Introduction
Hypertension is currently one of the most serious
public health challenges worldwide. It is a major risk factor for
cardiovascular diseases, myocardial infarction, stroke and chronic
renal failure (1). Vascular tone,
which represents the contractile activity of vascular smooth muscle
cells (VSMCs) in the small arteries and arterioles, is the major
determinant of vascular resistance to blood flow through the
circulation, rendering it a particularly important component of
hypertension (2). Enhanced
vascular contractility contributes to the long-term control of
systemic blood pressure (3), and
impaired cerebral vasoconstriction during hypertension affects
brain regional blood flow, which makes it a risk factor for stroke
(4). Ion channels have been
demonstrated to be critical in vascular tone and the development of
hypertension, and numerous investigations into the role of ion
channels in arterial smooth muscle contractility have been
conducted. In these studies, cation channels, including calcium
channel, voltage-dependent, L type, α 1C subunit, various
K+ channels (5) and
nonselective transient receptor potential (TRP) channels (6) have been found to regulate vascular
contraction (1). Anion channels,
such as chloride channels are also involved. Chloride channels
within the plasma membrane of VSMCs cause Cl− efflux and
membrane depolarization (7), and
Ca2+-activated chloride currents (CaCCs) may be involved
in increasing vascular contractility. CaCC-dependent depolarization
activates voltage-gated Ca2+ channels (8) and further increase intracellular
Ca2+ ([Ca2+]i), resulting in
signal amplification, leading to VSMC contraction and contractile
response of blood vessels. Transmembrane protein 16A (TMEM16A) was
recently identified to be responsible for the CaCCs in basilar
artery SMCs (BASMCs) and it is involved in the regulation of BASMCs
proliferation during hypertension (9). Previous research regarding cerebral
vasculature has revealed that pressure-induced membrane stretch may
activate TMEM16A channels and thus contribute to the myogenic tone
of basilar arteries (10). In
addition to blood pressure, constriction of resistance arteries is
also regulated by vasoconstrictors, such as angiotensin II (Ang
II), thromboxane A2 and 5-hydroxytryptamine; however, whether
TMEM16A channels are also involved in vasoconstrictor-induced
cerebral vasoconstriction remains unclear.
The renin-angiotensin system (RAS) is key in
cardiovascular and renal physiology, and its overactivation is
implicated in the induction and progression of hypertension
(11). Circulating Ang II, the
major contributing factor of the RAS, was significantly upregulated
during hypertension (12),
establishing it as an important and high-risk pathologic factor.
Ang II acts directly on vascular smooth muscle as a vasoconstrictor
in an either calcium-dependent or -independent manner; in the
latter case, Ang II activates the RhoA/Rho-associated protein
kinase (ROCK) signaling pathway and increases VSMC Ca2+
sensitization via Rho guanine nucleotide exchange factors (13), inducing phosphorylation of myosin
phosphatase-targeting subunit 1 (MYPT1) and myosin light chains
(MLCs), which finally causes SMC contraction. In a previous study,
it was found that basilar artery constriction in response to Ang II
was mediated by ROCK, while the vasoconstriction in response to KCl
was not significantly influenced by this signaling pathway
(14).
In the present study, the role of TMEM16A in Ang
II-induced basilar artery vasoconstriction was investigated using
its specific inhibitor, T16A-inhA01 (9,15).
The basilar arteries were obtained from hypertensive rats at
various time-points following 2-kidney, 2-clip (2k2c) surgery, and
the influence of ROCK inhibitor, Y-27632 was also detected.
Notably, the direct effect of Ang II on TMEM16A-mediated CaCC
channels was determined in cultured BASMCs, and it was further
revealed that the TMEM16A protein expression level affected Ang
II-induced phosphorylation of MYPT1 and MLC via the RhoA/ROCK
signaling pathway.
Materials and methods
Animal models and blood pressure
measurement
The present study was approved by the ethics
committee of Liuzhou People's Hospital (Liuzhou, China). All animal
experimental procedures were performed according to the policies of
the Second Medical University Animal Care and Use Committee and
conformed to the Guide for the Care and Use of Laboratory Animals
(16). Unless otherwise stated,
all materials were purchased from Sigma-Aldrich (St. Louis, MO,
USA). Hypertensive rat models (2k2c) were established, as described
previously (17). Briefly, 120
healthy male Sprague-Dawley rats (weight, 80–120 g; Laboratory
Animal Centre, Guangxi Medical University, Nanning, China) were
anaesthetized by sodium pentobarbital (40 mg/kg, i.p.), and
following midline laparotomy, ring-shaped silver clips (internal
diameter, 0.3 mm) were placed around the two renal arteries. All
animals were maintained in a pathogen-free room with controlled
temperature at ~25°C, under a 12-h light/dark cycle. Blood pressure
was measured in conscious rats by tail-cuff plethysmography
(Powerlab 4/30; ADInstruments Pty Ltd., Bella Vista, NSW,
Australia).
Basilar artery lumen diameter
measurement
Following induction of deep anesthesia
(pentobarbital; 200 mg/kg i.p.), the brain was removed from each of
the rats and placed in Krebs buffer containing: 137 mM NaCl, 5.4 mM
KCl, 2.0 mM CaCl2, 1.1 mM MgCl2, 0.4 mM
NaH2PO4, 5.6 mM glucose, 11.9 mM
NaHCO3, 105 U/l penicillin and 100 mg/l streptomycin, at
37°C. Basilar arteries were rapidly isolated and the connective
tissue was carefully removed. As described previously (18), arteries were mounted onto glass
micropipettes filled with Krebs buffer in an organ chamber, and
allowed to equilibrate and reach a stable diameter for ≥30 min at a
distending pressure of 60 mmHg. The stimuli and/or inhibitors were
subsequently added. Ang II (10−11–10−7 M) was
added for 5 min for measurement and T16A-inhA01 (10 µM) and
Y-27632 (10 µM) were added 5 min prior to Ang II
stimulation. Vessel images were captured using an AxioImager Z1
microscope (Zeiss GmbH, Jena, Germany) and an S-100 video camera
(Nikon Corporation, Tokyo, Japan), a VDA-10 electronic dimension
analyzer (Living Systems Instrumentation, St. Albans, VT, USA) was
then used to measure the lumen diameter.
Cell culture
Rat BASMCs were cultured from the rat basilar
arteries, as previously described (19). Briefly, healthy male Sprague-Dawley
rats (weight, 80–100 g) were anaesthetized with pentobarbital
sodium (200 mg/kg i.p.). Basilar arteries were harvested
immediately and immersed in iced Kreb's solution. The vessels were
isolated carefully and sliced into 0.2-mm rings in Dulbecco's
modified Eagle's medium/Ham's F-12 (DMEM/F12) medium and the vessel
segments were placed on the surface of the culture flask and
incubated in DMEM/F12 medium supplemented with 20% fetal bovine
serum (FBS; Thermo Fisher Scientific, Inc., Waltham, MA, USA) at
37°C, in an atmosphere of 5% CO2 for 5–7 days. Cells
from passage 3–6 were used in subsequent experiments.
Recording of CaCC currents in cultured
BASMCs
The Ca2+-activated Cl− current
was recorded from cultured BASMCs using an Axopatch™ 200B Amplifier
(Molecular Devices, LLC, Sunnyvale, CA, USA) according to a
previously described, whole-cell patch clamp technique (20). The current was elicited using
voltage steps from 60 to 100 mV in 20-mV increments for 250 msec
from a holding potential of −40 mV and stored on a computer
following digitalization at 5 kHz using a Digidata 1322A (Axon
Instrument). The bath solution contained 1.5 mM CaCl2, 1
mM MgCl2, 10 mM NaCl, 140 mM NMDG-Cl, 10 mM HEPES and 10
mM glucose, which was adjusted to pH 7.4 using HCl. The
Ca2+-buffered pipette solution contained 140 mM CsCl, 1
mM MgCl2, 5 mM EGTA, 10 mM HEPES, 4 mM
Na2-ATP, and 1.925 and 3.801 mM CaCl2 to
adjust the free Ca2+ concentration to 100 and 500 nM,
respectively (calculated using the CaBuf program; www.jgp.org/cgi/content/full/jgp.201411339/DC1),
which was adjusted to pH 7.2 using CsOH. The pipettes (Sutter
Instrument Co, Novato, CA, USA) were pulled from borosilicate glass
capillaries and had resistances of 3–5 MΩ after fire polishing. All
experiments were performed at room temperature (22–25°C).
TMEM16A small interfering (si)RNA and
adenovirus transfection
siRNA duplex (40 nM) against rat TMEM16A gene
(sense, 5′-GGAGUUAUCAUCUAUAGAATT-3′ and antisense,
5′-UUCUAUAGAUGAUAACUCCAA-3′; Qiagen GmbH, Hilden, Germany) was
transiently transfected with Hiperfect Transfection Reagent (Qiagen
GmbH), as previously described (19). A scrambled RNA (Qiagen GmbH) served
as the negative control. Briefly, the siRNA strand and Hiperfect
Transfection Reagent were diluted in serum- and antibiotic-free
DMEM/F12 and maintained at room temperature for 10 min to form the
transfection complexes. The complexes were added to BASMCs and
mixed gently to ensure uniform distribution. Following incubation
for 3 h at 37°C, transfection complexes were replaced with normal
DMEM/F12 medium containing 10% FBS for 48 h. TMEM16A adenovirus
(adv-TMEM16A) was constructed by Shengbo Biological Engineering
Co., Ltd. (Shanghai, China). The adv-TMEM16A (multiplicity of
infection, 200) was added into cultured BASMCs with serum- and
antibiotic-free DMEM/F12 for 6 h, then cells were washed twice with
phosphate-buffered saline and cultured in DMEM/F12 medium
containing 10% FBS for another 48 h. adv-lacz (Jikai Biological
Engineering Co., Ltd., Shanghai, China) served as a negative
control. Western blot analysis was used to examine the effect of
TMEM16A siRNA (si-TMEM16A) and adv-TMEM16A.
Western blot analysis
Samples were gathered and protein was sequentially
extracted according to the instructions of the Cell Lysis Buffer
for Western and IP protein extraction kit (Beyotime Institute of
Biotechnology, Shanghai, China). Protein concentrations in the
samples were quantified by bicinchoninic acid protein assay using
the BCA Protein assay kit (Beyotime Institute of Biotechnology).
Protein (50 µg) was loaded into each lane and separated by
10% SDS-PAGE (Beyotime Institute of Biotechnology) by
electrophoresis, and transferred at 200 mA for 1.5 h onto a
polyvinylidene fluoride membrane (EMD Millipore, Billerica, MA,
USA). After blocking in blocking buffer [PBS (pH 7.4) 5% skimmed
milk powder and 0.05% Tween-20] for 1 h at room temperature, the
membrane was incubated with primary and secondary antibodies.
Rabbit polyclonal TMEM16A antibody was obtained from Novus
Biologicals, LLC (Littleton, CO, USA; cat. no. NBP1-60076;
dilution, 1:500). Rabbit polyclonal MLC (cat. no. 3672; dilution,
1:1,000), mouse monoclonal phosphorylated (p)-MLC (cat. no. 3675;
dilution, 1:1,000), rabbit polyclonal MYPT1 (cat. no. 2634;
dilution, 1:1,000) and rabbit polyclonal p-MYPT1 (cat. no. 5163;
dilution, 1:1,000), were from Cell Signaling Technology, Inc.
(Danvers, MA, USA). Mouse monoclonal GAPDH as purchased from Santa
Cruz Biotechnology, Inc. (Dallas, TX, USA; cat. no. sc-365062;
dilution, 1:2,000). Horseradish peroxidase-conjugated goat
anti-rabbit (cat. no. sc-2004; dilution, 1:1,000) and goat
anti-mouse (cat. no. sc-2005; dilution, 1:1,000) immunoglobulin G
antibodies were purchased from Santa Cruz Biotechnology, Inc.
Detection was performed with the Immobilon Western Chemiluminescent
HRP Substrate system (EMD Millipore).
RhoA activity assay
The change in RhoA activity results from the shift
between the active GTP-bound state and the inactive guanosine
diphosphate (GDP)-bound state. A RhoA Activation assay kit (Abcam,
Cambridge, MA, USA) was used to detect the GTP-RhoA/RhoA ratio in
the present study. The experiment was conducted according to the
manufacturer's instructions. Briefly, BASMCs were washed twice with
ice-cold PBS and lysed in the supplied lysis buffer (1 mM
phenylmethanesulfonyl fluoride, 10 µg/ml leupeptin and 10
µg/ml aprotinin were added just prior to usage). Cell
lysates were quantified as described above and total RhoA detection
was performed using western blotting. For GTP-RhoA detection, the
lysates were incubated with anti-active RhoA mouse monoclonal
antibody and protein A/G agarose beads at 4°C in an end-over-end
mixer (Shanghai Xiyuan Technology Development Co., Ltd., Shanghai,
China) for 1 h. The beads were subsequently collected by
centrifugation at 5,000 × g for 1 min at 4°C and washed three times
with assay/lysis buffer, 10 sec at ~25°C. The pull-down supernatant
was then subjected to western blot analysis. An anti-RhoA antibody
served as the primary antibody and the secondary antibody was
anti-rabbit (included in the RhoA activation assay kit).
Statistical analysis
Statistical analyses were performed using GraphPad
Prism 5.0 (GraphPad Software, Inc., La Jolla, CA, USA). All data
were expressed as mean ± standard error of the mean, Student's
t-test was used to test for differences between two groups, and
one-way analysis of variance was used to test for differences among
the treatment groups followed by the Bonferroni multiple comparison
post hoc test. P<0.05 was considered to indicate a statistically
significant difference.
Results
TMEM16A activity inhibition decreases Ang
II-induced constriction in rat basilar arteries
Constriction of rat basilar artery in response to
Ang II has previously been observed (14). The effect of a specific TMEM16A
inhibitor, T16A-inhA01 on varying concentrations of Ang II induced
basilar artery constriction (diameter change, %) was assessed in
the present study. T16A-inhA01 (10 µM) partly reversed the
Ang II-induced reduction in diameter of basilar arteries at 10
(P<0.01) and 100 nM (P<0.01) and Rho kinase inhibitor,
Y-27632 abolished the effect of Ang II (Fig. 1A) at 0.01, 0.1 (P<0.01), 1, 10
and 100 nM (P<0.05). Ang II (100 nM) was used in subsequent
experiments. Similar results were observed in 2k2c hypertensive
rats, and Ang II-induced constriction was gradually decreased in
2k2c hypertension rats (Fig. 1B).
Furthermore, it was found that the T16A-inhA01-induced reduction in
basilar artery constriction (ΔDiameter; % change) in the presence
of 100 nM Ang II gradually decreased during the development of
hypertension at 8 weeks (P<0.05) and 12 weeks (P<0.05;
Fig. 1C), indicating less
involvement of TMEM16A during progression of hypertension, which
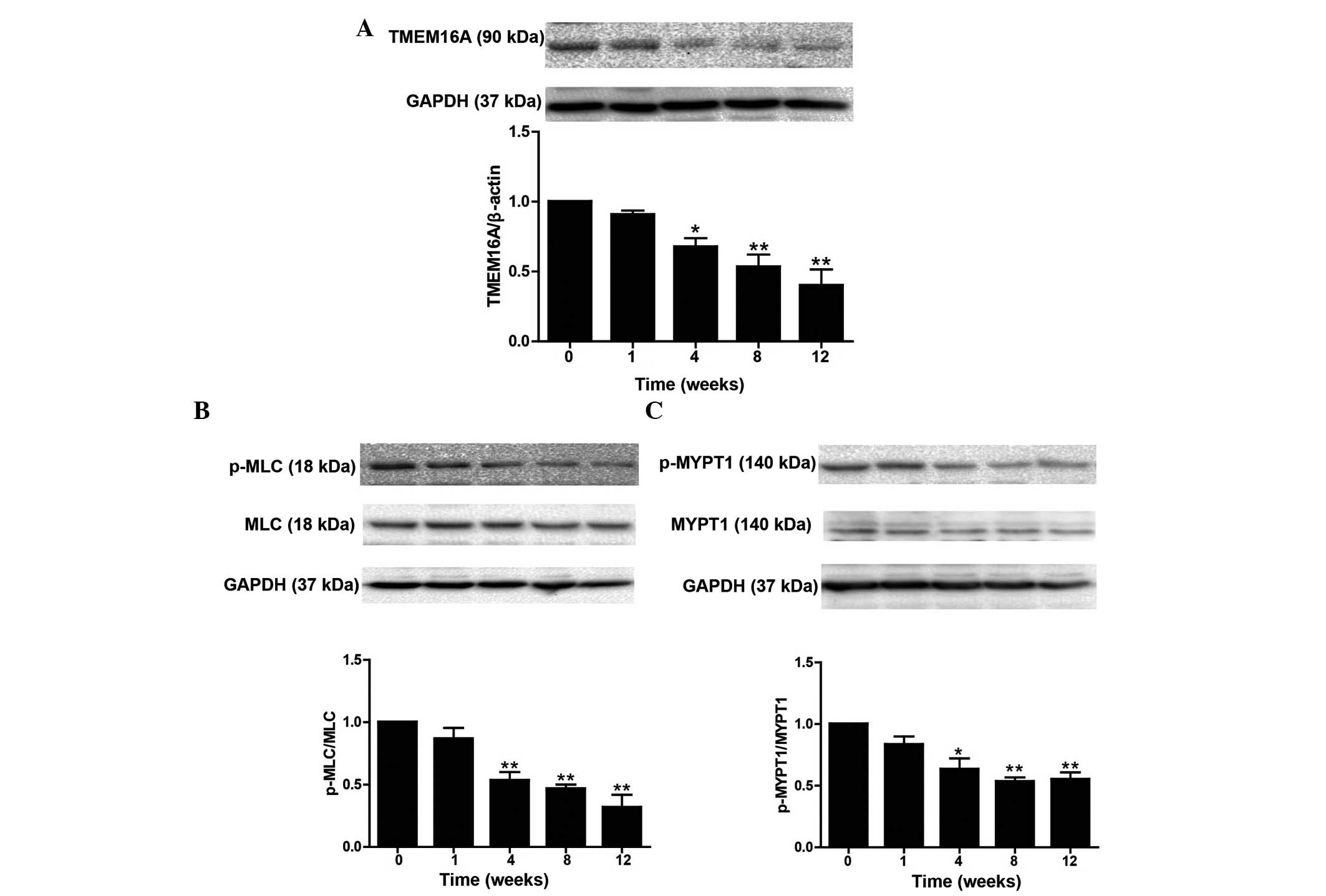
was consistent with the decreased level of TMEM16A protein
expression in basilar arteries at 4 (P<0.05), 8 (P<0.01) and
12 weeks (P<0.01; Fig. 2A).
Phosphorylation of MLC and MYPT1 in basilar arteries from the
hypertensive rats was also detected; western blot analysis
demonstrated that p-MLC/MLC was significantly downregulated in a
time-dependent manner at 4, 8 and 12 weeks (all P<0.01) and
p-MYPT1/MYPT1 was also significantly downregulated at 4
(P<0.05), 8 (P<0.01) and 12 weeks (P<0.01) with the
progression of hypertension (Fig. 2B
and C), presenting further evidence of the reduced
contractility of basilar arteries. These data strongly indicate
that TMEM16A is involved in cerebral vasoconstriction during
hypertension.
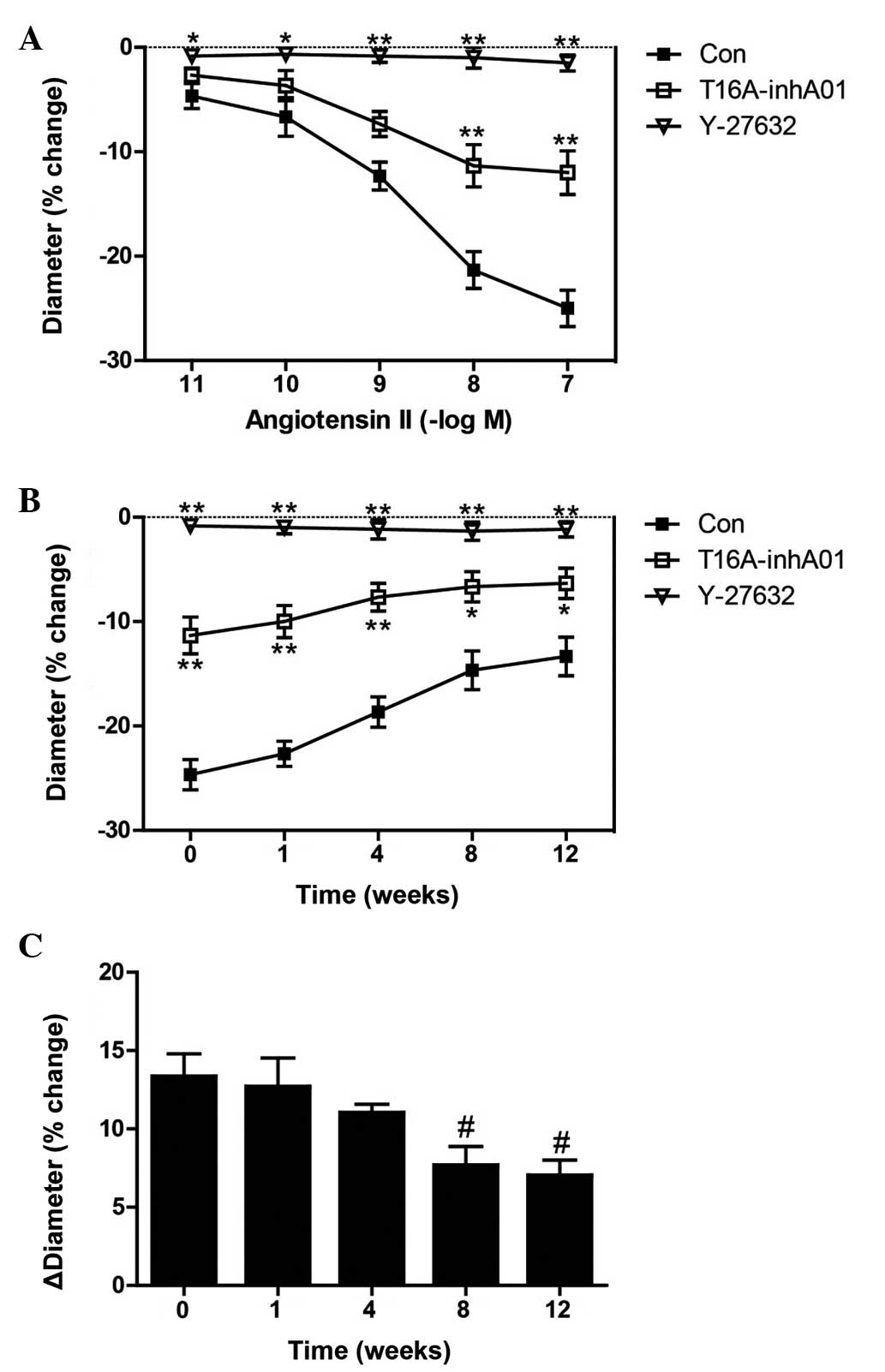 | Figure 1Inhibition of transmembrane protein
16A decreases rat basilar artery constriction in response to
varying concentrations of Ang II. (A) Constriction of the basilar
artery in response to Ang II in the absence and presence of
T16A-inhA01 (10 µM) or Y-27632 (10 µM). (B)
Constriction of the rat basilar arteries at week 0, and 1, 4, 8 and
12 weeks following 2-kidney, 2-clip surgery, in response to 100 nM
Ang II, and in the absence and presence of T16A-inhA01 or Y-27632.
(C) The bar graph indicates that T16A-inhA01 induced a decrease in
the ΔDiameter in the presence of 100 nM Ang II. *P<0.05,
**P<0.01 vs. Con; #P<0.05 vs. week 0
(n=6). ΔDiameter, basilar artery diameter; Ang II, angiotensin II;
Con, control. |
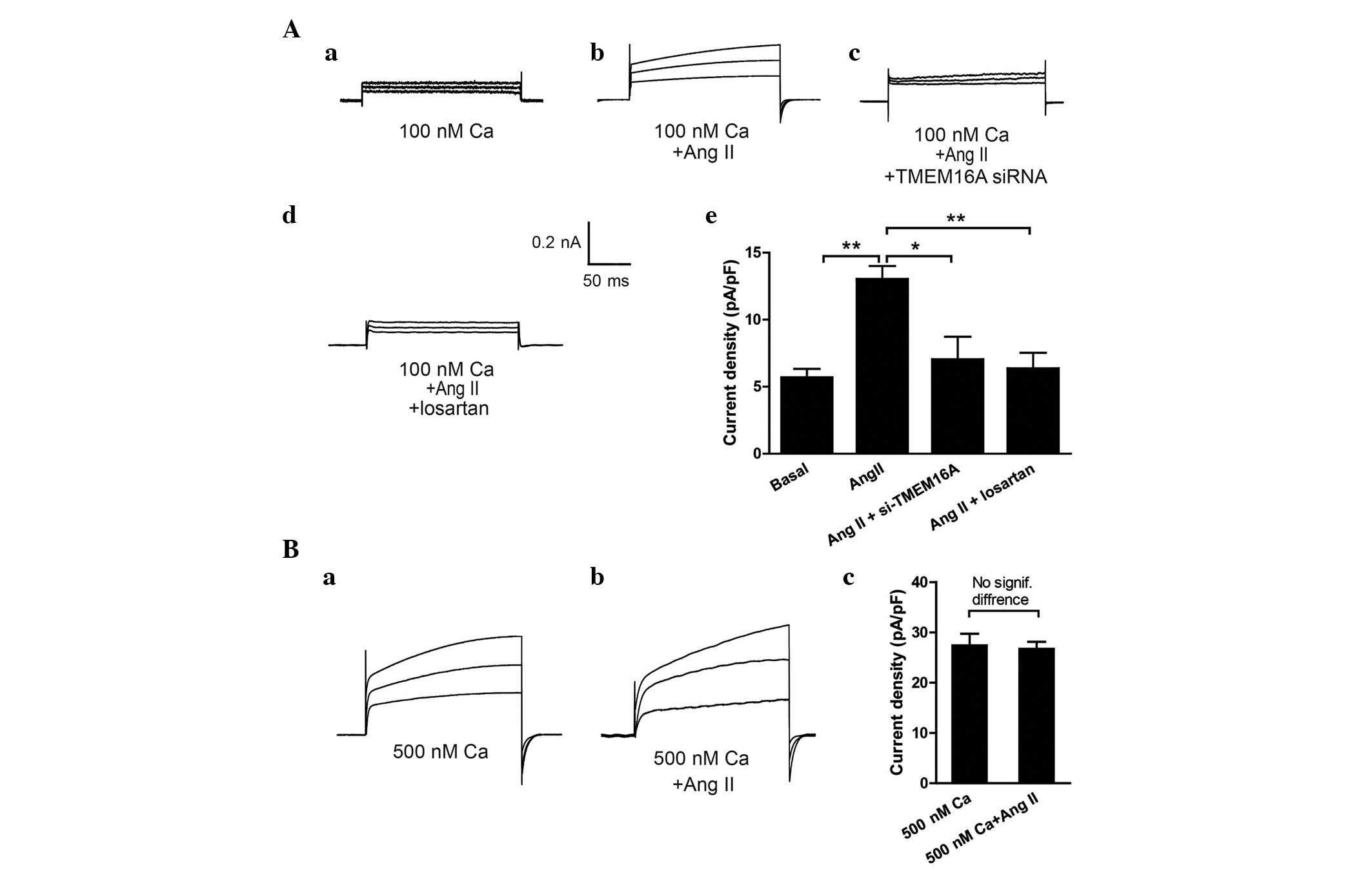
Ang II elicits a TMEM16A-mediated CaCC
current in BASMCs
In VSMCs, Ang II stimulates
Ca2+-dependent Cl− channel
(ICl.Ca) activity via increasing
[Ca2+]i (21,22).
Ca2+ -induced ICl.Ca (CaCCs) in BASMCs was
reported previously and TMEM16A was demonstrated to be a critical
component of CaCCs in basilar arteries (19). To investigate whether Ang II
directly elicits TMEM16A-mediated CaCC channels, the effect of 100
nM Ang II on 100 nM [Ca2+]i-induced
ICl.Ca (basal ICl.Ca) and 500 nM
[Ca2+]i-induced ICl.Ca in BASMCs
was analyzed. As shown in Fig. 3,
Ang II enhanced basal ICl.Ca to ~0.2 nA (P<0.01;
Fig. 3Aa and Ab) and this increase
was abolished when cells were pretreated with TMEM16A siRNA
(P<0.05) and 10 µM angiotensin type 1 (AT1) receptor
blocker, losartan (P<0.01; Fig. 3Ac
and Ad). In addition, it was demonstrated that Ang II did not
enhance the 500 nM [Ca2+]i-induced
ICl.Ca further (Fig. 3Ba
and Bb), indicating that this Ang II-elicited current was
contained in TMEM16A-mediated CaCCs.
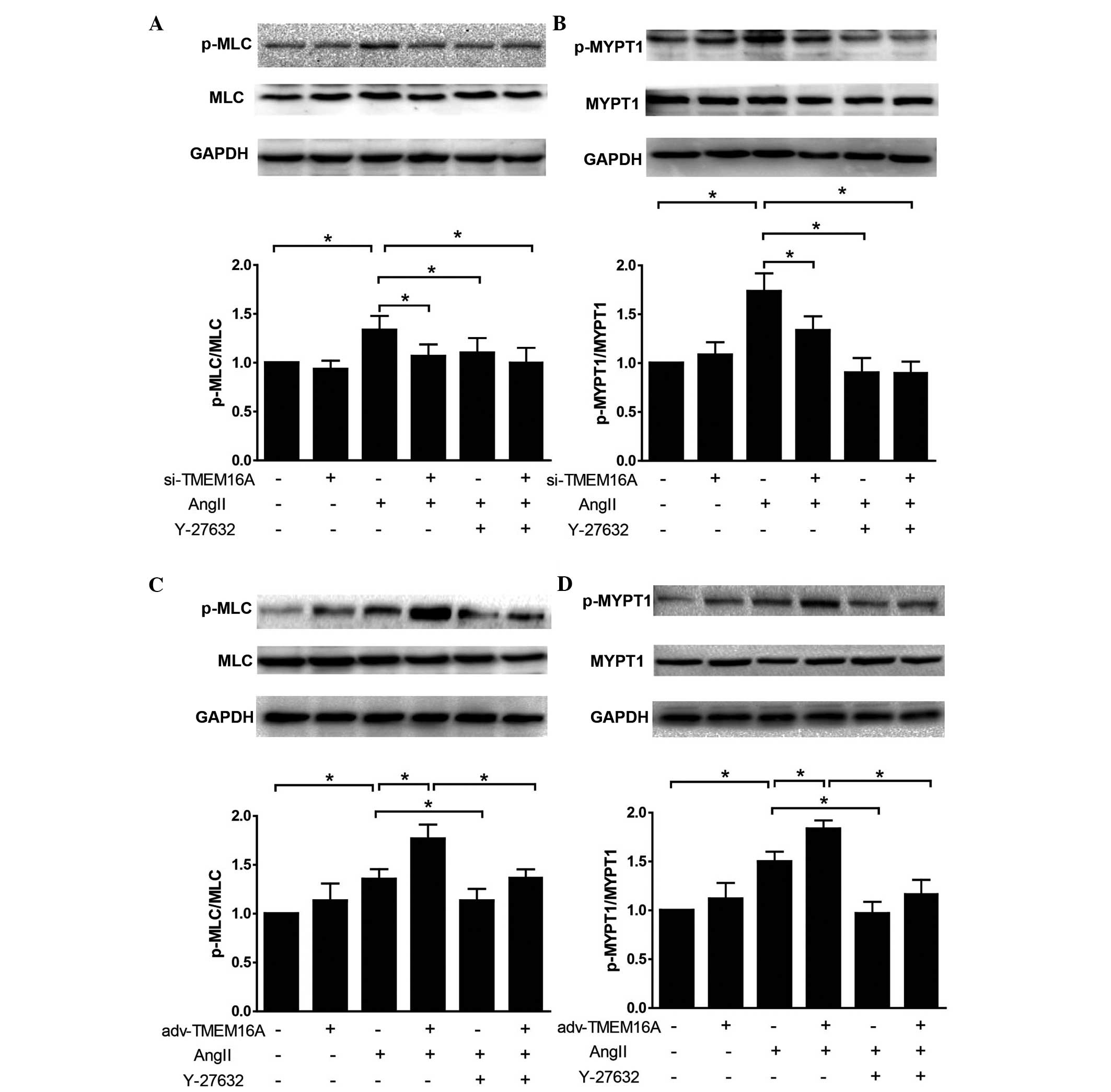
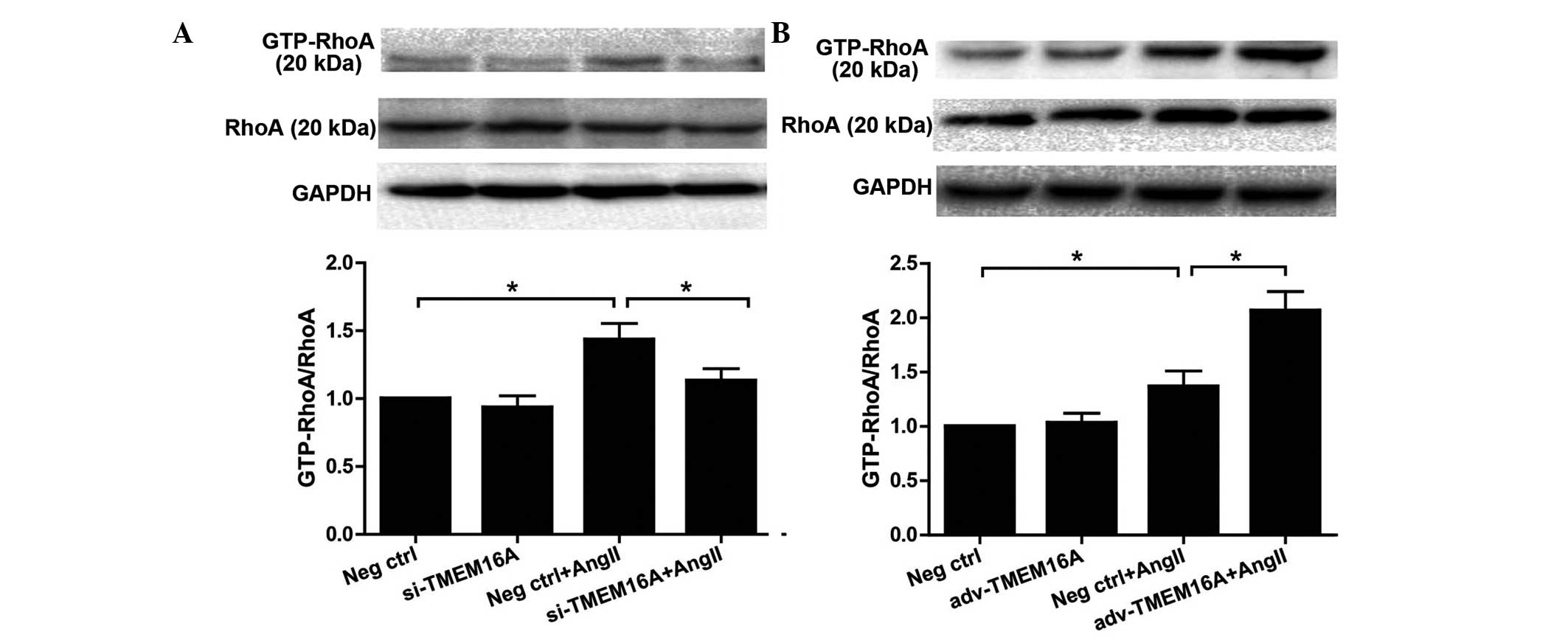
Effect of TMEM16A expression on
phosphorylation of MLC and MYPT1 in BASMCs in response to Ang II
and the influence of Y-27632
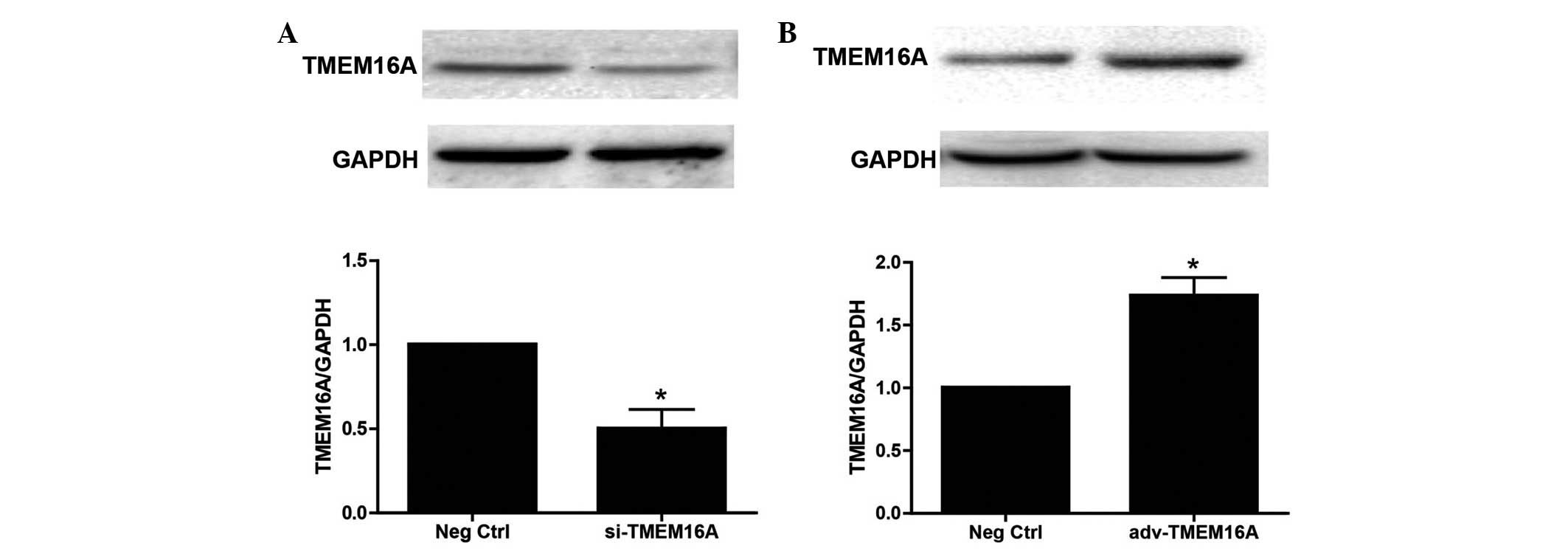
si-TMEM16A and adv-TMEM16A were evaluated in
cultured BASMCs (Fig. 4) TMEM16A
expression levels were significantly decreased by si-TMEM16A
(P<0.01) and increased by adv-TMEM16A. They were then used to
test the effect of TMEM16A expression on cell contractility in
response to Ang II. Western blot analysis demonstrated that
downregulation of TMEM16A by si-TMEM16A decreased Ang II-induced
phosphorylation of MLC and MYPT1 (Fig.
5A and B), while upregulation of TMEM16A by adv-TMEM16A
increased Ang II-induced phosphorylation of MLC (P<0.01) and
MYPT1 (P<0.01; Fig. 5C and D).
As shown in Fig. 1 and consistent
with a previous study (14), Ang
II-induced vasoconstriction is mediated to a great extent by ROCK.
To investigate whether TMEM16A affects Ang II cell contractility
via ROCK, a ROCK inhibitor, Y-27632 was applied to BASMCs treated
with si-TMEM16A or adv-TMEM16A, and the data demonstrated that
Y-27632 reversed the increase of contractility caused by
overexpression of TMEM16A (P<0.01; Fig. 5C and D), indicating that TMEM16A
regulates Ang II-induced contraction of SMCs by affecting the
RhoA/ROCK signaling pathway.
TMEM16A enhances Ang II-induced
phosphorylation of MYPT1 and MLC in BASMCs by increasing RhoA
activity
As shown in Fig. 1A and
B, and Fig. 5C and D, TMEM16A
regulated smooth muscle contraction in response to Ang II by
affecting the RhoA/ROCK signaling pathway, therefore, the direct
effect of TMEM16A on Ang II-induced RhoA activation was evaluated
in BASMCs. BASMCs were pretreated with si-TMEM16A or adv-TMEM16A
and then GTP-RhoA; the activated RhoA was detected following
treatment of 100 nM Ang II for 5 min. Western blotting showed that
downregulation of TMEM16A decreased the Ang II-induced GTP-RhoA
increase (P<0.01; Fig. 6A),
while upregulation of TMEM16A further increased Ang II-induced
augmentation of GTP-RhoA (P<0.01; Fig. 6B).
Discussion
The results of the present study demonstrate that
TMEM16A participates in basilar artery constriction via the
RhoA/ROCK signaling pathway. The following were evidenced by the
current study: i) Cerebral vascular contractility decreased in the
2k2c hypertensive rat model, which was indicated by the decreased
basilar artery constriction in response to Ang II, and by decreased
phosphorylation of MLC and MYPT1; ii) the TMEM16A inhibitor,
T16A-inhA01 partly inhibited Ang II-induced basilar artery
constriction, and the inhibition ratio was decreasing as the
hypertension progressed, in parallel with a decline in the level of
TMEM16A protein expression; iii) Ang II elicited CaCC in BASMCs,
which was blocked by si-TMEM16A and the AT1 receptor inhibitor,
losartan. Notably, Ang II did not further enhance the 500 nM
[Ca2+]i-activated ICl.Ca, which
has been demonstrated as a TMEM16A-mediated CaCC in BASMCs,
indicating that the Ang II-elicited current and the 500 nM
[Ca2+]i-induced current were mediated by the
same channel, TMEM16A. The data from the present study provide the
first direct evidence of the association between Ang II and
activation of the TMEM16A channel in cerebral SMCs; and iv) TMEM16A
overexpression increased Ang II-induced phosphorylation of MYPT1
and MLC by regulating RhoA activation in BASMCs. TMEM16A
downregulation decreased Ang II-induced phosphorylation of MYPT1
and MLC by regulating RhoA activation in BASMCs.
In the circulatory system, vascular resistance is
critical for regulating partial constriction. Small resistance
arteries (such as mesenteric, renal and basilar arteries) constrict
when subjected to KCl (depolarization), which increases
intraluminal pressure and levels of vasoconstrictors, such as
endothelin 1 and Ang II (23). The
mechanisms of various stimuli (including endothelin-1,
5-hydroxytryptamine and thromboxane A2) have been investigated in
past decades, and the present study focuses on cerebral
vasoconstriction induced by Ang II (24–26).
Ang II is the primary effector pleiotropic hormone of the RAS
cascade, which mediates physiological control of electrolyte
balance and blood pressure (27).
Ang II exerts its effect via activation of two receptor subtypes,
AT1 and AT2 (28,29); the AT1 receptor is noteworthy, as
it is critical in regulating vessel constriction and blood pressure
(30).
Earlier studies of cerebral arteries have
demonstrated the vasoconstrictor effects of Ang II on vascular tone
(31,32), and the effects are generally
considered to result from the activation of AT1 receptors, which in
turn stimulate phospholipase C (PLC) and phospholipase A2 (PLA2).
PLC and PLA2 then generate inositol trisphosphate/Ca2+
and diacylglycerol/protein kinase C, which activate multiple
downstream signaling cascades that result in SMC contraction
(33). Ang II has been reported to
affect numerous ion channels, such as the large conductance
Ca2+-activated K+ channel, a key determinant
of vascular tone (34), TRP cation
channels (35) and
voltage-dependent Ca2+ channels (36). In the present study, the influence
of Ang II on CaCCs was elucidated in BASMCs. Previous studies
hypothesized TMEM16A to be an important component of CaCCs
(20,37,38)
and its function was then widely investigated. It has been reported
that TMEM16A is critical in Cl− secretion of epithelial
cells of the airways (20,39), SMC proliferation and the nervous
system by controlling the excitability of various neurons. A strong
association between CaCC activity and the concentration of free
Ca2+ has been demonstrated in previous studies (40,41).
Ang II is involved in the regulation of intracellular
Ca2+, therefore, the present study hypothesized whether
Ang II influenced ICl.Ca in BASMCs. The data indicates
that Ang II directly evoked TMEM16A-mediated CaCCs and the
inhibition of which suppressed constriction of the basilar
arteries, indicating that TMEM16A may be involved in Ang II-induced
cerebral vasoconstriction. In addition, the effect of TMEM16A
inhibitor, T16A-inhA01 [a compound that inhibited CaCC currents in
TMEM16A-transfected Fisher rat thyroid cells with a half maximal
inhibitory concentration of ~1 µM (42)], on the constriction of basilar
arteries in response to Ang II was analyzed in the present study.
The data showed that inhibition of TMEM16A reduced Ang II-induced
basilar artery constriction, and the impact of the inhibitor was
abrogated in the hypertensive rats. Ang II acts as a
vasoconstrictor for acute stimulation and, in the long term, causes
vascular remodeling. The increase of plasma Ang II levels in
hypertensive rats is attributed to vascular remodeling (19) and TMEM16A expression levels
decrease during SMC proliferation. However, this is a different
process to vascular contraction, which is the focus of the current
study. The decline of TMEM16A observed during hypertension in the
present study illustrates our hypothesis of the role of TMEM16A in
vasoconstriction. Subsequent findings in cultured BASMCs
demonstrated that Ang II-induced phosphorylation of MYPT1 and MLC
were upregulated by TMEM16A, which confirmed our hypothesis that
TMEM16A is involved in cerebral vascular tone.
The small GTPase, Rho and ROCK regulate vascular
smooth muscle contraction and blood pressure (43). Once Rho is activated by agonists of
receptors coupled to cell membrane G proteins, such as Ang II and
phenylephrine, it then activates ROCK. Activated ROCK
phosphorylates MYPT1, a subunit of MLC phosphatase, which is then
inhibited, leading to phosphorylation of MLC stimulating vascular
smooth muscle contraction and cell migration (44). Ang II-induced vasoconstriction in
basilar arteries was demonstrated to be predominantly mediated by
ROCK using the ROCK inhibitor, Y-27632 (14). In a previous study, ROCK inhibitor,
Thiazovivin reversed Ang II-induced MYPT1 and MLC phosphorylation
(45); therefore, whether the
RhoA/ROCK signaling pathway mediated the effect of TMEM16A on Ang
II-induced BASMC contraction was investigated in the current study.
The results demonstrated that this was the case and, furthermore,
it was found that TMEM16A expression regulated Ang II-induced
activation of RhoA. These data are the first, to the best of our
knowledge, to provide a mechanism by which TMEM16A contributes to
Ang II-induced cerebral vascular constriction.
In conclusion, the current study reveals that
TMEM16A, the recently identified component of CaCCs, is involved in
Ang II-induced cerebral constriction. Decreased TMEM16A activity
and expression reduces basilar artery contractility, and this
effect of TMEM16A is mediated by affecting RhoA activation. These
data also indicate that Ang II activates TMEM16A-mediated CaCCs in
BASMCs. Therefore, TMEM16A has been demonstrated as a potent
therapeutic target for the control of vascular function,
hypertension and stroke. Future studies are required to determine
how TMEM16A affects the RhoA/ROCK signaling pathway, providing a
greater insight into the underlying mechanism.
References
|
1
|
Heinze C, Seniuk A, Sokolov MV, Huebner
AK, Klementowicz AE, Szijártó IA, Schleifenbaum J, Vitzthum H,
Gollasch M, Ehmke H, et al: Disruption of vascular Ca2+-activated
chloride currents lowers blood pressure. J Clin Invest.
124:675–686. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jackson WF: Ion channels and vascular
tone. Hypertension. 35:173–178. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mendelsohn ME: In hypertension, the kidney
is not always the heart of the matter. J Clin Invest. 115:840–844.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yonas H, Smith HA, Durham SR, Pentheny SL
and Johnson DW: Increased stroke risk preducted by compromised
cerebral blood flow reactivity. J Neurosurg. 79:483–489. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hill MA, Zou H, Potocnik SJ, Meininger GA
and Davis MJ: Invited review: Arteriolar smooth muscle
mechanotransduction: Ca(2+) signaling pathways underlying myogenic
reactivity. J Appl Physiol (1985). 91:973–983. 2001.
|
|
6
|
Earley S and Brayden JE: Transient
receptor potential channels and vascular function. Clin Sci (Lond).
119:19–36. 2010. View Article : Google Scholar
|
|
7
|
Rust MB, Faulhaber J, Budack MK, Pfeffer
C, Maritzen T, Didié M, Beck FX, Boettger T, Schubert R, Ehmke H,
et al: Neurogenic mechanisms contribute to hypertension in mice
with disruption of the K-Cl cotransporter KCC3. Circ Res.
98:549–556. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Leblanc N, Ledoux J, Saleh S, Sanguinetti
A, Angermann J, O'Driscoll K, Britton F, Perrino BA and Greenwood
IA: Regulation of calcium-activated chloride channels in smooth
muscle cells: A complex picture is emerging. Can J Physiol
Pharmacol. 83:541–556. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu MM, Lou J, Song BL, Gong YF, Li YC, Yu
CJ, Wang QS, Ma TX, Ma K, Hartzell HC, et al: Hypoxia augments the
calcium-activated chloride current carried by anoctamin-1 in
cardiac vascular endothelial cells of neonatal mice. Br J
Pharmacol. 171:3680–3692. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bulley S, Neeb ZP, Burris SK, Bannister
JP, Thomas-Gatewood CM, Jangsangthong W and Jaggar JH: TMEM16A/ANO1
channels contribute to the myogenic response in cerebral arteries.
Circ Res. 111:1027–1036. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kobori H, Nangaku M, Navar LG and
Nishiyama A: The intrarenal renin-angiotensin system: From
physiology to the pathobiology of hypertension and kidney disease.
Pharmacol Rev. 59:251–287. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang Z, Wang M, Xue SJ, Liu DH and Tang
YB: Simvastatin ameliorates angiotensin II-induced endothelial
dysfunction through restoration of Rho-BH4-eNOS-NO pathway.
Cardiovasc Drugs Ther. 26:31–40. 2012. View Article : Google Scholar
|
|
13
|
Hilgers RH, Todd J Jr and Webb RC:
Increased PDZ-RhoGEF/RhoA/Rho kinase signaling in small mesenteric
arteries of angiotensin II-induced hypertensive rats. J Hypertens.
25:1687–1697. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Faraci FM, Lamping KG, Modrick ML, Ryan
MJ, Sigmund CD and Didion SP: Cerebral vascular effects of
angiotensin II: New insights from genetic models. J Cereb Blood
Flow Metab. 26:449–455. 2006. View Article : Google Scholar
|
|
15
|
Davis AJ, Shi J, Pritchard HA, Chadha PS,
Leblanc N, Vasilikostas G, Yao Z, Verkman AS, Albert AP and
Greenwood IA: Potent vasorelaxant activity of the TMEM16A inhibitor
T16A (inh)-A01. Br J Pharmacol. 168:773–784. 2013. View Article : Google Scholar :
|
|
16
|
Committee for the Update of the Guide for
the Care and Use of Laboratory Animals: Guide for the care and use
of laboratory animals. 8th edition. National Academies Press;
Washington, DC, USA:
|
|
17
|
Shi XL, Wang GL, Zhang Z, Liu YJ, Chen JH,
Zhou JG, Qiu QY and Guan YY: Alteration of volume-regulated
chloride movement in rat cerebrovascular smooth muscle cells during
hypertension. Hypertension. 49:1371–1377. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lamping KG, Wess J, Cui Y, Nuno DW and
Faraci FM: Muscarinic (M) receptors in coronary circulation:
Gene-targeted mice define the role of M2 and M3 receptors in
response to acetylcholine. Arterioscler Thromb Vasc Biol.
24:1253–1258. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang M, Yang H, Zheng LY, Zhang Z, Tang
YB, Wang GL, Du YH, Lv XF, Liu J, Zhou JG and Guan YY:
Downregulation of TMEM16A calcium-activated chloride channel
contributes to cerebrovascular remodeling during hypertension by
promoting basilar smooth muscle cell proliferation. Circulation.
125:697–707. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang YD, Cho H, Koo JY, Tak MH, Cho Y,
Shim WS, Park SP, Lee J, Lee B, Kim BM, et al: TMEM16A confers
receptor-activated calcium-dependent chloride conductance. Nature.
455:1210–1215. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
White CR, Elton TS, Shoemaker RL and Brock
TA: Calcium-sensitive chloride channels in vascular smooth muscle
cells. Proc Soc Exp Biol Med. 208:255–262. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guibert C, Marthan R and Savineau JP:
Oscillatory Cl-current induced by angiotensin II in rat pulmonary
arterial myocytes: Ca2+ dependence and physiological implication.
Cell Calcium. 21:421–429. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bagi Z, Feher A, Cassuto J, Akula K,
Labinskyy N, Kaley G and Koller A: Increased availability of
angiotensin AT 1 receptors leads to sustained arterial constriction
to angiotensin II in diabetes-role for Rho-kinase activation. Br J
Pharmacol. 163:1059–1068. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kinlay S, Behrendt D, Wainstein M,
Beltrame J, Fang JC, Creager MA, Selwyn AP and Ganz P: Role of
endothelin-1 in the active constriction of human atherosclerotic
coronary arteries. Circulation. 104:1114–1118. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu JQ and Folz RJ: Extracellular
superoxide enhances 5-HT-induced murine pulmonary artery
vasoconstriction. Am J Physiol Lung Cell Mol Physiol.
287:L111–L118. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gao YJ and Lee RM: Hydrogen peroxide
induces a greater contraction in mesenteric arteries of
spontaneously hypertensive rats through thromboxane A(2)
production. Br J Pharmacol. 134:1639–1646. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hall JE: The kidney, hypertension, and
obesity. Hypertension. 41:625–633. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hein L: Genetic deletion and
overexpression of angiotensin II receptors. J Mol Med (Berl).
76:756–763. 1998. View Article : Google Scholar
|
|
29
|
Crowley SD, Tharaux PL, Audoly LP and
Coffman TM: Exploring type I angiotensin (AT1) receptor functions
through gene targeting. Acta Physiol Scand. 181:561–570. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ito K, Hirooka Y, Nakano M, Honda N,
Matsukawa R and Sunagawa K: Role of hypothalamic angiotensin type 1
receptors in pressure overload-induced mineralocorticoid receptor
activation and salt-induced sympathoexcitation. Hypertens Res.
36:513–519. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Stenman E and Edvinsson L: Cerebral
ischemia enhances vascular angiotensin AT1 receptor-mediated
contraction in rats. Stroke. 35:970–974. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Toda N, Ayaziki K and Okamura T:
Modifications by endogenous prostaglandins of angiotensin
II-induced contractions in dog and monkey cerebral and mesenteric
arteries. J Pharmacol Exp Ther. 252:374–379. 1990.PubMed/NCBI
|
|
33
|
Billington CK and Penn RB: Signaling and
regulation of G protein-coupled receptors in airway smooth muscle.
Respir Res. Mar 14–2003.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lu T, Zhang DM, Wang XL, He T, Wang RX,
Chai Q, Katusic ZS and Lee HC: Regulation of coronary arterial BK
channels by caveolae-mediated angiotensin II signaling in diabetes
mellitus. Circ Res. 106:1164–1173. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ilatovskaya DV, Palygin O,
Chubinskiy-Nadezhdin V, Negulyaev YA, Ma R, Birnbaumer L and
Staruschenko A: Angiotensin II has acute effects on TRPC6 channels
in podocytes of freshly isolated glomeruli. Kidney Int. 86:506–514.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Do KH, Kim MS, Kim JH, Rhim BY, Lee WS,
Kim CD and Bae SS: Angiotensin II-induced aortic ring constriction
is mediated by phosphatidylinositol 3-kinase/L-type calcium channel
signaling pathway. Exp Mol Med. 41:569–576. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Caputo A, Caci E, Ferrera L, Pedemonte N,
Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O and
Galietta LJ: TMEM16A, a membrane protein associated with
calcium-dependent chloride channel activity. Science. 322:590–594.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Schroeder BC, Cheng T, Jan YN and Jan LY:
Expression cloning of TMEM16A as a calcium-activated chloride
channel subunit. Cell. 134:1019–1029. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu B, Linley JE, Du X, Zhang X, Ooi L,
Zhang H and Gamper N: The acute nociceptive signals induced by
bradykinin in rat sensory neurons are mediated by inhibition of
M-type K+ channels and activation of Ca2+-activated Cl-channels. J
Clin Invest. 120:1240–1252. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Frings S, Reuter D and Kleene SJ: Neuronal
Ca2+-activated Cl-channels-homing in on an elusive channel species.
Prog Neurobiol. 60:247–289. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hartzell C, Putzier I and Arreola J:
Calcium-activated chloride channels. Annu Rev Physiol. 67:719–758.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Namkung W, Phuan PW and Verkman AS:
TMEM16A inhibitors reveal TMEM16A as a minor component of
calcium-activated chloride channel conductance in airway and
intestinal epithelial cells. J Biol Chem. 286:2365–2374. 2011.
View Article : Google Scholar :
|
|
43
|
Uehata M, Ishizaki T, Satoh H, Ono T,
Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M
and Narumiya S: Calcium sensitization of smooth muscle mediated by
a Rho-associated protein kinase in hypertension. Nature.
389:990–994. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sebbagh M, Renvoizé C, Hamelin J, Riché N,
Bertoglio J and Bréard J: Caspase-3-mediated cleavage of ROCK I
induces MLC phosphorylation and apoptotic membrane blebbing. Nat
Cell Biol. 3:346–352. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Cao X, Luo T, Luo X and Tang Z:
Resveratrol prevents AngII-induced hypertension via AMPK activation
and RhoA/ROCK suppression in mice. Hypertens Res. 37:803–810. 2014.
View Article : Google Scholar : PubMed/NCBI
|