Introduction
Non-alcoholic fatty liver disease (NAFLD), which
includes a broad spectrum of liver pathologies ranging from simple
steatosis to non-alcoholic steatohepatitis (NASH), can lead to
liver inflammation, cirrhosis and hepatocellular carcinoma
(1). NAFLD is one of the most
common chronic liver diseases worldwide, with an estimated
prevalence of 20–30% in Western countries, reaching 75–100% in
obese individuals (2). In China,
the incidence of NAFLD has also increased rapidly in recent years.
However, the exact mechanism underlying the progression of NAFLD
remains unclear and an effective treatment has not yet been
discovered.
The 'two-hit' mechanism is a classical theory used
to explain the pathogenesis of NAFLD (3). The first 'hit' leads to the
deposition of triglycerides and steatosis, and sensitizes the liver
to the second 'hit', which involves the release of inflammatory
cytokines and oxidative stress. Eventually, these factors lead to
the development of NASH and cirrhosis. However, there is increasing
evidence that NAFLD is the hepatic manifestation of the metabolic
syndrome (4). Hepatic insulin
resistance (IR) is the key pathophysiological hallmark of the
metabolic syndrome, which is important for the progression of NAFLD
(5). Insulin signaling pathways,
mediated by impaired tyrosine phosphorylation of insulin receptor
substrate (IRS) (6), have
previously been reported to be important for the development of IR.
The levels of tyrosine-phosphorylated IRS are regulated by
serine-threonine kinase and are negatively correlated with its
serine-phosphorylated form. In addition, inflammatory signals, such
as IkappaB kinase (IKK)/nuclear factor-κB (NF-κB) and c-Jun
N-terminal protein kinase 1, or proinflammatory cytokines,
including interleukin-6 (IL-6) and tumor necrosis factor α (TNF-α),
are involved in IR via the phosphorylation of IRS1 (7,8). A
previous study indicated that oxidative stress may also induce IR
(9). The interaction between
oxidative stress and IR may accelerate cellular injury,
inflammation and even lead to hepatic fibrosis (10). Furthermore, continual increase of
reactive oxygen species (ROS) levels may activate serine-threonine
kinase cascades, leading to reductions in tyrosine phosphorylated
IRS, consequently resulting in IR (11).
Nuclear erythroid 2-related factor 2 (Nrf2)
regulates the expression of various antioxidant genes and
detoxification enzymes against oxidative and electrophilic stress
(12), and has been regarded as a
novel therapeutic target in liver diseases, including NAFLD
(13). Under homeostatic
conditions, Nrf2 is sequestered in the cytosol by binding to
Kelch-like ECH-associated protein (Keap1) and forming a Nrf2-Keap1
complex. However, when exposed to oxidative or electrophilic
stress, Nrf2 dissociates with Keap1, translocates into the nucleus,
and activates numerous downstream genes (14). It has been demonstrated that Nrf2
deletion may aggravate inflammation and promote progression of
NAFLD to NASH by inducing the nuclear translocation of NF-κB p65
protein and the expression of IL-1β, TNF-α and cyclooxygenase-2 in
transgenic mice fed a methionine-choline deficient (MCD) diet
(15). However, to the best of our
knowledge, the effects of Nrf2 on hepatic IR in NAFLD have yet to
be elucidated.
The present study investigated the biological
function and underlying molecular mechanisms of Nrf2 in the
development and progression of nutritional steatohepatitis.
Initially, it was demonstrated that Nrf2 knockdown led to obesity
and steatohepatitis induced by a high-fat diet (HFD). Nrf2 deletion
may influence the initiation of NASH by promoting hepatic IR.
Finally, it was confirmed that activation of NF-κB and its
downstream effectors IL-6 and TNF-α, via Nrf2 deficiency-mediated
oxidative stress, contributed to the induction of hepatic IR.
Materials and methods
Animals and treatment
Wild-type (WT; n=16) and Nrf2-null (n=16) male mice
(6–8 weeks old; weight, 22–24 g) with an ICR background were
purchased from the Comparative Medicine Department of Nanjing
General Hospital (Nanjing, China). The mice were housed in a
specific facility with a 12-h light/dark cycle, temperature
maintained at 18–22°C and humidity at 50–60%. The mice were allowed
to acclimate to laboratory conditions for 1 week and were given
ad libitum access to water and an ordinary diet. Thereafter,
the mice were fed either a high-fat diet (HFD; 10% lard, 2%
cholesterol, 0.5% bile salt and 87.5% base forage) or a control
diet (100% base forage) for 8 weeks and were given ad
libitum access to water (n=8/group). During the experiment, the
body weight of each mouse was determined each week prior to being
sacrificed with intraperitoneal anesthetization by 1% pentobarbital
sodium (Sigma-Aldrich, St. Louis, MO, USA) at dose of 50 mg/kg.
Subsequently, serum samples, which were obtained following
centrifugation at 1,200 × g for 10 min at 4°C, and liver tissue
specimens were collected immediately for further examination. The
liver was divided as follows: A 1/3 was fixed in an aqueous
solution of 4% formaldehyde and the remaining tissue was stored at
−80°C. Throughout the investigation, all animal experiments were
performed following the Fourth Military Medical University animal
use guidelines, and the protocols were approved by the Fourth
Military Medical University Animal Care Committee (Xi'an,
China).
Determination of liver triglyceride (TG)
concentrations
TG concentration in liver tissues was determined by
the GPO-PAP method as previously described (16), and was examined enzymatically using
commercially available kits according to the manufacturer's
protocol (Nanjing Jiancheng Bioengineering Institute, Nanjing,
China).
Histopathology
Liver tissues were fixed in 4% paraformaldehyde (pH
7.4) at 4°C for 24 h, embedded in paraffin, cut into sections ~3×2
cm, stained with hematoxylin for 15 min and subsequently with eosin
for 3 min at room temperature. The sections were analyzed under a
conventional light microscope (Olympus Corporation, Tokyo,
Japan).
Fasting blood glucose (FBG) level
determination
FBG levels were determined spectrophotometrically
using a commercial kit according to the manufacturer's protocol
(Nanjing Jiancheng Bioengineering Institute).
Intraperitoneal glucose tolerance test
(iPGTT)
iPGTT was performed to estimate IR as previously
described (17). Briefly,
following fasting overnight for 12 h, mice were injected
intraperitoneally with glucose (20%, 2 g/kg) or insulin (5 U/kg).
Glucose level was measured at 0, 15, 30, 60, and 120 min time
points from tail-bleed samples and the area under the curve (AUC)
for blood glucose was calculated.
Measurement of lipid peroxidation and
glutathione (GSH) levels
Lipid peroxidation in the liver was determined by
measuring malondialdehyde (MDA) levels using the thiobarbituric
acid method (18), and oxidative
stress was estimated by measuring GSH levels using a colorimetric
assay. Briefly, 500 mg liver tissue was homogenized in 1 ml
ice-cold phosphate-buffered saline (PBS) and was centrifuged at 500
× g for 10 min at 4°C. The levels of MDA and GSH in the
supernatants were determined, according to the manufacturer's
protocols (Nanjing Jiancheng Bioengineering Institute).
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from homogenized tissues
using TRIzol® reagent according to the manufacturer's
protocol (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA,
USA). RNA concentration was determined using a spectrophotometer at
260 nm. The RT reaction was performed using PrimeScript RT reagent
kit (TaKaRa Biotechnology, Co. Ltd., Dalian, China), 1 µg
RNA was used in the reaction at 37°C for 15 min, followed by 85°C
for 5 sec. qPCR was conducted using a SYBR Premix Ex Taq II
kit (Takara Biotechnology, Co. Ltd.) in a 20 µl reaction.
The primer sequences used were as follows: TNF-α, forward (F)
5′-CCCAGGCAGTCAGATCATCTTC-3′, reverse (R) 5′-AGCTGCCCCTCAGCTTGA-3′;
IL-6, F 5′-GGTACATCCTCGACGGCATCT-3′-and R
5′-GTGCCTCTTTGCTGCTTTCAC-3′; and GAPDH, F
5′-AGGTCGGTGTGAACGGATTTG-3′-and R 5′-TGTAGACCATGTAGTTGAGGTCA-3′.
All primers were purchased from Guangzhou RiboBio Co., Ltd.
(Guangzhou, China). The mRNA expression levels of TNF-α and IL-6
were determined using the LightCycler 480 system (Roche
Diagnostics, Basel, Switzerland). The thermocycling conditions were
as follows: 95°C for 5 min; followed by 45 cycles at 95°C for 10
sec, 58°C for 10 sec and 72°C for 10 sec; and 1 cycle at 95°C for 5
sec, 50°C for 1 min, and finally 40°C for 30 sec. Glyceraldehyde
3-phosphate dehydrogenase (GAPDH) was used as an internal
reference. Relative expression level of TNF-α and IL-6 were
calculated with normalization to GAPDH values using the
2−∆∆Cq method (19).
All of the reactions were performed in triplicate.
Enzyme-linked immunosorbent assay
(ELISA)
Liver tissues from the WT and Nrf2-null mice fed
various diets were homogenized in ice-cold PBS, and were
centrifuged at 500 × g for 10 min at 4°C. The supernatants were
used to determine TNF-α and IL-6 levels by ELISA according to the
manufacturer's protocols (Westang Biotech Co. Ltd., Shanghai,
China).
Western blot analysis
Tissues were homogenized in radioimmunoprecipitation
assay lysis buffer (Beyotime Institute of Biotechnology, Haimen,
China). Cytoplasmic and nuclear proteins were prepared from the
liver tissues using the Nuclear and Cytoplasmic Extraction kits
(Nanjing Jiancheng Bioengineering Institute) according to the
manufacturer's protocol. Western blotting was performed as
previously described (20). The
primary antibodies used were as follows: Rabbit Nrf2 (1:400; Santa
Cruz Biotechnology, Santa Cruz, CA, USA; cat. no. sc-722), rabbit
NAD(P)H quinone dehydrogenase 1 (Nqo1; 1:400; Santa Cruz
Biotechnology, Inc.; cat. no. sc-25591), rabbit NF-κB (1:400; Santa
Cruz Biotechnology, Inc; cat. no. sc-372), rabbit IRS1 (1:500; Cell
Signaling Technology, Inc., Danvers, MA, USA; cat. no. 2390),
rabbit Akt (1:1,000; Cell Signaling Technology; cat. no. 4685),
goat phosphorylated (p)-Tyr IRS1 (1:300; Santa Cruz Biotechnology,
Inc.; cat. no. sc-17200), rabbit p-Akt (1:1,000; Cell Signaling
Technology, Inc.; cat. no. 4058), rabbit p-glycogen synthase kinase
3β (p-GSK-3β; 1:800; Cell Signaling Technology, Inc.; cat. no.
9322), rabbit p-forkhead box O1 (p-FoXO1; 1:800; Cell Signaling
Technology, Inc.; cat. no. 9461) and rabbit β-actin (1:1,000; Santa
Cruz Biotechnology, Inc.; cat. no. sc-130656). For detection,
horseradish peroxidase-conjugated secondary antibodies, including
goat anti-rabbit (1:3,000; Santa Cruz Biotechnology, Inc.; cat. no.
sc-2004) or donkey anti-goat (1:3,000; Santa Cruz Biotechnology,
Inc.; cat. no. sc-2020).
Statistical analysis
For statistical analysis, the results were evaluated
by SPSS 12.0 (SPSS, Inc., Chicago, IL, USA). Experimental data are
presented as the mean ± standard error. Student's t-test was used
to compare the means between two groups. One way analysis of
variance (ANOVA), followed by Duncan's multiple range test, was
used to compare the means between more than two groups. Repeated
measures ANOVA was used to compare the differences between groups,
which have been recorded through time. P<0.05 was considered to
indicate a statistically significant difference.
Results
Deletion of Nrf2 initiates obesity and
steatohepatitis in HFD-treated mice
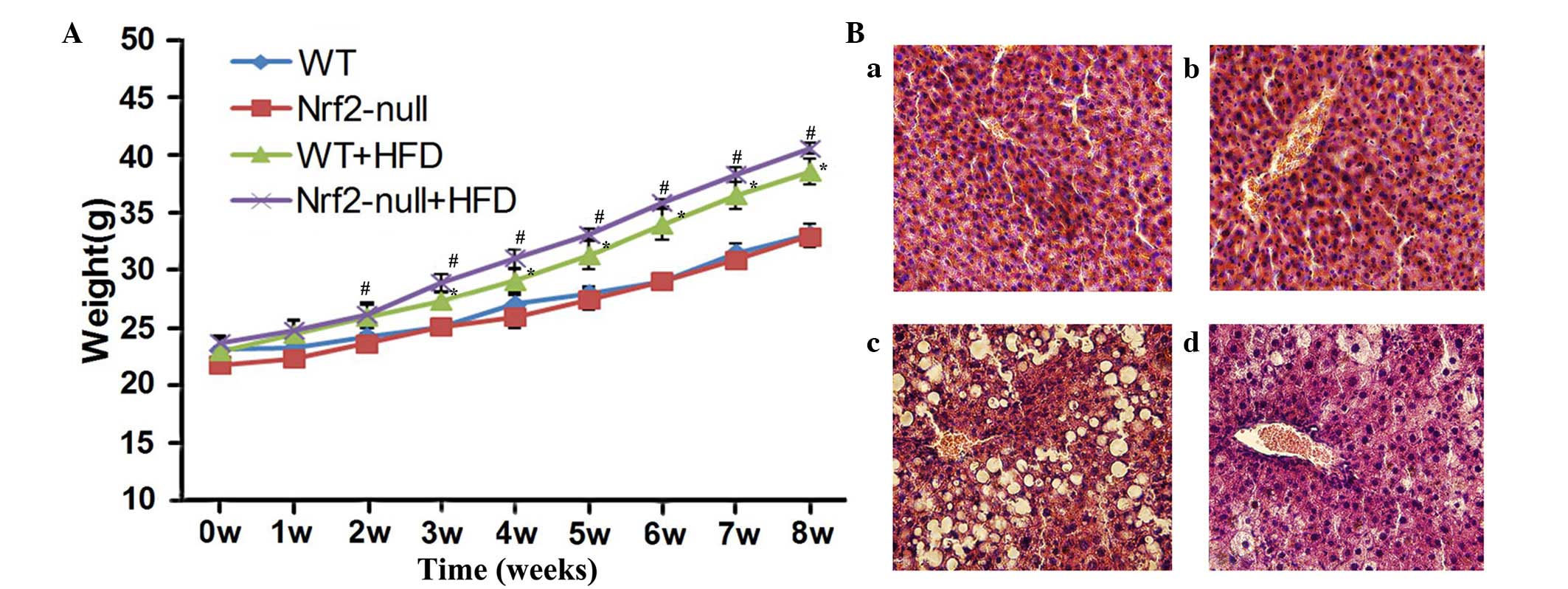
To observe the effect of HFD treatment on mice,
their body weight was measured weekly and hepatic weight was
measured after 8 weeks. The weight of WT and Nrf2-null mice fed a
HFD was significantly increased compared with the control group
(P<0.05; Fig. 1A). When fed a
HFD, Nrf2-null mice gained more weight than the WT mice; however,
there was no statistically significant difference between the two
groups (Fig. 1A). Similarly, the
weight of hepatic tissues in the Nrf2-null-HFD group was 2.60±0.20
g and was 1.92±0.07 g in the WT-HFD group, both of which were
significantly increased in comparison with their control
counterparts (P<0.05; Table I).
In addition, hepatic weight in the Nrf2-null-HFD mice was greater
than that in the WT-HFD mice (P<0.05; Table I). Conversely, the WT and Nrf2-null
mice fed a control diet experienced no significant increase in body
or hepatic weight.
 | Table ILiver weight, hepatic TG
concentration and FBG of WT and Nrf2-null groups fed a control or
HF diet for 8 weeks (n=8). |
Table I
Liver weight, hepatic TG
concentration and FBG of WT and Nrf2-null groups fed a control or
HF diet for 8 weeks (n=8).
| Parameter | Control
| HFD
|
|---|
| WT | Nrf2-null | WT | Nrf2-null |
|---|
| Liver weight
(g) | 1.00±0.08 | 0.94±0.11 | 1.92±0.07a | 2.60±0.20b,c |
| Liver TG
(mmol/l) | 3.38±0.22 | 3.59±0.46 | 6.50±0.30a | 13.16±0.94b,c |
| FBG (mmol/l) | 4.12±0.17 | 4.02±0.23 | 4.92±0.18a | 5.60±0.29b |
Histopathological analysis was performed and TG
concentration was examined in the various groups. Histopathological
examination revealed that there was a normal lobular structure and
no evidence of inflammation in the livers of mice fed the control
diet (Fig. 1B). However, hepatic
cells from WT HFD mice were evidently disordered, with increased
microvesicular lipid accumulation and minimal to mild inflammation
(Fig. 1B). Furthermore, liver
tissue samples from the Nrf2-null-HFD group exhibited disordered
hepatic structure, such as severe fat deposition, inflammatory
infiltration and hyaline degeneration in hepatic cells (Fig. 1B). However, no fibrosis was
observed in any of the groups. The liver TG concentration was
13.16±0.94 mmol/l in the Nrf2-null-HFD mice and 6.50±0.30 mmol/l in
the WT-HFD mice, which was significantly higher compared with both
control groups (P<0.05; Table
I). Notably, TG concentration in hepatic tissues from
Nrf2-null-HFD mice was much higher than that from WT-HFD mice
(P<0.05; Table I). These data
indicate that Nrf2 deficiency may promote the development and
progression of obesity and steatohepatitis in HFD-treated mice.
Nrf2 deletion promotes HFD-induced
hepatic IR
To explore the importance of Nrf2 deletion in IR,
FBG levels from the blood serum were determined, and an iPGTT was
performed in the treatment groups. There was a notable increase in
serum FBG levels in the WT mice (4.92±0.18 mmol/l) and Nrf2-null
mice (5.60±0.29 mmol/l) fed a HFD compared with mice fed the
control diet (P<0.05; Table I).
Although serum FBG levels in the Nrf2-null-HFD mice were slightly
higher than in the WT-HFD mice, no significant difference was
detected (Table I). In addition,
plasma glucose levels during iPGTT were significantly increased at
15, 30 and 60 min when the HFD-treated groups were compared with
the control groups (P<0.05; Fig.
2A). In addition, the AUC of iPGTT, was used as an indication
of impaired glucose tolerance level, and HFD induced a
significantly increased area (P<0.05; Fig. 2B). Glucose levels and HFD-induced
AUC of iPGTT exhibited greater significant increases in the
Nrf2-null group than in the WT group.
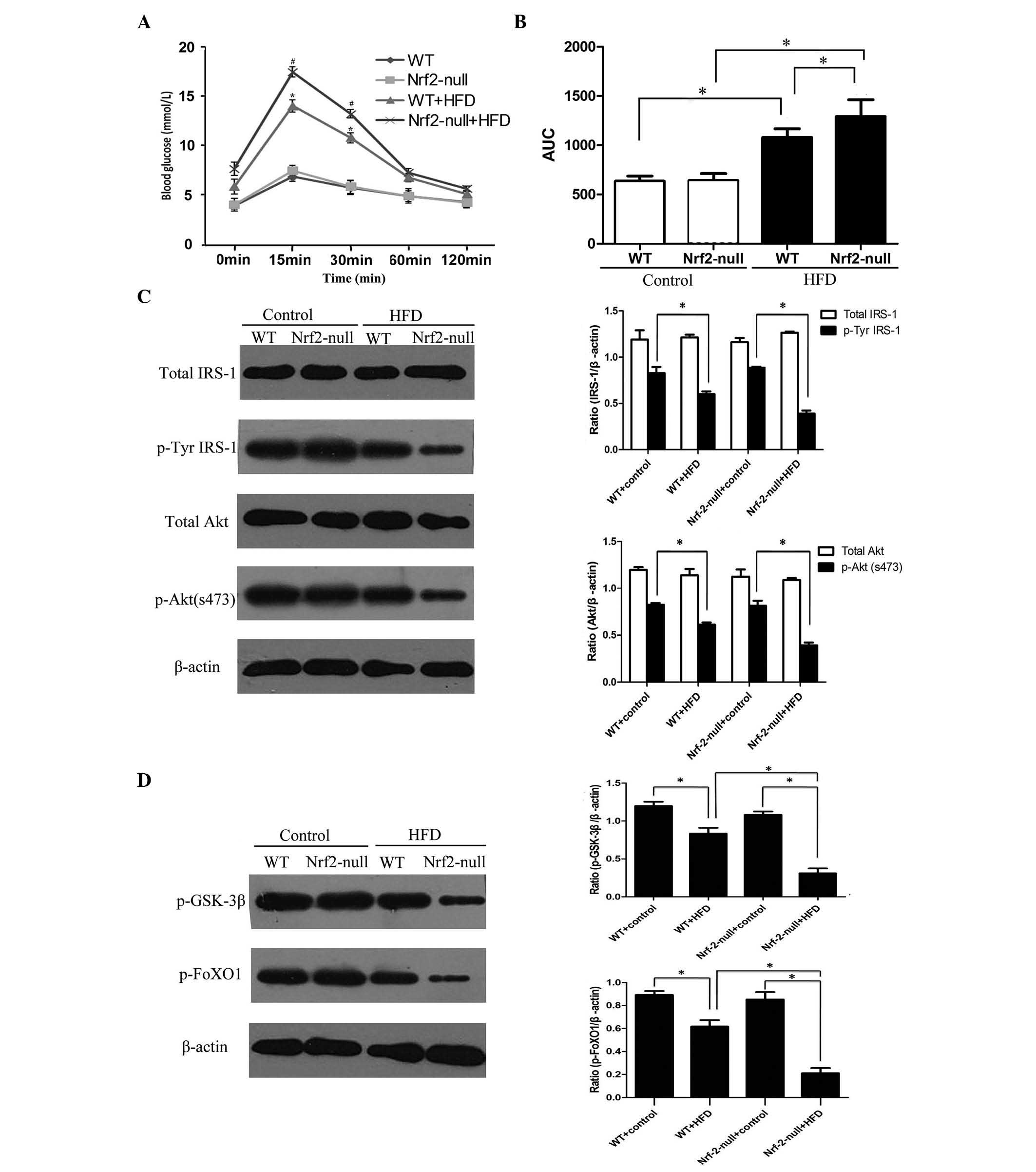 | Figure 2Nrf2 deletion led to insulin
resistance when mice were subjected to a HFD. (A) Blood glucose
levels at 0, 15, 30, 60 and 120 min were determined following
injection with glucose (2 g/kg body weight) in the four groups
using iPGTT. #P<0.05, Nrf2-null + HFD vs. Nrf2-null
mice groups; *P<0.05, WT + HFD vs WT mice groups. (B)
AUC was calculated from the results of the iPGTT at 15 min.
Expression levels of (C) total IRS1, p-Tyr IRS1, total Akt and
p-Akt (Ser473), (D) p-GSK3β and p-FoXO1 in liver tissues was
examined using western blot analysis. *P<0.05. Data
are presented as the mean ± standard error. WT, wild-type; Nrf2,
nuclear erythroid 2-related factor 2; HFD, high-fat diet; AUC, area
under curve; IRS, insulin receptor substrate 1; p-GSK-3β,
phosphorylated-glycogen synthase kinase 3β; p-FoXO1,
phosphorylated-forkhead box O1; iPGTT, intraperitoneal glucose
tolerance test. |
The expression levels of proteins involved in the
IRS1/PI3K/Akt pathway, which is important for the onset of IR, were
also detected. There was no change in the expression levels of
total IRS1 or Akt in liver tissues from any group. However, the
levels of p-Tyr-IRS1 and p-Akt were significantly decreased in mice
fed a HFD (Fig. 2C). Subsequently,
the expression levels of p-GSK-3β and p-FoXO1 in hepatic tissues
were determined, since they are key downstream effectors in the
IRS1/PI3K/Akt signaling pathway. The levels of p-GSK-3β and p-FoXO1
were notably reduced in HFD groups (Fig. 2D). Furthermore, Nrf2-null mice
exhibited reduced levels of p-GSK-3β and p-FoXO1 compared with WT
mice treated with a HFD. These results indicate that Nrf2
deficiency may lead to hepatic IR in mice fed a HFD.
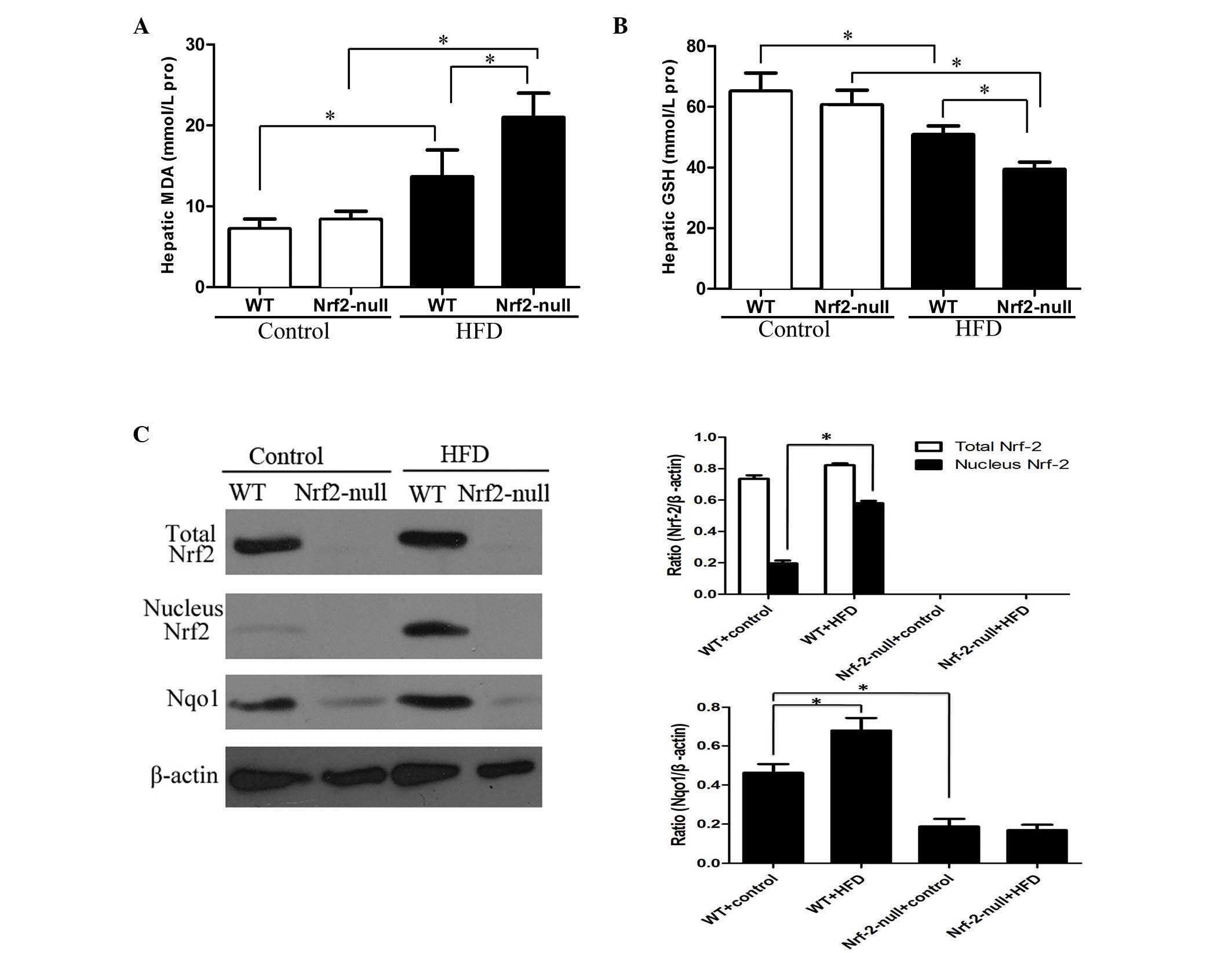
Nrf2 deletion leads to IR by activating
oxidative stress in the livers of HFD-treated mice
It has previously been confirmed that oxidative
stress is an important factor in IR (11). Therefore, in order to understand
the underlying mechanism by which Nrf2 deletion leads to the
induction of IR and to investigate if oxidative stress is involved
in the process, the concentration of hepatic MDA was determined, as
it is one of the products of lipid peroxidation. Hepatic GSH levels
were also determined, since GSH is an important endogenous
antioxidant, which reduces ROS levels. MDA concentration was
significantly enhanced in the livers of WT and Nrf2-null mice
following HFD feeding for 8 weeks compared with those fed a control
diet. In addition, MDA concentration was significantly greater in
Nrf2-null mice compared with in WT mice (P<0.05; Fig. 3A). Conversely, the decrease in GSH
levels was greater in the livers of HFD groups compared with those
from control groups. In addition, the magnitude of the decrease in
GSH concentration was greater in Nrf2-null-HFD mice compared with
in WT-HFD mice (P<0.05; Fig.
3B).
The expression levels of a typical Nrf2-regulated
gene, Nqo1 were subsequently detected in liver tissues. HFD feeding
significantly upregulated the expression levels of nuclear Nrf2 in
the livers of WT mice(P<0.05; Fig.
3C), without influencing total protein level. However, neither
total nor nuclear Nrf2 expression was observed in the Nrf2-null
mice fed any diet. Furthermore, the protein expression levels of
Nqo1 were significantly downregulated in the livers of
Nrf2-null-control mice compared with in the WT-control mice
(P<0.05; Fig. 3C). The protein
expression levels of Nqo1 were also significantly upregulated by
HFD feeding in WT mice but not in Nrf2-null mice (P<0.05;
Fig. 3C).
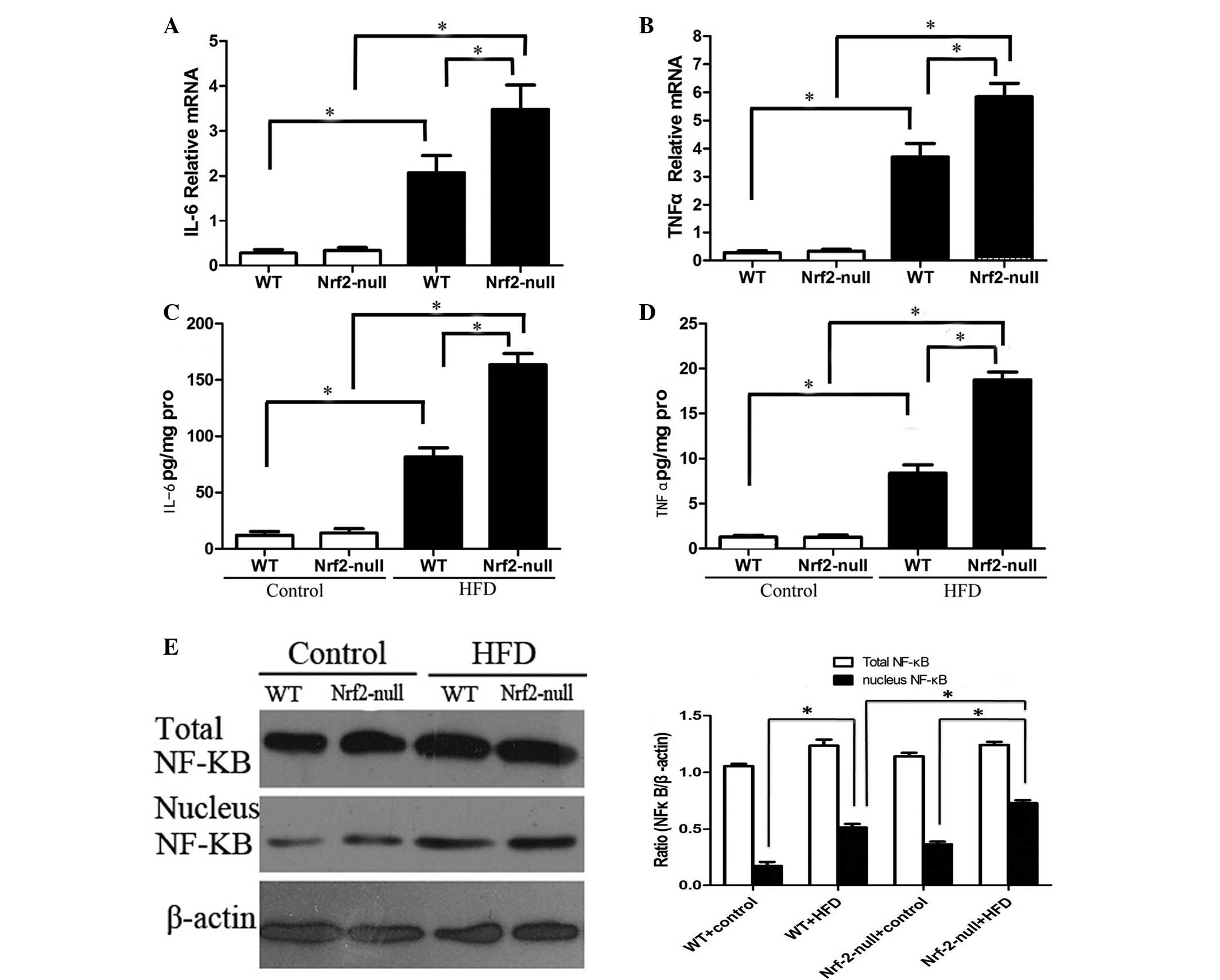
Nrf2 deficiency exacerbates activation of
NF-κB in the livers of HFD-treated mice
The IKK/NF-κB signaling pathway may be activated by
oxidative products, including MDA and ROS, and is associated with
oxidative stress. The release of TNF-α and IL-6 following
activation of the IKK/NF-κB signaling pathway has been demonstrated
to be associated with the pathogenesis of IR by interfering with
the IRS1/PI3K/Akt signaling pathway (21). Therefore, the expression levels of
TNF-α and IL-6 were detected in the livers of the different
treatment groups using RT-qPCR and ELISA. HFD feeding significantly
enhanced the mRNA expression levels of the two cytokines compared
with the control group (P<0.05; Fig. 4A and B). In addition, the mRNA
expression levels of TNF-α and IL-6 were significantly higher in
the livers of Nrf2-null mice compared with those of WT mice fed a
HFD (P<0.05; Fig. 4A and B).
The protein expression levels of TNF-α and IL-6 were also
correlated with the mRNA levels (P<0.05; Fig. 4C and D). Subsequently, the
expression levels of the upstream regulator of TNF-α and IL-6,
NF-κB, were determined in the livers of HFD-treated mice. The total
protein levels of NF-κB were not significantly different between
groups. However, the expression levels of nuclear NF-κB were
significantly upregulated in both WT-HFD and Nrf2-null-HFD mice
(P<0.05; Fig. 4E), the
magnitude of upregulation was greater in Nrf2-null mice compared
with WT mice (P<0.05; Fig. 4E).
These results indicate that Nrf2 deletion may induce hepatic IR by
activating the NF-κB signaling pathway in HFD-treated mice.
Discussion
NAFLD, which is associated with hepatic cirrhosis
and liver-related mortality, is a major public health issue
(22). Through extensive previous
research, it has become clear that oxidative stress, inflammation
and IR contribute to the pathogenesis of NAFLD (23), and several signaling pathways are
involved in the process. The present study determined that deletion
of the Nrf2 gene may lead to mice being more susceptible to the
development of NAFLD, partially due to hepatic IR induced by a
HFD.
Nrf2 is a member of the basic leucine zipper
transcription factor family, which mediates the expression of a
series of antioxidant and detoxification genes (24). It is expressed in various organs,
including the liver, lungs, kidneys, digestive tract and fat tissue
(25,26). It has previously been demonstrated
that Nrf2 is important for the onset of NAFLD alongside the
administration of various diets, and Nrf2-null mice have exhibited
rapid progression of steatohepatitis when fed an atherogenic HFD
(27). In addition, Sugimoto et
al (28) and Chowdhry et
al (15) demonstrated that
limitation of Nrf2 activity significantly promoted the progression
of NASH, from NAFLD to NASH, in mice placed on an MCD diet. In the
present study, a HFD was able to induce weight gain of body and
liver tissues in both WT and Nrf2-null mice compared with the
control diet. Furthermore, the increase observed was greater in the
livers of Nrf2-null mice compared with WT mice. TG accumulation in
the hepatic tissues of Nrf2-null mice fed a HFD was also greater
when compared with the control mice, which led to alterations in
liver structure accompanied by fat deposition, inflammatory
infiltration and hyaline degeneration.
IR has been identified as a major pathophysiological
feature in several metabolic diseases, including NAFLD (29). A previous study demonstrated that
hepatic IR may result in the development of metabolic dyslipidemia
and hepatic steatosis in AKT2-mutant individuals (30). Furthermore, another previous study
using tissue-specific knockout analysis demonstrated that knockdown
of hepatic insulin receptors may lead to IR, severe glucose
intolerance, and liver steatosis with a high level of fasting
hyperglycemia and postprandial blood glucose in mice (31). The process of IR is usually induced
by various factors, including genetic determinants, nutrition and
lifestyle. However, it has previously been demonstrated that
oxidative stress and inflammation are associated with hepatic IR
(32). The insulin signaling
pathway is of pivotal importance. Subsequent to binding insulin,
the insulin receptor is activated and autophosphorylated, resulting
in IRS phosphorylation of tyrosine residues (33). In response to IRS phosphorylation,
various downstream signaling molecules, such as the PI3K/AKT or
mitogen-activated protein kinase pathways, are activated. In the
majority of cases of IR, the aforementioned molecular event is
often impaired (31). The present
study determined that both serum FBG levels and plasma glucose
levels during iPGTT were significantly increased in mice following
administration of a HFD for 8 weeks, particularly in the
Nrf2-null-HFD group. Furthermore, the expression levels of
phosphorylated proteins associated with the IRS1/PI3K/Akt signaling
pathway were reduced in the livers of Nrf2-null mice fed a HFD.
These results indicated that Nrf2 deletion may lead to hepatic IR
in HFD models.
NF-κB is a primary transcription factor in
inflammatory diseases (34), which
also contributes to the pathology of IR (35). Activation of the hepatic NF-κB
pathway has been suggested to be directly responsible for
HFD-induced IR (36). Inactivated
NF-κB in the cytosol binds to inhibitor of κB (IκB) molecules and
its transcriptional function is prevented. When the IKK is
activated by extracellular stimuli or products of oxidative stress
it phosphorylates IκB, promoting the detachment of NF-κB from the
IKK complex and resulting in nuclear translocation (37). The activation of NF-κB consequently
leads to vast increases in the production of inflammatory
cytokines, including IL-6, TNF-α and IL-1β, which are considered
pathogenetic markers that have a pivotal role in IR (32). The present study confirmed that
Nrf2 deficiency may increase hepatic MDA levels and reduce GSH
levels in mice fed a HFD. The Nrf2-null-HFD mice suffered IR
through the activation of NF-κB and its downstream cytokines IL-6
and TNF-α.
The present study demonstrated that Nrf2 deficiency
may induce obesity, hepatic IR and alterations in liver tissue
structure in mice fed a HFD. Hepatic IR induced by Nrf2 deletion is
regulated by activation of the NF-κB signaling pathway, which is
associated with oxidative stress, as determined by increased
hepatic MDA concentrations and decreased GSH levels. However,
further investigations are required to clarify whether other
mechanisms are involved in IR in NAFLD mediated by Nrf2 deletion.
The findings of the present study present novel insights into the
mechanisms underlying IR and NAFLD, and a potential therapeutic
strategy for future treatment.
Abbreviations:
|
Nrf2
|
nuclear factor-erythroid 2-related
factor
|
|
NF-κB
|
nuclear factor-κB
|
|
HFD
|
high-fat diet
|
|
NAFLD
|
non-alcoholic fatty liver disease
|
|
IR
|
insulin resistance
|
|
iPGTT
|
intraperitoneal glucose tolerance
test
|
|
FGB
|
fasting blood glucose
|
|
MDA
|
malondialdehyde
|
|
GSH
|
glutathione
|
|
TNF-α
|
tumor necrosis factor α
|
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant nos. 30800515, 81270485
and 81170376) and the Natural Science Foundation of Shaanxi
Province (grant no. 2013JM4021).
References
|
1
|
Musso G, Gambino R, Cassader M and Pagano
G: Meta-analysis: Natural history of non-alcoholic fatty liver
disease (NAFLD) and diagnostic accuracy of non-invasive tests for
liver disease severity. Ann Med. 43:617–649. 2011. View Article : Google Scholar
|
|
2
|
Preiss D and Sattar N: Non-alcoholic fatty
liver disease: An overview of prevalence, diagnosis, pathogenesis
and treatment considerations. Clin Sci (Lond). 115:141–150. 2008.
View Article : Google Scholar
|
|
3
|
Day CP and James OF: Steatohepatitis: A
tale of two 'hits'? Gastroenterology. 114:842–845. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Musso G, Gambino R and Cassader M: Recent
insights into hepatic lipid metabolism in non-alcoholic fatty liver
disease (NAFLD). Prog Lipid Res. 48:1–26. 2009. View Article : Google Scholar
|
|
5
|
Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S,
Befroy D, Romanelli AJ and Shulman GI: Mechanism of hepatic insulin
resistance in non-alcoholic fatty liver disease. J Biol Chem.
279:32345–32353. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bugianesi E, Mccullough AJ and Marchesini
G: Insulin resistance: A metabolic pathway to chronic liver
disease. Hepatology. 42:987–1000. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wellen KE and Hotamisligil GS:
Inflammation, stress, and diabetes. J Clin Invest. 115:1111–1119.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Paz K, Hemi R, LeRoith D, Karasik A,
Elhanany E, Kanety H and Zick Y: A molecular basis for insulin
resistance. Elevated serine/threonine phosphorylation of IRS-1 and
IRS-2 inhibits their binding to the juxtamembrane region of the
insulin receptor and impairs their ability to undergo
insulin-induced tyrosine phosphorylation. J Biol Chem.
272:29911–29918. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Houstis N, Rosen ED and Lander ES:
Reactive oxygen species have a causal role in multiple forms of
insulin resistance. Nature. 440:944–948. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Videla LA, Rodrigo R, Araya J and
Poniachik J: Insulin resistance and oxidative stress
interdependency in non-alcoholic fatty liver disease. Trends Mol
Med. 12:555–558. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Evans JL, Maddux BA and Goldfine ID: The
molecular basis for oxidative stress-induced insulin resistance.
Antioxid Redox Signal. 7:1040–1052. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang DD and Hannink M: Distinct cysteine
in Keap1 are required for Keap1-dependent ubiquitination on Nrf2
and for stabilization of Nrf2 by chemopreventive agents and
oxidative stress. Mol Cell Biol. 23:8137–8151. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bataille AM and Manautou JE: Nrf2: A
potential target for new therapeutics in liver disease. Clin
Pharmacol Ther. 92:340–348. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Itoh K, Chiba T, Takahashi S, Ishii T,
Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, et
al: An Nrf2/small Maf heterodimer mediates the induction of phase
II detoxifying enzyme genes through antioxidant response elements.
Biochem Biophys Res Commun. 236:313–322. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chowdhry S, Nazmy MH, Meakin PJ,
Dinkova-Kostova AT, Walsh SV, Tsujita T, Dillon JF, Ashford ML and
Hayes JD: Loss of Nrf2 markedly exacerbates nonalcoholic
steatohepatitis. Free Radic Biol Med. 48:357–371. 2010. View Article : Google Scholar
|
|
16
|
Luley C, Ronquist G, Reuter W, Paal V,
Gottschling HD, Westphal S, King GL, Bakker SJ, Heine RJ and
Hattemer A: Point-of-care testing of triglycerides: Evaluation of
the Accutrend triglycerides system. Clin Chem. 46:287–291.
2000.PubMed/NCBI
|
|
17
|
Amaral ME, Oliveira HC, Carneiro EM,
Delghingaro-Augusto V, Vieira EC, Berti JA and Boschero AC: Plasma
glucose regulation and insulin secretion in hypertriglyceridemic
mice. Horm Metab Res. 34:21–26. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Angel MF, Ramasastry SS, Swartz WM,
Narayanan K, Kuhns DB, Basford RE and Futrell JW: The critical
relationship between free radicals and degrees of ischemia:
Evidence for tissue intolerance of marginal perfusion. Plast
Reconstr Surg. 81:233–239. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhong D, Zhang Y, Zeng YJ, Gao M, Wu GZ,
Hu CJ, Huang G and He FT: MicroRNA-613 represses lipogenesis in
HepG2 cells by downregulating LXRα. Lipids Health Dis. 12:322013.
View Article : Google Scholar
|
|
20
|
He L, Wang H, Jin H, Guo C, Xie H, Yan K,
Li X, Shen Q, Qiao T, Chen G, et al: CIAPIN1 inhibits the growth
and proliferation of clear cell renal cell carcinoma. Cancer Lett.
276:88–94. 2009. View Article : Google Scholar
|
|
21
|
Arkan MC, Hevener AL, Greten FR, Maeda S,
Li ZW, Long JM, Wynshaw-Boris A, Poli G, Olefsky J and Karin M:
IKK-beta links inflammation to obesity-induced insulin resistance.
Nat Med. 11:191–198. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nakamuta M, Kohjima M, Morizono S, Kotoh
K, Yoshimoto T, Miyagi I and Enjoji M: Evaluation of fatty acid
metabolism-related gene expression in nonalcoholic fatty liver
disease. Int J Mol Med. 16:631–635. 2005.PubMed/NCBI
|
|
23
|
Mantena SK, King AL, Andringa KK,
Eccleston HB and Bailey SM: Mitochondrial dysfunction and oxidative
stress in the pathogenesis of alcohol- and obesity-induced fatty
liver diseases. Free Radic Biol Med. 44:1259–1272. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kobayashi M and Yamamoto M: Nrf2-Keap1
regulation of cellular defense mechanisms against electrophiles and
reactive oxygen species. Adv Enzyme Regul. 46:113–140. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu M, Grigoryev DN, Crow MT, Haas M,
Yamamoto M, Reddy SP and Rabb H: Transcription factor Nrf2 is
protective during ischemic and nephrotoxic acute kidney injury in
mice. Kidney Int. 76:277–285. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chan K, Lu R, Chang JC and Kan YW: NRF2, a
member of the NFE2 family of transcription factors, is not
essential for murine erythropoiesis, growth, and development. Proc
Natl Acad Sci USA. 93:13943–13948. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Okada K, Warabi E, Sugimoto H, Horie M,
Gotoh N, Tokushige K, Hashimoto E, Utsunomiya H, Takahashi H, Ishii
T, et al: Deletion of Nrf2 leads to rapid progression of
steatohepatitis in mice fed atherogenic plus high-fat diet. J
Gastroenterol. 48:620–632. 2013. View Article : Google Scholar
|
|
28
|
Sugimoto H, Okada K, Shoda J, Warabi E,
Ishige K, Ueda T, Taguchi K, Yanagawa T, Nakahara A, Hyodo I, et
al: Deletion of nuclear factor-E2-related factor-2 leads to rapid
onset and progression of nutritional steatohepatitis in mice. Am J
Physiol Gastrointest Liver Physiol. 298:G283–G294. 2010. View Article : Google Scholar
|
|
29
|
Marchesini G and Forlani G: NASH: From
liver diseases to metabolic disorders and back to clinical
hepatology. Hepatology. 35:497–499. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hebbard L and George J: Animal models of
nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol.
8:35–44. 2011. View Article : Google Scholar
|
|
31
|
Biddinger SB and Kahn CR: From mice to
men: Insights into the insulin resistance syndromes. Annu Rev
Physiol. 68:123–158. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tilg H and Moschen AR: Insulin resistance,
inflammation, and non-alcoholic fatty liver disease. Trends
Endocrinol Metab. 19:371–379. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Leclercq IA, Da Silva Morais A, Schroyen
B, Van Hul N and Geerts A: Insulin resistance in hepatocytes and
sinusoidal liver cells: Mechanisms and consequences. J Hepatol.
47:142–156. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Barnes PJ and Karin M: Nuclear
factor-kappaB: A pivotal transcription factor in chronic
inflammatory diseases. N Engl J Med. 336:1066–1071. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Baker RG, Hayden MS and Ghosh S: NF-κB,
inflammation, and metabolic disease. Cell Metab. 13:11–22. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cai D, Yuan M, Frantz DF, Melendez PA,
Hansen L, Lee J and Shoelson SE: Local and systemic insulin
resistance resulting from hepatic activation of IKK-beta and
NF-kappaB. Nat Med. 11:183–190. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hayden MS and Ghosh S: Shared principles
in NF-kappaB signaling. Cell. 132:344–362. 2008. View Article : Google Scholar : PubMed/NCBI
|