Introduction
Chronic kidney disease (CKD), which affects up to
15% of adults in the USA, presents a significant threat to public
health (1–3). Diabetes mellitus, hypertension,
glomerulonephritis and idiopathic origins are the most common
causes. CKD is a primary risk factor for the development of
cardiovascular diseases (CVDs) (4–6).
Among various cardiovascular complications, atherosclerosis is
predictive of a markedly increased risk of cardiovascular mortality
in CKD patients (7). Notably,
coronary heart disease is prevalent in CKD patients, whereas the
survival rate following acute coronary syndrome (ACS) is decreased
compared with the general population (8). As reported in a population-based
prospective study of people lacking manifest vascular disease, even
the earliest stages of CKD are associated with an increased risk of
subsequent coronary heart disease (9). In addition, CKD patients suffer more
severe atherosclerotic lesions in coronary plaques as renal
function deteriorates (10).
However, the underlying mechanisms linking these two diseases
remain to be fully elucidated.
CKD is characterized by the retention of various
uremic toxins, including the protein-bound solutes p-cresyl
sulfate (PCS) and indoxyl sulfate (IS). Due to their
protein-binding properties, PCS and IS are not efficiently removed
from the circulation by traditional hemodialysis and continue to
accumulate in CKD patients (11,12).
PCS and IS have been associated with cardiovascular mortality
(13,14). However, whether these toxins
contribute to the process of CKD-associated ACS remains to be
elucidated. Although CKD patients have a greater incidence of, and
increased mortality due to, ACS, causal mechanisms underlying this
association remain to be precisely defined (15). Clinical evidence suggests that the
majority of acute cardiovascular events may be attributed to
vulnerable atherosclerotic plaques (16,17).
As the erosion or rupture of unstable atherosclerotic plaques is a
primary cause of ACS, it was hypothesized that protein-bound uremic
toxins may disturb the stability of atherosclerotic plaques.
Statins, a class of drugs that inhibit
3-hydroxy-3-methylglutaryl coenzyme A reductase, are a first-line
treatment for the primary and secondary prevention of
atherosclerosis-associated CVD (18). In addition to decreasing serum
cholesterol levels, statins have been demonstrated to be effective
at improving endothelial function, reducing inflammation and
promoting the stability of vulnerable atherosclerotic plaques
(19). Therefore, it was
hypothesized that atorvastatin, a member of the statin family, may
attenuate the detrimental effects of protein-bound uremic toxins in
the development of atherosclerosis.
The aim of the present study was to investigate the
effect of excess PCS on the formation and stability of
atherosclerotic plaques, using an apolipoprotein E knockout (ApoE
KO) mouse model. In addition, the therapeutic potential of
atorvastatin was carefully assessed.
Materials and methods
Reagents and antibodies
PCS was synthesized using the method previously
described by Feigenbaum and Neuberg (20). The identity and purity (>99%) of
PCS were confirmed using nuclear magnetic resonance spectroscopy.
Atorvastatin was purchased from Pfizer, Inc. (Shanghai, China). Rat
anti-mouse vascular cell adhesion molecule-1 (VCAM-1; catalog no.
sc-18864), rat anti-mouse intercellular cell adhesion molecule-1
(ICAM-1; catalog no. sc-71303) and rabbit anti-mouse
glyceraldehyde-3-phosphate dehydrogenase (GAPDH; catalog no.
sc-25778) antibodies were purchased from Santa Cruz Biotechnology,
Inc. (Dallas, TX, USA). Horseradish peroxidase (HRP)-conjugated
rabbit anti-rat IgG (catalog no. 7077) and goat anti-rabbit IgG
(catalog no. 7074) secondary antibodies were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA).
Radioimmunoprecipitation assay buffer (RIPA lysis buffer),
phenylmethanesulfonyl fluoride (PMSF) and bicinchoninic acid (BCA)
protein assay kit were purchased from Beyotime Institute of
Biotechnology (Shanghai, China).
Animals
Pathogen-free ApoE KO mice (C57BL/6 background,
male, 8 weeks of age) were purchased from the Model Animal Research
Center of Nanjing University (Nanjing, China). ApoE KO mice were
maintained in air-filtered units at 21±2°C and 50±15% relative
humidity under a 12-h light/dark cycle. Animals were provided with
a high-fat diet (0.25% cholesterol and 15% cocoa butter) and
sterile water ad libitum. Mice were randomly divided into
three groups: i) Control group (n=16), in which mice received water
by oral gavage; ii) PCS group (n=16), in which mice received 0.4%
PCS in water (17 µl/g) by oral gavage, to give a daily
intake of 100 mg/kg, as previously described (21,22);
and iii) PCS + atorvastatin group (n=16), in which mice received
0.4% PCS (17 µl/g) by oral gavage and atorvastatin dissolved
in isosmotic saline and infused via a stomach tube at a dose of 10
mg/kg/day, as previously described (23). Control and PCS groups were infused
with equal volumes of isosmotic saline. Following 20 weeks of
treatment, mice were sacrificed for further analyses. The study
protocol was approved by the Committee on the Ethics of Animal
Experiments of the Shanghai Jiao Tong University School of Medicine
[Shanghai, China; permit no. (2012)-86]. Standards from the Guide
for the Care and Use of Laboratory Animals by the National
Institutes of Health (Bethesda, MD, USA; publication no. 85–23,
revised 1996) were followed.
Tissue collection
Prior to collection of blood samples, animals were
fasted overnight and anesthetized with 1.5% pentobarbital sodium
(60 mg/kg i.p.; Shanghai XiTang Biotechnology Co., Ltd., Shanghai,
China). Blood was collected from the inferior vena cava using a
syringe and needle. Mice were perfused with ice-cold normal saline.
The heart and arteries were dissected out. A portion of the samples
was embedded in optimal cutting temperature compound, frozen and
cut into 5-µm cryosections. For western blot analysis,
tissue samples were immediately placed in liquid nitrogen. The
samples were kept at −80°C until use.
Biochemical investigations
The serum lipid profile of triglycerides (TG), total
cholesterol (TC), low-density lipoprotein cholesterol (LDL-C) and
high-density lipoprotein cholesterol (HDL-C) were detected
enzymatically using kits (catalog nos. 10010303 and 10007640;
Cayman Chemical Company, Ann Arbor, MI, USA) according to the
manufacturer's instructions.
Western blot analysis
Total protein was extracted from the aortas of ApoE
KO mice. Briefly, protein extracts of the ascending aorta were
homogenized in RIPA lysis buffer containing 1% PMSF. Following
centrifugation of the homogenates at 14,000 × g for 30 min at 4°C,
supernatants were collected and protein concentrations assessed
using a BCA protein assay kit. The supernatant was mixed with
loading buffer and heated in a boiling water bath for 5 min. Equal
quantities (50 µg) of prepared protein were subjected to 10%
SDS-PAGE (100 mV, 2 h) and transferred onto polyvinylidene
difluoride membranes. Membranes were blocked with 5% non-fat milk
in Tris-buffered saline with Tween-20 (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) and probed overnight at 4°C with antibodies
recognizing ICAM-1 (1:1,000), VCAM-1 (1:1,000) and GAPDH (1:2,000)
as previously described (24,25).
Membranes were then incubated with HRP-conjugated secondary
antibodies at a dilution of 1:5,000 for 1 h at room temperature.
Immunoreactive bands were detected using an Enhanced
Chemiluminescence system (EMD Millipore, Billerica, MA, USA). The
expression of ICAM-1 and VCAM-1 was normalized to GAPDH and
quantified in Image-Pro Plus software version 6 (Media Cybernetics,
Inc., Rockville, MD, USA).
Histology
The aorta was dissected and an aortic lesion en face
assay was performed by staining with oil red O as previously
described (26,27). The percentage of lesion coverage
was calculated by dividing the stained area by the total aortic
surface area (28). In addition,
aortic root analysis was performed. The collagen contents of the
vascular intima were evaluated with Sirius red staining, using
aortic root sections (10 µm). Images were viewed and
captured with an Olympus microscope (Olympus Corporation, Tokyo,
Japan) and quantified in Image-Pro Plus software version 6.0.
Intravital microscopy
Following 20 weeks of treatment, an in vivo
adhesion assay was performed as previously described (25,29).
Briefly, mice were anesthetized by i.p. injection of 60 mg/kg
pentobarbital sodium. Leukocytes were labeled by retro-orbital
injection of 50 µl 0.05% Rhodamine 6G (Sigma-Aldrich, St.
Louis, MO, USA). Mesenteric venules were exteriorized and rolling
leukocytes were monitored using an inverted fluorescent microscope
(Nikon Eclipse Ti; Nikon Corporation, Tokyo, Japan) equipped with a
stage warmer (Thermo Plate; Tokai Hit Co., Ltd., Fujinomiya,
Japan), a QuantEM:512SC camera (Photometrics, Tucson, AZ, USA), and
a Nikon S Plan Fluor ELWD 20X objective (Nikon Corporation).
Image-Pro Plus software version 6.2 (Media Cybernetics, Inc.) was
used to automatically track real-time moving leukocytes. Leukocytes
were classified as adherent or rolling, according to the quality or
duration of their interaction with the venular wall. Leukocytes
that were stationary for >30 sec were defined as adherent. The
leukocyte rolling flux was defined as the total number of
leukocytes crossing the 100-µm venular segment in 1 min at a
velocity that was significantly decreased compared with the
centerline velocity. Leukocyte rolling velocity was determined by
measuring the time required for a leukocyte to roll along a 100
µm length of venule. Leukocyte adhesion is expressed as the
number of cells/100 µm of venular length.
Statistical analysis
Data are expressed as the mean ± standard error and
were analyzed by paired or unpaired t-test unless otherwise
stated. Differences between groups were analyzed by one-way
analyses of variance followed by Student-Newman-Keuls post-hoc
tests. P<0.05 was considered to indicate a statistically
significant difference. Data were analyzed in GraphPad Prism
software version 5 (GraphPad Software, Inc., La Jolla, CA,
USA).
Results
Atorvastatin attenuates PCS-induced
atherogenesis in ApoE KO mice
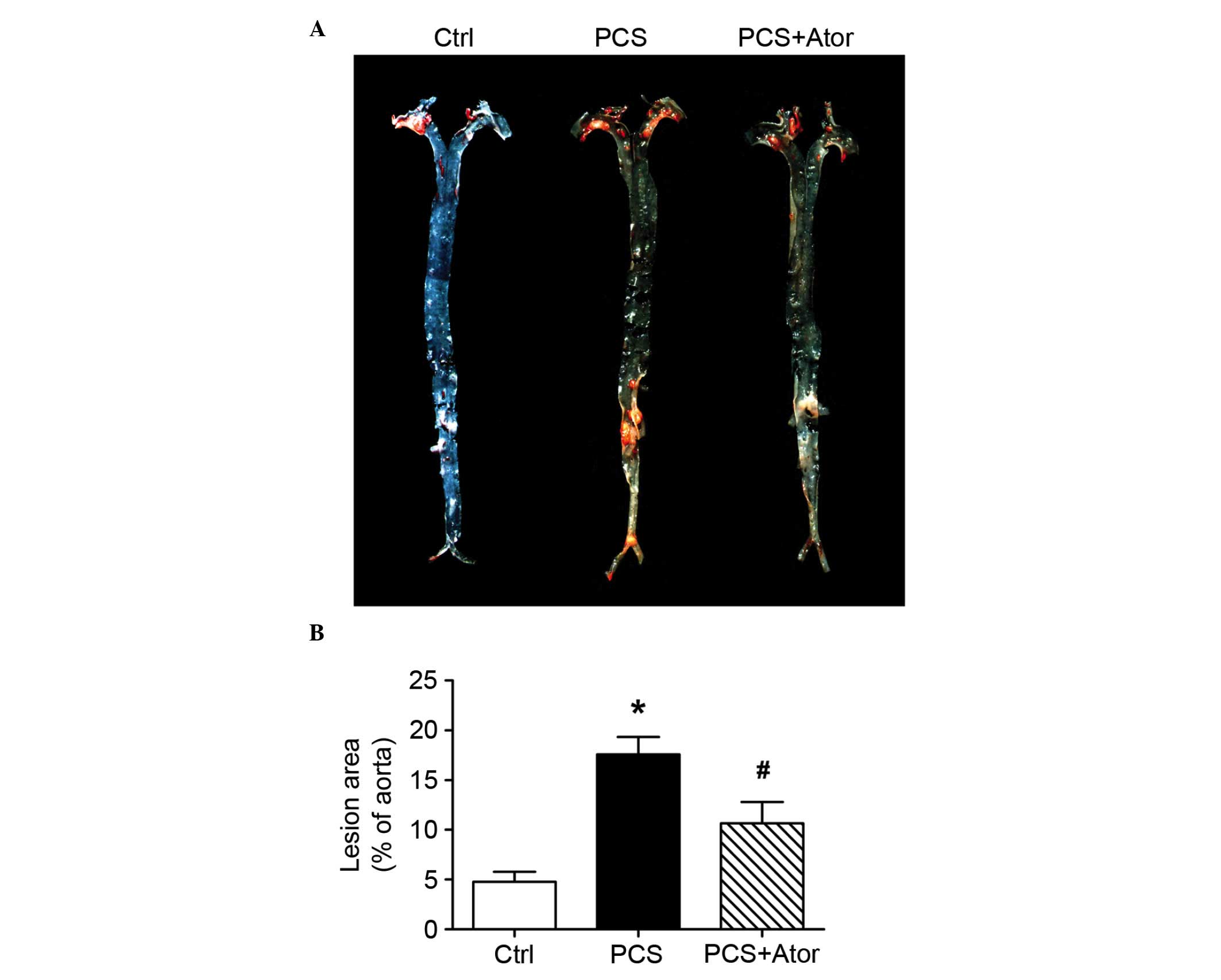
To evaluate the impact of PCS on atherogenesis in
ApoE KO mice, aortas were dissected and en face gross examination
by general oil red O staining was performed. Compared with control
mice, increased atherosclerotic lesion sizes were observed in
aortas from PCS-treated mice (17.60±1.74 vs. 4.75±1.03; P=0.001;
Fig. 1). Atorvastatin treatment
significantly abrogated the PCS-induced atherosclerotic plaque
growth in aortas (10.63±2.15; P=0.03 vs. PCS alone). This
therapeutic effect was independent of its lipid-reducing capacity,
as low doses of atorvastatin did not significantly alter serum
lipid profiles in experimental mice (Table I).
 | Table IBody weight and serum lipid profiles
in the three mouse groups. |
Table I
Body weight and serum lipid profiles
in the three mouse groups.
| Group
|
|---|
| Parameter | Ctrl
(n=8) | PCS
(n=8) | PCS+Ator
(n=8) |
|---|
| Body weight, g | 29.88±0.81 | 31.25±0.80 | 30.50±0.76 |
| TC, mmol/l | 20.55±0.66 | 20.33±0.54 | 19.96±0.59 |
| TG, mmol/l | 1.34±0.07 | 1.32±0.06 | 1.29±0.07 |
| LDL-C, mmol/l | 5.17±0.18 | 5.21±0.18 | 5.18±0.19 |
| HDL-C, mmol/l | 1.69±0.06 | 1.71±0.07 | 1.74±0.08 |
Atorvastatin inhibits the PCS-induced
decrease in collagen expression levels in atherosclerotic plaques
from ApoE KO mice
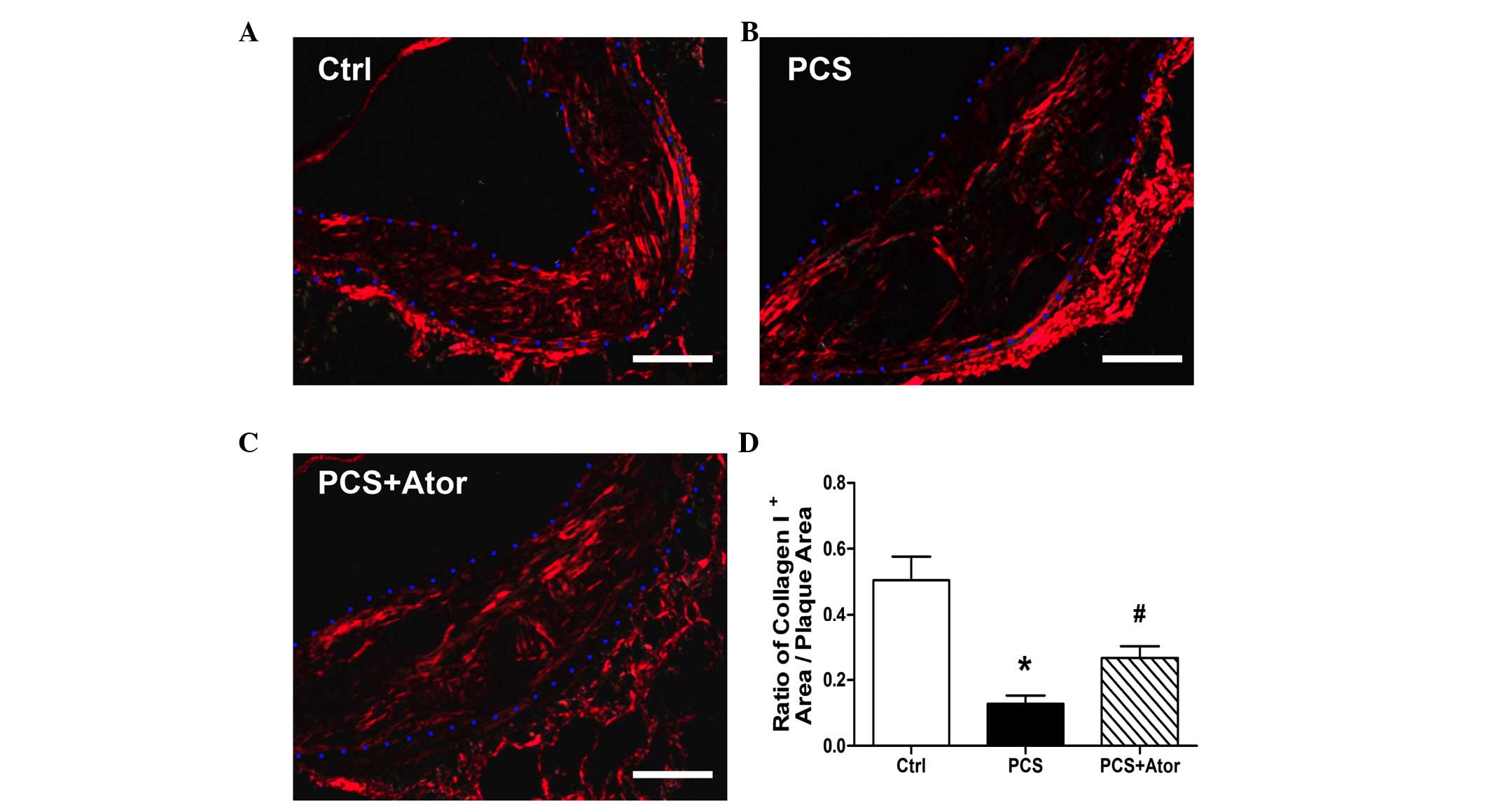
Histological analysis revealed that Sirius
red-positive collagen I structures in atherosclerotic plaques were
significantly decreased following 20 weeks of PCS administration
(Ctrl, 0.51±0.07; PCS, 0.13±0.02; P=0.002; Fig. 2). This decrease was partially
reversed by treatment with atorvastatin, which significantly
augmented the ratio of collagen I+ area/plaque area
(0.27±0.03; P=0.005 vs. PCS).
Atorvastatin abrogates the PCS-induced
increase in leukocyte-endothelium interaction in ApoE KO mice
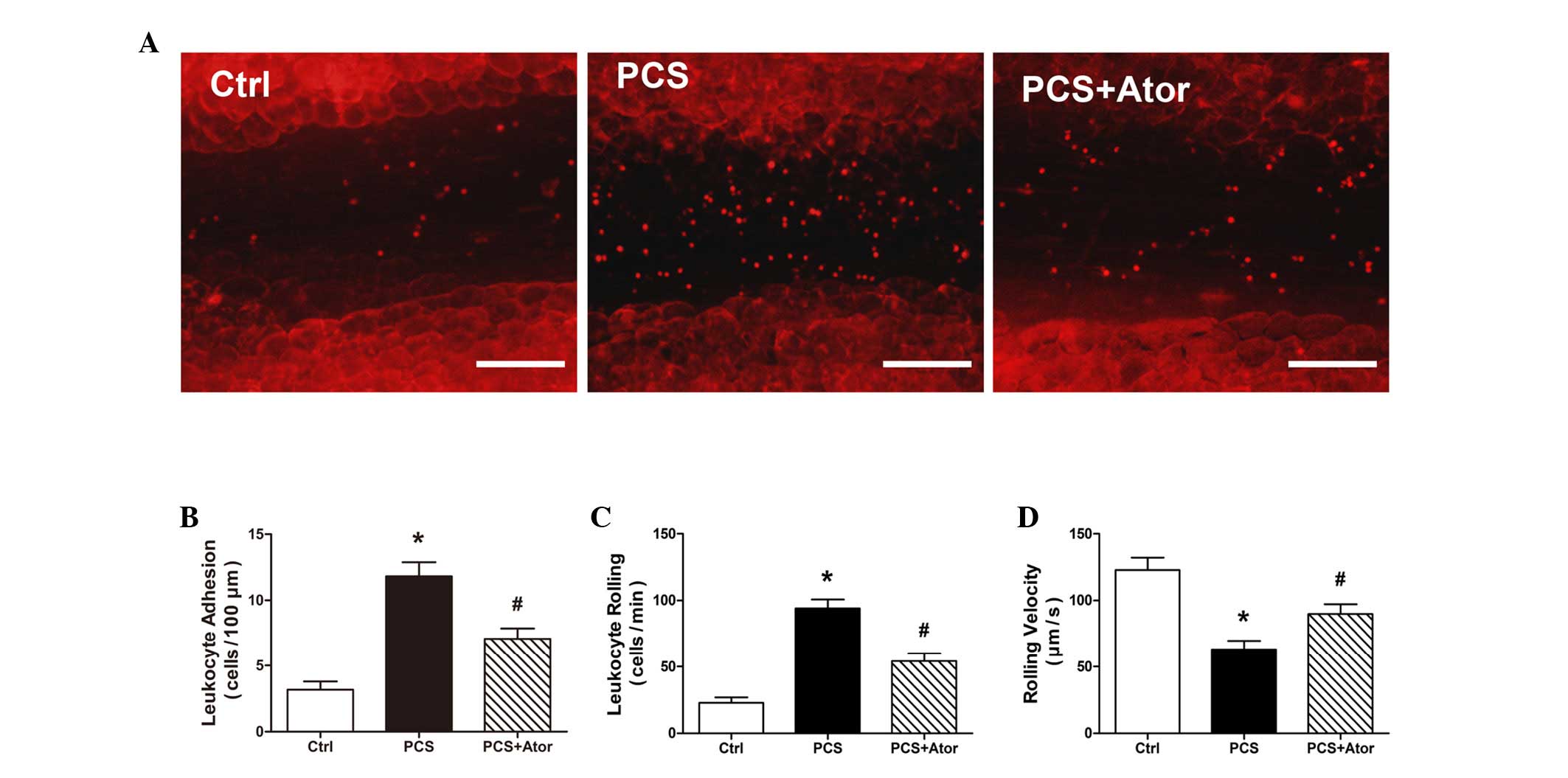
Leukocyte adhesion to the endothelium is crucial for
the early stages of atherogenesis. The impact of PCS on the
leukocyte-endothelium interaction in the mesenteric venules of ApoE
KO mice was therefore investigated. Compared with control mice, PCS
mice demonstrated an increased leukocyte-endothelium adhesiveness,
manifested by the increased rolling and adhering of leukocytes, and
their decreased rolling velocity (all P<0.001; Fig. 3). In mice treated with
atorvastatin, however, the increased interaction between leukocytes
and the endothelium was significantly abrogated (leukocyte
adhesion, P=0.006; leukocyte rolling, P=0.001; and rolling
velocity, P=0.021).
Atorvastatin inhibits PCS-induced
upregulated protein expression levels of VCAM-1 and ICAM-1 in
aortas of ApoE KO mice
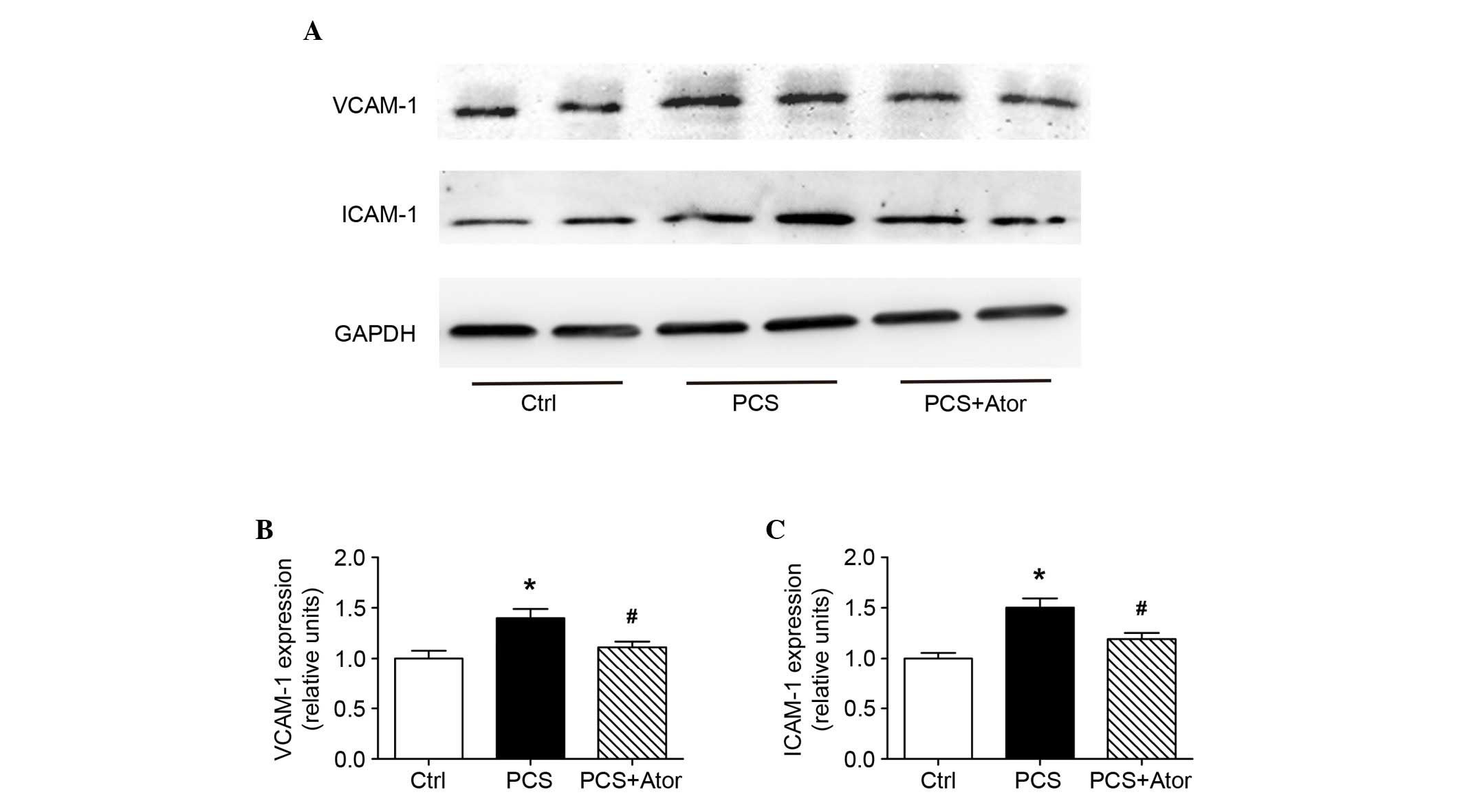
As adhesion molecules, VCAM-1 and ICAM-1 are
involved in the interaction between circulating leukocytes and
vascular endothelia. In the present study, the protein expression
levels of these two molecules were significantly upregulated by PCS
administration [by 1.402±0.089-fold (P=0.001 vs. Ctrl) and
1.505±0.089-fold (P=0.007 vs. Ctrl), respectively; Fig. 4]; treatment with atorvastatin,
however, protected against this [increases were 1.110±0.057-fold
(P=0.016 vs. PCS) and 1.192±0.061-fold (P=0.020 vs. PCS),
respectively].
Discussion
In the present study, PCS was demonstrated to
promote endothelial-leukocyte adhesion, thereby accelerating
atherogenesis and causing instability of atherosclerotic plaques in
ApoE KO mice. However, atorvastatin partially reversed these
PCS-induced effects.
Atherosclerosis is a complex process characterized
by accumulation of lipids and fibrous elements in large arteries
(30,31). Increasing evidence suggests that
the occurrence of acute cardiovascular events is facilitated by the
erosion or rupture of vulnerable atherosclerotic plaques, which
feature atrophic fibrous caps, large necrotic cores, accumulation
of inflammatory cells and an imbalance between extracellular matrix
synthesis and degradation (32–34).
In the present study, PCS administration exacerbated the
atherosclerotic burden of aortas in ApoE KO mice. In addition,
collagen expression levels were significantly decreased in the
atherosclerotic plaques of mice treated with PCS. PCS
administration accelerated the progression of atherosclerosis and
undermined plaque stability.
Leukocyte recruitment and vascular inflammation into
lesions contribute to the initiation and progression of
atherosclerosis (35–37). As a natural barrier, the vascular
endothelium is crucial in inhibiting the adhesion of circulating
inflammatory cells to the vessel wall (38). Endothelial cells express various
adhesion molecules on their cell surface, including VCAM-1 and
ICAM-1, the upregulation of which promotes leukocyte-endothelium
interactions and facilitates the transendothelial migration of
leukocytes (39). PCS, as an
atherogenic stimulus, was predicted to promote leukocyte
recruitment to vessel walls. The present study confirmed the
enhanced leukocyte-endothelium adhesiveness using intravital
microscopy. A potential mechanism underlying this phenomenon may be
the upregulation of adhesion molecules, including VCAM-1 and
ICAM-1.
Statins have been widely used in clinical practice
for the management of dyslipidemia management in CKD patients and
their potential in vessel protection has been confirmed (40,41).
In the present study, statin therapy significantly reversed the
atherogenic effects of PCS. In addition, plaque stability was
rescued by atorvastatin. Notably, these therapeutic effects are
independent of its lipid-reduction effect. These findings emphasize
the potential of statins as preventive and therapeutic treatments
for atherosclerosis-associated CVD in CKD patients, particularly
those with high PCS levels.
In conclusion, the results of the present study
demonstrate that the uremic toxin PCS accelerates the progression
of atherosclerosis and disturbs the stability of formed plaques. In
addition, PCS upregulated the protein expression levels of adhesion
molecules and enhanced the adhesiveness between leukocytes and
endothelia. Furthermore, statin therapy was effective in abrogating
these PCS-induced effects. The results of the present study provide
novel insights into protein-bound uremic toxins, and the potential
of statin therapy for cardiovascular complications in CKD. Patients
with high levels of PCS may benefit from the direct vessel
protection effects of statin treatment.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81300178, 81370401
and 81500196).
References
|
1
|
McMahon GM, Hwang SJ and Fox CS: Residual
lifetime risk of chronic kidney disease. Nephrol Dial Transplant.
pii: gfw253. 2016. View Article : Google Scholar
|
|
2
|
Stenvinkel P: Chronic kidney disease: A
public health priority and harbinger of premature cardiovascular
disease. J Intern Med. 268:456–467. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stojceva-Taneva O, Otovic NE and Taneva B:
Prevalence of diabetes mellitus in patients with chronic kidney
disease. Open Access Maced J Med Sci. 4:79–82. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Foley RN, Parfrey PS and Sarnak MJ:
Clinical epidemiology of cardiovascular disease in chronic renal
disease. Am J Kidney Dis. 32(5 Suppl 3): S112–S119. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Matsui M, Takeda Y, Uemura S, Matsumoto T,
Seno A, Onoue K, Tsushima H, Morimoto K, Soeda T, Okayama S, et al:
Suppressed soluble Fms-like tyrosine kinase-1 production aggravates
atherosclerosis in chronic kidney disease. Kidney Int. 85:393–403.
2014. View Article : Google Scholar
|
|
6
|
Go AS, Chertow GM, Fan D, McCulloch CE and
Hsu CY: Chronic kidney disease and the risks of death,
cardiovascular events, and hospitalization. N Engl J Med.
351:1296–1305. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gluba-Brzózka A, Michalska-Kasiczak M,
Franczyk B, Nocuń M, Toth P, Banach M and Rysz J: Markers of
increased atherosclerotic risk in patients with chronic kidney
disease: A preliminary study. Lipids Health Dis. 15:222016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Roberts JK and McCullough PA: The
management of acute coronary syndromes in patients with chronic
kidney disease. Adv Chronic Kidney Dis. 21:472–479. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Di Angelantonio E, Chowdhury R, Sarwar N,
Aspelund T, Danesh J and Gudnason V: Chronic kidney disease and
risk of major cardiovascular disease and non-vascular mortality:
Prospective population based cohort study. BMJ. 341:c49862010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hage FG, Venkataraman R, Zoghbi GJ, Perry
GJ, DeMattos AM and Iskandrian AE: The scope of coronary heart
disease in patients with chronic kidney disease. J Am Coll Cardiol.
53:2129–2140. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Martinez AW, Recht NS, Hostetter TH and
Meyer TW: Removal of P-cresol sulfate by hemodialysis. J Am Soc
Nephrol. 16:3430–3436. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lekawanvijit S, Adrahtas A, Kelly DJ,
Kompa AR, Wang BH and Krum H: Does indoxyl sulfate, a uraemic
toxin, have direct effects on cardiac fibroblasts and myocytes? Eur
Heart J. 31:1771–1779. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu IW, Hsu KH, Lee CC, Sun CY, Hsu HJ,
Tsai CJ, Tzen CY, Wang YC, Lin CY and Wu MS: p-Cresyl sulphate and
indoxyl sulphate predict progression of chronic kidney disease.
Nephrol Dial Transplant. 26:938–947. 2011. View Article : Google Scholar :
|
|
14
|
Liabeuf S, Barreto DV, Barreto FC, Meert
N, Glorieux G, Schepers E, Temmar M, Choukroun G, Vanholder R and
Massy ZA; European Uraemic Toxin Work Group (EUTox): Free
p-cresylsulphate is a predictor of mortality in patients at
different stages of chronic kidney disease. Nephrol Dial
Transplant. 25:1183–1191. 2010. View Article : Google Scholar
|
|
15
|
Gansevoort RT, Correa-Rotter R, Hemmelgarn
BR, Jafar TH, Heerspink HJ, Mann JF, Matsushita K and Wen CP:
Chronic kidney disease and cardiovascular risk: Epidemiology,
mechanisms, and prevention. Lancet. 382:339–352. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kaluski E, Waller A, Patel A, Gerula C,
Maher J, Haider B and Klapholz M: Clinical applicability of
coronary atherosclerotic lesion characterization. Minerva
Cardioangiol. 59:255–270. 2011.PubMed/NCBI
|
|
17
|
Ruggeri ZM: Platelets in atherothrombosis.
Nat Med. 8:1227–1234. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nakamura K, Sasaki T, Cheng XW, Iguchi A,
Sato K and Kuzuya M: Statin prevents plaque disruption in
apoE-knockout mouse model through pleiotropic effect on acute
inflammation. Atherosclerosis. 206:355–361. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Werner N, Nickenig G and Laufs U:
Pleiotropic effects of HMG-CoA reductase inhibitors. Basic Res
Cardiol. 97:105–116. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Feigenbaum J and Neuberg CA: Simplified
method for the preparation of aromatic sulfuric acid esters. J Am
Chem Soc. 63:3529–3530. 1941. View Article : Google Scholar
|
|
21
|
Han H, Zhu J, Zhu Z, Ni J, Du R, Dai Y,
Chen Y, Wu Z, Lu L and Zhang R: p-Cresyl sulfate aggravates cardiac
dysfunction associated with chronic kidney disease by enhancing
apoptosis of cardiomyocytes. J Am Heart Assoc. 4:e0018522015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Vanholder R, Schepers E, Pletinck A,
Nagler EV and Glorieux G: The uremic toxicity of indoxyl sulfate
and p-cresyl sulfate: A systematic review. J Am Soc Nephrol.
25:1897–1907. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nie P, Li D, Hu L, Jin S, Yu Y, Cai Z,
Shao Q, Shen J, Yi J, Xiao H, et al: Atorvastatin improves plaque
stability in ApoE-knockout mice by regulating chemokines and
chemokine receptors. PLoS One. 9:e970092014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Meng X, Zhang K, Li J, Dong M, Yang J, An
G, Qin W, Gao F, Zhang C and Zhang Y: Statins induce the
accumulation of regulatory T cells in atherosclerotic plaque. Mol
Med. 18:598–605. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang XQ, Nigro P, World C, Fujiwara K, Yan
C and Berk BC: Thioredoxin interacting protein promotes endothelial
cell inflammation in response to disturbed flow by increasing
leukocyte adhesion and repressing Kruppel-like factor 2. Circ Res.
110:560–568. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang XQ, Wan HQ, Wei XJ, Zhang Y and Qu P:
CLI-095 decreases atherosclerosis by modulating foam cell formation
in apolipoprotein E-deficient mice. Mol Med Rep. 14:49–56.
2016.PubMed/NCBI
|
|
27
|
Dong M, Yang X, Lim S, Cao Z, Honek J, Lu
H, Zhang C, Seki T, Hosaka K, Wahlberg E, et al: Cold exposure
promotes atherosclerotic plaque growth and instability via
UCP1-dependent lipolysis. Cell Metab. 18:118–129. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Akhmedov A, Rozenberg I, Paneni F, Camici
GG, Shi Y, Doerries C, Sledzinska A, Mocharla P, Breitenstein A,
Lohmann C, et al: Endothelial overexpression of LOX-1 increases
plaque formation and promotes atherosclerosis in vivo. Eur Heart J.
35:2839–2848. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ouedraogo R, Gong Y, Berzins B, Wu X,
Mahadev K, Hough K, Chan L, Goldstein BJ and Scalia R: Adiponectin
deficiency increases leukocyte-endothelium interactions via
upregulation of endothelial cell adhesion molecules in vivo. J Clin
Invest. 117:1718–1726. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhou M, Ma C, Liu W, Liu H, Wang N, Kang Q
and Li P: Valsartan promoting atherosclerotic plaque stabilization
by upregulating renalase: A potential-related gene of
atherosclerosis. J Cardiovasc Pharmacol Ther. 20:509–519. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ross R: Atherosclerosis-an inflammatory
disease. N Engl J Med. 340:115–126. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Haft JI: Multiple atherosclerotic plaque
rupture in acute coronary syndrome. Circulation. 107:e65–e66;
author reply e65–e66. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bergström G, Fagerberg B, Sallsten G,
Lundh T and Barregard L: Is cadmium exposure associated with the
burden, vulnerability and rupture of human atherosclerotic plaques?
PLoS One. 10:e01212402015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang K, Meng X, Kong J, Liu FF, Yang JM,
Gao F, Zhang Y and Zhang C: Simvastatin increases
Prolyl-4-Hydroxylase α1 expression in atherosclerotic plaque and
ox-LDL-stimulated human aortic smooth muscle cells via p38 MAPK and
ERK1/2 signaling. J Mol Cell Cardiol. 65:43–50. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Erbel C, Wolf A, Lasitschka F, Linden F,
Domschke G, Akhavanpoor M, Doesch AO, Katus HA and Gleissner CA:
Prevalence of M4 macrophages within human coronary atherosclerotic
plaques is associated with features of plaque instability. Int J
Cardiol. 186:219–225. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Molina-Sánchez P, Chèvre R, Rius C, Fuster
JJ and Andrés V: Loss of p27 phosphorylation at Ser10 accelerates
early atherogenesis by promoting leukocyte recruitment via
RhoA/ROCK. J Mol Cell Cardiol. 84:84–94. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hartwig H, Silvestre Roig C, Daemen M,
Lutgens E and Soehnlein O: Neutrophils in atherosclerosis. A brief
overview. Hamostaseologie. 35:121–127. 2015. View Article : Google Scholar
|
|
38
|
Inoue M, Ishida T, Yasuda T, Toh R, Hara
T, Cangara HM, Rikitake Y, Taira K, Sun L, Kundu RK, et al:
Endothelial cell-selective adhesion molecule modulates
atherosclerosis through plaque angiogenesis and
monocyte-endothelial interaction. Microvasc Res. 80:179–187. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fu C, He J, Li C, Shyy JY and Zhu Y:
Cholesterol increases adhesion of monocytes to endothelium by
moving adhesion molecules out of caveolae. Biochim Biophys Acta.
1801:702–710. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Strippoli GF, Navaneethan SD, Johnson DW,
Perkovic V, Pellegrini F, Nicolucci A and Craig JC: Effects of
statins in patients with chronic kidney disease: Meta-analysis and
meta-regression of randomised controlled trials. BMJ. 336:645–651.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rysz J, Gluba-Brzózka A, Banach M and
Więcek A: Should we use statins in all patients with chronic kidney
disease without dialysis therapy? The current state of knowledge.
Int Urol Nephrol. 47:805–813. 2015. View Article : Google Scholar : PubMed/NCBI
|