Introduction
Anal cancer is a rare type of digestive tract
disease, which has had an increasing incidence in a number of
regions (1–3). It is estimated a total of >7,200
new cases were diagnosed in the United States in 2015, with ~1,000
anal cancer-associated mortalities (4). Tumors in this site are classified,
according to World Health Organization (WHO), as intraepithelial
neoplasias, carcinomas and carcinoid tumors (5). Carcinomas are most frequently
identified, with squamous cell carcinoma (SCC) comprising~95% of
all the anal tumors (6), and ~5%
of the lesions are adenocarcinomas (7). The age-standardized incidence is
<1/100,000 people, and the mortality is 0.2/100,000 (1). In men who practice anal receptive
intercourse, the incidence of anal cancer increases up to
35/100,000 (5,8). This is predominantly due to increased
risk of human papillomavirus (HPV) infection (1), HPV16 is most frequently observed in
anal SCC (9). HPV infection leads
to intraepithelial neoplasia that progresses from low-grade to
high-grade dysplasia and, finally, to invasive cancer. The
regression of high-grade dysplastic lesions is rare (5). HPV infection results in high
expression of cyclin-dependent kinase inhibitor 2A (p16), and
disrupts the association between retinoblastoma protein and the E2F
family of transcription factors, ultimately leading to cellular
proliferation (10). Recently, it
was demonstrated that a high frequency of women with cervical
cancer also have infection of the anal canal by HPV16 (11). In addition to HPV infection, other
known risk factors of anal cancer are immunodeficiency due to human
immunodeficiency virus seropositivity, low cluster of
differentiation 4 T cell count, immunosuppression following solid
organ transplantation, and tobacco smoking (1,5).
Previous studies have indicated that patients
presenting with no HPV infection and no p16 expression (HPV-/p16-)
have a poorer outcome than patients that are HPV+/p16+, and suggest
an optimization in the therapy to the former (12,13),
and that improved molecular characterization should be performed. A
previous study demonstrated that patients with SCC of the anal
canal and HPV were irresponsive to standard chemoradiotherapy
treatment and frequently presented with TP53 mutations
(13). Furthermore, an additional
study using immunohistochemistry (IHC), fluorescence in situ
hybridization and next generation sequencing, observed alterations
in molecular markers that may aid in the understanding of failure
of certain therapeutic strategies, including overexpression of
multidrug resistance-associated protein 1, DNA excision repair
protein ERCC-1 and thymidylate synthetase, and suggest potential
therapeutic targets, including the tyrosine kinase receptor,
epidermal growth factor receptor (EGFR) (14). Molecular therapies targeting EGFR,
such as cetuximab and panitumumab are currently used in colorectal
cancer treatment, and tumor genetic make-up, including mutational
status of KRAS and BRAF may predict patient response
(15).
The aim of the present study is to evaluate the
mutational status of two important oncogenes, KRAS and
BRAF in a series of SCC, adenocarcinomas and high-grade
squamous intra-epithelial lesions (HSILs), and whether they are
associated with patient's clinicopathological features, and HPV
status.
Materials and methods
In the current study, resected tumors of the anal
canal from 43 patients were evaluated. The present study was
approved by the ethics committee of Barretos Cancer Hospital
(Barretos, Brazil) and informed consent was obtained from each
patient. Histological review of the slides was performed by an
expert pathologist (Dr Cristovam Scapulatempo-Neto), who confirmed
the diagnosis and delimited the area of the slide containing the
neoplastic lesion. Clinical data of the patients was obtained, and
is summarized in Table I. HPV16
and HPV18 status, and immunohistochemical analysis of β-globin,
p16, antigen Ki67 (Ki67), minichromosome maintenance protein
complex (MCM) and DNA topoisomerase 2-α (TOP2A) were retrieved from
a previous study (16).
 | Table IClinicopathological features of
patients with anal lesions. |
Table I
Clinicopathological features of
patients with anal lesions.
| Parameter | Frequency (%)
|
|---|
| HSIL | Adenocarcinoma | SCC |
|---|
| Gender |
| Female | 6 (20.7) | 7 (24.1) | 16 (55.2) |
| Male | 3 (21.4) | 4 (28.6) | 7 (50.0) |
| Ethnicity |
| Caucasian | 8 (21.6) | 8 (21.6) | 21 (56.8) |
| Non-caucasian | 1 (16.7) | 3 (50) | 2 (33.3) |
| History of previous
disease |
| No | 2 (11.1) | 7 (38.9) | 9 (50.0) |
| Yes | 7 (29.2) | 3 (12.5) | 14 (58.3) |
| NA | 0 | 1 (100) | 0 |
| History of tumor in
the family |
| No | 6 (21.4) | 6 (21.4) | 16 (57.1) |
| Yes | 3 (21.4) | 4 (28.6) | 7 (50.0) |
| NA | 0 | 1 (100) | 0 |
| Tobacco
consumption |
| No | 1 (5.6) | 7 (38.9) | 10 (55.6) |
| Yes | 7 (33.3) | 3 (14.3) | 11 (52.4) |
| NA | 1 (25.0) | 1 (25.0) | 2 (50.0) |
| Surgery |
| No | 6 (26.1) | 6 (26.1) | 11 (47.8) |
| Yes | 3 (16.7) | 5 (27.8) | 10 (55.6) |
| NA | 0 | 0 | 2 (100) |
| Radiotherapy |
| No | 2 (40.0) | 1 (20.0) | 2 (40.0) |
| Yes | 7 (19.4) | 10 (27.8) | 19 (52.8) |
| NA | 0 | 0 | 2 (100) |
| Chemotherapy |
| No | 2 (25.0) | 3 (37.5) | 3 (37.5) |
| Yes | 7 (21.2) | 8 (24.2) | 18 (54.5) |
| NA | 0 | 0 | 2 (100) |
| Response to the
treatment |
| No response | 4 (23.5) | 6 (35.3) | 7 (41.2) |
| Complete
response | 1 (8.3) | 2 (16.7) | 9 (75.0) |
| Progression | 2 (22.2) | 2 (22.2) | 5 (55.6) |
| NA | 2 (40.0) | 1 (20.0) | 2 (40.0) |
| Recurrence |
| No | 7 (22.6) | 6 (19.4) | 18 (58.1) |
| Yes | 2 (16.7) | 5 (41.7) | 5 (41.7) |
| Status |
| Death by
cancer | 3 (23.1) | 4 (30.8) | 6 (46.2) |
| Death by other
cause | 0 | 2 (66.7) | 1 (33.3) |
| Alive, free of
disease | 6 (27.3) | 3 (13.6) | 13 (59.1) |
| Alive, with the
disease | 0 | 2 (40.0) | 3 (60.0) |
| Age (years) |
| <48 | 3 (33.3) | 1 (11.1) | 5 (55.6) |
| 48–66 | 5 (20.0) | 6 (24.0) | 14 (56.0) |
| >66 | 1 (11.1) | 4 (44.4) | 4 (44.4) |
The histological slides (10 µm) were
processed, and DNA was isolated from macrodissected tumor area of
one unstained section as previously described (17). The slides were placed at 80°C for
deparaffinization for 10 min and hydrated with xylene and graded
ethanol (100, 70 and 50%). DNA was isolated using QIAamp DNA Micro
kit (Qiagen GmbH, Hilden, Germany), following the manufacturer's
protocols, and quantified using NanoDrop 2000 (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). The samples were diluted to a
final concentration of 50 ng/µl and stored at −20°C for
further analysis.
The hotspots of KRAS (codons 12 and 13) and
BRAF (codon 600) were amplified using PCR and sequenced, as
previously described (17).
Amplification of KRAS was performed in a final reaction
volume of 15 µl containing 1.5 µl buffer (Qiagen
GmbH), 2 mM MgCl2 (Qiagen GmbH), 100 mM dNTPs
(Invitrogen; Thermo Fisher Scientific, Inc.), 0.2 mM sense and 0.2
mM anti-sense primers (Sigma-Aldrich, St. Louis, MO, USA), 1 unit
HotStarTaq DNA polymerase (Qiagen GmbH) and 1 µl DNA. The
KRAS region was amplified using the following primers:
Sense, 5′-GTGTGACATGTTCTAATATAGTCA-3′ and anti-sense,
5′-GAATGGTCCTGCACCAGTAA-3′. The BRAF amplification reaction
was performed as described above, with 0.3 mM sense and anti-sense
primers used. The region was amplified using the following primers:
Sense, 5′-TCATAATGCTTG CTCTGATAGGA-3′ and anti-sense,
5′-GGCCAAAAATTTAATCAGTGGA-3′. The following cycling conditions were
used: Initial denaturation at 96°C for 15 min, followed by 40
cycles of denaturation at 96°C for 45 sec, annealing at 55.5°C for
45 sec, then extension at 72°C for 10 min, all using a Veriti
96-Well thermal cycler (Applied Biosystems; Thermo Fisher
Scientific, Inc.)
The PCR products were purified with EXO-SAP (GE
Healthcare Life Sciences, Chalfont, UK), and sequenced using 1
µl BigDye (Applied Biosystems; Thermo Fisher Scientific,
Inc.), 1.5 µl sequencing buffer (Applied Biosystems; Thermo
Fisher Scientific, Inc.) and 1 µl primer (Thermo Fisher
Scientific, Inc.). The sequencing reaction, which consisted of 30
cycles of denaturation at 96°C for 10 sec, annealing at 50°C for 5
sec and extension at 60°C for 4 min, was followed by
post-sequencing purification with EDTA, alcohol and sodium citrate.
The products of PCR were eluted in Hi-Di formamide (Thermo Fisher
Scientific, Inc.) and incubated at 95°C for 5 min and at −4°C for
at least 5 min. Direct sequencing was performed in a 3500 Genetic
Analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc.). The
mutations were confirmed with two independent reactions.
Survival analysis was performed considering the
three different histology types using Kaplan-Meier plots and log
rank statistical analysis using R.
Results
Histological review of the slides demonstrated that,
from the 43 patients, 9 were diagnosed with HSIL, 11 patients were
diagnosed with adenocarcinomas, and 23 with SCC. The mean age at
diagnosis varied from 51 (HSIL) to 64 (adenocarcinoma) years.
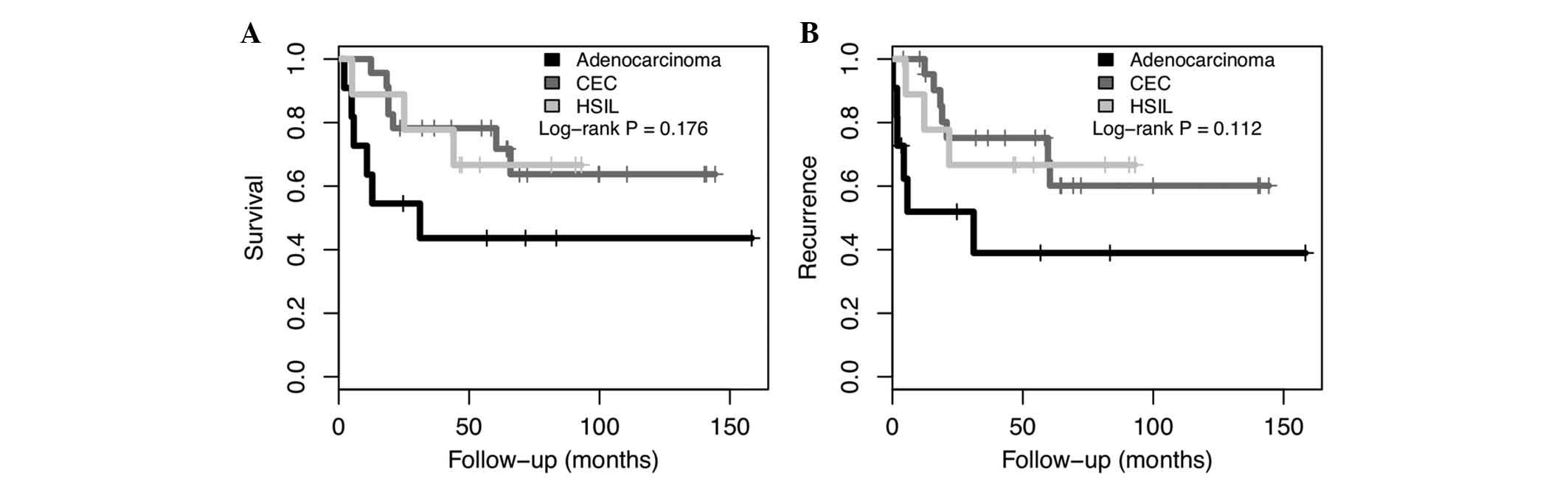
Overall survival (OS) and disease-free survival (DFS), based on the
histologic type is presented in Fig.
1. There was a trend of poorer prognosis in adenocarcinoma
compared with HSIL and SCC (P=0.176 and P=0.112 for OS and DFS,
respectively).
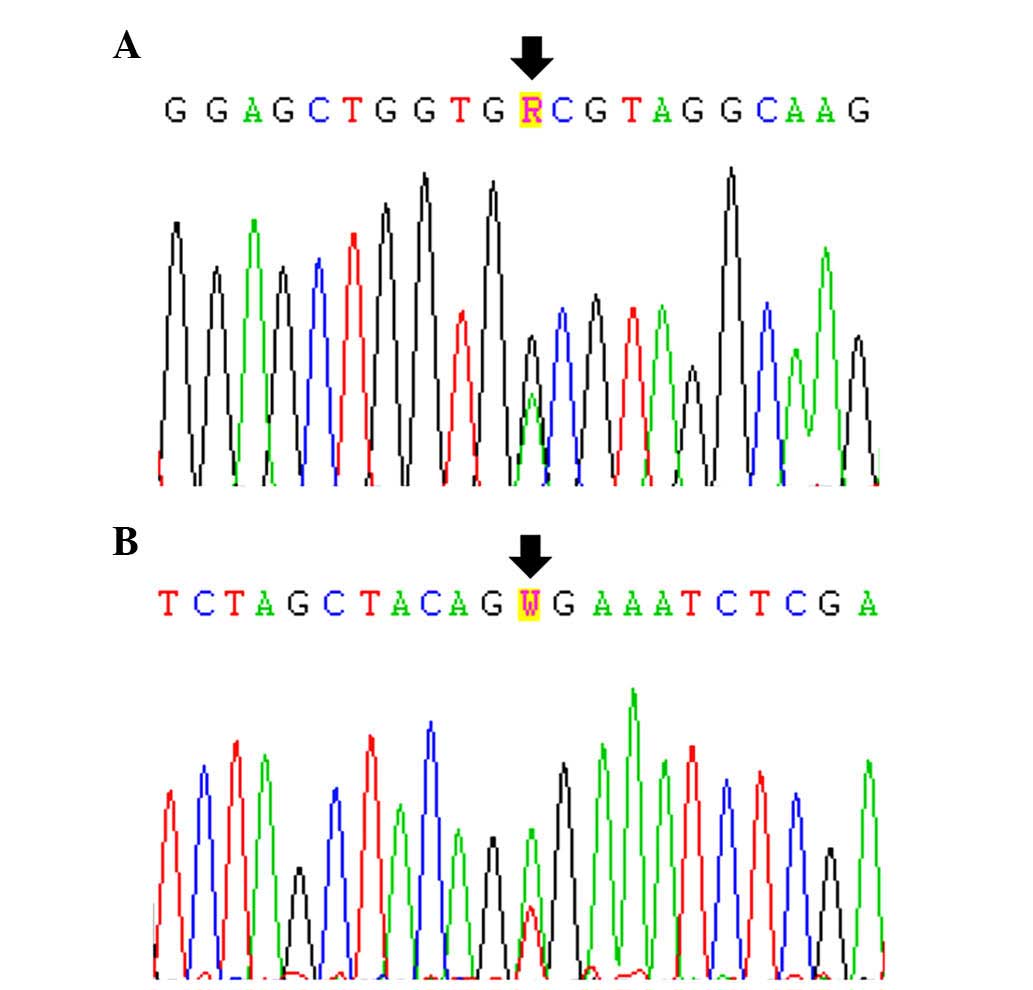
Of the 43 patient samples examined, 1 (2.3%),
exhibited a KRAS mutation, which was p.G13D (Fig. 2A and Table II). This case was a female SCC
patient (age, 58) with previous gynecological or anal disease. The
SCC was positive for β-globin and p16 using IHC, as well as 4+ Ki67
labeling. Notably, this patient also presented with HPV16 anal
infection (Table II). Following
surgery, radiotherapy and chemotherapy treatment, the patient
exhibited complete response, and was free of disease at the last
follow up of 140 months (Table
II).
| Table IIMolecular features of anal lesion
patients. |
Table II
Molecular features of anal lesion
patients.
| Histological
type | Gender | Age (years) | HPV16a | HPV18a |
Immunohistochemistrya
| Mutation
| Treatment | Oncological
response | DFS (months) | Survival
(months) | Status |
|---|
| β-globin | p16 | Ki67 | MCM | TOP2A | KRAS | BRAF |
|---|
| HSIL | M | 50 | + | − | Ok | + | 4+ | 3+ | 3+ | wt | wt | Surgery + RDT +
QT | Progression | 21.74 | 25.10 | D |
| HSIL | F | 57 | + | − | Weak | + | 4+ | 2+ | 2+ | wt | wt | RDT + QT | No evidence | 47.17 | 47.17 | A |
| HSIL | F | 57 | + | − | Ok | + | 4+ | 3+ | 2+ | wt | wt | RDT + QT | No evidence | 5.13 | 5.13 | D |
| HSIL | F | 47 | + | − | Ok | + | 4+ | 1+ | 2+ | wt | wt | Surgery + RDT +
QT | Progression | 12.20 | 44.05 | D |
| HSIL | M | 38 | + | + | Ok | + | 4+ | 3+ | 2+ | wt | wt | RDT + QT | No evidence | 54.11 | 54.11 | A |
| HSIL | F | 64 | + | − | Ok | − | 4+ | 2+ | 3+ | wt | wt | No treatment | No evidence | 46.38 | 46.38 | A |
| HSIL | F | 69 | + | − | Ok | + | 4+ | − | NA | wt | wt | RDT + QT | NA | 93.03 | 93.03 | A |
| HSIL | F | 53 | + | − | Ok | + | 4+ | 1+ | NA | wt | wt | RDT + QT | NA | 90.79 | 90.79 | A |
| HSIL | M | 27 | + | − | Ok | − | 4+ | 2+ | NA | wt | wt | Surgery | CR | 81.51 | 81.51 | A |
| ADC | F | 35 | + | − | Ok | − | 4+ | − | 3+ | wt | wt | Surgery + RDT +
QT | Progression | 1.74 | 12.86 | D |
| ADC | M | 69 | + | − | Ok | + | 4+ | − | 3+ | wt | wt | RDT + QT | No evidence | 4.38 | 10.82 | D |
| ADC | M | 66 | − | − | Ok | − | NA | 1+ | 2+ | wt | wt | RDT | No evidence | 1.88 | 4.97 | D |
| ADC | F | 86 | + | − | Ok | − | 3+ | − | 2+ | wt | wt | RDT | No evidence | 31.15 | 31.15 | D |
| ADC | F | 60 | + | − | Ok | − | 4+ | − | 2+ | wt | wt | RDT + QT | No evidence | 5.76 | 5.76 | D |
| ADC | M | 70 | + | − | Ok | + | 4+ | 3+ | 3+ | wt | V600E | Surgery | No evidence | 0.13 | 2,11 | D |
| ADC | F | 59 | + | − | Ok | + | 4+ | 3+ | 2+ | wt | wt | RDT + QT | NA | 56.78 | 56,78 | A |
| ADC | M | 58 | + | − | Ok | + | 1+ | +CB | 1+ | wt | wt | Surgery + RDT +
QT | CR | 24.70 | 24.70 | A |
| ADC | F | 63 | + | − | Ok | − | 4+ | − | NA | wt | wt | RDT + QT | Progression | 3.42 | 71.61 | A |
| ADC | F | 52 | − | − | Ok | − | 4+ | 1+ | 2+ | wt | wt | Surgery + RDT +
Q | No evidence | 83.42 | 83.42 | A |
| ADC | F | 81 | + | − | Ok | − | 2+ | +CB | 2+ | wt | wt | Surgery + RDT +
QT | CR | 158.36 | 158.36 | A |
| SCC | M | 85 | + | − | Ok | + | 4+ | 3+ | 3+ | wt | wt | Surgery + RDT | No evidence | 19.05 | 19.05 | D |
| SCC | M | 64 | + | − | Ok | + | 3+ | 3+ | 2+ | wt | wt | Surgery + RDT +
QT | No evidence | 59.64 | 65.86 | D |
| SCC | M | 50 | + | − | Ok | NA | 4+ | 2+ | 2+ | wt | wt | RDT + QT | Progression | 4.14 | 23.49 | A |
| SCC | F | 43 | + | _ | Weak | + | 4+ | − | 2+ | wt | wt | RDT + QT | Progression | 58.42 | 58.42 | A |
| SCC | M | 43 | + | − | Ok | − | 1+ | − | 1+ | wt | wt | Surgery + RDT +
QT | Progression | 31.97 | 31.97 | A |
| SCC | F | 44 | + | − | Ok | + | 4+ | 3+ | 3+ | wt | wt | NA | CR | 36.61 | 36.61 | A |
| SCC | F | 48 | + | − | Ok | + | 4+ | 2+ | 3+ | wt | wt | RDT + QT | No evidence | 18.36 | 18.36 | D |
| SCC | F | 62 | + | − | Ok | + | 4+ | 1+ | 2+ | wt | wt | Surgery +QT | Progression | 15.86 | 19.05 | D |
| SCC | F | 56 | + | − | Ok | + | 4+ | 3+ | 2+ | wt | wt | RDT + QT | CR | 64.34 | 64.34 | A |
| SCC | F | 64 | + | − | Weak | + | 4+ | 3+ | 3+ | wt | wt | Surgery + QT | No evidence | 60.39 | 60.39 | D |
| SCC | F | 68 | − | − | Ok | + | 4+ | 2+ | 2+ | wt | wt | Surgery + RDT +
QT | No evidence | 12.37 | 12.37 | D |
| SCC | F | 58 | + | − | Ok | + | 4+ | 3+ | 2+ | wt | wt | NA | NA | 69.31 | 69.31 | A |
| SCC | M | 56 | + | − | Ok | + | 4+ | 2+ | NA | wt | wt | RDT + QT | No evidence | 12.70 | 99.61 | A |
| SCC | M | 34 | − | − | Ok | + | 4+ | 3+ | 3+ | wt | wt | RDT + QT | CR | 54.77 | 54.77 | A |
| SCC | F | 44 | + | − | Ok | NA | 2+ | 1+ | 1+ | wt | wt | RDT + QT | NA | 43.16 | 43.16 | A |
| SCC | F | 48 | + | − | Ok | − | 3+ | 2+ | 1+ | wt | wt | No treatment | No evidence | 20.82 | 20.82 | D |
| SCC | F | 56 | + | − | Ok | + | 4+ | 2+ | 2+ | wt | wt | Surgery + RDT +
QT | CR | 64.80 | 64.80 | A |
| SCC | F | 79 | + | − | Ok | + | 4+ | 3+ | 2+ | wt | wt | RDT + QT | CR | 72.27 | 72.27 | A |
| SCC | F | 60 | + | − | Ok | + | 4+ | +CB | 2+ | wt | wt | Surgery + RDT | Progression | 10.46 | 110.53 | A |
| SCC | M | 57 | + | − | Ok | + | 4+ | 2+ | 3+ | wt | wt | Surgery + RDT +
QT | CR | 144.31 | 144.31 | A |
| SCC | F | 57 | + | − | Weak | + | 3+ | 2+ | 2+ | wt | wt | RDT + QT | CR | 140.95 | 140.95 | A |
| SCC | F | 58 | + | − | Ok | + | 4+ | − | NA | G13D | wt | Surgery + RDT +
QT | CR | 140.23 | 140.23 | A |
| SCC | F | 80 | + | − | Ok | + | 4+ | − | 2+ | wt | wt | RDT + QT | CR | 99.93 | 99.93 | A |
A BRAF mutation, p.V600E, was also observed
in only 1/43 patients (2.3%; Fig.
2B and Table II). This case
was an adenocarcinoma of a male 70 year-old tobacco smoking
patient. Similarly to the case described above, the patient also
presented with HPV16 anal infection, and IHC indicated positivity
for β-globin and p16 IHC labeling, as well as 4+ Ki67 labeling
(Table II). In addition, the
sample presented 3+ MCM and 3+ TOP2A IHC labeling. The patient
relapsed and succumbed to the condition 2 months following surgery
(Table II).
Discussion
The present study aimed to evaluate the mutational
status of KRAS and BRAF in tumors arising in the anal
canal, and to associate these findings with clinicopathological
data. To the best of our knowledge, the majority of the mutational
screening of KRAS and BRAF in anal tumors focused on
SCC samples due to the predominance of this histological type in
anal tumors.
Although tumors located in the anal region may be
anatomically close to colorectal tumors, they exhibit different
histological patterns, distinct features, and therefore distinct
etiologies (18). Previous studies
have observed a high incidence of KRAS mutation in
colorectal cancer (CRC) worldwide (19), and in Brazilian populations
(17,20). Overall, among 8,234 Brazilian CRC
cases analyzed, the KRAS mutation frequency of 31.9%, with
the majority of these samples exhibiting a p.G12D mutation
(20). KRAS mutations
generally arise in codons 12 or 13, and constitutively activate its
pathway. The protein generated by the mutated gene is capable of
transmitting the signal independently of tyrosine kinase receptor
activation (21). Of the 43
patients analyzed, 1 patient with SCC was observed to have a
KRAS mutation (2.3%). This low mutation rate is consistent
with previous studies, which identified no KRAS mutation in
SCC of the anal canal samples [n=36 samples (22), n=89 samples (23), n=53 samples (24) and n=66 samples (25)]. Additional studies observed a total
of 1.6% (n=193 samples) (26), and
5% (n=84 samples) (27) of SCC to
have a KRAS mutation. Furthermore, BRAF, another
mitogen-activated protein kinase pathway gene, which was identified
as mutated in ~50% of melanomas (28), presented a low mutation rate in the
samples in the present study (2.3%). This gene was observed with a
low percentage of mutation in anal tumors, varying from 0%
(24,25,27)
to 4.7% (26) consistent with the
findings of the current study. In addition, this gene was also
found mutated in a low percentage of precursor lesions of
colorectal cancer (17) and
colorectal cancer (29). It is
important to highlight that no mutation was observed in patients
with HSIL, which suggest that these mutations may occur
preferentially in tumors with an invasive phenotype.
The standard treatment of anal SCC in the majority
of patients is chemotherapy and radiotherapy, with a response rate
of up to 80% (30). Metastatic and
refractory cases are rare, although, it has been demonstrated that
cetuximab-based treatment results in disease progression of
KRAS-mutated tumors, while those with wild type KRAS
exhibited partial or minor remission (31). This data is consistent with further
studies in colorectal cancer that demonstrated KRAS and
BRAF mutational status predicted tumor response to targeted
therapies (15). Thus, an
understanding of KRAS and BRAF mutational status is
key in personalized medicine.
Using the mutational rate of KRAS and
BRAF, the present study evaluated the anatomical association
between anal and rectal tumors, tumors arising in the anus have
different KRAS mutation percentage of the rectal
counterparts, thus, the molecular differences require elucidation.
To the best of our knowledge, the current study is the first to
evaluate the percentage of KRAS and BRAF mutations in
adenocarcinomas and HSIL of the anal canal, in addition to SCC.
Furthermore, to date, the present study is the first to describe
these mutations in tumors of anal canal of the Brazilian
population. In addition to the well-known risk factor of HPV
infection that drives anal cancer tumorigenesis, there are patients
who develop these tumors in the absence of this infection. The
present study evaluated the mutation percentage of two well-known
drivers of colorectal cancer (KRAS) and melanoma
(BRAF) to further elucidate other risk factors in anal
cancer development. In conclusion, a low percentage of mutation was
identified in SCCs, adenocarcinomas and HSIL, however, these tumors
may exhibit other molecular alterations that result in anal cancer
development, which require elucidation in future studies.
Acknowledgments
The present study was partially supported by the São
Paulo Research Foundation (grant no. 2010/16795-4 to Dr Adhemar
Longatto-Filho) and the Ministério da Ciência e
Tecnologia/Financiadora de Estudos e Projetos (grant no.
CT-INFRA-PROINFRA 01/2011). Dr Lucas Tadeu Bidinotto received a São
Paulo Research Foundation fellowship (grant no. 2011/08523-7).
References
|
1
|
Bosman FT; World Health Organization and
International Agency for Research on Cancer: WHO Classification of
Tumours of the Digestive System. IARC Press; Lyon: 2010
|
|
2
|
Lampejo T, Kavanagh D, Clark J, Goldin R,
Osborn M, Ziprin P and Cleator S: Prognostic biomarkers in squamous
cell carcinoma of the anus: A systematic review. Br J Cancer.
103:1858–1869. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rousseau DL Jr, Thomas CR Jr, Petrelli NJ
and Kahlenberg MS: Squamous cell carcinoma of the anal canal. Surg
Oncol. 14:121–132. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Leonard D, Beddy D and Dozois EJ:
Neoplasms of anal canal and perianal skin. Clin Colon Rectal Surg.
24:54–63. 2011. View Article : Google Scholar :
|
|
6
|
Deans GT, McAleer JJ and Spence RA:
Malignant anal tumours. Br J Surg. 81:500–508. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Franklin RA, Giri S, Valasareddy P, Lands
LT and Martin MG: Comparative survival of patients with anal
adenocarcinoma, squamous cell carcinoma of the anus, and rectal
adenocarcinoma. Clin Colorectal Cancer. 15:47–53. 2016. View Article : Google Scholar
|
|
8
|
Franceschi S and De Vuyst H: Human
papillomavirus vaccines and anal carcinoma. Curr Opin HIV AIDS.
4:57–63. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Serup-Hansen E, Linnemann D,
Skovrider-Ruminski W, Høgdall E, Geertsen PF and Havsteen H: Human
papillo-mavirus genotyping and p16 expression as prognostic factors
for patients with American joint committee on cancer stages I to
III carcinoma of the anal canal. J Clin Oncol. 32:1812–1817. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Doorbar J: Molecular biology of human
papillomavirus infection and cervical cancer. Clin Sci (Lond).
110:525–541. 2006. View Article : Google Scholar
|
|
11
|
Veo CA, Saad SS, Fregnani JH,
Scapulatempo-Neto C, Tsunoda AT, Resende JC, Lorenzi AT, Mafra A,
Cinti C, Cotrim ID, et al: Clinical characteristics of women
diagnosed with carcinoma who tested positive for cervical and anal
high-risk human papillomavirus DNA and E6 RNA. Tumour Biol.
36:5399–5405. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mai S, Welzel G, Ottstadt M, Lohr F,
Severa S, Prigge ES, Wentzensen N, Trunk MJ, Wenz F, von
Knebel-Doeberitz M and Reuschenbach M: Prognostic relevance of HPV
infection and p16 overexpression in squamous cell anal cancer. Int
J Radiat Oncol Biol Phys. 93:819–827. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Meulendijks D, Tomasoa NB, Dewit L, Smits
PH, Bakker R, van Velthuysen ML, Rosenberg EH, Beijnen JH,
Schellens JH and Cats A: HPV-negative squamous cell carcinoma of
the anal canal is unresponsive to standard treatment and frequently
carries disruptive mutations in TP53. Br J Cancer. 112:1358–1366.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Smaglo BG, Tesfaye A, Halfdanarson TR,
Meyer JE, Wang J, Gatalica Z, Reddy S, Arguello D and Boland PM:
Comprehensive multiplatform biomarker analysis of 199 anal squamous
cell carcinomas. Oncotarget. 6:43594–43604. 2015.PubMed/NCBI
|
|
15
|
De Roock W, Claes B, Bernasconi D, De
Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V,
Papamichael D, Laurent-Puig P, et al: Effects of KRAS, BRAF, NRAS,
and PIK3CA mutations on the effcacy of cetuximab plus chemotherapy
in chemotherapy-refractory metastatic colorectal cancer: A
retrospective consortium analysis. Lancet Oncol. 11:753–762. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Scapulatempo-Neto C, Veo CA, Fregnani JH,
Lorenzi A, Mafra A, Melani A, Loaiza E, Rosa L, de Oliveira C, Levi
J and Longatto-Filhø A: Characterization of topoisomerase II alpha
(TOP2A) and minichromosome maintenance protein (MCM)2 expression in
anal carcinoma. Oncol Lett. 2016.
|
|
17
|
Yamane LS, Scapulatempo-Neto C, Alvarenga
L, Oliveira CZ, Berardinelli GN, Almodova E, Cunha TR, Fava G,
Colaiacovo W, Melani A, et al: KRAS and BRAF mutations and MSI
status in precursor lesions of colorectal cancer detected by
colonoscopy. Oncol Rep. 32:1419–1426. 2014.PubMed/NCBI
|
|
18
|
Matalon SA, Mamon HJ, Fuchs CS, Doyle LA,
Tirumani SH, Ramaiya NH and Rosenthal MH: Anorectal cancer:
Critical anatomic and staging distinctions that affect use of
radiation therapy. Radiographics. 35:2090–2107. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ta n C and Du X: KRAS mutation testing in
metastatic colorectal cancer. World J Gastroenterol. 18:5171–5180.
2012.
|
|
20
|
Gil Ferreira C, Aran V, Zalcberg-Renault
I, Victorino AP, Salem JH, Bonamino MH, Vieira FM and Zalis M: KRAS
mutations: Variable incidences in a Brazilian cohort of 8,234
metastatic colorectal cancer patients. BMC Gastroenterol.
14:732014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Okumura S and Jänne PA: Molecular
pathways: The basis for rational combination using MEK inhibitors
in KRAS-mutant cancers. Clin Cancer Res. 20:4193–4199. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gilbert DC, Williams A, Allan K, Stokoe J,
Jackson T, Linsdall S, Bailey CM and Summers J: p16INK4A, p53, EGFR
expression and KRAS mutation status in squamous cell cancers of the
anus: Correlation with outcomes following chemo-radiotherapy.
Radiother Oncol. 109:146–151. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Paliga A, Onerheim R, Gologan A, Chong G,
Spatz A, Niazi T, Garant A, Macheto D, Alcindor T and Vuong T: EGFR
and K-ras gene mutation status in squamous cell anal carcinoma: A
role for concurrent radiation and EGFR inhibitors? Br J Cancer.
107:1864–1868. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Casadei Gardini A, Capelli L, Ulivi P,
Giannini M, Freier E, Tamberi S, Scarpi E, Passardi A, Zoli W,
Ragazzini A, et al: KRAS, BRAF and PIK3CA status in squamous cell
anal carcinoma (SCAC). PLoS One. 9:e920712014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Prigge ES, Urban K, Stiegler S, Müller M,
Kloor M, Mai S, Ottstadt M, Lohr F, Wenz F, Wagner S, et al: No
evidence of oncogenic KRAS mutations in squamous cell carcinomas of
the anogenital tract and head and neck region independent of human
papillomavirus and p16 (INK4a) status. Hum Pathol. 45:2347–2354.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Serup-Hansen E, Linnemann D, Høgdall E,
Geertsen PF and Havsteen H: KRAS and BRAF mutations in anal
carcinoma. APMIS. 123:53–59. 2015. View Article : Google Scholar
|
|
27
|
Martin V, Zanellato E, Franzetti-Pellanda
A, Molinari F, Movilia A, Paganotti A, Deantonio L, De Dosso S,
Assi A, Crippa S, et al: EGFR, KRAS, BRAF, and PIK3CA
characterization in squamous cell anal cancer. Histol Histopathol.
29:513–521. 2014.
|
|
28
|
Davies H, Bignell GR, Cox C, Stephens P,
Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W,
et al: Mutations of the BRAF gene in human cancer. Nature.
417:949–954. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
El-Deiry WS, Vijayvergia N, Xiu J,
Scicchitano A, Lim B, Yee NS, Harvey HA, Gatalica Z and Reddy S:
Molecular profiling of 6,892 colorectal cancer samples suggests
different possible treatment options specific to metastatic sites.
Cancer Biol Ther. 16:1726–1737. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bartelink H, Roelofsen F, Eschwege F,
Rougier P, Bosset JF, Gonzalez DG, Peiffert D, van Glabbeke M and
Pierart M: Concomitant radiotherapy and chemotherapy is superior to
radiotherapy alone in the treatment of locally advanced anal
cancer: Results of a phase III randomized trial of the European
Organization for Research and Treatment of Cancer Radiotherapy and
Gastrointestinal Cooperative groups. J Clin Oncol. 15:2040–2049.
1997.PubMed/NCBI
|
|
31
|
Lukan N, Ströbel P, Willer A, Kripp M,
Dinter D, Mai S, Hochhaus A and Hofheinz RD: Cetuximab-based
treatment of metastatic anal cancer: Correlation of response with
KRAS mutational status. Oncology. 77:293–299. 2009. View Article : Google Scholar : PubMed/NCBI
|