Introduction
Crouzon syndrome, a type of craniosynostosis, is a
dominantly inherited disorder, which is predominantly caused by
mutations in the fibroblast growth factor receptor 2 (FGFR
2) gene and is characterized by craniosynostosis, shallow
orbits, ocular proptosis, midface hypoplasia and a curved,
beak-like nose (1–4).
The most common genetic mutations of FGFR 2,
located at chromosome 10q26, are localized at exons IIIa (exon 8)
and IIIc (exon 10) that encode the extracellular
immunoglobulin-like III (IgIII) domain of the protein (5–7).
Although Crouzon syndrome is inherited as an
autosomal dominant trait, several cases present as de novo
mutations arising from unaffected parents (8–11).
The present study reported the mutational analyses of three
patients with Crouzon syndrome from separate families at the gene
level and reported its associated clinical features, resulting in
the identification of two heterozygous mutations.
Patients and methods
Crouzon syndrome families
Two probands in two Chinese families were diagnosed
as having Crouzon syndrome at the Zhongshan Ophthalmic Center
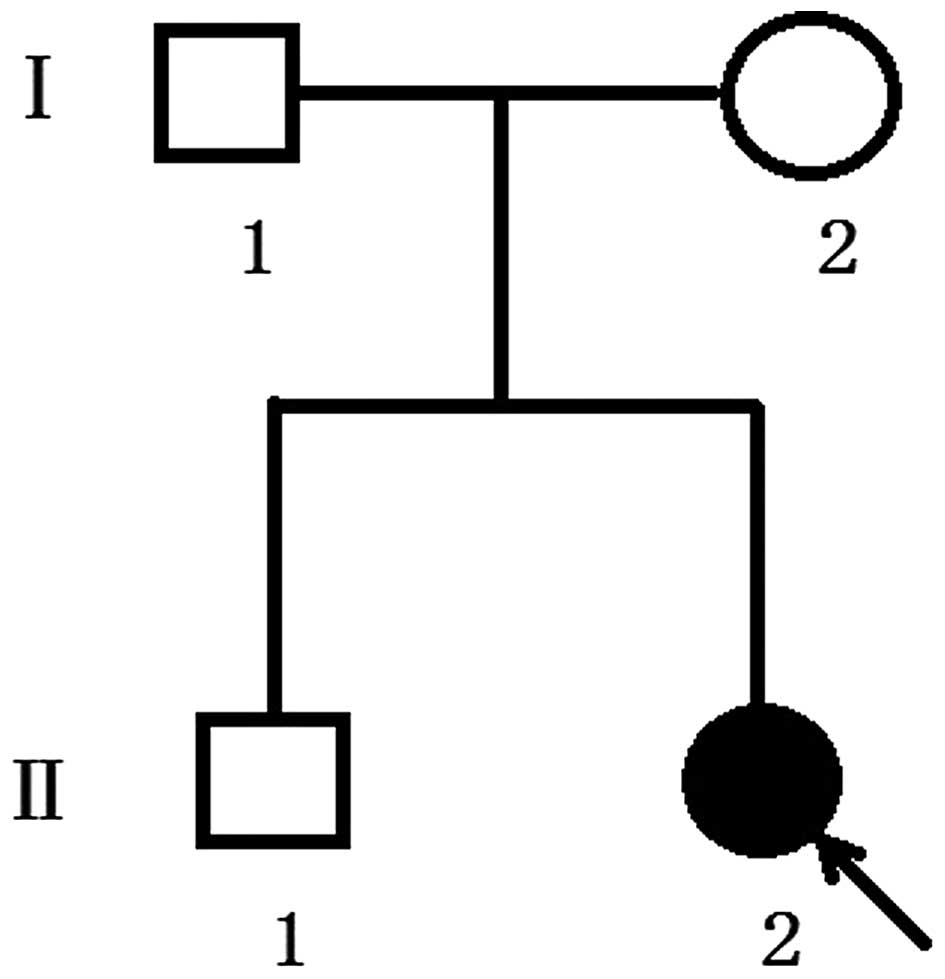
(Guangzhou, China). The proband of family 1 (Fig. 1) was a one-year-old girl and was
the second child of healthy unrelated parents, vaginally delivered
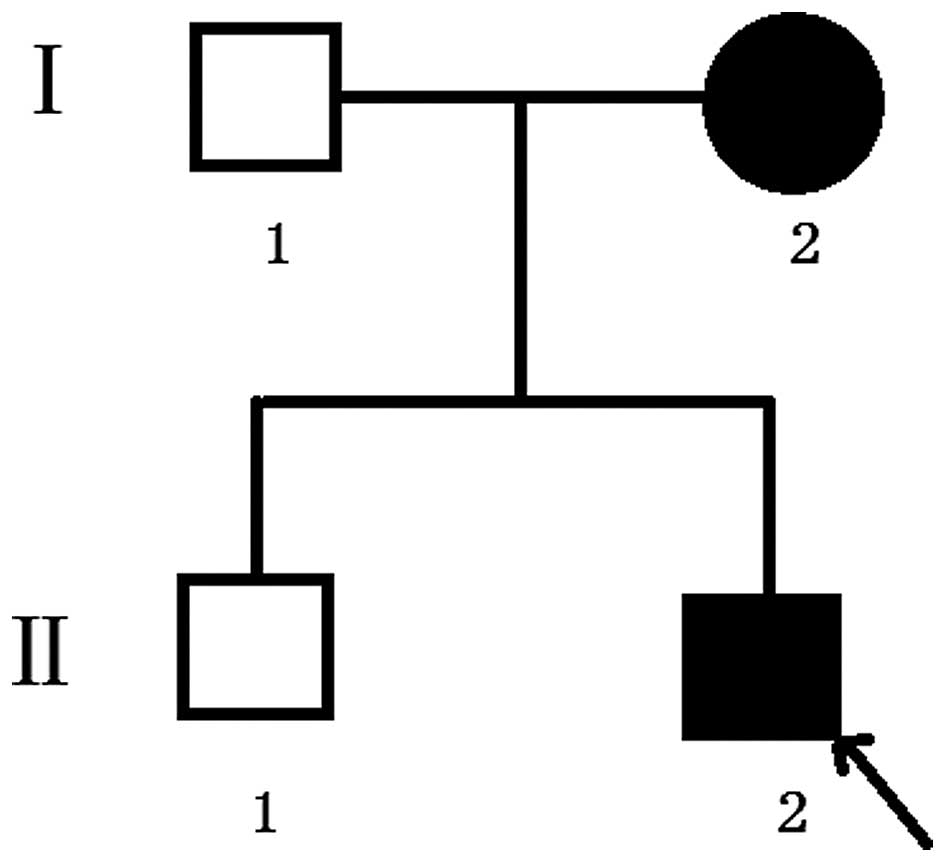
maturely at 39 weeks. The second proband, in family 2, (Fig. 2) was a three-year-old boy and was
also the second child of his family. For the present study,
ophthalmic examinations of these two families were performed, as
follows: Visual acuity was examined using the Early Treatment
Diabetic Retinopathy Study chart (Precision Vision, LaSalle, IL,
USA). Anterior segment images were captured using a BX 900 Slit
Lamp (Haag-StreitAG, Köniz, Switzerland). Anterior segment
measurements were obtained using Pentacam® HR version
70700 (OCULUS Optikgeräte GmbH, Wetzlar, Germany). Fundus imaging
was performed using a Heidelberg Retina Angiograph (Heidelberg
Engineering GmbH, Heidelberg, Germany). In addition, physical
examinations, including blood examination, a urine test,
electrocardiogram, chest X-ray, blood biochemistry test, blood
lipid and blood coagulation tests, were performed to exclude
systemic diseases. The study was approved by the ethics committee
of Zhongshan Ophthalmic Center, Sun Yat-Sen University.
Sample collection
The affected families were identified at Zhongshan
Ophthalmic Center. In addition, 200 subjects from the same
population, but without diagnostic features of Crouzon syndrome,
were recruited to serve as normal controls. Informed consent was
obtained from all participating individuals and, in accordance with
the principles of the Declaration of Helsinki, venous blood samples
were collected from the two families and 200 controls for genomic
DNA extraction from peripheral blood leukocytes, using the a DNA
extraction kit (Qiagen, Inc., Valencia, CA, USA) with standard
protocols.
Mutation detection
Exons 8 and 10 of the FGFR 2 gene were
amplified using polymerase chain reaction (PCR) analysis with the
following primers: FGFR2-8 (IIIa), forward
5′-GGTCTCTCATTCTCCCATCCC-3′, reverse 5′-CCAACAGGAAATCAAAGAACC-3′
(product size, 325 bp); FGFR2-10 (IIIc), forward
5′-CCTCCACAATCATTCCTGTGTC-3′, reverse 5′-ATAGCAGTCAACCAAGAAAAGGG-3′
(product size, 257 bp) (9-10) (Beijing Genomics Institute,
Guangzhou, China). Briefly, PCR was performed with a 50 μl
reaction volume with 2 μl each primer, 2 μl DNA, 25
μl buffer mix and 19 μl H2O. All reagents
used for PCR were purchased from (Takara Bio, Inc., Tokyo,
Japan).
The cycling profile included one cycle at 94°C for 5
min, followed by 40 cycles at 94°C for 45 sec, 61°C for 45 sec, and
72°C for 45 sec, with one cycle at 72°C for 10 min. The PCR
products were sequenced from both directions using an ABI3730
automated sequencer (PE Biosystems, Foster City, CA, USA). The
sequencing results were analyzed using Chromas (version 2.3;
Technelysium Pty., Ltd., Brisbane, QLD, Australia), and they were
compared with the reference sequences in the database at the
National Center for Biotechnology Information (NCBI;
NC_000010).
Results
Clinical data
The Chinese families included in the present study
were from the southern area of China. In two successive
generations, one individual was found to have a congenital disease.
The proband of family 1 was referred by her local pediatrician at
the age of 2 months, owing to concerns about her elongated head
shape and a possible diagnosis of sagittal synostosis. The proband
had otherwise been developing well with normal feeding and steady
weight gain following birth. There was no history of learning
difficulties or genetic problems in the family. The parents
considered her development to be equivalent to her age-matched
peers.
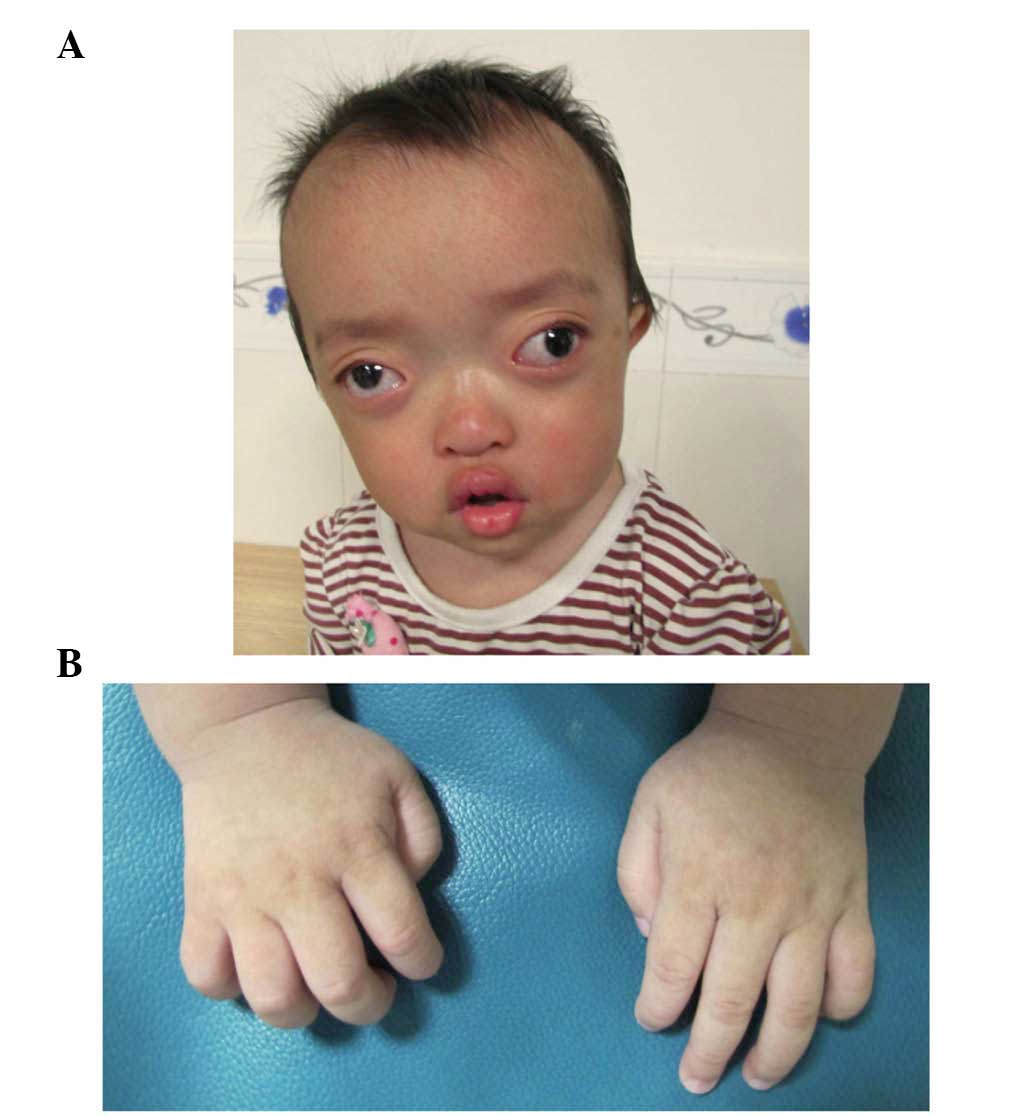
On examination, the proband exhibited shallow orbits
and ocular proptosis, accompanied by midface hypoplasia,
craniosynostosis and a curved, beak-like nose (Fig. 3A), with clinically normal hands and
feet (Fig. 3B). The proband also
showed exotropia of the left eye, although the corneas were normal
in size and transparency, and the lenses were positioned normally
and remained clear. As the child was just 1 year old, it was not
possible to assess visual acuity, however, visual tracking was
present. The parents and the old brother of the proband showed
normal visual acuity and eye examinations.
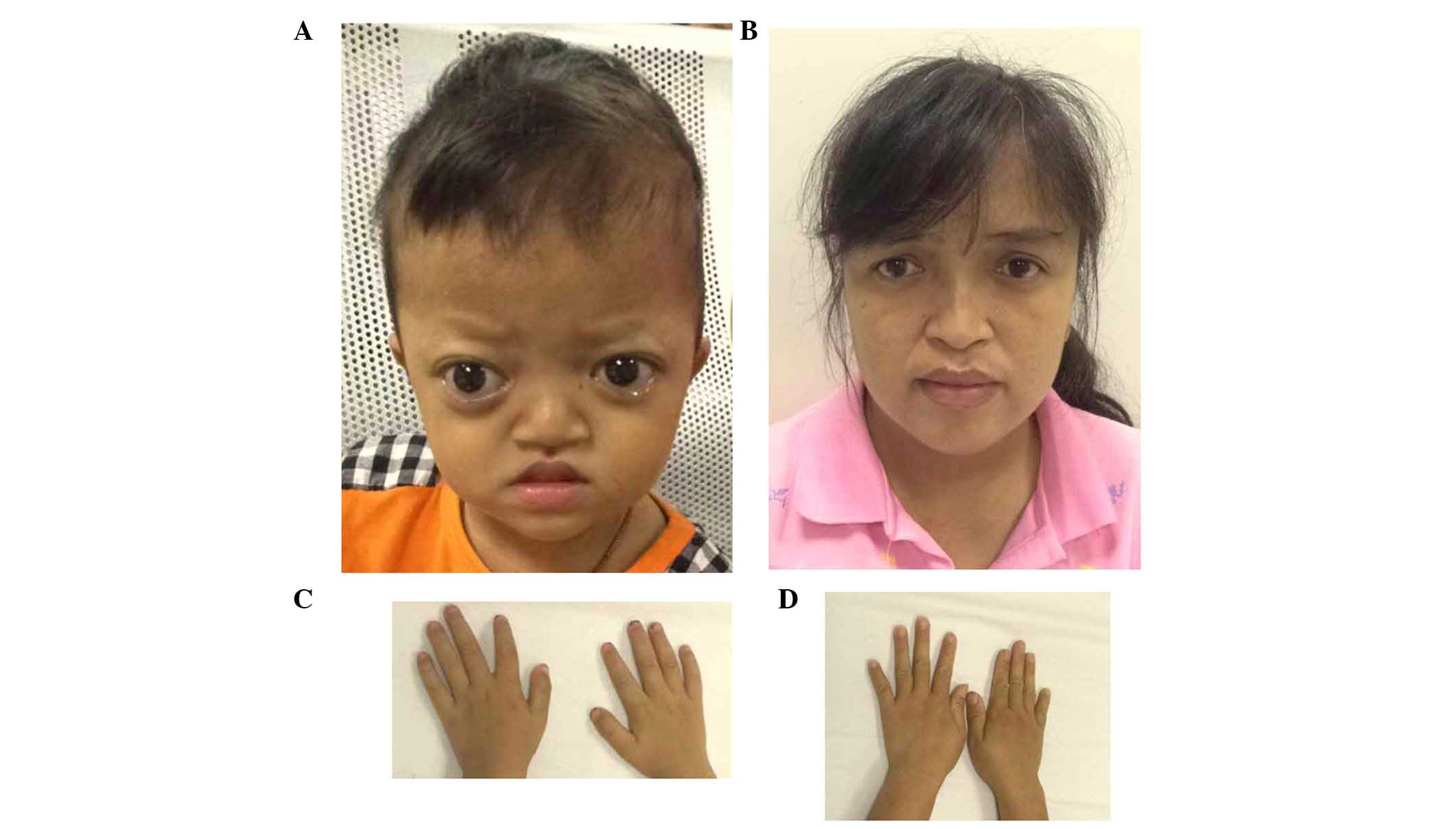
The proband of family 2 had midface hypoplasia and
craniosynostosis (Fig. 4A). His
mother's condition was less severe, with normal visual acuity
(Fig. 4B). The proband and his
mother had clinically normal hands and feet (Fig. 4C and D). The proband had
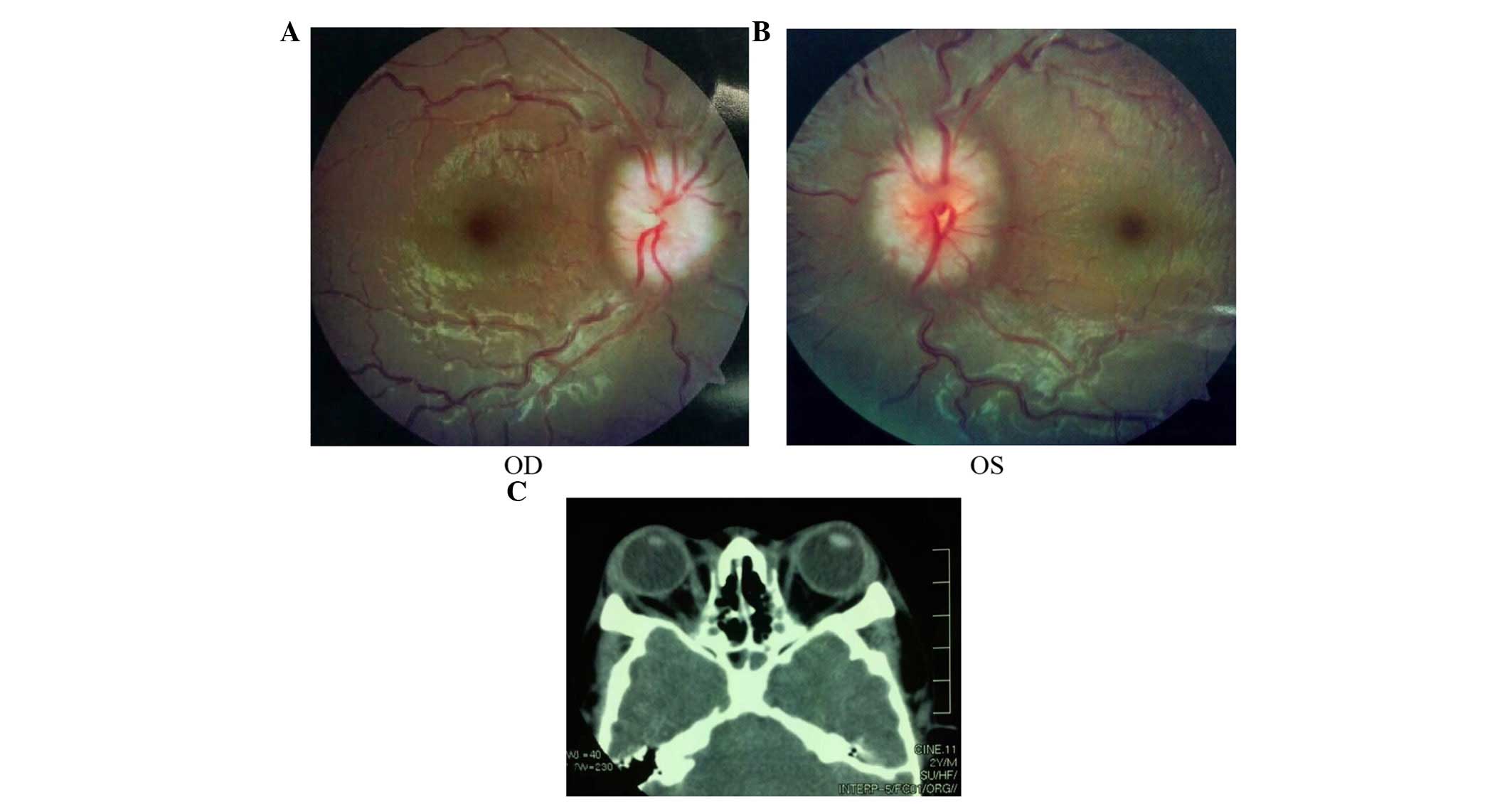
papilloedema of both eyes (Fig. 5A and
B) and had a history of intracranial hypertension (1 year
previously). The Computed Tomography examination revealed shallow
orbits (Fig. 5C). No abnormalities
were detected in the corneas or lens.
Mutation screening
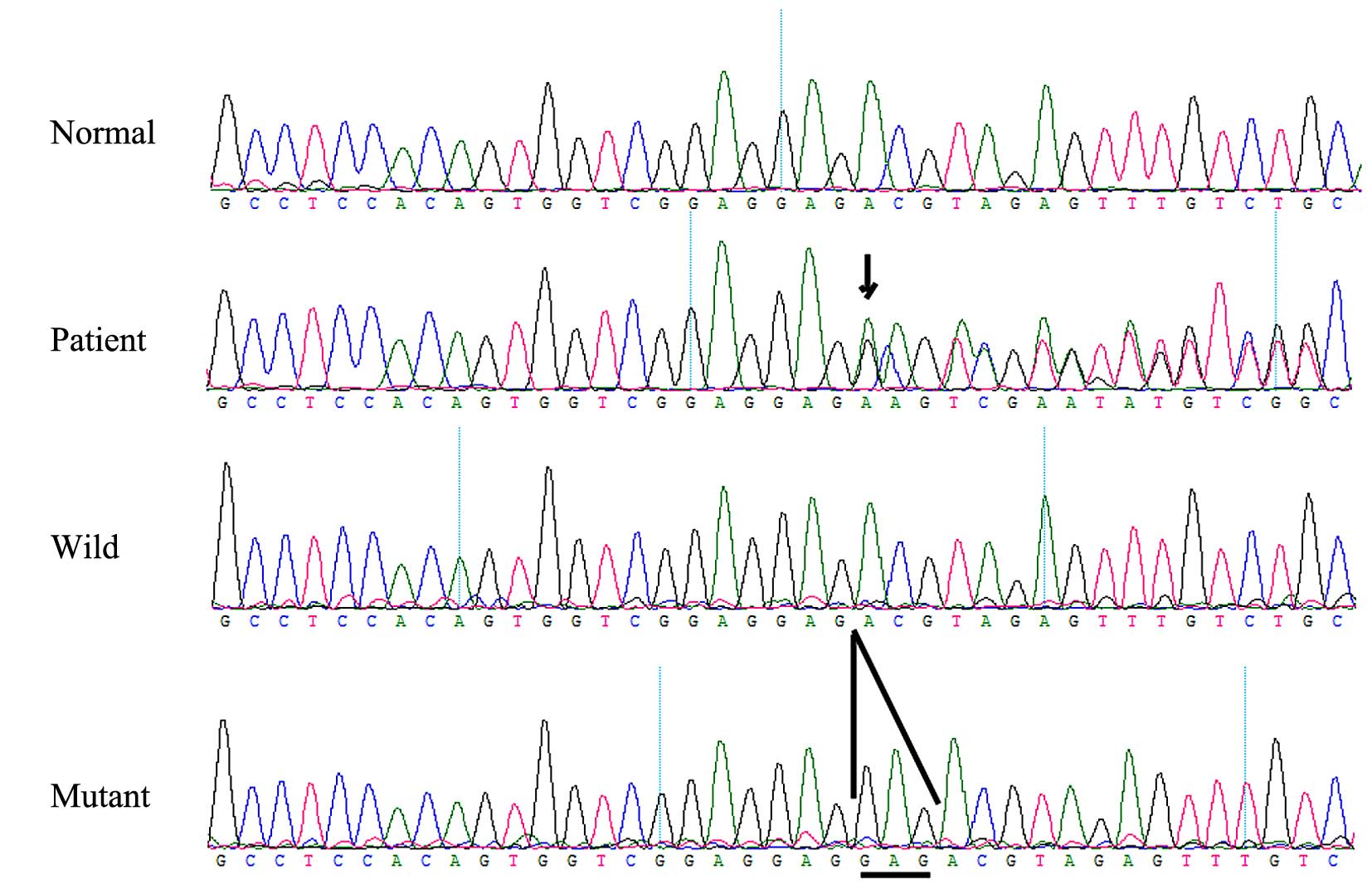
A heterozygous FGFR 2 missense mutation,
c.811-812insGAG (p.273insGlu), in exon 8 (Fig. 6) was identified in the affected
individual, but was not identified in any of the unaffected family
members or the normal control individuals. This mutation causes the
insertion of glutamic acid in position of 273 of the FGFR 2
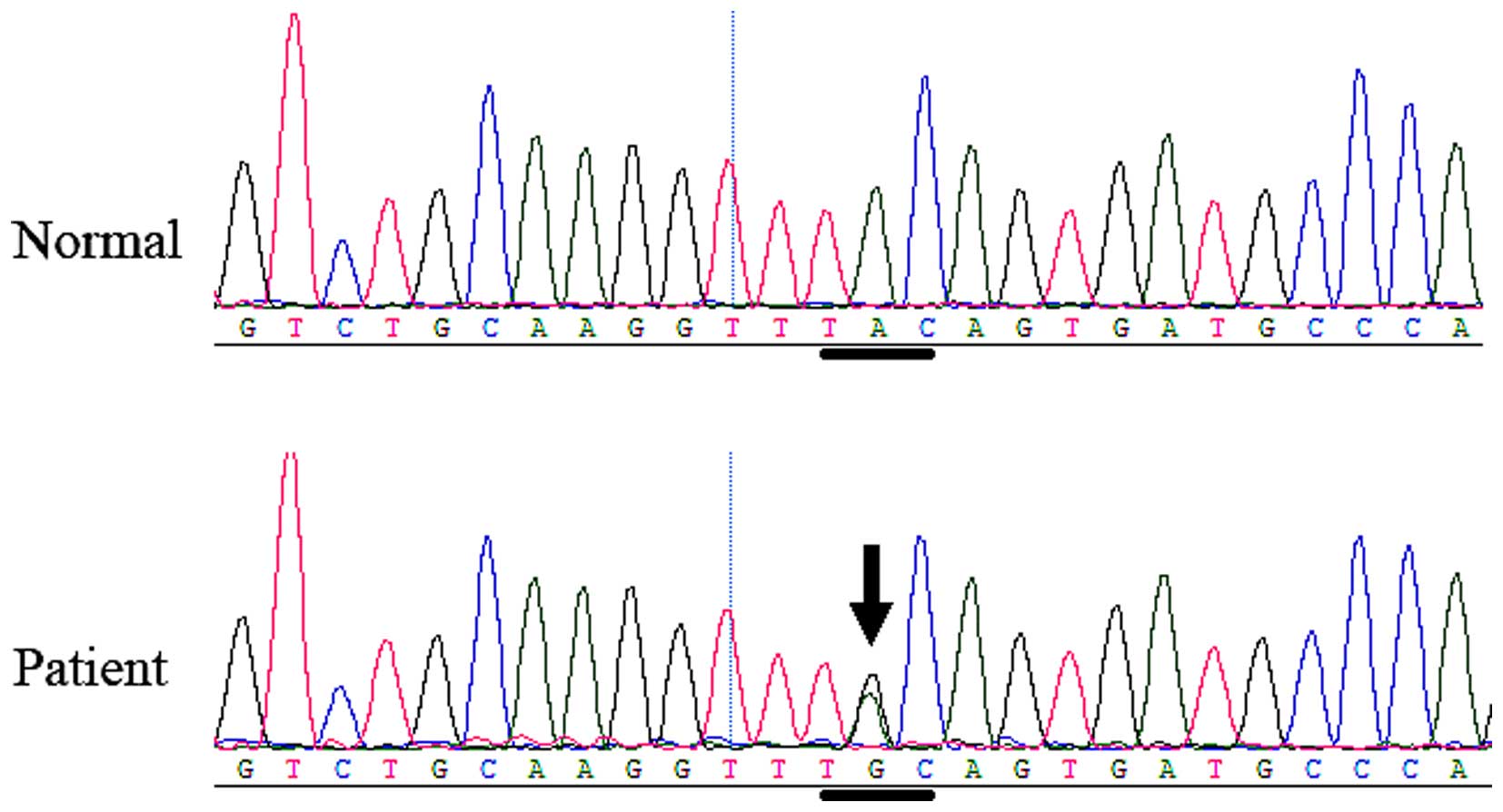
gene. In family 2, another heterozygous FGFR 2 missense
mutation, c.842A>G (P.Tyr281Cys or Y281C), in exon 8 (Fig. 7) was identified in the affected boy
and his mother, but was not identified in any of the unaffected
family members or the normal control individuals. The mutation
causes the tyrosine 281 codon to change to a cysteine codon.
Discussion
In the present study, two mutations in exon 8 of the
FGFR 2 gene were identified, which were associated with
Crouzon syndrome: A de novo mutation, c.811-812insGAG
(p.273insGlu), in family 1 and a familial mutation, c.842A>G
(p.Tyr281Cys or Y281C), in family 2. These mutations, rather than
representing a rare polymorphism in the normal population, were the
causative mutations in these two families.
The c.811-812insGAG (p.273insGlu) mutation was
identified for the first time in the FGFR 2 gene in Chinese
patients, and, to the best of our knowledge, has not previously
been reported either in China or elsewhere. However, the
c.842A>G (P.Tyr281Cys or Y281C) mutation, identified in family
2, has been previously reported outside of China (12). Although Crouzon syndrome is
inherited as an autosomal dominant trait, several cases are
sporadic and present as de novo mutations arising from
unaffected parents, as observed in family 1 in the present
study.
The most common genetic mutation of FGFR 2
has been localized in the third Ig-like domain and in the flanking
linker regions, coded by exons IIIa (exon 8) and IIIc (exon 10)
(13,14). The two mutations found in the two
families in the present study were in exon IIIa. The mutations may
disrupt the intra-Ig domain disulfide bond, thus leading to changes
in FGF signaling, and may induce the activation or downregulation
of FGFR 2 (15–20).
Okajima et al (21) reported that certain patients with
an FGFR 2 mutation may have Peters anomaly, optic nerve
hypoplasia, scleralization of the cornea and corectopia in
craniosynostosis syndromes. In the present study, the proband of
family 2 had papilloedema, which expands the clinical
manifestations of Crouzon syndrome.
In conclusion, the present study identified two
mutations of FGFR 2 in two Chinese families with Crouzon
syndrome. This finding expands the mutation spectrum of FGFR
2, and is useful and valuable for genetic counseling and
prenatal diagnosis in families with Crouzon syndrome with ocular
disorders.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (grant no. 81500709), the Project of
Fundamental Research Funds of State Key Laboratory of Ophthalmology
(grant nos. 2011Q09, 2012KF03 and 2014QN04), and the Medical
Scientific Research Foundation of Guangdong Province (grant no.
A201646).
References
|
1
|
Reardon W, Winter RM, Rutland P, Pulleyn
LJ, Jones BM and Malcolm S: Mutations in the fibroblast growth
factor receptor 2 gene cause Crouzon syndrome. Nat Genet. 8:98–103.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gorry MC, Preston RA, White GJ, Zhang Y,
Singhal VK, Losken HW, Parker MG, Nwokoro NA, Post JC and Ehrlich
GD: Crouzon syndrome: Mutations in two spliceoforms of FGFR2 and a
common point mutation shared with Jackson-Weiss syndrome. Hum Mol
Genet. 4:1387–1390. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Oldridge M, Lunt PW, Zackai EH,
McDonald-McGinn DM, Muenke M, Moloney DM, Twigg SR, Heath JK,
Howard TD, Hoganson G, et al: Genotype-phenotype correlation for
nucleotide substitutions in the IgII-IgIII linker of FGFR2. Hum Mol
Genet. 6:137–143. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Murano I: Crouzon syndrome. Nihon Rinsho.
(Suppl 3): S416–S417. 2006.In Japanese.
|
|
5
|
Steinberger D, Reinhartz T, Unsöld R and
Müller U: FGFR2 mutation in clinically nonclassifiable autosomal
dominant craniosynostosis with pronounced phenotypic variation. Am
J Med Genet. 66:81–86. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Park WJ, Meyers GA, Li X, Theda C, Day D,
Orlow SJ, Jones MC and Jabs EW: Novel FGFR2 mutations in Crouzon
and Jackson-Weiss syndromes show allelic heterogeneity and
phenotypic variability. Hum Mol Genet. 4:1229–1233. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Meyers GA, Day D, Goldberg R, Daentl DL,
Przylepa KA, Abrams LJ, Graham JM Jr, Feingold M, Moeschler JB,
Rawnsley E, et al: FGFR2 exon IIIa and IIIc mutations in Crouzon,
Jackson-Weiss and Pfeiffer syndromes: Evidence for missense
changes, insertions, and a deletion due to alternative RNA
splicing. Am J Hum Genet. 58:491–498. 1996.PubMed/NCBI
|
|
8
|
Steinberger D, Collmann H, Schmalenberger
B and Müller U: A novel mutation (a886 g) in exon 5 of FGFR2 in
members of a family with Crouzon phenotype and plagiocephaly. J Med
Genet. 34:420–422. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lin Y, Ai S, Chen C, Liu X, Luo L, Ye S,
Liang X, Zhu Y, Yang H and Liu Y: Ala344Pro mutation in the FGFR2
gene and related clinical findings in one Chinese family with
Crouzon syndrome. Mol Vis. 18:1278–1282. 2012.PubMed/NCBI
|
|
10
|
Lin Y, Liang X, Ai S, Chen C, Liu X, Luo
L, Ye S, Li B, Liu Y and Yang H: FGFR2 molecular analysis and
related clinical findings in one Chinese family with Crouzon
syndrome. Mol Vis. 18:449–454. 2012.PubMed/NCBI
|
|
11
|
Hollway GE, Suthers GK, Haan EA, Thompson
E, David DJ, Gecz J and Mulley JC: Mutation detection in FGFR2
craniosynostosis syndromes. Hum Genet. 99:251–255. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kan SH, Elanko N, Johnson D,
Cornejo-Roldan L, Cook J, Reich EW, Tomkins S, Verloes A, Twigg SR,
Rannan-Eliya S, et al: Genomic screening of fibroblast
growth-factor receptor 2 reveals a wide spectrum of mutations in
patients with syndromic craniosynostosis. Am J Hum Genet.
70:472–486. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tartaglia M, Valeri S, Velardi F, Di Rocco
C and Battaglia PA: Trp290Cys mutation in exon IIIa of the
fibroblast growth factor receptor 2 (FGFR2) gene is associated with
Pfeiffer syndrome. Hum Genet. 99:602–606. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ke R, Yang X, Tianyi C, Ge M, Lei J and Mu
X: The C342R mutation in FGFR2 causes Crouzon syndrome with elbow
deformity. J Craniofac Surg. 26:584–586. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Padmanabhan V, Hegde AM and Rai K:
Crouzon's syndrome: A review of literature and case report. Contemp
Clin Dent. 2:211–214. 2011. View Article : Google Scholar
|
|
16
|
Robin NH, Falk MJ and Haldeman-Englert CR:
FGFR-Related Craniosynostosis Syndromes. Pagon RA, Adam MP,
Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT,
Mefford HC, Smith RJH and Stephens K: GeneReviews®. http://www.ncbi.nlm.nih.gov/books/NBK1455/.
University of Washington; Seattle, WA: 1993
|
|
17
|
Snyder-Warwick AK, Perlyn CA, Pan J, Yu K,
Zhang L and Ornitz DM: Analysis of a gain-of-function FGFR2 Crouzon
mutation provides evidence of loss of function activity in the
etiology of cleft palate. Proc Natl Acad Sci USA. 107:2515–2520.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Piccione M, Antona V, Niceta M, Fabiano C,
Martines M, Bianchi A and Corsello G: Q289P mutation in the FGFR2
gene: First report in a patient with type 1 Pfeiffer syndrome. Eur
J Pediatr. 168:1135–1139. 2009. View Article : Google Scholar
|
|
19
|
Lapunzina P, Fernández A, Sánchez Romero
JM, Delicado A, Sáenz de Pipaon M, López Pajares I and Molano J: A
novel insertion in the FGFR2 gene in a patient with Crouzon
phenotype and sacrococcygeal tail. Birth Defects Res A Clin Mol
Teratol. 73:61–64. 2005. View Article : Google Scholar
|
|
20
|
Gong SG: The Fgfr2 W290R mouse model of
Crouzon syndrome. Childs Nerv Syst. 28:1495–1503. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Okajima K, Robinson LK, Hart MA, Abuelo
DN, Cowan LS, Hasegawa T, Maumenee IH and Jabs EW: Ocular anterior
chamber dysgenesis in craniosynostosis syndromes with a fibroblast
growth factor receptor 2 mutation. Am J Med Genet. 85:160–170.
1999. View Article : Google Scholar : PubMed/NCBI
|