1. Introduction
Adenosine triphosphate-sensitive potassium channels
(KATP), which are distributed throughout the body in
tissue types including smooth muscle, brain, skeletal muscle and
cardiac muscle, have been known for decades (1). The basic biological function of
KATP is to adjust cell activities to the metabolic
status, and KATP is situated at the crosstalk site
between cell metabolism and membrane excitability. When
encountering insufficient energy levels, the inwardly rectifying
potassium channels of KATP are activated by nucleotides
in the presence of magnesium ions. Opening of the channels would
result in hyperpolarization of the membrane, which was found to be
cytoprotective under various pathophysiological conditions.
Cardiomyopathy is among one of the leading causes of deterioration
of cardiac function and even heart failure; however, to date,
knowledge regarding the etiology and underlying mechanisms has
remained limited. As cardiomyopathies are associated with metabolic
disorders, studies on KATP may provide novel basic
knowledge and treatment strategies for cardiomyopathies. The
present review briefly summarized the functions of KATP
with a focus on the current understanding of its role in
cardiomyopathies.
2. Molecular structural properties of
KATP
KATP is generally accepted as a
hetero-octameric complex composed of inward-rectifier potassium ion
channel (Kir)6 and sulfonylurea receptor (SUR) subunits. Kir6 is a
pore-forming unit, and is encoded by the KCNJ8 (for Kir6.1)
(2) and KCNJ11 (for Kir6.2)
genes (3). The regulatory SUR
subunits belong to the family of the ATP binding cassette (ABC),
which are encoded by genes including ABCC8 (for SUR1) and
ABCC9 (for SUR2) (4).
Post-transcriptional modification by RNA splicing generates mainly
two molecular variants of SUR, namely SUR2A and SUR2B, whose
biophysiological characteristics vary distinctly (5,6).
Biochemical and physiological studies suggested that
the normal functional KATP is supported and maintained
by a 4:4 stoichiometric co-assembly of Kir6.2 and SUR1, or Kir6.2
and SUR2A (SUR2B) subunits (7,8).
This octamer arrangement implies that the genes of Kir and SUR may
be co-regulated (9). Indeed, it
was found that KCNJ11 and ABCC8 share neighboring
locations on human chromosome 11p15.1 (10); similarly, KCNJ8 and
ABCC9 were located on human chromosome 12p12.1 adjacently
(11).
The understanding of the structure of
KATP is mainly based on crystallographic studies on
bacteria and eukaryotic cells (12). It was demonstrated that the main
structure of the Kir channel was composed of two transmembrane M1
and M2 helices, which were connected by a bridge-like loop,
favoring ion selection control and the generation of a narrow
porous architecture (13). TMD1
and TMD2, which are six-helix trans-membrane domains, put up the
primary structure of the SUR sub-units (14). An accessory five-helix
transmembrane TMD0 domain was found at the N-terminus of SURs,
having a role in gating and trafficking of the Kir6 sub-unit
(15). Between TMD1 and TMD2,
nucleotide binding fold (NBF), comprising NBF1 and NBF2, was
identified in previous studies (16). An octameric structure composed of
four Kir6.x and four SUR subunits was proposed (17); however, the specific physical
contact, connection and interaction of the sub-units have remained
to be fully elucidated.
3. Biological function and regulation of
KATP
The signature sequence of potassium ion
(K+) channels, which is ubiquitous among the
K+ channel family, is highly conserved in Kir sub-units,
eliciting K+-selective properties (18). Rapid and reversible closure and
activation accommodating to the metabolic status is the
characteristic biological property of KATP (19). In the presence of ATP,
non-hydrolysable ATP analogues or even adenosine diphosphate (ADP)
with the absence of magnesium ions (Mg2+), the channel
activity is blocked and the channel is closed (20,21),
suggesting that the inactivation of KATP does not rely
on phosphorylation and the binding relies on the gamma-phosphate of
the ATP molecule (22). A binding
pocket is formed by the C- and N-termini residues in the plasma
with three-dimensional folding (23,24).
There are four binding pockets for the octameric structure - one
for each channel at each kir6 sub-unit (25,26).
Channel gating and ATP binding are linked via a helical structure,
which was proposed to lie parallel to the interface of the membrane
(27). The location of the contact
point was suggested at the junction of the inner helix bundle
(28,29) (Fig.
1).
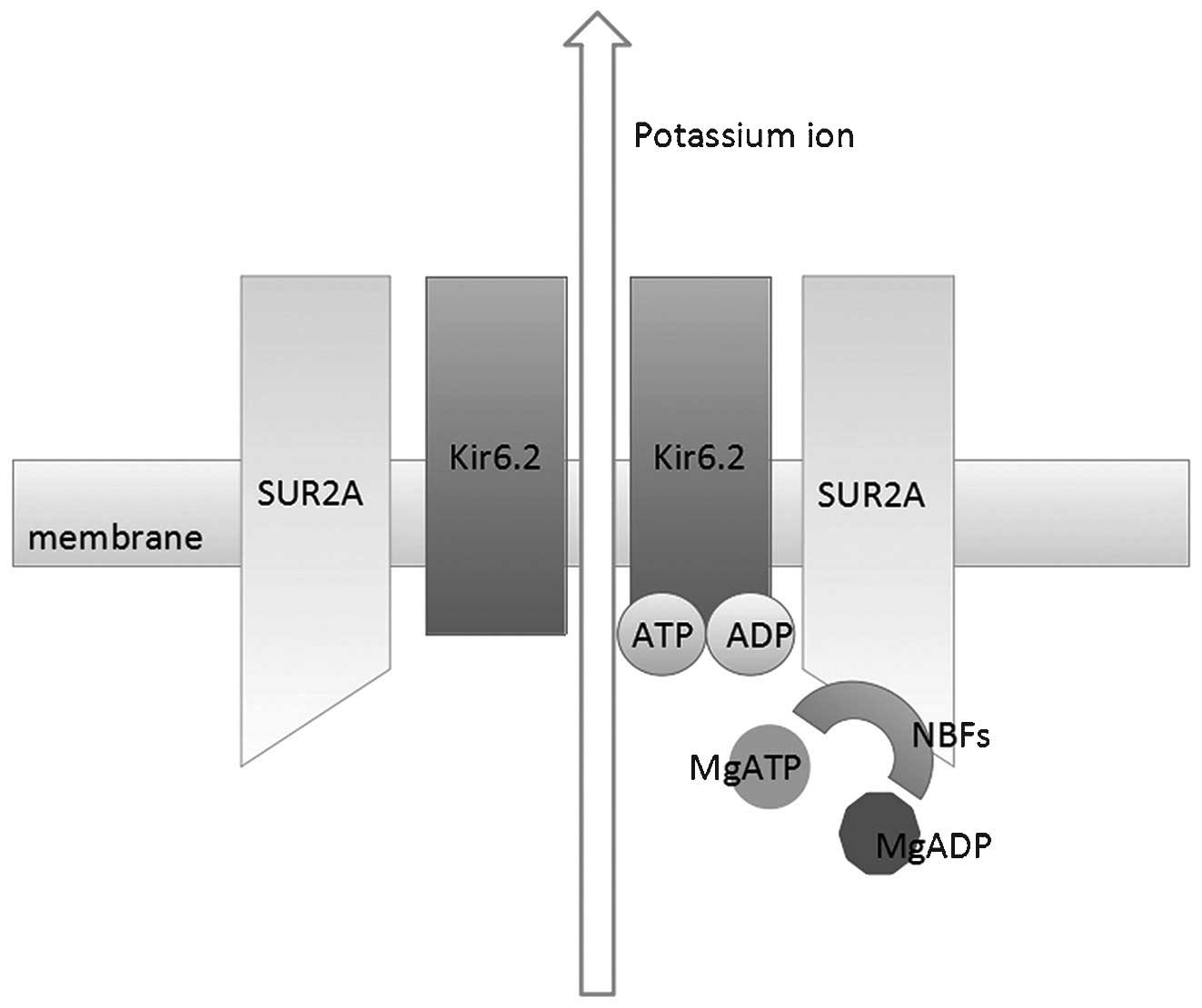 | Figure 1Schematic demonstration of the
molecular structure and function of cardiac KATP
channels. 4 Kir6.2 subunits and 4 SUR2A subunits constitute the
heterooctameric complex. KATP activity is inhibited by
ATP by direct binding to Kir6.2. In the absence of magnesium ions,
ADP also inhibits channel activity even with lower binding affinity
to Kir6.2. However, in the presence of magnesium ions, cytoplasmic
ADP and ATP serve to stimulate channel activity by binding to NBF
domains, which are located on SUR2A subunits. ATP, adenosine
triphosphate; ADP, adenosine diphosphate; KATP,
ATP-sensitive potassium channels; NBF, nucleotide binding fold;
Kir, inward-rectifier potassium ion channel; SUR, sulfonylurea
receptor. |
The SUR sub-unit regulates KATP activity
by interacting with Mg2+ adenosine nucleotides: ATP and
ADP stimulate channel opening in the presence of Mg2+,
while the nucleotides deactivate channel activity in the absence of
Mg2+ (30). In the
cytoplasm, composed of nucleotide-binding motifs, the NBFs (NBF1
and NBF2) have the main regulatory effect on KATP
function. It was suggested that the dimerization of two NBFs was
required for Mg2+-dependent ATP hydrolysis by SUR
(31). The inhibition induced by
ATP on the Kir6 sub-unit may be overcome by the hydrolytic activity
of dimeric NBFs on ATP in the presence of Mg2+ (32). These regulatory effects of SUR on
Kir6 gating are supported by a connecting structure named L0
linker, which is situated between the SUR TMD0 domain and the
Kir6.2 cytoplasmic N-terminus (33).
Briefly, KATP regulation is characterized
by fast and reversible deactivation and closure induced by
cytoplasmic nucleotide diphosphates and triphosphates. In intact
cells, the KATP is almost permanently inhibited by ATP,
whose concentration is steadily maintained at a millimolar level
(1–5 mmol/l), even while the cells undergo metabolic changes
(34). Under such circumstance, by
interaction with the SUR subunit, the channel is effectively
activated by exogenous Mg2+ nucleotides, particularly
MgADP (19). Nucleotide regulation
is currently considered the key mechanism in controlling
KATP opening, though several other regulators were also
proposed in certain KATP-associated diseases.
4. Distribution of KATP in
cardiac muscle
KATP in cardiac
sarcolemma
Previous studies confirmed that in hearts of
rodents, SUR1 and Kir6.2 constitute atrial sarcolemmal
KATP (35), while
ventricular sarcolemmal KATP is mainly composed of SUR2A
and Kir6.2 (36). Variants of SUR1
and SUR2A were identified in atrial as well as ventricular cardiac
muscles in humans. Generally, under normal physiological
conditions, the sarcolemmal KATP remains a static status
unless it encounters severe metabolic challenges, including anoxia,
ischemia and metabolic toxic drugs (37,38).
Activated sarcolemmal KATP serves a cardioprotective
role by inhibiting calcium overload, recovering contractility,
preserving energy supply and stabilizing the membrane potential
(39). Treatment with
KATP openers, such as diazoxide, resulted in a decrease
in the incidence of arrhythmias, including tachycardia and
ventricular fibrillation (40).
KATP in cardiac
mitochondria
Except for the sarcolemmal KATP,
KATP distributed in cardiac mitochondria
(mitoKATP) are also considered important in cardiac
pathophysiology. To date, the molecular composition of
mitoKATP has remained elusive. It was proposed that the
heterogenous integration of SUR1 and Kir6.1 properly represents the
properties of mitoKATP (41); however, in Kir6.1 and Kir6.2
knockout animals, the activity of mitoKATP remained
unaffected (42). Several studies
have assessed the canonical composition of SUR and Kir6 molecules
in the mitoKATP structure. In mitochondrial extracts,
protein detected with anti-Kir6.1 antibody was proved not to be
Kir6.1 by subsequent mass spectrometric analysis (43). In another study, an NBF1 domain,
which was specifically localized to mitochondria, and the lack of a
SUR2 sub-unit protein were identified in myocytes (44).
Unlike the indeterminacy of its structure, the basic
function of mitoKATP is relatively clear in the heart,
though it is not completely understood. Under stress induced by
multiple stimuli, efficient energy transfer from mitochondria to
cytosol is guaranteed by mitoKATP activation. Extrinsic
stressful signals, including reactive oxygen species, transduced
across the cytosol to the mitochondria, may induce the activation
of mitoKATP, whose opening would decrease opening of the
mitochondrial permeability transition (MPT) pore, which would
result in myocyte death (45).
5. KATP and cardiomyopathies
Cardiomyopathy was defined by the World Health
Organization as cardiac diseases accompanied by cardiac
dysfunction. Dilated cardiomyopathy (DCM), hypertrophic
cardiomyopathy (HCM), restrictive cardiomyopathy (RCM),
arrhythmogenic right ventricular cardiomyopathy and secondary
cardiomyopathy are accepted as types of cardiomyopathy. It is
considered that energetic and metabolic disorders are involved in
pathophysiological processes of cardiomyopathy, which is highly
associated with cardiac KATP as mentioned above.
KATP and HCM
HCM is characterized by unexplained and asymmetric
left ventricular hypertrophy without explicit causes and includes
coronary heart disease, arterial stenosis, hypertension, valvular
heart disease and further systemic diseases that induce left
ventricular hypertrophy (46).
Heart failure, sudden cardiac death (SCD) and stroke are the common
clinical manifestations in patients with HCM, which is often
diagnosed by echocardiography showing a maximal left ventricular
wall thickness of ≥15 mm. Myocyte malalignment, myocyte hypertrophy
and myocardial interstitial fibrosis are the main histological
features of HCM (47).
Imbalances in energy metabolism were suggested to be
the underlying cause of the occurrence and development of HCM,
corresponding with mitochondrial dysfunction and biophysical
disorganization in HCM. In response to energetic metabolic
deficiency, myocyte hypertrophy may be the compensatory
consequence. As KATP are highly involved in energy
metabolism, they may be implicated in HCM development (48).
Mechanistic assays using KATP antagonists
or activators were performed to testify the role of KATP
in HCM. Hypertrophic myocytes were acquired from spontaneously
hypertensive rats (SHR) by Sodder et al (49). Trypsin, which was able to
re-activate KATP, only increased KATP channel
activity by 29% in hypertrophic myocytes as opposed to 63% in the
control, indicating that KATP activity loss was involved
in the pathogenesis of cardiac hypertrophy. In another study by
Rajesh et al (50),
ischemic pre-conditioning (IP) was demonstrated to have protective
effects against supra-renal transverse abdominal aortic
constriction-induced cardiac hypertrophy. 5-hydroxydecanoic acid
(5-HD), a specific KATP antagonist, was applied to
animals after IP. The results showed that 5-HD pre-treatment
impaired the protective effects of IP during sustained cardiac
ischemia in hypertrophied hearts (50). In another study, the induction of
hypertrophy in cultured ventricular myocytes by alpha1 adrenoceptor
agonist phenylephrine (PE) was evidenced by increased cell size,
elevated expression of myosin light chain-2 and atrial natriuretic
peptide (51). Diazoxide, as one
of the canonical mitoKATP openers, almost completely
prevented the hypertrophic inductive effects of PE.
Numerous previous studies provided direct evidence
for the protective role of KATP in cardiac hypertrophy.
By partial ligation of the ascending aorta, Yuan et al
(52) created an animal model of
left ventricular hypertrophy. Responsiveness of KATP to
ATP (exogenous as well as locally generated ATP) in isolated
myocytes from hypertrophic hearts was found to be markedly
decreased in a patch clamp assay (52). In a study investigating hearts from
SUR2-knockout mice, a significantly greater heart size and
ventricular mass were identified (53). Shimokawa et al (54) found that in endocardial cells
isolated from hypertrophied hearts of SHR, the KATP
channel currents were significantly smaller and the time required
to reach peak currents after the onset of KATP channel
opening was significantly longer than that in the control group.
Furthermore, the dysfunctional KATP failed to respond
rapidly to exogenous ATP. These results indicated that
biophysiologically dysfunctional KATP may contribute to
cardiac hypertrophy.
The possible underlying mechanisms of
KATP impairment and HCM were investigated by several
studies. Heart hypertrophy was achieved in a rat model of pressure
overload, which was achieved by abdominal aortic banding (55). A KATP opener, iptakalim,
was applied orally to rats, which reversed the deteriorating
cardiac function hemodynamically and histologically, as well as the
serum content of B-type natriuretic peptide. After KATP
activation, the potassium efflux facilitated calcium influx to
increase calcium concentration, which activated endothelial nitric
oxide synthase (eNOS) via the calcium-calmodulin pathway. eNOS then
catalyzed the biological synthesis of endogenous NO. Thus,
indirectly, the activation of KATP led to the
maintenance of cardiac function and hemodynamic homeostasis by
modulation of NO production (55).
In chronic transverse aortic constriction-induced cardiac
hypertrophied KATP-disrupted rats, the expression of
PPAR gamma co-activator-1 alpha (PGC-1alpha) was significantly
decreased (56). It was thought
that PGC-1alpha had an important role in regulating energetic
metabolism through mitochondrial enzymes during exposure to cardiac
pressure overload. The transcription of PGC-1 alpha was activated
by phosphorylated forkhead box protein O1 (FOXO1), whose
phosphorylation was reported to proceed through activation of Akt.
In addition, it was observed that the KATP channel
dysfunction induced by SUR1 disruption and Kir6.2 knockout resulted
in an overall decrease in FOXO1 expression (56). The study indicated that
FOXO1/PGC-1alpha signaling was one of the possible mechanisms of
sarcolemmal KATP-associated cardiac hypertrophy.
KATP and dilated
cardiomyopathy
As another important type of cardiomyopathy, DCM is
clinically characterized by ventricular dilation and impaired
contractility, often leading to heart failure and SCD (3). As myocardial mass and volume
increase, the ventricular wall often becomes thin and stretched
(57). To date, the etiology of
DCM has remained to be fully elucidated. DCM may occur secondary to
heart diseases, including congenital heart disease, valvular heart
disease, ischemic heart disease, viral myocarditis and Chagas
disease (58). Of note, it is now
widely accepted that DCM is highly genetic. Mutations of
KATP-associated genes were confirmed to be involved in
the etiology of DCM.
Bienengraeber et al (59) identified two mutations in the
ABCC9 gene encoding the KATP sub-unit SUR2A by
genomic DNA scanning in patients with dilated cardiomyopathy with
tachycardia (DCM10). One mutation was described as a three-base
pair deletion and a four-base pair insertion mutation
(4,570–4,572delTTAinsAAAT), which introduced four abnormal terminal
residues followed by a premature stop codon and caused a frameshift
at Leu1524 (Fs1524) after translation. Another mutation was
suggested as a missense mutation (4,537G>A), causing an A1,513T
amino acid substitution as occurring at the C terminus of SUR2A and
leading to a disruption of the normal organization of the NBD2
pocket. Reduced KATP channel trafficking, aberrant
KATP channel gating and an anomalous intrinsic ATP
hydrolysis cycle were observed when the SUR2 sub-unit was defective
and co-expressed with the Kir6.2 sub-unit (59). Thus, the mediation between
energetic and electrical signals by KATP is impaired in
DCM. Patients with the abovementioned genetic mutations may
therefore be considered to have an increased susceptibility to
DCM.
A study using Langendorff hearts extracted from
patients diagnosed with DCM revealed that the expression of the
Kir6 sub-unit (Kir6.1 as well as Kir6.2) changed correspondingly
with that of the SUR sub-unit (SUR1 as well as SUR2A) in the
endocardium and epicardium (60).
This result indicated that the other sub-unit, Kir6, may also have
a role as one of the etiological factors of DCM. The results of a
study on KCNJ11 gene knockout hearts exposed to hemodynamic
overload showed that these hearts were more susceptible to
maladaptive remodeling and congestive heart failure (59). When under imposed overload stress,
KCNJ11-null mutant hearts were markedly dilated and
inefficient regarding their contractility, sharing common features
with CMD10 (61). Indeed, after
the deficiency of Kir6.2 was compensated by embryonic stem cell
therapy, the cardiac function was partially restored (62). Recently, KCNJ11 gene
mutation was also suggested to be one of the causes of DCM. A gene
polymorphism called E23K, which is a non-synonymous mutation
occurring at codon 23 of the KCNJ11 gene (634G>A), led to
the replacement of a glutamic acid residue by a lysine at this
polymorphic site (rs5219) at the Kir6.2 sub-unit (63). By analyzing the blood of patients
with DCM, Xi et al (64)
discovered that this mutation was highly associated with the left
ventricular end diastolic dimension (LVEDD) and left atrial
dimension (LAD), which markedly increases in DCM (64) (Fig.
2).
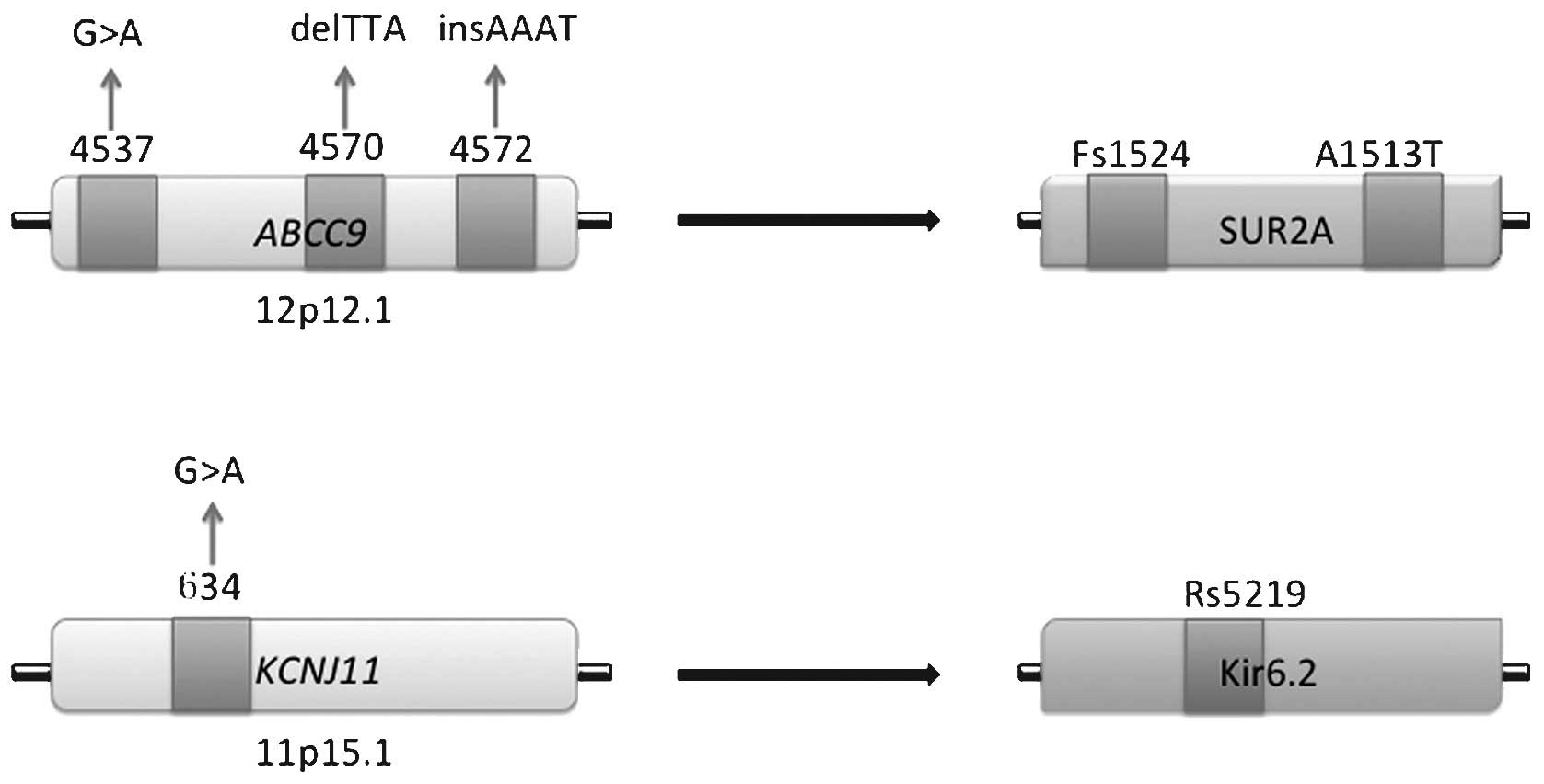 | Figure 2Mutations of KATP sub-unit
genes correlated with dilated cardiomyopathy. Mutations in
ABCC9 gene comprise a mis-sense mutation at site 4,537, a
three-base pair deletion at site 4,572 and a four-base pair
insertion mutation at site 4,570–4,572. These mutations lead to a
frameshift at site 1,524 and amino acid substitution at site 1,513
of the SUR2A sub-unit. In the KCNJ11 gene, a non-synonymous
mutation at site 634 causes amino acid replacement at site 5,219 of
the Kir6.2 sub-unit. Fs, frameshift; A, amino acid substitution;
Rs, acid replacement; Kir, inward-rectifier potassium ion channel;
SUR, sulfonylurea receptor. |
6. KATP and secondary
cardiomyopathies
KATP and ischemic
cardiomyopathy (ICM)
Due to the high and increasing morbidity of coronary
heart disease, ICM is now considered to be one of the most common
underlying causes of heart failure in modern-day society (65). As a result of sustained myocardial
ischemia, ICM is characterized by marked loss of contractile units
in the myocardium. Ischemia and accompanied re-perfusion injury may
lead to myocyte apoptosis and myocardial necrosis.
Several previous studies have examined the
correlation between myocyte apoptosis and KATP under
ischemia/re-perfusion conditions. They posed the hypothesis that
KATP exerts its anti-apoptotic effects upon activation.
Indeed, the role of KATP in cellular calcium signal
regulation may have a preventive effect against cardiac apoptosis
(66). Calcium overload, which
refers to the accumulation of calcium ions in the cell matrix, is
one of the mechanisms triggering apoptosis. As the concentration of
calcium ions rises in the mitochondria, the opening of the MPT pore
becomes irreversible. Pro-apoptotic proteins, such as cytochrome C,
are subsequently released to induce apoptosis. However, after being
activated, KATP opens to allow potassium ion influx into
the cell matrix to depolarize the inner membrane, thus reducing the
calcium uptake of the matrix (67).
Furthermore, previous studies implied that
inflammation is involved in coronary artery disease and myocardial
ischemia. During this process, inflammatory cytokines, including
interleukin-1 (IL-1), IL-6 and tumor necrosis factor-alpha
(TNF-alpha) were released (68).
Through interacting with TNF receptors, TNF-alpha potently induced
cell apoptosis through death receptor- or caspase cascade-mediated
apoptotic pathways (69). Zhou
et al (70) reported that
the opening of KATP reduced TNF-alpha production by
inhibiting its downstream protein mitogen-activated protein kinase
(70). Thus, the anti-apoptotic
effects of cardiac KATP in ICM may be based on its
abilities to modulate inflammation.
KATP and cardiomyopathy in
Duchenne muscular dystrophy (DMD)
It is generally accepted that DMD is an X-linked
progressive neuromuscular disorder with the manifestation of
generalized muscular weakness and wasting. Patients with DMD are
often diagnosed at 6–8 years of age and die of respiratory or
cardiac failure by the age of 30 years in most cases (71). The dystrophin gene, which is
located at the short arm of the X chromosome at cytogenetic band 21
(Xp21), was suggested to be the leading cause of DMD. Mutations of
this gene lead to the expression of mutant dystrophin protein
within myofibers throughout all types of muscle cell, including
smooth, skeletal and cardiac muscle cells. A lethal form of
cardiomyopathy occurs in the majority of DMD patients, which is
characterized by ventricular wall thickness reduction and cardiac
chamber enlargement (72).
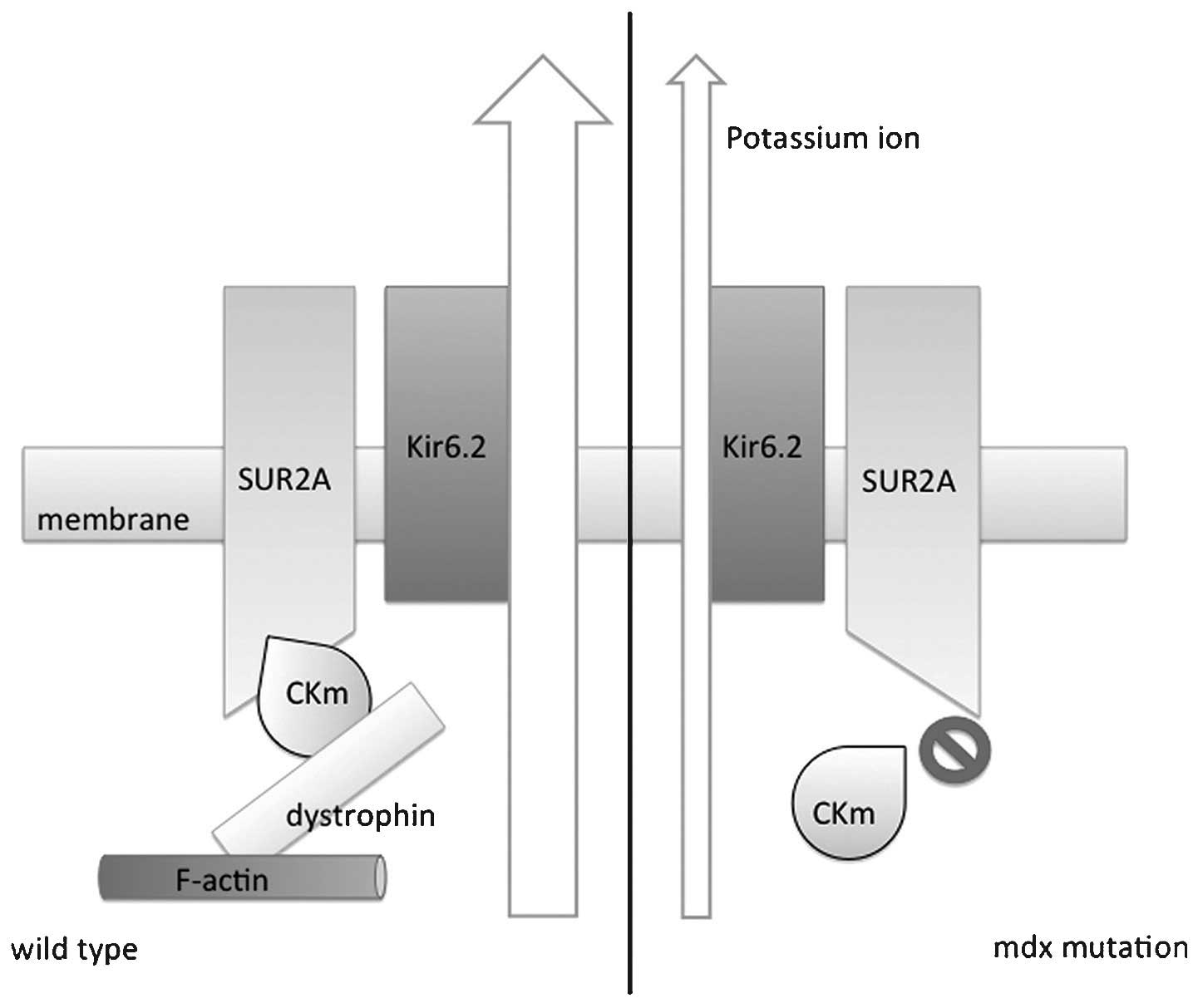
Graciotti et al (73) reported that cardiac KATP
has an important role in cardiomyopathy in DMD and proposed a
possible regulatory mechanism. Importantly, the study reported that
in myocytes from normal mice, KATP sub-unit Kir6.2 and
dystrophin were physically connected, sharing the same location on
the t-tube. Furthermore, a metabolic enzyme, creatine kinase muscle
isoform (CKm), which was described as a regulator of
KATP activity, was also in physical contact with
dystrophin. In mdx mutant mice, which were deficient of the full
length of dystrophin, CKm membrane localization was disrupted
(73). This result suggested that
dystrophyin may act as a scaffold allowing KATP and its
regulatory proteins to form a complex, coordinating metabolic
regulation. Thus, when dystrophin was absent in DMD, loss of CKm
interaction led to the disruption of its modulation of
KATP channel activity, resulting in a disability of
KATP in sensing the intracellular ATP concentration
(Fig. 3).
KATP and diabetic
cardiomyopathy (DbCM)
In patients with diabetes mellitus, almost every
tissue type is affected by metabolic disorders, resulting in vital
organ dysfunctions. It was reported that cardiovascular diseases
take responsibility for ~65% of diabetes-associated mortality
(74). Myocardial dysfunction
occurring without evidence of any other primary heart disease,
including coronary artery disease and valvular heart disease, is
now generally defined as DbCM.
The association between diabetes and KATP
has been known for several years. Gloyn et al (75) launched a case-control study on
2,486 diabetes patients in the United Kingdom, showing that
KCNJ11 gene polymorphism E23K was associated with type 2
diabetes. In addition, in the Walker B motif of NBD2 of SUR
sub-units, a mutation of the conserved glutamate catalytic residue
(E1506) to lysine (E1506K) resulted in reduced KATP
channel activation in beta cells, which was detected in patients
with neonatal diabetes. This mutation was therefore thought to be
one of the causes of neonatal diabetes (76).
Based on these results, studies on KATP
in cardiomyocytes may be of potential significance to reveal the
underlying mechanisms of DbCM. To date, only a few studies on this
association are available; however, the results are of importance.
Fancher et al (77)
evaluated the function and expression of mitoKATP in the
hearts of mice with type 1 diabetes. The expression of Kir6.1 and
SUR1 was found decreased in interfibrillar mitochondria, while the
expression of Kir6.1 was found to be reduced in sub-sarcolemmal
mitochondria in diabetic rat hearts (77). Furthermore, the expression of
Kir6.2 and SUR2A was significantly decreased in diabetic rats,
which could be restored by correction of hyperglycemia. Of note,
diazoxide, a KATP opener, showed cardioprotective
effects (78).
KATP and Keshan disease
(KD)
KD initially drew attention in 1930s by its outbreak
in Keshan County in northeast China. The heart is the primary
target organ of KD (79). Enlarged
heart, cardiac arrhythmia, cardiogenic shock and congestive heart
failure are the clinical manifestations of KD. It was recognized as
a form of cardiomyopathy, which was histologically characterized by
multifocal necrosis and cardiac fibrosis (80). KD is endemic as it is limited to
certain geographical areas and with seasonal variations. Though the
etiology of KD still remains to be fully elucidated, selenium
deficiency is considered the major cause, as selenium deficiency in
local residents and food were significantly associated with the
geographical distribution of KD (81).
A previous study by our group reported that cardiac
function was significantly impaired in selenium-deficient rats
(82). At the same time, the
expression of the two sub-units of KATP, Kir6.2 and
SUR2A, was inhibited in myocytes, accompanied by a decrease of
glutathione peroxidase, which indicated the occurrence of oxidative
stress (82). After introduction
of oxidative stress, the activity of mitoKATP was
upregulated according to a study by Pereira et al (83). They concluded that KATP
acted as a molecular sensor for oxidative stress, whose activation
helped to reduce free-radical generation in the mitochondrial
respiratory chain. However, the study did not continue to observe
the activity of mitoKATP during sustained and severe
oxidative stress, which may have induced significant mitochondrial
dysfunction, and the activity and expression of mitoKATP
may have been jeopardized under these conditions. Further study
regarding oxidative stress, KATP and cardiac dysfunction
in KD is still required.
7. Summary and perspectives
As a mediator in cellular metabolism,
KATP couples the energetic status to the excitability of
the cell membrane, sensing metabolic changes and leading to
morphological changes as well as secondary signaling.
KATP channels are distributed in the cytosol and
mitochondria of cardiomyocytes. As one of the leading causes of
heart failure, cardiomyopathy is characterized by metabolic
challenges, which could be alleviated by activation of
KATP. Dysfunction and deficiency of cardiac
KATP were suggested to have important roles in primary
cardiomyopathies, including hypertrophic cardiomyopathy and dilated
cardiomyopathy, as well as secondary cardiomyopathies, including
ischemic cardiomyopathy, diabetic cardiomyopathy, endemic
cardiomyopathy and cardiomyopathy in Duchenne muscular
dystrophy.
Due to the lack of sufficient knowledge regarding
KATP in cardiomyopathies, numerous questions remain: Do
KATP channels share unitary features in the occurrence
and development of different types of cardiomyopathies? Are there
any unique changes of KATP specific for each type of
cardiomyopathy? What are the polymorphisms of the gene encoding
KATP in other primary cardiomyopathies, including
restrictive cardiomyopathy and arrhythmogenic right ventricular
cardiomyopathy? Is KATP involved in gene-environmental
interactions in endemic cardiomyopathies? To address these
questions, further studies on KATP in cardiomyopathies
should be implemented in the future.
Acknowledgments
The present review was fully supported by the
National Natural Science Foundation of China (grant nos. 81371473
and 81171262).
References
|
1
|
Rapposelli S: Novel adenosine
5′-triphosphate-sensitive potassium channel ligands: a patent
overview (2005–2010). Expert Opin Ther Pat. 21:355–379. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Delaney JT, Muhammad R, Blair MA, Kor K,
Fish FA, Roden DM and Darbar D: A KCNJ8 mutation associated with
early repolarization and atrial fibrillation. Europace.
14:1428–1432. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yamada S, Kane GC, Behfar A, Liu XK, Dyer
RB, Faustino RS, Miki T, Seino S and Terzic A: Protection conferred
by myocardial ATP-sensitive K+ channels in pressure
overload-induced congestive heart failure revealed in KCNJ11
Kir6.2-null mutant. J Physiol. 577:1053–1065. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fatima N, Schooley JF Jr, Claycomb WC and
Flagg TP: Promoter DNA methylation regulates murine SUR1 (Abcc8)
and SUR2 (Abcc9) expression in HL-1 cardiomyocytes. PLoS One.
7:e415332012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shi Y, Wu Z, Cui N, Shi W, Yang Y, Zhang
X, Rojas A, Ha BT and Jiang C: PKA phosphorylation of SUR2B subunit
underscores vascular KATP channel activation by
beta-adrenergic receptors. Am J Physiol Regul Integr Comp Physiol
ATP. 293:R1205–R1214. 2007. View Article : Google Scholar
|
|
6
|
Zhou M, He HJ, Suzuki R, Liu KX, Tanaka O,
Sekiguchi M, Itoh H, Kawahara K and Abe H: Localization of
sulfonylurea receptor subunits, SUR2A and SUR2B, in rat heart. J
Histochem Cytochem. 55:795–804. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shyng S and Nichols CG: Octameric
stoichiometry of the KATP channel complex. J Gen
Physiol. 110:655–664. 1997. View Article : Google Scholar
|
|
8
|
Nakaya H: Role of ATP-sensitive
K+ channels in cardiac arrhythmias. J Cardiovasc
Pharmacol Ther. 19:237–243. 2014. View Article : Google Scholar
|
|
9
|
Kang Y, Ng B, Leung YM, He Y, Xie H,
Lodwick D, Norman RI, Tinker A, Tsushima RG and Gaisano HY:
Syntaxin-1A actions on sulfonylurea receptor 2A can block acidic
pH-induced cardiac K(ATP) channel activation. J Biol Chem.
281:19019–19028. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Damaj L, le Lorch M, Verkarre V, Werl C,
Hubert L, Nihoul-Fékété C, Aigrain Y, de Keyzer Y, Romana SP,
Bellanne-Chantelot C, et al: Chromosome 11p15 paternal isodisomy in
focal forms of neonatal hyperinsulinism. J Clin Endocrinol Metab.
93:4941–4947. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Inagaki N, Gonoi T, Clement JP IV, Namba
N, Inazawa J, Gonzalez G, Aguilar-Bryan L, Seino S and Bryan J:
Reconstitution of IKATP: an inward rectifier subunit
plus the sulfonylurea receptor. Science. 270:1166–1170. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Park S and Terzic A: Quaternary structure
of KATP channel SUR2A nucleotide binding domains resolved by
synchrotron radiation X-ray scattering. Struct Biol. 169:243–251.
2010. View Article : Google Scholar
|
|
13
|
Durell SR and Guy HR: A family of putative
Kir potassium channels in prokaryotes. BMC Evol Biol. 1:142001.
View Article : Google Scholar
|
|
14
|
Aggarwal NT, Shi NQ and Makielski JC:
ATP-sensitive potassium currents from channels formed by Kir6 and a
modified cardiac mitochondrial SUR2 variant. Channels (Austin).
7:493–502. 2013. View Article : Google Scholar
|
|
15
|
Chan KW, Zhang H and Logothetis DE:
N-terminal trans-membrane domain of the SUR controls trafficking
and gating of Kir6 channel subunits. EMBO J. 22:3833–3843. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mori H and Ito K: The long alpha-helix of
SecA is important for the ATPase coupling of translocation. J Biol
Chem. 281:36249–36256. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Baczko I, Husti Z, Lang V, Leprán I and
Light PE: Sarcolemmal KATP channel modulators and cardiac
arrhythmias. Curr Med Chem. 18:3640–3661. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nichols CG, Singh GK and Grange DK:
KATP channels and cardiovascular disease: Suddenly a
syndrome. Circ Res. 112:1059–1072. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nichols CG: KATP channels as
molecular sensors of cellular metabolism. Nature. 440:470–476.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Haissaguerre M, Chatel S, Sacher F,
Weerasooriya R, Probst V, Loussouarn G, Horlitz M, Liersch R,
Schulze-Bahr E, Wilde A, et al: Ventricular fibrillation with
prominent early repolarization associated with a rare variant of
KCNJ8/KATP channel. J Cardiovasc Electrophysiol.
20:93–98. 2009. View Article : Google Scholar
|
|
21
|
Fox JE, Magga J, Giles WR and Light PE:
Acyl coenzyme A esters differentially activate cardiac and
beta-cell adenosine triphosphate-sensitive potassium channels in a
side-chain length-specific manner. Metabolism. 52:1313–1319. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
John SA, Weiss JN and Ribalet B: ATP
sensitivity of ATP-sensitive K+ channels: Role of the
gamma phosphate group of ATP and the R50 residue of mouse Kir6.2. J
Physiol. 568:931–940. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Antcliff JF, Haider S, Proks P, Sansom MS
and Ashcroft FM: Functional analysis of a structural model of the
ATP-binding site of the KATP channel Kir6.2 subunit.
EMBO J. 24:229–239. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Enkvetchakul D and Nichols CG: Gating
mechanism of KATP channels: Function fits form. J Gen
Physiol. 122:471–480. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ribalet B, John SA and Weiss JN: Molecular
basis for Kir6.2 channel inhibition by adenine nucleotides. Biophys
J. 84:266–276. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Trapp S, Haider S, Jones P, Sansom MS and
Ashcroft FM: Identification of residues contributing to the ATP
binding site of Kir6.2. EMBO J. 22:2903–2912. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Enkvetchakul D, Jeliazkova I,
Bhattacharyya J and Nichols CG: Control of inward rectifier K
channel activity by lipid tethering of cytoplasmic domains. J Gen
Physiol. 130:329–334. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ribalet B, John SA, Xie LH and Weiss JN:
ATP-sensitive K+ channels: Regulation of bursting by the
sulphonylurea receptor, PIP2 and regions of Kir6.2. J Physiol.
571:303–317. 2006. View Article : Google Scholar
|
|
29
|
Pegan S, Arrabit C, Zhou W, Kwiatkowski W,
Collins A, Slesinger PA and Choe S: Cytoplasmic domain structures
of Kir2.1 and Kir3.1 show sites for modulating gating and
rectification. Nat Neurosci. 8:279–287. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hund TJ and Mohler PJ: Differential roles
for SUR subunits in KATP channel membrane targeting and
regulation. Am J Physiol Heart Circ Physiol. 300:H33–H35. 2011.
View Article : Google Scholar
|
|
31
|
Cui N, Kang Y, He Y, Leung YM, Xie H,
Pasyk EA, Gao X, Sheu L, Hansen JB, Wahl P, et al: H3 domain of
syntaxin 1A inhibits KATP channels by its actions on the
sulfonylurea receptor 1 nucleotide-binding folds-1 and -2. J Biol
Chem. 279:53259–53265. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ueda K, Inagaki N and Seino S: MgADP
antagonism to Mg2+-independent ATP binding of the sulfonylurea
receptor SUR1. J Biol Chem. 272:22983–22986. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Babenko AP and Bryan J: Sur domains that
associate with and gate KATP pores define a novel
gatekeeper. J Biol Chem. 278:41577–41580. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tsounapi P, Satio M, Dimitriadis F,
Kitatani K, Kinoshita Y, Shomori K, Takenaka A and Satoh K: The
role of K ATP channels on ischemia-reperfusion injury in the rat
testis. Life Sci. 90:649–656. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Glukhov AV, Flagg TP, Fedorov VV, Efimov
IR and Nichols CG: Differential K (ATP) channel pharmacology in
intact mouse heart. J Mol Cell Cardiol. 48:152–160. 2010.
View Article : Google Scholar
|
|
36
|
Glukhov AV, Uchida K, Efimov IR and
Nichols CG: Functional roles of KATP channel subunits in
metabolic inhibition. J Mol Cell Cardiol. 62:90–98. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Marinovic J, Ljubkovic M, Stadnicka A,
Bosnjak ZJ and Bienengraeber M: Role of sarcolemmal ATP-sensitive
potassium channel in oxidative stress-induced apoptosis:
Mitochondrial connection. Am J Physiol Heart Circ Physiol.
294:H1317–H1325. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang DM, Chai Y, Erickson JR, Brown JH,
Bers DM and Lin YF: Intracellular signalling mechanism responsible
for modulation of sarcolemmal ATP-sensitive potassium channels by
nitric oxide in ventricular cardiomyocytes. J Physiol. 592:971–990.
2014. View Article : Google Scholar :
|
|
39
|
Voitychuk OI, Strutynskyi RB, Yagupolskii
LM, Tinker A, Moibenko OO and Shuba YM: Sarcolemmal cardiac K(ATP)
channels as a target for the cardioprotective effects of the
fluorine-containing pinacidil analogue, flocalin. Br J Pharmacol.
162:701–711. 2011. View Article : Google Scholar :
|
|
40
|
Xie C, Hu J, Motloch LJ, Karam BS and Akar
FG: The classically cardioprotective agent diazoxide elicits
arrhythmias in type 2 diabetes mellitus. J Am Coll Cardiol.
66:1144–1156. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu Y, Ren G, O'Rourke B, Marban E and
Seharaseyon J: Pharmacological comparison of native mitochondrial K
(ATP) channels with molecularly defined surface K (ATP) channels.
Mol Pharmacol. 59:225–230. 2001.PubMed/NCBI
|
|
42
|
Suzuki M, Sasaki N, Miki T, Sakamoto N,
Ohmoto-Sekine Y, Tamagawa M, Seino S, Marbán E and Nakaya H: Role
of sarcolemmal K(ATP) channels in cardioprotection against
ischemia/reperfusion injury in mice. J Clin Invest. 109:509–516.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Foster DB, Rucker JJ and Marban E: Is
Kir6.1 a subunit of mitoK(ATP)? Biochem Biophys Res Commun.
366:649–656. 2008. View Article : Google Scholar :
|
|
44
|
Pu JL, Ye B, Kroboth SL, McNally EM,
Makielski JC and Shi NQ: Cardiac sulfonylurea receptor short
form-based channels confer a glibenclamide-insensitive
KATP activity. J Mol Cell Cardiol. 44:188–200. 2008.
View Article : Google Scholar
|
|
45
|
Mykytenko J, Reeves JG, Kin H, Wang NP,
Zatta AJ, Jiang R, Guyton RA, Vinten-Johansen J and Zhao ZQ:
Persistent beneficial effect of postconditioning against infarct
size: Role of mitochondrial K(ATP) channels during reperfusion.
Basic Res Cardiol. 103:472–484. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hensley N, Dietrich J, Nyhan D, Mitter N,
Yee MS and Brady M: Hypertrophic cardiomyopathy: a review. Anesth
Analg. 120:554–569. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Teekakirikul P, Padera RF, Seidman JG and
Seidman CE: Hypertrophic cardiomyopathy: Translating cellular cross
talk into therapeutics. J Cell Biol. 199:417–421. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Vakrou S and Abraham MR: Hypertrophic
cardiomyopathy: a heart in need of an energy bar? Front Physiol.
19:3092014.
|
|
49
|
Sodder VH, Bowie LD and Cameron JS:
Trypsin alters ATP sensitivity of KATP channels in
control and hypertrophied myocytes. Eur J Pharmacol. 315:115–118.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Rajesh KG, Sasaguri S, Suzuki R, Xing Y
and Maeda H: Ischemic preconditioning prevents reperfusion heart
injury in cardiac hypertrophy by activation of mitochondrial
KATP channels. Int J Cardiol. 96:41–49. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Xia Y, Rajapurohitam V, Cook MA and
Karmazyn M: Inhibition of phenylephrine induced hypertrophy in rat
neonatal cardio-myocytes by the mitochondrial KATP
channel opener diazoxide. J Mol Cell Cardiol. 37:1063–1067. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yuan F, Brandt NR, Pinto JM, Wasserlauf
BJ, Myerburg RJ and Bassett AL: Hypertrophy decreases cardiac
KATP channel responsiveness to exogenous and locally
generated (glycolytic) ATP. J Mol Cell Cardiol. 29:2837–2848. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Stoller D, Kakkar R, Smelley M, Chalupsky
K, Earley JU, Shi NQ, Makielski JC and McNally EM: Mice lacking
sulfonylurea receptor 2 (SUR2) ATP-sensitive potassium channels are
resistant to acute cardiovascular stress. J Mol Cell Cardiol.
43:445–454. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Shimokawa J, Yokoshiki H and Tsutsui H:
Impaired activation of ATP-sensitive K+ channels in
endocardial myocytes from left ventricular hypertrophy. Am J
Physiol Heart Circ Physiol. 293:H3643–H3649. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Gao S, Long CL, Wang RH and Wang H: K
(ATP) activation prevents progression of cardiac hypertrophy to
failure induced by pressure overload via protecting endothelial
function. Cardiovasc Res. 83:444–456. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hu X, Xu X, Huang Y, Fassett J, Flagg TP,
Zhang Y, Nichols CG, Bache RJ and Chen Y: Disruption of sarcolemmal
ATP-sensitive potassium channel activity impairs the cardiac
response to systolic overload. Circ Res. 103:1009–1017. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Sanbe A: Dilated cardiomyopathy: A disease
of the myocardium. Biol Pharm Bull. 36:18–22. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Luk A, Ahn E, Soor GS, Soor GS and Butany
J: Dilated cardiomyopathy: a review. J Clin Pathol. 62:219–225.
2009. View Article : Google Scholar
|
|
59
|
Bienengraeber M, Olson TM, Selivanov VA,
Kathmann EC, O'Cochlain F, Gao F, Karger AB, Ballew JD, Hodgson DM,
Zingman LV, et al: ABCC9 mutations identified in human dilated
cardiomyopathy disrupt catalytic KATP channel gating.
Nat Genet. 36:382–387. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
60
|
Farid TA, Nair K, Massé S, Azam MA, Maguy
A, Lai PF, Umapathy K, Dorian P, Chauhan V, Varró A, et al: Role of
KATP channels in the maintenance of ventricular
fibrillation in cardiomyopathic human hearts. Circ Res.
109:1309–1318. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Grover GJ and Garlid KD: ATP-Sensitive
potassium channels: A review of their cardioprotective
pharmacology. J Mol Cell Cardiol. 32:677–695. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Yamada S, Nelson TJ, Crespo-Diaz RJ,
Perez-Terzic C, Liu XK, Miki T, Seino S, Behfar A and Terzic A:
Embryonic stem cell therapy of heart failure in genetic
cardiomyopathy. Stem Cells. 26:2644–2653. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Cheung CY, Tso AW, Cheung BM, Xu A, Fong
CH, Ong KL, Law LS, Wat NM, Janus ED, Sham PC, et al: The KCNJ11
E23 K polymorphism and progression of glycaemia in Southern
Chinese: A long-term prospective study. PLoS One. 6:e285982011.
View Article : Google Scholar
|
|
64
|
Xi HL, Liu JF, Li L and Wan J:
Relationship between dilated cardiomyopathy and the E23K and I337V
polymorphisms in the Kir6.2 subunit of the KATP channel.
Genet Mol Res. 12:4383–4392. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kalogeropoulos A, Georgiopoulou V,
Kritchevsky SB, Psaty BM, Smith NL, Newman AB, Rodondi N,
Satterfield S, Bauer DC, Bibbins-Domingo K, et al: Epidemiology of
incident heart failure in a contemporary elderly cohort: The
health, aging and body composition study. Arch Intern Med.
169:708–715. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Elrod JW and Molkentin JD: Physiologic
functions of cyclophilin D and the mitochondrial permeability
transition pore. Circ J. 77:1111–1122. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Rousou AJ, Ericsson M, Federman M,
Levitsky S and McCully JD: Opening of mitochondrial KATP
channels enhances cardioprotection through the modulation of
mitochondrial matrix volume, calcium accumulation and respiration.
Am J Physiol Heart Circ Physiol. 287:H1967–H1976. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
McPherson R and Davies RW: Inflammation
and coronary artery disease: Insights from genetic studies. Can J
Cardiol. 28:662–666. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Safranow K, Dziedziejko V, Rzeuski R,
Czyzycka E, Wojtarowicz A, Bińczak-Kuleta A, Jakubowska K,
Olszewska M, Ciechanowicz A, Kornacewicz-Jach Z, et al: Plasma
concentrations of TNF-alpha and its soluble receptors sTNFR1 and
sTNFR2 in patients with coronary artery disease. Tissue Antigens.
74:386–392. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zhou F, Yao HH, Wu JY, Ding JH, Sun T and
Hu G: Opening of microglial K(ATP) channels inhibits
rotenone-induced neuroinflammation. J Cell Mol Med. 12:1559–1570.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Kaspar RW, Allen HD and Montanaro F:
Current understanding and management of dilated cardiomyopathy in
Duchenne and Becker muscular dystrophy. J Am Acad Nurse Pract.
21:241–249. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Townsend D, Yasuda S and Metzger J:
Cardiomyopathy of Duchenne muscular dystrophy: Pathogenesis and
prospect of membrane sealants as a new therapeutic approach. Expert
Rev Cardiovasc Ther. 5:99–109. 2007. View Article : Google Scholar
|
|
73
|
Graciotti L, Becker J, Granata AL,
Procopio AD, Tessarollo L and Fulgenzi G: Dystrophin is required
for the normal function of the cardio-protective K(ATP) channel in
cardiomyocytes. PLoS One. 6:e270342011. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Pappachan JM, Varughese GI, Sriraman R and
Arunagirinathan G: Diabetic cardiomyopathy: Pathophysiology,
diagnostic evaluation and management. World J Diabetes. 4:177–189.
2013.PubMed/NCBI
|
|
75
|
Gloyn AL, Weedon MN, Owen KR, Turner MJ,
Knight BA, Hitman G, Walker M, Levy JC, Sampson M, Halford S, et
al: Large-scale association studies of variants in genes encoding
the pancreatic beta-cell KATP channel subunits Kir6.2
(KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is
associated with type 2 diabetes. Diabetes. 52:568–572. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Mannikko R, Flanagan SE, Sim X, Segal D,
Hussain K, Ellard S, Hattersley AT and Ashcroft FM: Mutations of
the same conserved glutamate residue in NBD2 of the sulfonylurea
receptor 1 subunit of the KATP channel can result in
either hyperinsulinism or neonatal diabetes. Diabetes.
60:1813–1822. 2011. View Article : Google Scholar
|
|
77
|
Fancher IS, Dick GM and Hollander JM:
Diabetes mellitus reduces the function and expression of
ATP-dependent K(+) channels in cardiac mitochondria. Life Sci.
92:664–668. 2013. View Article : Google Scholar :
|
|
78
|
Chen ZC, Cheng YZ, Chen LJ, Cheng KC, Li Y
and Cheng J: Increase of ATP-sensitive potassium (K(ATP)) channels
in the heart of type-1 diabetic rats. Cardiovasc Diabetol.
11:82012. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Li GS, Wang F, Kang D and Li C: Keshan
disease: An endemic cardiomyopathy in China. Hum Pathol.
16:602–609. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Lei C, Niu X, Ma X and Wei J: Is selenium
deficiency really the cause of Keshan disease? Environ Geochem
Health. 33:183–188. 2011. View Article : Google Scholar
|
|
81
|
Chen J: An original discovery: Selenium
deficiency and Keshan disease (an endemic heart disease). Asia Pac
J Clin Nutr. 21:320–326. 2012.PubMed/NCBI
|
|
82
|
Liu ZW, Niu XL, Chen KL, Xing YJ, Wang X,
Qiu C and Gao DF: Selenium attenuates adriamycin-induced cardiac
dysfunction via restoring expression of ATP-sensitive potassium
channels in rats. Biol Trace Elem Res. 153:220–228. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Pereira SP, Pereira GC, Pereira CV,
Carvalho FS, Cordeiro MH, Mota PC, Ramalho-Santos J, Moreno AJ and
Oliveira PJ: Dioxin-induced acute cardiac mitochondrial oxidative
damage and increased activity of ATP-sensitive potassium channels
in Wistar rats. Environ Pollut. 180:281–290. 2013. View Article : Google Scholar : PubMed/NCBI
|