection of ISO (0.5 mg/kg), and a
percentage of the animals was co-injected with 3-MA (22.4 mg/kg) or
CQ (10 mg/kg) (27); each
experimental group contained six mice. Tumor volumes were measured
every three days with a caliper and calculated according to the
formula V=1/2(L × W2), where L and W stand for length
and width, respectively. All mice were sacrificed 13 days after
tumor inoculation by CO2 inhalation following anesthesia
by isofluorane inhalation and the tumors were excised and
weighed.
Immunohistochemistry
Tumor specimens were immediately removed from
sacrificed mice and prepared for histological examination. Tumors
were fixed in 4% neutral buffered formalin overnight, embedded in
paraffin and sectioned to 6 μm thickness. The tumor sections
were immobilized and de-paraffinized by immersing in xylene,
dehydrated in a graded series of ethanol and washed with distilled
water. For antigen retrieval, the tumor sections were boiled in 10
mM sodium citrate buffer (pH 6.0) for 10 min and cooled to room
temperature. After washing with Tris-buffered saline (TBS),
endogenous peroxidase activity was blocked by incubation in 3%
H2O2-methanol for 10 min at room temperature.
The sections were stained with antibodies against proliferating
cell nuclear antigen (PCNA; Santa Cruz Biotechnology, Inc.) and
caspase-3 overnight at 4°C using Avidin-Biotin Complex and
Diaminobenzidine kits (Vector Laboratories, Inc., Burlingame, CA,
USA) and also counterstained with Mayer's hematoxylin solution
(Sigma-Aldrich). Terminal deoxynucleotidyl transferase-mediated
dUTP nick end labeling (TUNEL) staining was performed using a
TACS® TdT kits (R&D Systems, Minneapolis, MN, USA)
and counterstained with methyl green (Sangon Biotech). Images of
all stained sections were captured using an Axiovert S 100 light
microscope (Carl Zeiss, Inc., Oberkochen, Germany) at 400x
magnification. At least four tumors were analyzed per group, and at
least four fields of view each of at least six slices of each tumor
were analyzed. Quantitative evaluation of the images was performed
using PixeLink Capture OEM (Ottawa, ON, Canada) software and
indexes were calculated as follows: Proliferative index (%) =
(Number of PCNA positive cells/total cells) ×100, Apoptotic index
(%) = (Number of TUNEL-positive cells/total cells) ×100, and
cleaved caspase-3 index: (Number of caspase-3 positive cells/total
cells) ×100.
Statistical analysis
Values are expressed as the mean ± standard
deviation of three independent experiments. Comparisons were
performed using a two-tailed paired Student's t-test. GraphPad
Prism version 5.01 (GraphPad Software, Inc., La Jolla, CA, USA) was
used to conduct statistical analysis. P<0.05 was considered to
indicate statistically significant differences, which was denoted
in figures as *P<0.05, **P<0.01 and
***P<0.001.
Results
ISO inhibits proliferation and induces
apoptosis of NSCLC cells
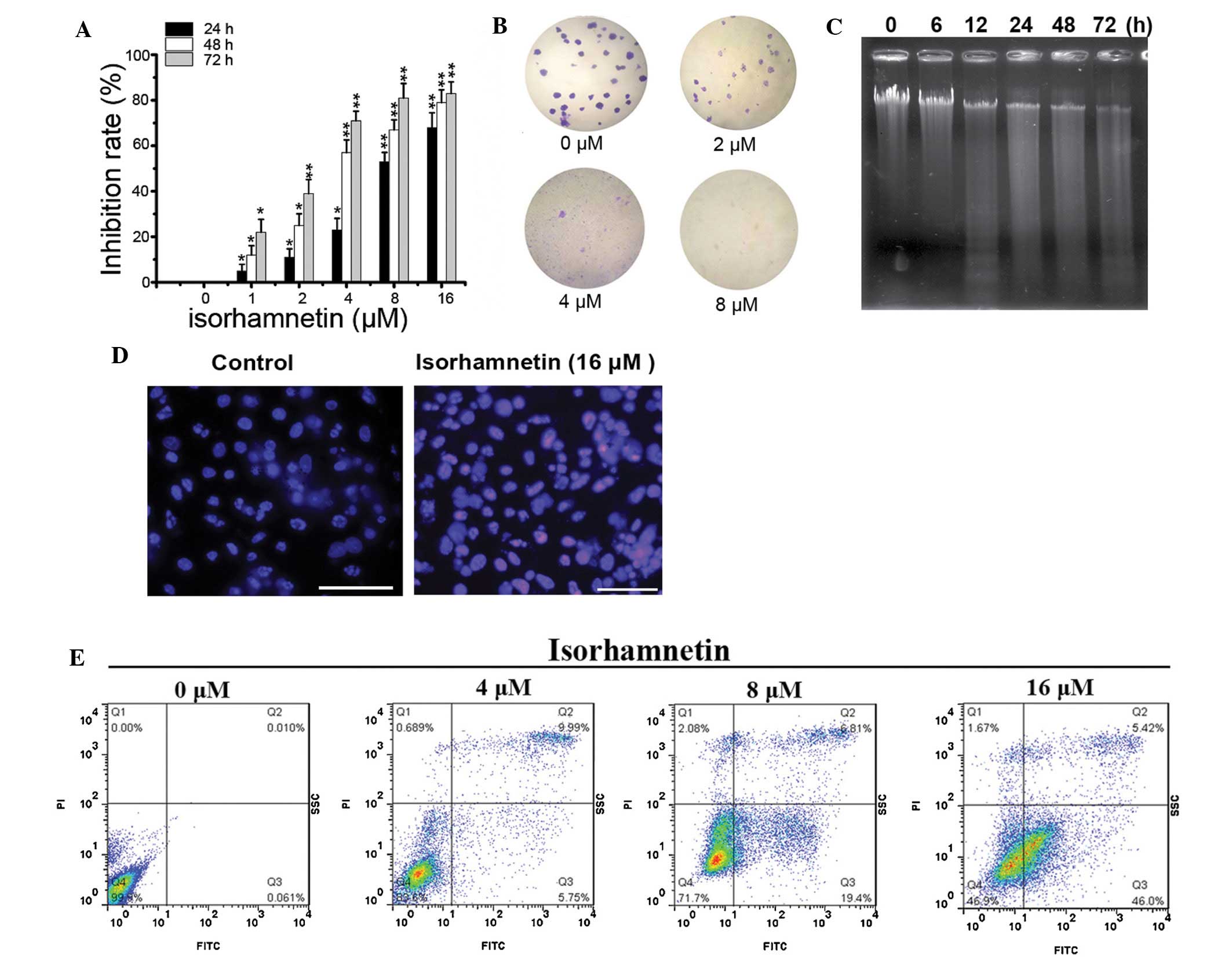
To investigate the proliferation inhibition effect
of ISO, an MTT assay was performed. As shown in Fig. 1A, ISO treatment significantly
inhibited the proliferation of A549 cells in a dose- and
time-dependent manner. In addition, ISO also significantly
suppressed the colony formation ability of A549 cells in a
dose-dependent manner (Fig.
1B).
In addition, to determine whether the growth
inhibition is accompanied by an induction of apoptosis in A549
cells, apoptotic DNA fragmentation was assessed by gel
electrophoresis. As shown in Fig.
1C, a ladder pattern of discontinuous DNA fragments was
detected at 24 h after exposure to ISO, and the extent of DNA
fragmentation was significantly elevated at 72 h. Similarly, a
TUNEL assay also showed a significant increase in the number of
apoptotic bodies following treatment with ISO (Fig. 1D). Furthermore, apoptosis was
assessed using Annexin V/PI staining and flow cytometric analysis.
As expected, ISO treatment resulted in a significant increase in
the number of apoptotic cells with Annexin V-positive staining in a
dose-dependent manner (Fig. 1E).
All of these results demonstrated that ISO treatment significantly
suppressed cell proliferation and colony formation, and induced
apoptotic cell death in NSCLC cells in a time- and dose-dependent
manner.
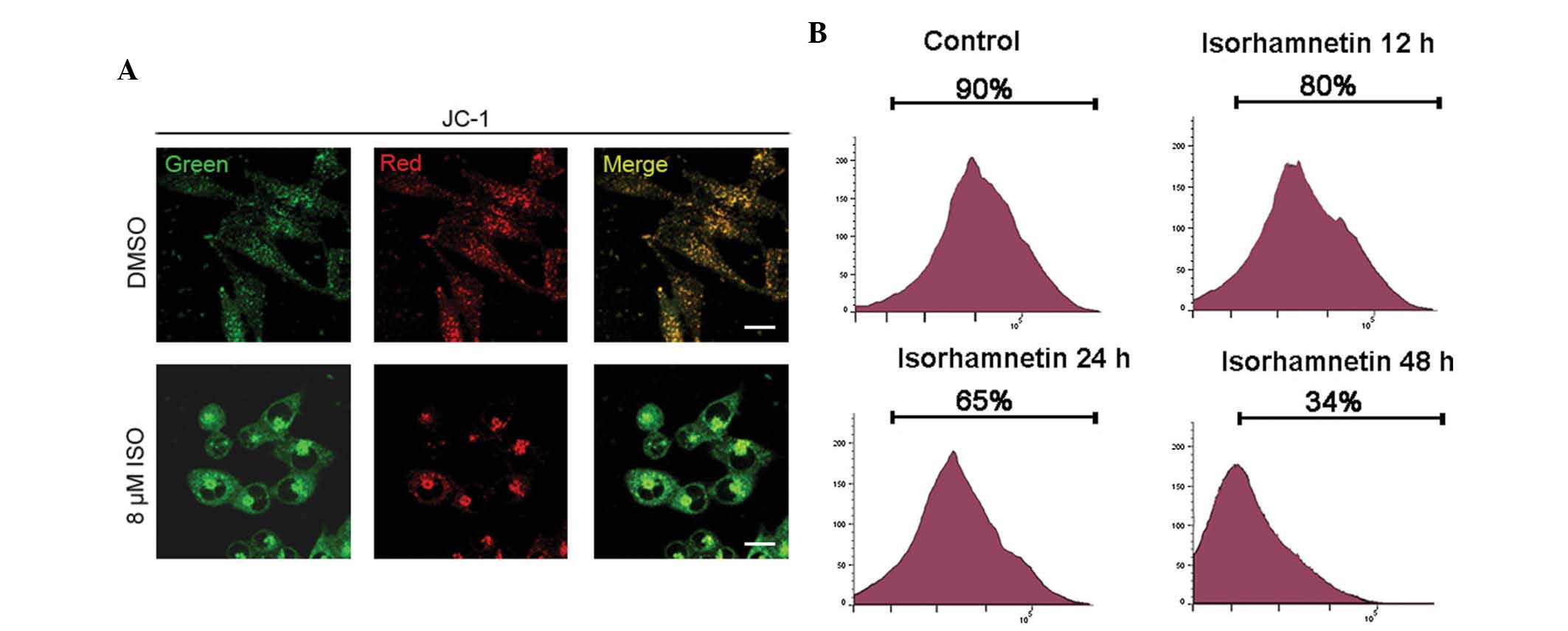
ISO induces mitochondria-dependent
caspase activation
With regard to apoptosis signaling, mitochondria are
a central sensor and integration point for diverse apoptotic
signals; furthermore, they function as the storehouse of cytochrome
C and Smac/Diablo, which binds and disables inhibitors of
apoptosis-associated proteins (IAPs) (28,29).
The 'apoptosome' cascade or intrinsic pathway involves activation
of pro-caspase-9 by cytochrome C released from the mitochondria,
leading to the activation of the executioner pro-caspases
(caspase-3, -6 and -7) that cleave poly (adenosine diphosphate
ribose) polymerase (PARP) and other apoptotic protein substrates
(30). To investigate whether
ISO-induced apoptosis was mitochondrial-dependent, mitochondrial
membrane potential and caspase assays were performed. The
permeabilization of mitochondria is one of the most important
events during apoptosis (31,32).
Mitochondrial de-polarization in apoptotic cells can be detected by
a decrease in the red/green fluorescence intensity ratio of the dye
JC-1 as a result of its disaggregation into monomers. As shown in
Fig. 2A, a significantly higher
red/green fluorescence rate was observed in cells treated with DMSO
only compared with that in ISO-treated cells, suggesting that ISO
treatment resulted in the de-polarization and permeabilization of
mitochondria of A549 cells. To further verify the depolarization of
the mitochondrial membrane potential after ISO treatment (16
μM), A549 cells were stained with TMRE followed by flow
cytometric analysis at the indicated times. As shown in Fig. 2B, ISO treatment resulted in a
left-shift of the TMRE fluorescence at as early as 12 h of
incubation with ISO. The ratio of cells with intact mitochondrial
membrane potential decreased from 90.2% in the control cells to
80.6, 65.5, and 34.2% at 12, 24, and 48 h of ISO treatment,
respectively (Fig. 2B).
Furthermore, the ISO-induced alterations in the mRNA
expression of apoptosis marker genes in A549 cells were examined.
RT-qPCR analysis showed a significant (P<0.01) upregulation in
the expression of caspase-3 (9.6±0.53-fold), caspase-9
(9.4±0.65-fold), Bax (1.6±0.19-fold), p53 (5.89±0.21-fold), p21
(2.7±0.33-fold) and Puma (2.22±0.23-fold) at 12 h of treatment with
8 μM ISO (Fig. 3A). In
addition, the protein expression of cleaved-caspase-3, cleaved-PARP
and pro-caspase-9 were detected by western blotting. As shown in
Fig. 3B, the expression levels of
cleaved-caspase-3 and cleaved-PARP were significantly increased
following ISO treatment in a dose-dependent manner, whereas the
levels of pro-caspase-9 were obviously decreased, indicating that
ISO treatment resulted in the activation of the caspase-dependent
apoptotic pathway. As it is known that caspase activation involves
changes in mitochondrial permeability and the release of cytochrome
C, the levels of cytochrome C in the cytosolic
fraction were then examined. As shown in Fig. 3C, a signifi-cant increase of
released cytochrome C was detected at 12 h after treatment
with 16 μM ISO. To further determine whether the ISO-induced
apoptosis of NSCLC cells was caspase-mediated, A549 cells were
incubated with caspase-9 inhibitor Z-LEHD-FMK or caspase-3/7
inhibitor Z-DEVD-FMK for 2 h followed by treatment of the cells
with ISO for 48 h (Fig. 3D and E).
These caspase inhibitors completely blocked the ISO-induced sub-G1
fractions in the cell cycle distribution. These results therefore
suggested that ISO-induced apoptosis was mediated by
mitochondria-dependent caspase activation.
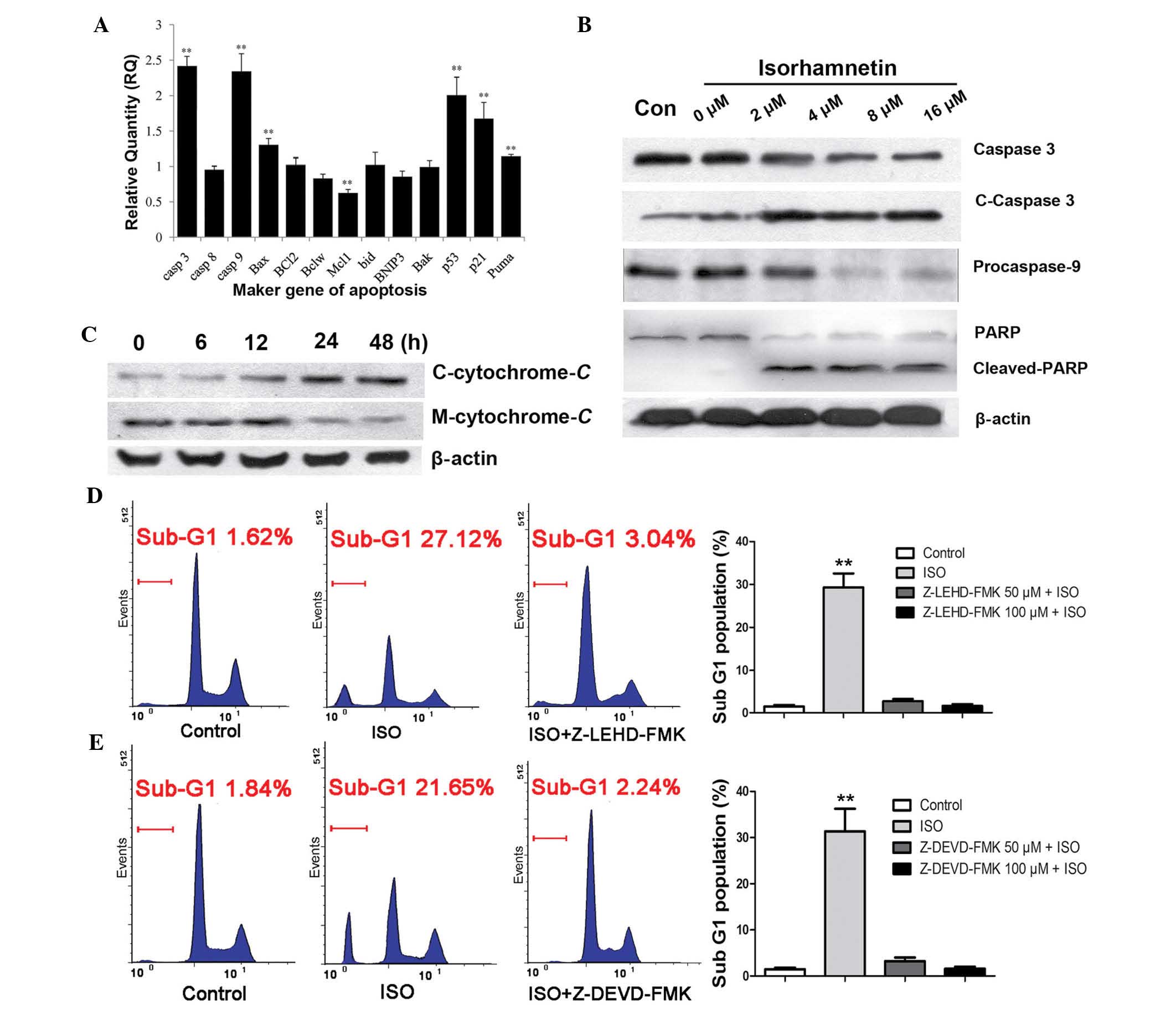 | Figure 3ISO induces mitochondria-dependent
caspase activation (A) Treatment with 8 μM ISO for 12 h
induced alterations in the mRNA expression of marker genes
associated with apoptosis in A549 cells. (B) Cleaved-caspase-3,
cleaved-PARP and pro-caspase-9 were detected after treatment with
the indicated concentrations of ISO for 24 h. (C) A significant
increase of cytochrome C release was detected at 12 h after
16-μM ISO treatment. (D and E) Caspase-9 inhibitor
Z-LEHD-FMK or caspase-3/7 inhibitor Z-DEVD-FMK significantly
blocked the ISO-induced sub-G1 peaks. Cells were treated
with 8 μM ISO for 48 h. Values are expressed the mean ±
standard deviation (n=4). **P<0.01. C-cytochrome
C, cytosolic cytochrome C; M-cytochrome C,
mitochondrial cytochrome C; ISO, isorhamnetin; casp,
caspase; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X protein;
Bclw, Bcl-2-like protein 2; Mcl1, myeloid cell leukemia 1; bid, BH3
interacting-domain death agonist; BNIP3, Bcl-2/adenovirus E1B 19
kDa protein-interacting protein 3; Bak, Bcl-2 homologous
antagonist/killer; Puma, p53-upregulated modulator of apoptosis;
Con, control; PARP, poly(adenosine diphosphate ribose)
polymerase. |
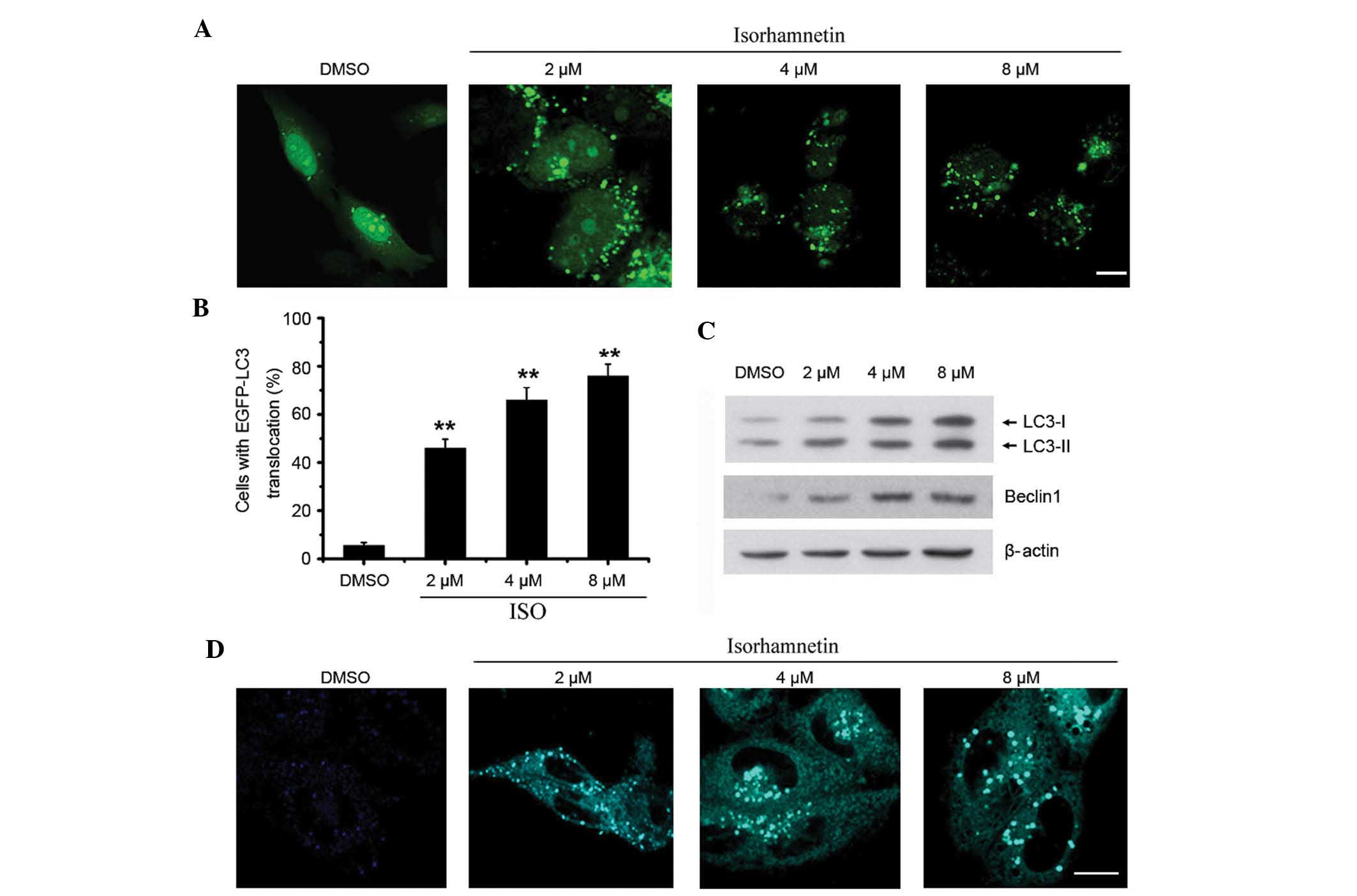
ISO induces autophagy in NSCLC cells
Numerous chemotherapeutic agents designed to kill
cancer cells are also known to induce autophagy (18). When autophagy is initiated,
microtubule-associated protein 1 light chain 3 (LC3) is cut on the
C-terminal to produce LC3-II protein. The resulting LC3-II is then
preferentially translocated to the membranes of autophagosomes and
shows a punctate staining pattern in the cytosol (33). In order to detect autophagy,
EGFP-LC3 was overexpressed in A549 cells. As shown in Fig. 4A and B, the number of
autophagosomes with a punctate staining pattern was significantly
increased in ISO-treated cells compared with that in untreated
cells. Accordingly, the protein levels of LC3-II were also
significantly increased in A549 cells treated with ISO in a
dose-dependent manner (Fig. 4C).
Beclin1 has been shown to be the activator of the Class III
phosphoinositide 3-kinase (PI3K) complex that has an important role
in the regulation of autophagy induction (34). In the present study, it was found
that the expression of Beclin1 was upregulated following ISO
treatment (Fig. 4C).
To further confirm that ISO indeed induced
autophagosome formation, an MDC staining assay was performed.
Similar to the results of the EGFP-LC3-II translocation assay, ISO
treatment also induced apparent accumulation of MDC in the
cytoplasmic vacuoles compared to that in the control cells
(Fig. 4D).
Inhibition of autophagy enhances the
growth inhibition and pro-apoptotic effect of ISO
3-MA is a PI3K inhibitor, which suppresses autophagy
formation by blocking the activity of the the Class III PI3K
complex at an early stage of autophagy (35,36).
CQ inhibits autophagy by interfering with the fusion of
autophagosomes and lysosomes at a late stage of autophagy (37,38).
As shown in Fig. 5A, pre-treatment
with 3-MA significantly inhibited the ISO-induced autophagosome
formation in the A549 cells (Fig. 5A
and B). By contrast, an increased amount of ISO-induced
autophagosomes was accumulated in the A549 cells pre-treated with
CQ compared with that in cells treated with ISO only (Fig. 5A and B). Next, the protein levels
of endogenous LC3-II were detected. As expected, 3-MA pre-treatment
significantly inhibited the formation of endogenous LC3-II protein
in ISO-treated cells, compared to that in cells treated with ISO
only. However, there was no obvious difference in endogenous LC3-II
levels between cells treated with ISO only and those co-treated
with CQ (Fig. 5C).
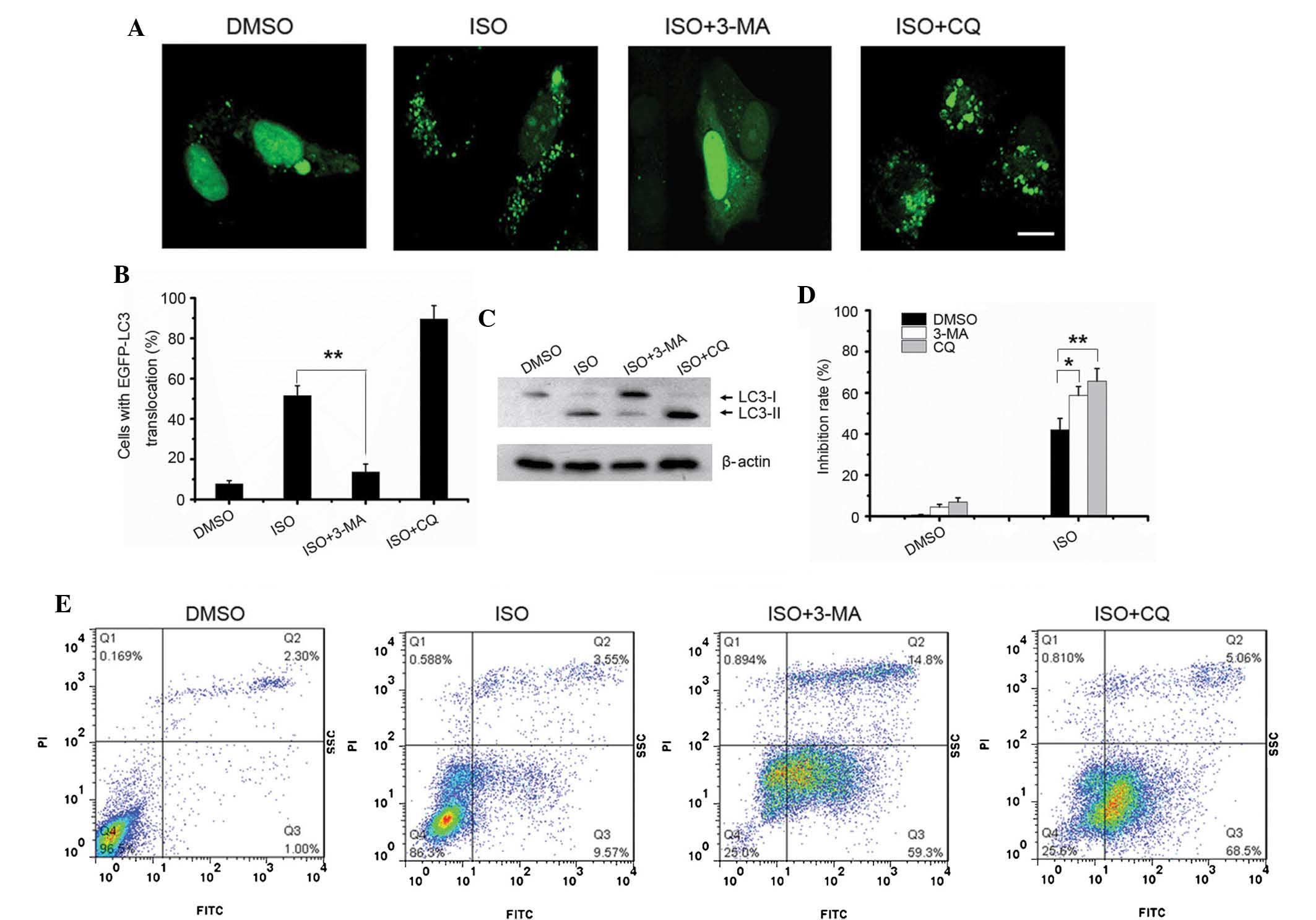 | Figure 5Inhibition of autophagy sensitizes
A549 cells to ISO-induced growth inhibition and apoptosis. (A)
pEGFP-LC3-transfected A549 cells were treated with 4 μM
isorhamnetin alone or in combination with 10 mM 3-MA or 50
μM CQ for 24 h, and autophagy was observed using confocal
microscopy (scale bar, 10 μm). (B) The percentages of cells
with LC3 translocation as indicated by the formation of dots were
counted (n=250 cells/sample). Values are expressed as the mean ±
standard deviation of three independent experiments
(**P<0.01). (C) Endogenous LC3-II levels in cells
with the same treatment as in A were detected by western blotting
using the LC3B antibody. (D) A549 cells were treated as in C, and
an MTT cell viability assay was conducted. Values are expressed as
the mean ± standard deviation of three independent experiments
(*P<0.05; **P<0.01). (E) A549 cells
were treated as in C and apoptosis was detected using Annexin V/PI
staining followed by flow cytometric analysis. ISO, isorhamnetin;
DMSO, dimethyl sulfoxide; LC3, microtubule-associated protein 1
light chain 3; PI, propidium iodide; MA, methyladenine; EGFP,
enhanced fluorescence protein; CQ, hydroxychloroquine; FITC,
fluorescein isothiocyanate. |
Next, the effect of autophagy inhibition on
ISO-induced growth inhibition and apoptosis was investigated. An
MTT assay showed that pre-treatment with 3-MA or CQ markedly
enhanced the growth inhibition induced by ISO treatment (Fig. 5D). Consistent with the MTT assay,
the number of ISO-induced apoptotic cells was also markedly
increased in the cells pre-treated with 3-MA and CQ compared with
that in cells treated with ISO only (Fig. 5E). Collectively, these results
suggested that inhibition of autophagy sensitized the A549 cells to
ISO-induced growth inhibition and apoptosis.
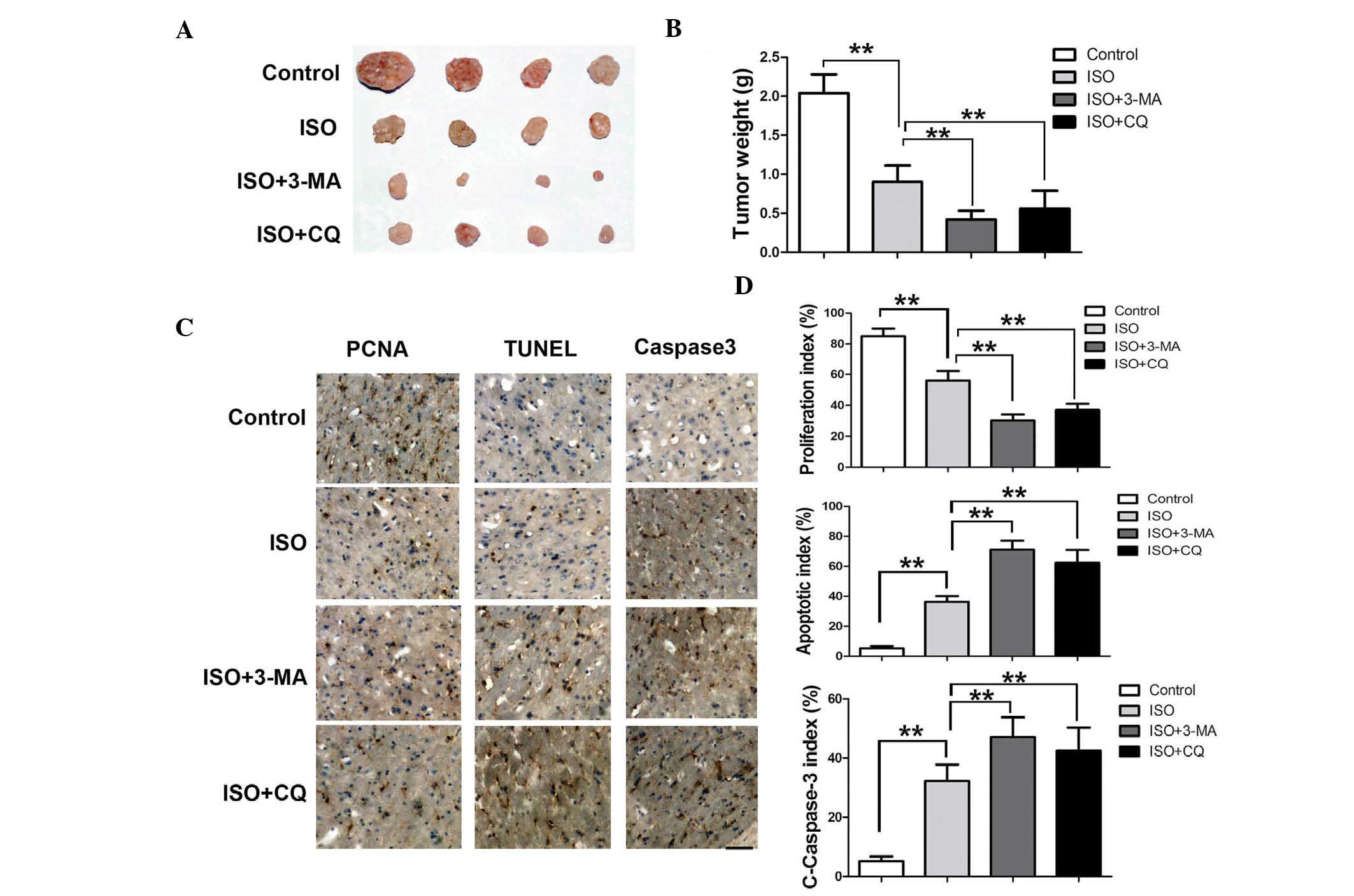
Inhibitors of autophagy significantly
enhance the inhibitory effect of ISO on mouse xenograft tumors
Confirmed by the marked anti-proliferative and
apoptosis-inducing activities observed in cell culture experiments,
the present study examined the anti-tumor activity of ISO in
vivo. BALB/c nu/nu mice bearing A549 NSCLC xenografts were
given a daily intraperitoneal injection of ISO. In a preliminary
study, ISO showed an in vivo anti-tumor activity at 0.5
mg/kg/day, and this dose was therefore used in the present study.
The growth of xenografts was monitored every three days over two
weeks. Side effects, including body weight loss, mortality and
lethargy were not observed in mice treated by ISO for two weeks.
The final tumor size was markedly lower in the majority of the 0.5
mg/kg ISO-treated mice compared with that in the control group. Of
note, the tumor size was significantly lower in the group
co-injected with 3-MA (22.4 mg/kg) or CQ (10 mg/kg) (Fig. 6A), compared with that in the mice
injected with ISO only. The tumor weight was 2.11±0.35 g in the
control mice, 0.91±0.27 g in ISO-treated mice, 0.42±0.12 g in ISO
and 3-MA co-injected mice and 0.58±0.16 in ISO and CQ co-injected
mice, respectively (Fig. 6B). The
results therefore indicated that autophagy inhibition markedly
promoted the inhibitory effect of ISO on the NSCLC xenograft
tumors.
Suppression of autophagy decreases
ISO-induced growth suppression and enhances apoptosis of NSCLC in
vivo
To assess apoptosis in the experimental groups,
TUNEL-positive cells were detected in the tumor tissue.
Quantitative evaluation showed that the apoptotic index was 7±3% in
the control tumors, while it was 33±5% in the ISO-treated tumors.
As expected, the apoptotic index was increased to 65±8% in the ISO
and 3-MA co-treated tumors, and 60±9% in the ISO and CQ co-treated
tumors (Fig. 6C and D). In
addition, the levels of cleaved caspase-3 showed a similar trend to
that of the apoptotic rate in the different experimental groups
(Fig. 6C and D). The proliferative
indices in the groups were also assessed; as shown in Fig. 6D, in the control group, the
proliferative index was 81±7%, whereas in all treatment groups, the
proliferation was markedly decreased to 51±4% in the ISO-treated
group, 23±5% in the ISO- and 3-MA-treated group and 32±4% in the
ISO and CQ co-injected group. These results therefore supported
that caspase-mediated apoptosis is a key contributor to tumor
growth suppression and that suppression of autophagy markedly
promoted the inhibitory effect of ISO on NSCLC.
Discussion
ISO, an immediate 3′-O-methylated metabolite of
quercetin, has been studied in recent years for its marked
anti-cancer activity in several human cancer types (6-8).
However, to date, the anti-cancer effects of ISO have not been
investigated in lung cancer. In the present study, the
anti-proliferative and pro-apoptotic effects of ISO on human NSCLC
cells were investigated in vitro and in vivo. The
results showed that ISO efficiently inhibited the proliferation and
induced apoptosis of NSCLS cells in a time- and dose-dependent
manner, indicating that ISO may be a potential candidate for a
novel anti-lung cancer drug.
Numerous chemotherapeutic drugs designed to kill
cancer, most likely by inducing apoptosis, including fluorouracil,
arsenic trioxide, tamoxifen, paclitaxel, adriamycin and cisplatin,
have been shown to also induce autophagy (39,40).
The present study found that ISO treatment of A549 cells resulted
in the upregulated expression of endogenous LC3-II and Beclin1, the
translocation to autophagosomes of GFP-LC3-II, and the accumulation
of MDC in cytoplasmic vacuoles, suggesting that ISO also induced
autophagy in lung cancer cells.
The in vitro and in vivo experiments
of the present study as described above significantly enhanced the
mechanistic understanding of the signaling events involved in the
induction of apoptosis in lung cancer cells by ISO as well as their
relevance to its in vivo tumor-inhibitory efficacy.
Mechanistically, the in vitro results suggested that the
induction of apoptosis by ISO proceeds through a mitochondrial
pathway. This was indicated by loss of the transmembrane potential
as cytochrome C was released into cytosolic fraction,
decreased pro-caspase-9 levels (through cleavage), increased
cleaved caspase-3 and PARP levels as well as DNA fragmentation,
TUNEL positivity and sub-G1 apoptotic bodies. The critical role of
the mitochondria/cytochrome C/caspase-9 cascade was
supported by the complete blockage of apoptosis by the caspase-9
inhibitor Z-LEHD-FMK and caspase-3 inhibitor Z-DEVD-FMK. The
detailed mechanisms of how ISO affects the mitochondria to initiate
apoptosis signaling as well as a possible involvement of
mitogen-associated protein kinase (MAPK) pathways (extracellular
signal-regulated kimase, c-Jun N-terminal kinase and p38MAPK)
pathways or the PI3K-AKT survival pathway require further
investigation.
Autophagy has major protective roles in maintaining
the homeostasis in the cells by clearing damaged organelles, such
as mitochondria, and toxic proteins (40,41).
However, the functions and mechanisms of drug-induced autophagy in
cancer cells have remained to be fully elucidated (42,43).
Most studies indicated that drug-induced autophagy can be divided
into two types: Pro-survival autophagy and pro-death autophagy.
Pro-survival autophagy induced by chemotherapeutic drugs was found
to reduce the anti-tumor effects of the drugs, which indicated the
presence of a cross-talk between apoptotic signaling and autophagy
(44-46). The present study investigated the
contributions of autophagy to ISO-induced apoptosis of lung cancer
cells, and the results showed that ISO was able to induce
autophagy. Pre-treatment with autophagy inhibitors 3-MA and CQ
efficiently suppressed ISO-induced autophagy and enhanced
ISO-induced growth inhibition as well as apoptosis in lung cancer
cells. The results therefore indicated that ISO induced a
pro-survival-type autophagy, and that co-treatment with autophagy
inhibitors can be used to improve the therapeutic effects of ISO in
lung cancer. The analyses of the mouse xenograft tumors showed a
decreased PCNA index (~20% reduction), as well as >four-fold
increases of TUNEL- and cleaved-caspase-3-positive tumor cells when
the animals were co-treated with 3-MA or CQ compared with those
following treatment with ISO only. This demonstrated that autophagy
inhibition markedly promoted the suppressive effect of ISO on the
growth of NSCLC xenograft tumors.
In conclusion, the present study demonstrated that
ISO may serve as an anti-tumor agent to significantly suppress the
cell viability and promote apoptosis of lung cancer cells. In
addition, ISO treatment also induced a pro-survival-type autophagy
and inhibition of this type of autophagy may enhance ISO-induced
growth inhibition and apoptosis in lung cancer cells. Thus, the
present study suggested the combined treatment with ISO and
inhibitors of autophagy as an efficient strategy for anti-lung
cancer therapy.
Acknowledgments
The authors would like to thank Mr. Wugen Zhan for
the assistance with the flow cytometric analysis, and Mr. Yu Bing
for the assistance with the microscopic analysis (both Division of
Life and Health Sciences, Shenzhen Graduate School of Tsighua
University, Shenzhen, China). The present study was supported by
the Guangdong Natural Science Foundation (no. 2014A030313758), a
Doctoral Fund of the Ministry of Education of China (no.
20120002120020) and the Science, Technology & Innovation
Commission of Shenzhen Municipality (no.
JCYJ20140417115840285).
References
|
1
|
Molina JR, Yang P, Cassivi SD, Schild SE
and Adjei AA: Non-small cell lung cancer: Epidemiology, risk
factors, treatment, and survivorship. Mayo Clin Proc. 83:584–594.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tada H, Tsuchiya R, Ichinose Y, Koike T,
Nishizawa N, Nagai K and Kato H: A randomized trial comparing
adjuvant chemotherapy versus surgery alone for completely resected
pN2 non-small cell lung cancer (JCOG9304). Lung Cancer. 43:167–173.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Asano N, Kuno T, Hirose Y, Yamada Y,
Yoshida K, Tomita H, Nakamura Y and Mori H: Preventive effects of a
flavonoid myricitrin on the formation of azoxymethane-induced
prema-lignant lesions in colons of rats. Asian Pac J Cancer Prev.
8:73–76. 2007.PubMed/NCBI
|
|
4
|
Khacha-ananda S, Tragoolpua K,
Chantawannakul P and Tragoolpua Y: Antioxidant and anti-cancer cell
proliferation activity of propolis extracts from two extraction
methods. Asian Pac J Cancer Prev. 14:6991–6995. 2013. View Article : Google Scholar
|
|
5
|
Manu KA, Shanmugam MK, Ramachandran L, et
al: Isorhamnetin augments the anti-tumor effect of capeciatbine
through the negative regulation of NF-κB signaling cascade in
gastric cancer. Cancer Lett. 363:28–36. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Saud SM, Young MR, Jones-Hall YL, Ileva L,
Evbuomwan MO, Wise J, Colburn NH, Kim YS and Bobe G:
Chemopreventive activity of plant flavonoid isorhamnetin in
colorectal cancer is mediated by oncogenic Src and beta-catenin.
Cancer Res. 73:5473–84. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li C, Yang X, Chen C, Cai S and Hu J:
Isorhamnetin suppresses colon cancer cell growth through the
PI3K-Akt-mTOR pathway. Mol Med Rep. 9:935–940. 2014.PubMed/NCBI
|
|
8
|
Ramachandran L, Manu KA, Shanmugam MK, Li
F, Siveen KS, Vali S, Kapoor S, Abbasi T, Surana R, Smoot DT, et
al: Isorhamnetin inhibits proliferation and invasion and induces
apoptosis through the modulation of peroxisome
proliferator-activated receptor gamma activation pathway in gastric
cancer. J Biol Chem. 287:38028–38040. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fulda S and Debatin KM: Debatin, extrinsic
versus intrinsic apoptosis pathways in anticancer chemotherapy.
Oncogene. 25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao J, Wang SZ, Tang XF, Liu N, Zhao D
and Mao ZY: Analysis of thermochemotherapy-induced apoptosis and
the protein expressions of Bcl-2 and Bax in maxillofacial squamous
cell carcinomas. Med Oncol. 28(Suppl 1): S354–S359. 2011.
View Article : Google Scholar
|
|
11
|
Kim R: Recent advances in understanding
the cell death pathways activated by anticancer therapy. Cancer.
103:1551–1560. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fang K, Chen Z, Liu M, Peng J and Wu P:
Apoptosis and calcifi-cation of vascular endothelial cell under
hyperhomocysteinemia. Med Oncol. 32(403)2015. View Article : Google Scholar
|
|
13
|
Choi KS: Autophagy and cancer. Exp Mol
Med. 44:109–120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu D, Yang Y, Liu Q and Wang J:
Inhibition of autophagy by 3-MA potentiates cisplatin-induced
apoptosis in esophageal squamous cell carcinoma cells. Med Oncol.
28:105–111. 2011. View Article : Google Scholar
|
|
15
|
Mathew R, Karantza-Wadsworth V and White
E: Role of autophagy in cancer. Nat Rev Cancer. 7:961–967. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mathew R and White E: Why sick cells
produce tumors: The protective role of autophagy. Autophagy.
3:502–505. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
White E: Deconvoluting the
context-dependent role for autophagy in cancer. Nat Rev Cancer.
12:401–410. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mah LY and Ryan KM: Autophagy and cancer.
Cold Spring Harb Perspect Biol. 4:a0088212012. View Article : Google Scholar
|
|
19
|
Karantza-Wadsworth V and White E: Role of
autophagy in breast cancer. Autophagy. 3:610–613. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Maycotte P, Aryal S, Cummings CT, Thorburn
J, Morgan MJ and Thorburn A: Chloroquine sensitizes breast cancer
cells to chemotherapy independent of autophagy. Autophagy.
8:200–212. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Maclean KH, Dorsey FC, Cleveland JL and
Kastan MB: Targeting lysosomal degradation induces p53-dependent
cell death and prevents cancer in mouse models of lymphomagenesis.
J Clin Invest. 118:79–88. 2008. View
Article : Google Scholar
|
|
22
|
Zhang X, Dong Y, Zeng X, Liang X, Li X,
Tao W, Chen H, Jiang Y, Mei L and Feng SS: The effect of autophagy
inhibitors on drug delivery using biodegradable polymer
nanoparticles in cancer treatment. Biomaterials. 35:1932–1943.
2014. View Article : Google Scholar
|
|
23
|
Gibson UE, Heid CA and Williams PM: A
novel method for real time quantitative RT-PCR. Genome Res.
6:995–1001. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Høyer-Hansen M, Bastholm L, Szyniarowski
P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N,
Elling F, Rizzuto R, et al: Control of macroautophagy by calcium,
calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell.
25:193–205. 2013. View Article : Google Scholar
|
|
25
|
Abnosi MH, Solemani Mehranjani M, Momeni
HR, Mahdiyeh Najafabadi M, Barati M and Shojafar E: The induction
of apoptosis and autophagy in rat bone marrow mesenchymal stem
cells following in vitro treatment with p-nonylphenol. IJST A.
3:239–244. 2012.
|
|
26
|
Estavillo GM, Verhertbruggen Y, Scheller
HV, et al: Isolation of the plant cytosolic fraction for proteomic
analysis. Methods Mol Biol. 1072:453–467. 2014. View Article : Google Scholar
|
|
27
|
Kalani K, Agarwal J, Alam S, Khan F, Pal A
and Srivastava SK: In silico and in vivo anti-malarial studies of
18β glycyrrhetinic acid from Glycyrrhiza glabra. PLoS One.
8:e747612013. View Article : Google Scholar
|
|
28
|
Hasenjäger A, Gillissen B, Müller A,
Normand G, Hemmati PG, Schuler M, Dörken B and Daniel PT: Smac
induces cytochrome c release and apoptosis independently from
Bax/Bcl-x(L) in a strictly caspase-3-dependent manner in human
carcinoma cells. Oncogene. 23:4523–4535. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Suzuki S, Higuchi M, Proske RJ, Oridate N,
Hong WK and Lotan R: Implication of mitochondria-derived reactive
oxygen species, cytochrome C and caspase-3 in N-(4-hydroxyphenyl)
retinamide-induced apoptosis in cervical carcinoma cells. Oncogene.
18:6380–6387. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cuvillier O, Nava VE, Murthy SK, Edsall
LC, Levade T, Milstien S and Spiegel S: Sphingosine generation,
cytochrome c release, and activation of caspase-7 in
doxorubicin-induced apoptosis of MCF7 breast adenocarcinoma cells.
Cell Death Differ. 8:162–171. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang W, Wang Z and Chen T: Curcumol
induces apoptosis via caspases-independent mitochondrial pathway in
human lung adenocarcinoma ASTC-a-1 cells. Med Oncol. 28:307–314.
2011. View Article : Google Scholar
|
|
32
|
Li XY, Lin YC, Huang WL, Lin W, Wang HB,
Lin WZ and Lin SL: Zoledronic acid inhibits human nasopharyngeal
carcinoma cell proliferation by activating mitochondrial apoptotic
pathway. Med Oncol. 29:3374–3380. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kim MS, Jeong EG, Ahn CH, Kim SS, Lee SH
and Yoo NJ: Frameshift mutation of UVRAG, an autophagy-related
gene, in gastric carcinomas with microsatellite instability. Hum
Pathol. 39:1059–1063. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hoare M, Young AR and Narita M: Autophagy
in cancer: Having your cake and eating it. Semin Cancer Biol.
21:397–404. 2011.PubMed/NCBI
|
|
36
|
Liu F, Liu D, Yang Y and Zhao S: Effect of
autophagy inhibition on chemotherapy-induced apoptosis in A549 lung
cancer cells. Oncol Lett. 5:1261–1265. 2013.PubMed/NCBI
|
|
37
|
Pellegrini P, Strambi A, Zipoli C,
Hägg-Olofsson M, Buoncervello M, Linder S and De Milito A: Acidic
extracellular pH neutralizes the autophagy-inhibiting activity of
chloroquine: Implications for cancer therapies. Autophagy.
10:562–571. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zou Y, Ling YH, Sironi J, Schwartz EL,
Perez-Soler R and Piperdi B: The autophagy inhibitor chloroquine
overcomes the innate resistance of wild-type EGFR non-small-cell
lung cancer cells to erlotinib. J Thorac Oncol. 8:693–702. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lozy F and Karantza V: Autophagy and
cancer cell metabolism. Semin Cell Dev Biol. 23:395–401. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Giuliani CM and Dass CR: Autophagy and
cancer: Taking the 'toxic' out of cytotoxics. J Pharm Pharmacol.
65:777–789. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gewirtz DA: Autophagy and senescence in
cancer therapy. J Cell Physiol. 229:6–9. 2014.
|
|
42
|
He W, Ma X, Yang X, Zhao Y, Qiu J and Hang
H: A role for the arginine methylation of Rad9 in checkpoint
control and cellular sensitivity to DNA damage. Nucleic Acids Res.
39:4719–4727. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
He W, Zhao Y, Zhang C, An L, Hu Z, Liu Y,
Han L, Bi L, Xie Z, Xue P, et al: Rad9 plays an important role in
DNA mismatch repair through physical interaction with MLH1. Nucleic
Acids Res. 36:6406–6417. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Eng CH and Abraham RT: The autophagy
conundrum in cancer: Influence of tumorigenic metabolic
reprogramming. Oncogene. 30:4687–4696. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jain K, Paranandi KS, Sridharan S and Basu
A: Autophagy in breast cancer and its implications for therapy. Am
J Cancer Res. 3:251–265. 2013.PubMed/NCBI
|
|
46
|
Lorin S, Hamaï A, Mehrpour M and Codogno
P: Autophagy regulation and its role in cancer. Semin Cancer Biol.
23:361–379. 2013. View Article : Google Scholar : PubMed/NCBI
|