Introduction
Pancreatic cancer is the fourth leading cause of
cancer-associated mortality for men and women in the USA, with a
5-year survival rate of <5% (1). To date, the sole potential cure for
pancreatic cancer is surgery (2).
However, ~80% patients present when the cancer is too advanced for
surgery (3). For patients with
locally advanced or metastatic inoperative pancreatic cancer,
chemotherapy and radiotherapy are the standard treatments.
Gemcitabine (GEM) is currently the first-line therapeutic agent
prescribed for advanced pancreatic cancer; however, its efficacy is
limited, with a median survival extension of just six weeks
(4). Therefore, adjuvant
strategies have been increasingly investigated to improve these
outcomes.
Metformin (MET) is widely used in the treatment of
type 2 diabetes mellitus and has been investigated due to its
anti-tumor effects (5). Previous
studies have suggested that MET has an effect on various cancer
types (6), including pancreatic
cancer. Certain in vitro and in vivo studies using
mouse xenograft models have indicated that MET may exert direct
anti-tumor activity via the inhibition of cancer cell
proliferation, migration and invasion, promotion of cancer cell
apoptosis, and suppression of tumor growth (7–9).
However, the underlying mechanism of these effects and the
combination of MET with conventional chemotherapy remains to be
clarified.
The aim of the present study was to investigate the
underlying anti-tumor mechanism of MET on the growth of the CFPAC-1
human pancreatic cell line, and to evaluate the effects of
combination therapy with MET and GEM in vitro and in
vivo.
Materials and methods
Cell lines and animals
The human CFPAC-1 pancreatic ductal adenocarcinoma
cell line was donated by the Institute of Hematology of Soochow
University (Suzhou, China). The cells were incubated in
75-cm2 cell culture flasks and maintained in RPMI-1640
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA),
supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml
streptomycin in a humidified atmosphere composed of 5%
CO2 at 37°C. A total of 20 specific-pathogen-free female
athymic BALB/c nu/nu mice (age, 4–5 weeks; weight, 15–16 g) were
purchased from the Shanghai Laboratory Animal Center (Shanghai,
China). Mice were allowed free access to sterilized water and food,
and were housed in individual ventilated cages at 23±5°C under a
12-h light/dark cycle. The experimental protocol was approved by
the Animal Care and Use Committee of Soochow University.
Cell counting kit (CCK)-8 assay
The CFPAC-1 cells were trypsinized with 0.05%
trypsin and then plated in 96-well plates with 100 µl medium
per well. Following an overnight incubation in 5% CO2 at
37°C, cells were treated with normal saline (NS) or MET (0, 1, 2.5,
5, 10, 20, 40 or 60 mmol/l; Sigma-Aldrich, St. Louis, MO, USA),
with six replicates per concentration. Following incubations of 24,
48 or 72 h at 37°C, cells were incubated for 1–4 h with 10
µl CCK-8 reagent per well. Cell viability was measured with
CCK-8 according to the manufacturer's instructions (Peptide
Institute, Inc., Osaka, Japan), and the survival and growth
inhibition rates were calculated as follows: Survival rate =
absorbance of MET (1, 2.5, 5, 10, 20, 40 or 60 mmol/l) / absorbance
of MET (0 mmol/l) × 100; inhibition rate = [absorbance of MET (0
mmol/l) - absorbance of MET (1, 2.5, 5, 10, 20, 40 or 60 mmol/l)] /
absorbance of MET (0 mmol/l) × 100. In the combination therapy
experiment, cells were treated with MET (20 mmol/l) and GEM (5
µmol/l) alone or in combination and cell viability was
measured as above.
Cell cycle analysis and annexin
V/propidium iodide (PI) assay
The CFPAC-1 cells were placed in 6-well plates at
5×104 cells/well and treated with NS or MET (10, 20 or
40 mmol/l). Separate cell cultures were collected and trypsinized
48 h later, then washed twice with cold phosphate-buffered saline
(PBS). The cells were subsequently incubated with an annexin V/PI
double staining solution (Sigma-Aldrich) at room temperature.
Fifteen min later, the stained cells were analyzed by flow
cytometry and the percentage of apoptotic cells (those in the lower
right quadrant) was calculated with ModFit LT software version 4.0
(Verity Software House, Inc., Topsham, ME, USA). Cell cycle
analysis was performed by flow cytometry using CellQuest Pro
software version 5.1 (BD Biosciences, Franklin Lakes, NJ, USA). In
combination therapy experiments, cells were treated with MET (20
mmol/l) and GEM (5 µmol/l), alone or in combination for 48
h. The percentage of apoptotic cells was calculated as above.
Mice xenograft model
The CFPAC-1 cells were resuspended in serum-free
RPMI-1640 at a concentration of 1×107 cells/ml. A total
volume of 0.2 ml cell suspension (2×106 cells) was then
injected subcutaneously into the right anterior armpit of nude
mice, to establish a xenograft model. The 20 injected mice were
randomly divided into four groups: NS-treated control, MET-treated,
GEM-treated and combination therapy (n=5 per group). MET (200
mg/kg, daily), GEM (50 mg/kg, twice/week) or combination therapy
[MET (200 mg/kg, daily)+GEM (50 mg/kg, twice/week)] were
administered intraperitoneally for four weeks. The tumor volume was
measured with calipers every second day and calculated as follows:
Volume (mm3) = 4π / 3× (width / 2)2 × (length
/ 2). The mice were weighed weekly then sacrificed by cervical
dislocation following four weeks of treatment and the tumors were
collected.
Reverse transcription-polymerase chain
reaction (RT-PCR) assay
Total RNA was extracted from CFPAC-1 cells and tumor
tissues using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). The first strand cDNA was synthesized with the
Omniscript RT kit (Qiagen GmbH, Hilden, Germany), using 2,000 ng
RNA per 20 µl reaction and an oligo (dT) primer. PCR
reactions were performed for 35 cycles. Each cycle was performed as
follows: Denaturation for 30 sec at 94°C, annealing for 30 sec at
60°C and polymerization for 30 sec at 72°C. Primers used in the PCR
reaction were as follows: Forward, 5′-CGGGCATTCAGTGACCTGAC-3′ and
reverse, 5′-TCAGGAACCAGCGGTTGAAG-3′ for B-cell lymphoma-extra large
(Bcl-xL); forward, 5′-GCGTCCACCAAGAAGCTGA-3′ and reverse,
5′-ACCACCCTGGTCTTGGATCC-3′ for Bcl2 associated X protein (Bax);
forward, 5′-GTCTCAATGCCACAGTCCAGT-3′ and reverse,
5′-AGCAAACCTCAGGGAAACATT-3 for caspase-3; forward,
5′-TACGCCTGTAATACCAGCAC-3′ and reverse, 5′-TCTCCGCAGTTTCCTCAA-3′
for survivin; forward, 5′-TGTTCGTGGCCTCTAAGATG-3′ and reverse,
5′-ACTCCAGAAGGGCTTCAATC-3′ for cyclin D1; and forward,
5′-AGCGGGAAATCGTGCGTG-3′ and reverse, 5′-CAGGGTACATGGTGGTGCC-3′ for
β-actin. The PCR products were detected using Molecular Analyst
software version 2.10e (BioRad Laboratories, Inc., Hercules, CA,
USA). β-actin served as an internal standard.
Western blot assay
Cells or tumor tissue were lyzed with
radioimmunoprecipitation assay buffer on ice for 30 min and
centrifuged at 12,000 × g, 4°C for 10 min. The protein samples (20
µg) were separated on a 10% SDS-PAGE gel, at 80 V (stacking
gel) and 100 V (separating gel), and transferred onto
nitrocellulose membranes. The membranes were blocked with PBS-Tween
(PBS-T) containing 5% skimmed milk powder for 1 h at room
temperature. The membranes were then incubated at 4°C overnight
with primary mouse monoclonal antibodies, diluted 1:1,000, against
Bcl-xL (catalog no. sc-8392), Bax (catalog no. sc-7480), caspase-3
(catalog no. sc-65497), survivin (catalog no. sc-374616) and cyclin
D1 (catalog no. sc-8396), purchased from Santa Cruz Biotechnology,
Inc. (Dallas, TX, USA). Subsequently, the membranes were washed
with PBS-T three times and incubated for 1 h with a goat anti-mouse
IgG antibody conjugated to horseradish peroxidase (HRP) (1:1,000;
catalog no. sc-2031; Santa Cruz Biotechnology, Inc.). Membranes
were then washed three times with PBS-T, and protein bands were
visualized using an enhanced chemiluminescence (ECL) -detection
system (Beyotime Institute of Biotechnology, Haimen, China).
Immunohistochemistry
Tumor xenografts were fixed in formalin and embedded
in paraffin blocks, which were sliced into 4-µm thick
sections for immunohistochemical staining.
Following deparaffinization, the sections were
incubated at 4°C overnight with primary mouse monoclonal
antibodies, diluted 1:200, against caspase-3 (catalog no. 65497) or
proliferating cell nuclear antigen (PCNA; catalog no. sc-25280),
purchased from Santa Cruz Biotechnology, Inc. The sections were
washed three times with PBS-T, and incubated for 1 h at room
temperature with a goat anti-mouse IgG antibody conjugated to HRP
(1:200; catalog no. sc-2031; Santa Cruz Biotechnology, Inc.). The
slices were washed with PBS-T three times, and incubated in
diaminobenzidine (Sangon Biotech Co., Ltd., Shanghai, China) and
counterstained with hematoxylin (Sangon Biotech Co., Ltd.) for 1
min. Images were captured using a light microscope (magnification,
×400; model CKX41-A32RC; Olympus Corporation, Tokyo, Japan).
Statistical analysis
Data were expressed as the mean ± standard error and
analyzed using SPSS version 18.0 (SPSS, Inc., Chicago, IL, USA).
Statistical analysis was performed using one-way analysis of
variance. The Student-Newman-Keuls (SNK) method was used as a
post-hoc test. The Kruskal-Wallis test was used to evaluate the
differences of categorical values, followed by the Mann-Whitney U
test as a post-hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Effect of MET on growth of CFPAC-1
cells
The human CFPAC-1 pancreatic ductal adenocarcinoma
cells were treated with various concentrations of MET for 24, 48 or
72 h, and survival and growth inhibition rates were evaluated with
the CCK-8 assay. The proliferation and survival of CFPAC-1 cells
was inhibited in a dose-and time-dependent manner (Fig. 1A and B). Cell apoptosis was
determined by annexin V/PI assay. The percentage of apoptotic cells
increased in a dose-dependent manner following a 48 h incubation
with MET (Fig. 1C; Table I). Cell cycle analysis revealed
that cells in the G0/G1 phase increased
significantly following MET treatment in a dose-dependent manner
(Fig. 1D and E; Table II). Furthermore, RT-PCR and
western blot analysis demonstrated that the expression levels of
Bcl-xL, survivin and cyclin D1 were reduced following 48 h of MET
treatment (Fig. 1F and G). By
contrast, the expression levels of Bax and caspase-3 increased
following MET treatment.
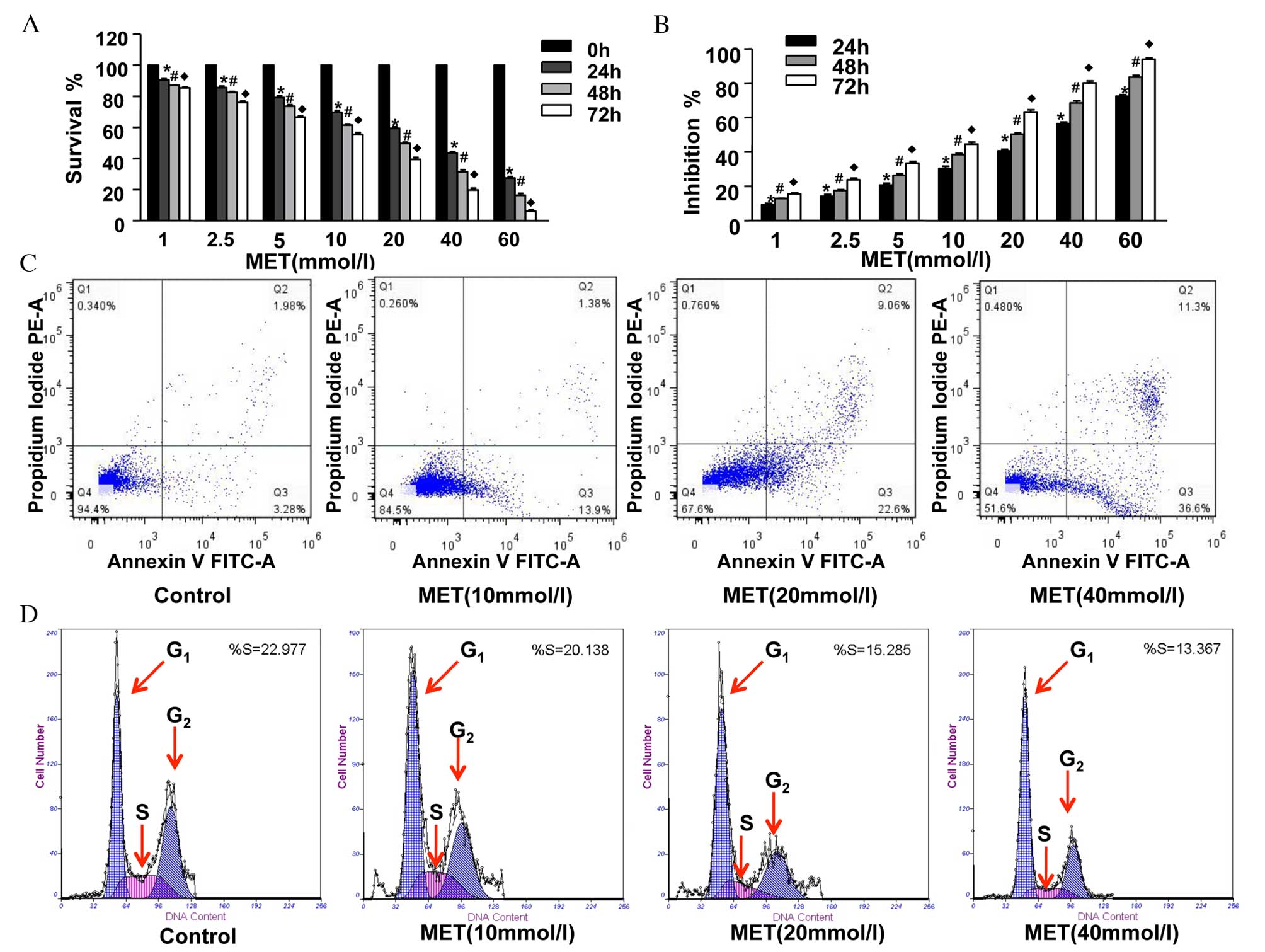 | Figure 1Inhibitory effect of MET on the growth
of the human CFPAC-1 pancreatic cancer cell line in vitro.
CFPAC-1 cells were incubated with various concentrations of MET,
from 0–60 mmol/l, for 0, 24, 48 or 72 h. The cell counting kit-8
assay was then conducted to analyze (A) cell viability and (B)
growth inhibition rates. MET treatment decreased survival and
inhibited growth of CFPAC-1 cells. Data are presented as the mean ±
standard error. (C) The percentage of apoptotic CFPAC-1 cells (the
lower right quadrant, annexin V+ PI−) was
analyzed using an annexin V/propidium iodide assay. (D) Cell cycle
analysis was conducted by flow cytometry. (E) Percentage of cells
in G0/G1 phases increased following MET
treatment. Subsequent to treatment with MET, CFPAC-1 cells were
harvested and the (F) mRNA and (G) protein expression levels of
Bax, Bcl-xL, caspase-3, survivin and cyclin D1 were analyzed by
reverse transcription-polymerase chain reaction and western
blotting, respectively. β-actin served as an internal standard. The
expression levels of Bax and caspase-3 increased following MET
treatment, while Bcl-xL, survivin and cyclin D1 were reduced.
*P<0.05 vs. 0 h; #P<0.05 vs. 24 h;
♦P<0.05 vs. 48 h. MET, metformin; Bcl-xL, B-cell
lymphoma-extra large; Bax, Bcl2 associated X protein. |
 | Table IImpact of MET on viability of CFPAC-1
cells in vitro. |
Table I
Impact of MET on viability of CFPAC-1
cells in vitro.
| Parameter | MET concentration
(mmol/l)
|
|---|
| 0 | 10 | 20 | 40 |
|---|
| Apoptosis, % | 3.01±0.49 | 13.77±1.31a | 22.63±1.45a | 32.97±3.19a |
| Table IIImpact of MET on cell cycle of
CFPAC-1 cells in vitro. |
Table II
Impact of MET on cell cycle of
CFPAC-1 cells in vitro.
| Cell cycle
phase | MET concentration
(mmol/l)
|
|---|
| 0 | 10 | 20 | 40 |
|---|
|
G0/G1, % | 42.89±1.02 | 49.59±3.15a | 56.03±1.49a | 65.93±0.27a |
| G2/M,
% | 38.28±4.93 | 28.72±1.32a | 30.70±2.75a | 22.01±2.95a |
| S, % | 20.12±3.38 | 21.68±4.18 | 13.27±1.78a | 13.58±0.43a |
Effect of MET combined with GEM on growth
of CFPAC-1 cells
The CFPAC-1 cells were treated with MET, GEM or a
combination of the two agents. CCK-8 assay revealed that the
proliferation and survival of CFPAC-1 cells were inhibited
significantly following treatment with MET in combination with GEM,
compared with MET or GEM alone (combined treatment vs. MET
treatment at 24, 48 and 72 h, P<0.001; combined treatment vs.
GEM treatment at 24, 48 and 72 h, P<0.001; Fig. 2A and B). In addition, the
percentage of apoptotic cells increased following combination
therapy compared with MET or GEM treatment alone (Fig. 2C; Table III). The mRNA and protein
expression levels of Bcl-xL, survivin and cyclin D1 were decreased
in MET and GEM alone treatment groups, and a greater decrease was
observed in the combination therapy group (Fig. 2D and E). Similarly, a greater
increase in Bax and caspase-3 expression levels was observed in the
combination therapy group compared with the single treatment
groups.
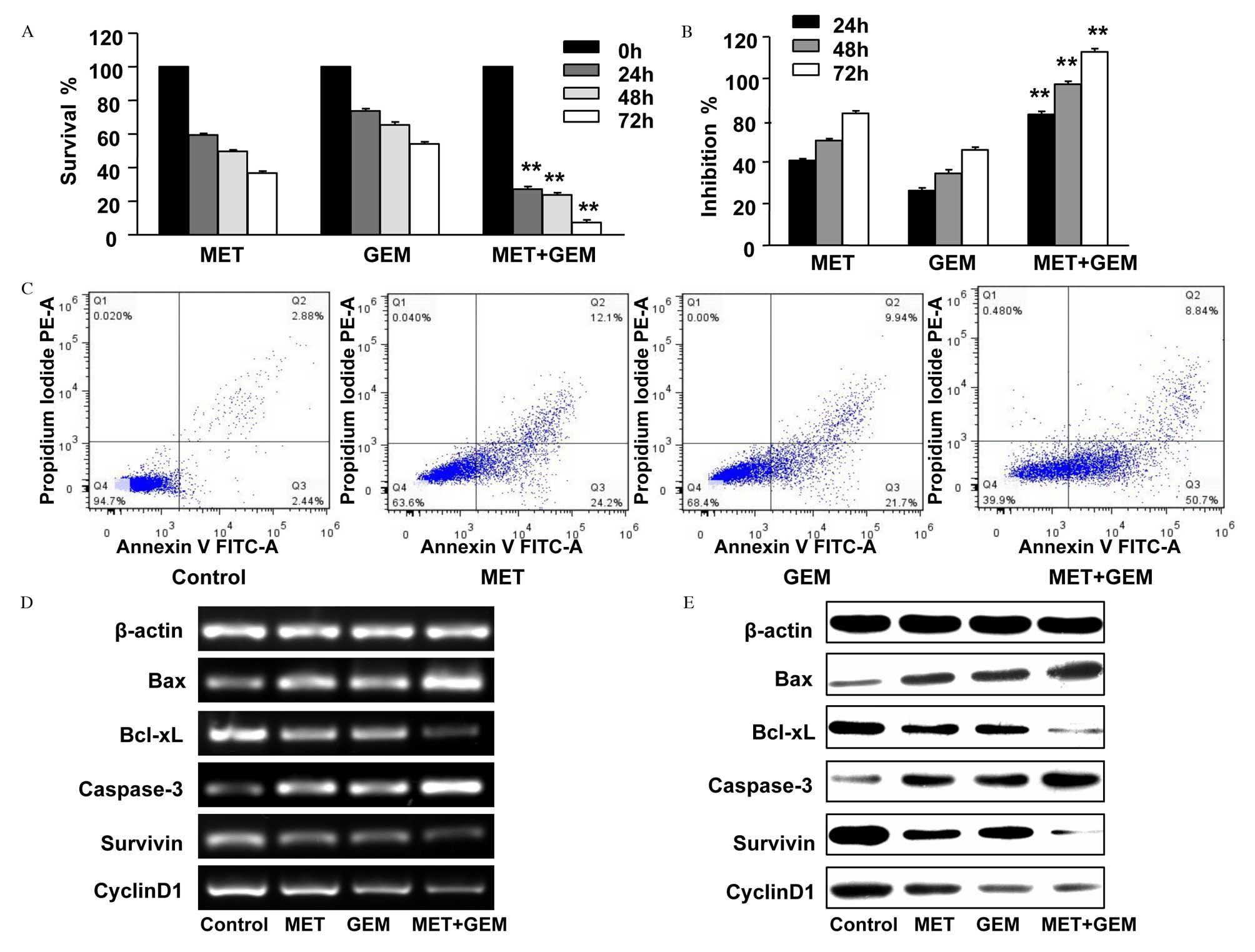 | Figure 2Inhibitory effect of MET combined with
GEM on the growth of the human CFPAC-1 pancreatic cancer cell line
in vitro. CFPAC-1 cells were incubated with 20 mmol/l MET, 5
µmol/l GEM or MET and GEM in combination for 0, 24, 48 or 72
h. The cell counting kit-8 assay was conducted to analyze (A) cell
viability and (B) growth inhibition rates. Combination therapy
significantly increased the effectiveness of MET and GEM. The data
are presented as the mean ± standard error. (C) The percentage of
apoptotic CFPAC-1 cells (the lower right quadrant, annexin
V+ PI−) was analyzed using an annexin
V/propidium iodide assay. Subsequent to treatment with 20 mmol/l
MET, 5 µmol/l GEM or MET and GEM combined, CFPAC-1 cells
were harvested and the (D) mRNA and (E) protein expression levels
of Bax, Bcl-xL, caspase-3, survivin and cyclin D1 were analyzed by
reverse transcription-polymerase chain reaction and western
blotting, respectively. **P<0.05 vs. MET or GEM
treatment alone. MET, metformin; GEM, gemcitabine; Bcl-xL, B-cell
lymphoma-extra large; Bax, Bcl2 associated X protein. |
| Table IIIImpact of MET combined with GEM on
the viability of CFPAC-1 cells in vitro. |
Table III
Impact of MET combined with GEM on
the viability of CFPAC-1 cells in vitro.
| Parameter | Treatment group
|
|---|
| Control | 20 mmol/l MET | 5 µmol/l
GEM | 20 mmol/l MET+5
µmol/l GEM |
|---|
| Apoptosis, % | 3.01±0.49 | 24.53±2.18a | 22.37±1.61a | 52.07±2.81a,b |
Growth inhibitory effect of MET in
vivo
All mice survived for the duration of the experiment
with no observable toxic effect, such as weight loss. Mice were
sacrificed at 28 days and the weights of the xenografts were
measured. In comparison to the NS-treated control group, xenografts
from mice treated with MET, GEM or combination therapy were smaller
in size and weight (MET vs. normal saline treatment, P=0.007; GEM
vs. normal saline treatment, P=0.001; combined vs. normal saline
treatment, P<0.001; Fig. 3A–C).
Furthermore, the tumor volume and weight were decreased
significantly following combined treatment (combined vs. MET
treatment, P<0.001; combined vs. GEM treatment, P=0.001).
Bcl-xL, survivin, Bax, caspase-3 and cyclin D1 mRNA and protein
expression levels followed a similar pattern to that of the in
vitro experiment (Fig. 3D and
E). The protein expression level of caspase-3 was markedly
upregulated in the combination therapy group, and this was
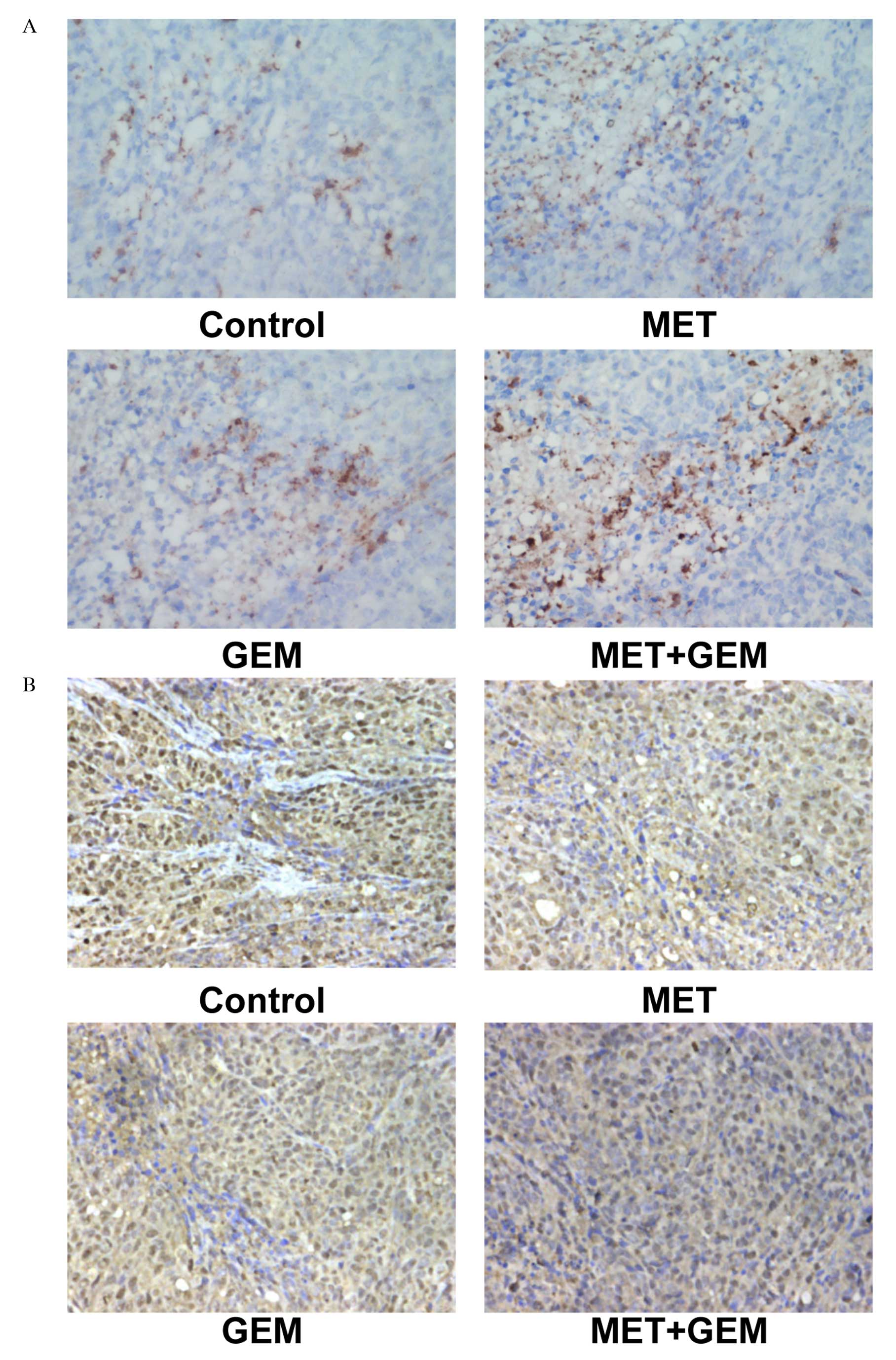
confirmed by immunohistochemistry (Fig. 4A). However, expression of PCNA
protein was decreased following treatment (Fig. 4B).
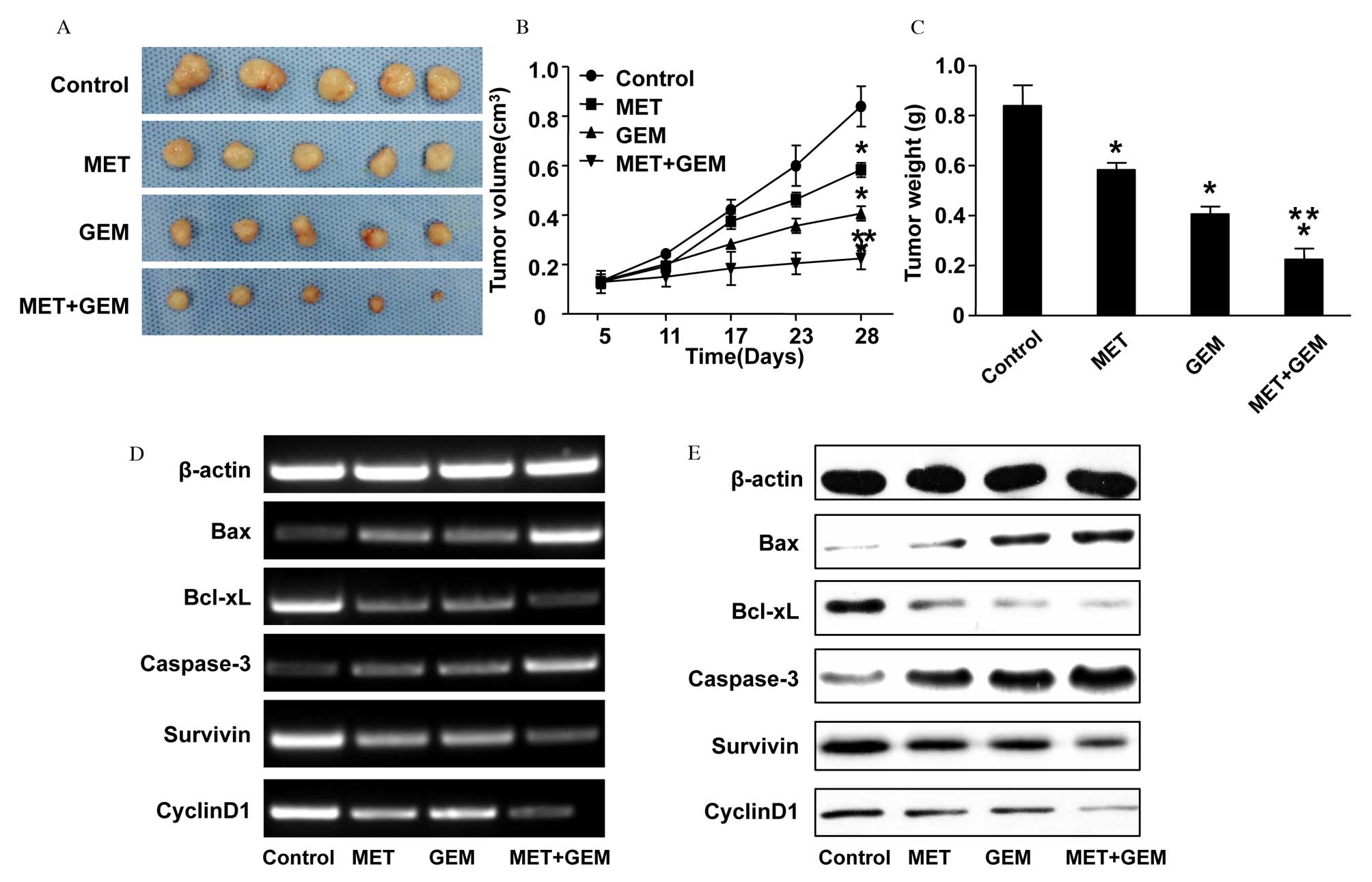 | Figure 3Inhibitory effect of combined
treatment with MET and GEM on the growth of the human CFPAC-1
pancreatic cancer cell line in vivo. CFPAC-1 cells were
injected into nude mice, which were then treated for 28 days with
200 mg/kg MET, 50 mg/kg GEM or MET and GEM in combination. (A)
Gross morphology of tumors at sacrifice. (B) Volume changes in the
xenografts. Treatment with MET, GEM or a combination of the two
significantly inhibited tumor growth, compared with the control
group. The data are presented as the mean ± SE. (C) Following 28
days of treatment, the tumor weights were significantly decreased
in all three treatment groups compared with the control group. The
data are presented as the mean ± SE. (D) mRNA and (E) protein
expression levels of Bax, Bcl-xL, caspase-3, survivin and cyclin D1
in nude mice xenografts were analyzed by reverse
transcription-polymerase chain reaction and western blotting,
respectively. *P<0.05 vs. normal saline-treated
control group; **P<0.05 vs. MET or GEM treatment
alone. MET, metformin; GEM, gemcitabine; Bcl-xL, B-cell
lymphoma-extra large; Bax, Bcl2 associated X protein; SE, standard
error. |
Discussion
The glucose-lowering effect of MET is associated
with an increase in insulin sensitivity in vivo, resulting
in increased glucose uptake, decreased gluconeogenesis and reduced
plasma glucose concentrations. Emerging evidence from in
vitro, in vivo and epidemiological studies suggests an
anti-tumor role of MET (10–12).
The underlying mechanisms are complex and require further
investigation. It has been demonstrated that MET activates the 5′
adenosine mono-phosphate-activated protein kinase/mammalian target
of rapamycin signaling pathway through inhibition of complex 1 of
the mitochondrial respiratory chain (13,14).
In addition, MET has indirect anti-tumor effects via the reduction
of circulating insulin and insulin growth factor 1 levels (15). In vitro, MET induces cell
cycle arrest, thus inhibiting proliferation and inducing cell
apoptosis. Furthermore, MET affects the proliferation of cancer
stem cells, reduces DNA damage and inhibits the inflammatory
response (16–18).
In the present study, the influence of MET on the
apoptosis and proliferation of the human CFPAC-1 pancreatic ductal
adenocarcinoma cell line was investigated in vitro and in
vivo. The mechanism underlying induction of apoptosis in cancer
cells by MET may be associated with altered expression of
pro-apoptotic and anti-apoptotic molecules. In addition, MET
arrested cells in the G0/G1 phase and
downregulated the expression of cyclin D1 and PCNA, demonstrating
anti-proliferative activity. Furthermore, the pro-apoptotic and
anti-proliferative activities of MET were enhanced when it was
administered in combination with GEM.
Appropriate apoptotic signaling is crucial for
preserving tissue homeostasis by maintaining a healthy balance
between cell death and cell survival. The ratio of pro- and
anti-apoptotic molecules regulates cell death. Carcinogenesis may
occur if the balance is disturbed and apoptosis in malignant cells
is reduced. Bcl-xL and Bax are members of the Bcl-2 protein family
and, as such, are important in apoptosis. Bax mediates the
permeabilization of the outer mitochondrial membrane and the
release of cytochrome c into the cytoplasm, which activates
caspase-3 to induce chromosome cleavage and apoptosis (19). However, the effect triggered by Bax
is blocked by Bcl-xL (20). It has
been reported that >50% of cancers are associated with excessive
expression of Bcl-xL (21).
Survivin belongs to the inhibitor of apoptosis proteins (IAPs)
family and is considered a node protein, inhibiting apoptosis and
regulating cell mitosis (22).
Promising cancer treatment strategies that target apoptotic
inhibitors, including Bcl-2 family proteins and IAPs are currently
under investigation (23).
The cell cycle is divided into three phases:
G0, the interphase (G1, S and G2)
and M. Cells are quiescent in the G0 phase. The
G1 checkpoint control mechanism ensures that the cell is
prepared for DNA synthesis. Cyclin D1 is an important cell cycle
regulatory protein, which performs a positive role during the
crucial restriction point of the G1/S transition. Cyclin
D1 expression and accumulation are induced by growth factors and
occur at multiple levels, including increased transcription,
translation and protein stability (24). PCNA is a DNA clamp that acts as a
processivity factor for DNA polymerase δ in eukaryotic cells, and
the presence of PCNA is a specific marker of cell proliferation
(25).
Pancreatic cancer remains one of the most fatal
types of cancer despite the oncological advances achieved over the
past two decades. Patients suffering from pancreatic cancer have a
median survival time of 4–6 months (26). GEM, the first-line chemotherapeutic
agent prescribed in unresectable cases, only marginally improves
the outcome. GEM-based combination therapies, including cisplatin,
capecitabine and exatecan have failed to make any significant
improvements (27–29). Furthermore, combination therapies
may be more toxic and, therefore, less well tolerated (27–29).
The lack of effective and less toxic treatment options for
pancreatic cancer has prompted investigations into novel combined
treatment strategies.
There are complex associations between diabetes
mellitus and pancreatic cancer, as diabetes may be a risk factor
for, or a result of, pancreatic cancer (30). Previous studies have demonstrated
that MET is beneficial for pancreatic cancer prognosis (31,32).
A borderline significant relative survival benefit was observed in
MET-treated patients compared with non-MET-treated patients [hazard
ratio (HR), 0.80; 95% confidence interval (CI), 0.62–1.03] in a
pooled analysis of four publications containing 1,429 patients
(33). However, few clinical
trials of MET in pancreatic cancer treatment have been
reported.
Between 2010 and 2014, Kordes et al (34) presented a randomized,
placebo-controlled trial of MET in the treatment of patients with
advanced pancreatic cancer. Despite the laboratory evidence of
anti-tumor activity, MET addition did not improve the clinical
outcome for these patients (median survival of 7.6 months in the
placebo group vs. 6.8 months in the MET group; HR, 1.056; 95% CI,
0.72–1.55). The authors hypothesized that the conventional
anti-diabetic dose of MET may fail to accumulate to a sufficient
concentration to cause energetic stress (34). Blood MET concentrations were in the
micromolar range, significantly lower than that of in vitro
studies, which demonstrated the direct anti-tumor effects of MET.
In addition, only patients with advanced pancreatic cancer were
included in the trial (34).
Future studies focusing on patients with hyperinsulinaemia or
patients with tumors expressing markers of sensitivity to energetic
stress are required.
In conclusion, MET inhibited the growth of
pancreatic cancer cells via the induction of apoptosis and the
reduction of proliferation. In addition, the efficacy of MET was
significantly improved when administered in combination with GEM.
The findings of the present study may provide an alternative
strategy for the treatment of pancreatic cancer, but requires
validation in clinical trials. In addition, the mechanisms
underlying the effects of apoptosis induction and proliferation
reduction require additional investigation. Furthermore, future
studies are required to identify inhibitors of oxidative
phosphorylation, which are more potent than MET.
Acknowledgments
The present study was supported by the Natural
Science Foundation of Jiangsu Province (grant no. BK20131157).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xu Q, Zhang TP and Zhao YP: Advances in
early diagnosis and therapy of pancreatic cancer. Hepatobiliary
Pancreat Dis Int. 10:128–135. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stathis A and Moore MJ: Advanced
pancreatic carcinoma: Current treatment and future challenges. Nat
Rev Clin Oncol. 7:163–172. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Burris HR, Moore MJ, Andersen J, Green MR,
Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM,
Tarassoff P, et al: Improvements in survival and clinical benefit
with gemcitabine as first-line therapy for patients with advanced
pancreas cancer: A randomized trial. J Clin Oncol. 15:2403–2413.
1997.PubMed/NCBI
|
|
5
|
He H, Ke R, Lin H, Ying Y, Liu D and Luo
Z: Metformin, an old drug, brings a new era to cancer therapy.
Cancer J. 21:70–74. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Morales DR and Morris AD: Metformin in
cancer treatment and prevention. Annu Rev Med. 66:17–29. 2015.
View Article : Google Scholar
|
|
7
|
Kisfalvi K, Moro A, Sinnett-Smith J, Eibl
G and Rozengurt E: Metformin inhibits the growth of human
pancreatic cancer xenografts. Pancreas. 42:781–785. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bao B, Wang Z, Ali S, Ahmad A, Azmi AS,
Sarkar SH, Banerjee S, Kong D, Li Y, Thakur S and Sarkar FH:
Metformin inhibits cell proliferation, migration and invasion by
attenuating CSC function mediated by deregulating miRNAs in
pancreatic cancer cells. Cancer Prev Res (Phila). 5:355–364. 2012.
View Article : Google Scholar
|
|
9
|
Wang LW, Li ZS, Zou DW, Jin ZD, Gao J and
Xu GM: Metformin induces apoptosis of pancreatic cancer cells.
World J Gastroenterol. 14:7192–7198. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gao ZY, Liu Z, Bi MH, Zhang JJ, Han ZQ,
Han X, Wang HY, Sun GP and Liu H: Metformin induces apoptosis via a
mitochondria-mediated pathway in human breast cancer cells in
vitro. Exp Ther Med. 11:1700–1706. 2016.PubMed/NCBI
|
|
11
|
Fujimori T, Kato K, Fujihara S, Iwama H,
Yamashita T, Kobayashi K, Kamada H, Morishita A, Kobara H, Mori H,
et al: Antitumor effect of metformin on cholangiocarcinoma: In
vitro and in vivo studies. Oncol Rep. 34:2987–2996. 2015.PubMed/NCBI
|
|
12
|
Ramjeesingh R, Orr C, Bricks CS, Hopman WM
and Hammad N: A retrospective study on the role of diabetes and
metformin in colorectal cancer disease survival. Curr Oncol.
23:e116–e122. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Owen MR, Doran E and Halestrap AP:
Evidence that metformin exerts its anti-diabetic effects through
inhibition of complex 1 of the mitochondrial respiratory chain.
Biochem J. 348:607–614. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
El-Mir MY, Nogueira V, Fontaine E, Averet
N, Rigoulet M and Leverve X: Dimetforminhylbiguanide inhibits cell
respiration via an indirect effect targeted on the respiratory
chain complex I. J Biol Chem. 275:223–228. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gallagher EJ and LeRoith D: Minireview:
IGF, insulin, and cancer. Endoscopy. 152:2546–2255. 2011.
|
|
16
|
Algire C, Moiseeva O, Deschênes-Simard X,
Amrein L, Petruccelli L, Birman E, Viollet B, Ferbeyre G and Pollak
MN: Metformin reduces endogenous reactive oxygen species and
associated DNA damage. Cancer Prev Res (Phila). 5:536–543. 2012.
View Article : Google Scholar
|
|
17
|
Hirsch HA, Iliopoulos D and Struhl K:
Metformin inhibits the inflammatory response associated with
cellular transformation and cancer stem cell growth. Proc Natl Acad
Sci USA. 110:972–977. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gou S, Cui P, Li X, Shi P, Liu T and Wang
C: Low concentrations of metformin selectively inhibit CD133 (+)
cell proliferation in pancreatic cancer and have anticancer action.
Plos One. 8:e639692013. View Article : Google Scholar
|
|
19
|
Breckenridge DG and Xue D: Regulation of
mitochondrial membrane permeabilization by BCL-2 family proteins
and caspases. Curr Opin Cell Biol. 16:647–652. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang J, Liu X, Bhalla K, Kim CN, Ibrado
AM, Cai J, Peng TI, Jones DP and Wang X: Prevention of apoptosis by
Bcl-2: Release of cytochrome c from mitochondria blocked. Science.
275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Amundson SA, Myers TG, Scudiero D, Kitada
S, Reed JC and Fornace AJ Jr: An informatics approach identifying
markers of chemosensitivity in human cancer cell lines. Cancer Res.
60:6101–6610. 2000.PubMed/NCBI
|
|
22
|
Mobahat M, Narendran A and Riabowol K:
Survivin as a preferential target for cancer therapy. Int J Mol
Sci. 15:2494–2516. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hassan M, Watari H, AbuAlmaaty A, Ohba Y
and Sakuragi N: Apoptosis and molecular targeting therapy in
cancer. Biomed Res Int. 2014:1508452014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim JK and Diehl JA: Nuclear cyclin D1: An
oncogenic driver in human cancer. J Cell Physiol. 220:292–296.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Juríková M, Danihel Ľ, Polák Š and Varga
I: Ki67, PCNA, and MCM proteins: Markers of proliferation in the
diagnosis of breast cancer. Acta Histochem. 30084–30088. 2016.Epub
ahead of print.
|
|
26
|
Vincent A, Herman J, Schulick R, Hruban RH
and Goggins M: Pancreatic cancer. Lancet. 378:607–620. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Colucci G, Labianca R, Di Costanzo F,
Gebbia V, Cartenì G, Massidda B, Dapretto E, Manzione L, Piazza E,
Sannicolò M, et al: Randomized phase III trial of gemcitabine plus
cisplatin compared with single-agent gemcitabine as first-line
treatment of patients with advanced pancreatic cancer: The GIP-1
study. J Clin Oncol. 28:1645–1651. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Abou-Alfa GK, Letourneau R, Harker G,
Modiano M, Hurwitz H, Tchekmedyian NS, Feit K, Ackerman J, De Jager
RL, Eckhardt SG and O'Reilly EM: Randomized phase III study of
exatecan and gemcitabine compared with gemcitabine alone in
untreated advanced pancreatic cancer. J Clin Oncol. 24:4441–4447.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cunningham D, Chau I, Stocken DD, Valle
JW, Smith D, Steward W, Harper PG, Dunn J, Tudur-Smith C, West J,
et al: Phase III randomized comparison of gemcitabine versus
gemcitabine plus capecitabine in patients with advanced pancreatic
cancer. J Clin Oncol. 27:5513–5518. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gong J, Robbins LA, Lugea A, Waldron RT,
Jeon CY and Pandol SJ: Diabetes, pancreatic cancer, and metformin
therapy. Front Physiol. 5:4262014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Choi Y, Kim TY, Oh DY, Lee KH, Han SW, Im
SA, Kim TY and Bang YJ: The impact of diabetes mellitus and
metformin treatment on survival of patients with advanced
pancreatic cancer undergoing chemotherapy. Cancer Res Treat.
48:171–179. 2016. View Article : Google Scholar :
|
|
32
|
Sadeghi N, Abbruzzese JL, Yeung SC, Hassan
M and Li D: Metformin use is associated with better survival of
diabetic patients with pancreatic cancer. Clin Cancer Res.
18:2905–2912. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang JW and Sun Q: Metformin may improve
the prognosis of patients with pancreatic cancer. Asian Pac J
Cancer Prev. 16:3937–3940. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kordes S, Pollak MN, Zwinderman AH, Mathôt
RA, Weterman MJ, Beeker A, Punt CJ, Richel DJ and Wilmink JW:
Metformin in patients with advanced pancreatic cancer: A
double-blind, randomised, placebo-controlled phase 2 trial. Lancet
Oncol. 16:839–847. 2015. View Article : Google Scholar : PubMed/NCBI
|