Introduction
The hallmark of type 2 diabetes (T2D) is associated
with low-grade systemic inflammation, characterized by upregulated
cytokine production and the activation of inflammatory signaling
pathways (1). Interleukin (IL)-1β
is one of the major inflammatory cytokines involved in T2D. IL-1β
is a risk factor for T2D by inducing insulin resistance in
insulin-sensitive cells (2,3).
Hepatic inflammation is a complex process and originates in
response to a variety of stresses (4), which are associated with systemic
metabolic conditions, including non-alcoholic streatohepatitis,
obesity, diabetes and metabolic syndrome (5).
Lipopolysaccharide (LPS) is known to be an
endogenous danger mediator of the host inflammatory response to
infection. It has been reported that LPS promotes the inflammatory
responses by directly engaging Toll-like receptor-4 (TLR-4) and
inducing the nuclear translocation of nuclear factor (NF)-κB
(6). In addition to TLRs, members
of the nucleotide-binding oligomerization domain (NOD)-like
receptor (NLR) family are important in inflammation and metabolism
(7). NLR pyrin domain-containing 3
(NLRP3) inflammasome is one molecule of the NLR family, along with
apoptotic speck protein containing a caspase recruitment domain and
pro-caspase-1; these form molecular platforms termed inflammasomes,
which mediate caspase-1 activation, followed by the cleavage and
release of the proinflammatory cytokines, interleukin (IL)-1β and
IL-18 (8,9). It has been demonstrated that a
reduction in the adipose tissue expression of NLRP3 is coupled with
decreased inflammation and improved insulin-sensitivity in obese
patients with T2D (10). The
ablation of NLRP3 prevents the obesity-induced inflammsome
activation in fat depots and the liver, and enhances
insulin-signaling (10). However,
the mechanism underlying how the NLRP3-dependent inflammatory
effects are generated and their role in LPS-induced HepG2 apoptosis
remain to be fully elucidated.
To avoid biological and chemical inflammatory
stimuli, cells rely on robust adaptive responses for the
maintenance of cellular and histological homeostasis. Key processes
are the endoplasmic reticulum (ER) stress response, autophagy and
redox stress (11). ER stress
signaling, referred to as the unfolded response (UPR), is triggered
by three downstream proteins: PKR-like eukaryotic initiation factor
2 kinase, activating transcription factor 6 and inositol requiring
1α. Mild ER stress activates the UPR responsible for the recovery
of homeostasis (12). By contrast,
protracted and excessive signaling via ER stress sensors is
associated with the initiation of a mitochondrial pathway of
apoptosis, which involves the transcription factor, C/EPB
homologous protein (CHOP) (13). A
causal link between the ER stress response and the apoptosis of
hepatocytes has been established in a wide range of hepatic
disorders. For example, ER stress-induced hepatocyte apoptosis in
acute liver failure (14),
increased ER stress during the development of non-alcoholic fatty
liver disease and reduced ER stress decreased hepatic cell death,
together with recovery of autophagic flux (15).
Autophagy is an evolutionarily conserved,
lysosome-dependent system in eukaryotes, which regulates the
turnover of cellular proteins and organelles. In hepatocytes,
autophagy contributes to the removal of damaged mitochondria
(16) and controls intracellular
lipid metabolism (17). The
selective autophagic degradation of mitochondria is known as
mitophagy (18). Autophagy has
been shown to control a variety of functions, including the control
of innate and adaptive immune responses by regulating cytokine
production (19,20) and combatting persistent ER stress
(21). It has been demonstrated
that the downregulation of hepatic autophagy in obesity results in
increased ER stress and insulin resistance (22). Macrophages derived from
autophagy-related gene (ATG)16L1-deficient mice have been shown to
produce higher levels of IL-1β (23), whereas mice with a conditional
deletion of ATG7 in the intestinal epithelium show enhanced
expression of IL-1β (24). The
role of autophagy in hepatocyte inflammation remains to be fully
elucidated, however, the NLRP3 inflammasome contributes to
hepatocyte injury and inflammation (25). In the present study, the activation
of ER stress and NLRP3-dependent inflammation were examined in
LPS-induced HepG2 cells, and the role of autophagy in response to
this stress was investigated in order to interpret the association
between autophagy, ER stress and inflammation in hepatocytes.
Materials and methods
Reagents and antibodies
Fetal bovine serum (FBS) was purchased from (GE
Healthcare Life Sciences, Coelbe, Germany). Dulbecco's modified
Eagle's medium (DMEM) and 1% (v/v) streptomycin/penicillin were
purchased from Gibco; Thermo Fisher Scientific, Inc, (Waltham, MA,
USA). LPS, 4-phenyl butyrate (4-PBA), 3-methyladenine (3-MA) and
N-acetyl-L-cysteine (NAC) were purchased from Sigma-Aldrich;
Thermo Fisher Scientific, Inc. Complete protease inhibitor mixture
and immunoblot polyvinylidene difluorid (PVDF) membranes were
purchased from Roche Diagnostics (Barcelona, Spain). RIPA lysis
buffer and the BCA protein assay kit were purchased from Beijing
ComWin Biotech Co., Ltd. (Beijing, China). Western Chemiluminescent
Horseradish Peroxidase (HRP) substrate was purchased from EMD
Millipore (Billerica, MA, USA). Rabbit anti-microtubule-associated
protein 1 light chain 3 (LC3; 1:1,000; cat. no. 2775), mouse
anti-C/EBP homologous protein (CHOP; 1:1,000; cat. no. 2895),
rabbit anti-caspase-1 (1:1,000; cat. no. 2225), rabbit anti-IL-1β
(cat. no. 2022) and rabbit β-actin (1:1,000; cat. no. 8475) were
purchased from Cell Signaling Technology, Inc. (Beverly, MA,
USA).
Cell culture and stimulation
HepG2 cells (ATCC, Manassas, VA, USA) were cultured
in DMEM, supplemented with 10% FBS. The cells were maintained at
37°C in an atmosphere of 5% CO2 and 100% humidity. The
cells (1×104) were pretreated with 3-MA (2.5 mM), 4-PBA
(0.1 mg/ml) or NAC (5 mM), respectively, for 1 h, followed by
treatment with 0.1 mg/ml LPS for 24 h at 37°C.
Transmission electron microscopy
(TEM)
The HepG2 cells were fixed in phosphate buffer (pH
7.4) containing 2.5% glutaraldehyde and 2% paraformaldehyde at room
temperature for 60 min. The cells were post-fixed in 1%
OsO4 at room temperature for 60 min, dehydrated through
graded ethanol solutions and embedded in Quetol 812 (Nisshin EM
Co., Ltd., Tokyo, Japan). The regions containing the cells were
block-mounted and cut into 70 nm sections, which were stained with
uranyl acetate (saturated aqueous solution) and lead citrate, and
examined using TEM (H-7100; Hitachi, Ibaraki, Japan).
Western blot analysis
The cells were washed with phosphate-buffered saline
(PBS) and lysed in lysis buffer comprising 0.5% Triton X-100, 10 mM
HEPES (pH 7.9), 50 mM NaCl, 100 mM EDTA and 0.5 M sucrose, with
0.1% protease inhibitor cocktail (Roche Diagnostics). The lysates
were then incubated on ice for 30 min and centrifuged at 8,000 × g
for 10 min at 4°C. The total protien was quantified using a Bio-Rad
Protein Assay kit (Bio-Rad Laboratories, Inc., Hertfordshire, UK).
Equal quantities of protein were subjected to sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (10–15%) and then
transferred onto PVDF membranes. The molecular weights were
estimated by comparison with a pre-stained protein ladder.
Non-specific binding was blocked using 5% skim milk. The membranes
were then incubated with specific primary antibodies, as noted
above, overnight at 4°C. The membranes were then washed with
PBS-Tween-20 and incubated with HRP-conjugated secondary
antibodies: Anti-rabbit IgG (1:5,000; cat. no. 7074; Cell Signaling
Technology, Inc.). The protein bands were detected using Western
Chemiluminescent HRP substrate (EMD Millipore). The immunoblots
were quantified by densitometric analysis using ImageTool 3.0
software (Adobe Photoshop). The quantification of protein
phosphorylation was normalized to the corresponding total protein
expression, and the relative expression level of a specific protein
was normalized to β-actin.
Flow cytometry for the analysis of
apoptosis
The rates of apoptosis of the HepG2 cells were
examined by flow cytometry using annexin V-fluorescein
isothiocyanate (FITC)/propidium iodide (PI) staining. Briefly, the
cells were treated, as described above, in each group for 24 h. The
cells were then harvested, washed and resuspended in PBS at a
density of 1×106 cells. Apoptotic cell death was
measured by double staining with annexin V-FITC and PI using an
annexin V-FITC apoptosis detection kit (Beyotime Institute of
Biotechnology, Shanghai, China) according to the manufacturer's
protocol. Flow cytometric analysis was performed immediately
following staining. Data acquisition and analysis were performed by
flow cytometry using Cell Quest software.
ROS detection
The formation of intracellular ROS in the HepG2
cells was measured by flow cytometry using the peroxide-sensitive
fluorescent probe: 2′, 7′-dichlorofluorescin diacetate (DCFH-DA).
The cells (~1×106 cells/ml) were cultured in 6-well
plates and incubated for 24 h in eight groups. Subsequently, the
cells were incubated with medium containing 10 μM DCFH-DA
for 20 min at 37°C. Following incubation with the dye, the cells
were harvested and washed three times with serum-free medium to
remove the extracellular dye. The cells were then resuspended in
ice-cold serum-free medium and placed on ice in the dark. The
levels of intracellular peroxide were measured using a flow
cytometer (BD FACSAria; BD Biosciences, Franklin Lakes, NJ, USA).
The peak excitation wavelength for oxidized DCF-DA was 488 nm and
the emission was 525 nm.
Statistical analysis
The results are expressed as the mean ± standard
error of the mean. Comparisons of a single variable in more than
two groups were analyzed using one-way analysis of variance
followed by Tukey's multiple comparison test (GraphPad Prism;
GraphPad Software Inc., La Jolla, CA, USA). Statistical analysis
was performed using a paired and unpaired t-test between two
groups, using SPSS 12.0 software (SPSS, Inc., Chicago, IL, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
LPS activates the ROS-dependent NLRP3
inflammasome
The production of mature caspase-1, which is
required for the processing and production of IL-1β and IL-18,
requires two signals. The first, for the transcription and
translation of pro-IL-1β and pro-IL-18, can be achieved by a number
of stimuli, including LPS (26).
The second signal is required to activate the inflammasome to cause
the autocatalytic cleavage of pro-caspase-1 to caspase-1. The
present study examined whether LPS induced the production of IL-1β
in HepG2 cells with 24 h treatment (Fig. 1A). To biochemically assess
inflammasome activation, the cleavage maturation of pro-IL-1β and
pro-caspase-1 were determined using immunoblot analysis. The
cleavage of pro-IL-1β to IL-1β and pro-caspase-1 to caspase-1 were
increased by exposure to LPS for 24 h (Fig. 1A).
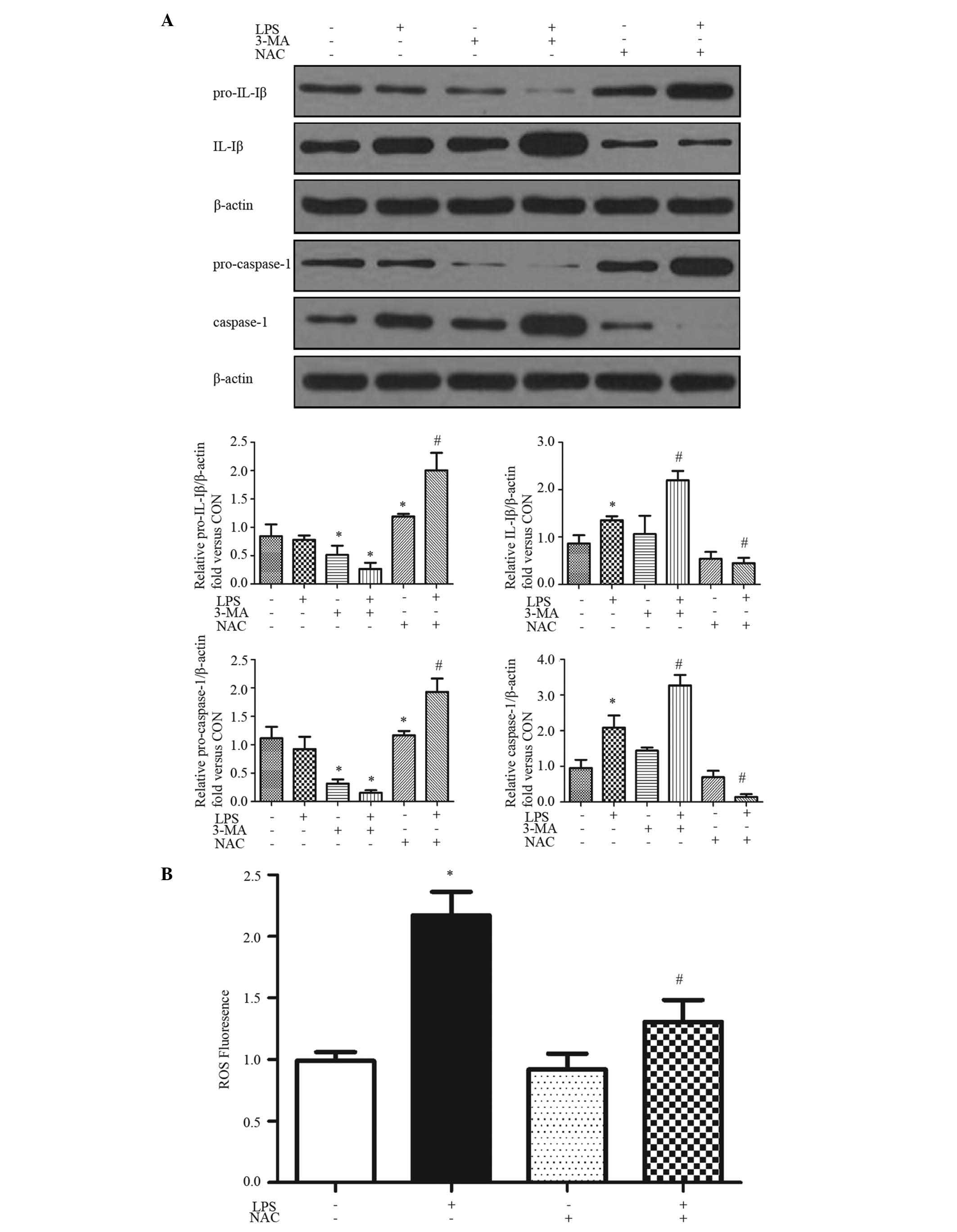 | Figure 1LPS activates ROS-dependent NLRP3
inflammasome. HepG2 cells were pretreated with or without 3-MA (2.5
mM) and/or NAC (5 mM) for 1 h, followed with LPS (0.1 mg/ml) for 24
h. (A) Western blot analysis was performed with the indicated
antibodies. Proteins were quantified and normalized to β-actin.
Representative blots and quantifications are shown. (B) ROS
generation following treatment was evaluated using 2′,
7′-dichlorofluorescin diacetate fluorescence (excitation, 488 nm;
emission, 525 nm). Results are presented as the mean ± standard
error of the mean of 3–5 separate experiments.
*P<0.05, vs. control group; #P<0.05,
vs. LPS-treated group. LPS, lipopolysaccharide; ROS, reactive
oxygen species; NLRP3, NLR pyrin domain-containing protein 3; 3-MA,
3-methyladenine; NAC, N-acetyl-L-cysteine; IL, interleukin;
CON, control (Dulbecco's modified Eagle's medium). |
ROS has an essential role in inflammasome activation
(9). The present study examined
ROS formation using DCF-DA as the detection reagent (27). Treatment of the HepG2 cells with
LPS induced ROS activation (Fig.
1B). The generation of ROS in HepG2 cells was significantly
enhanced by ~2-fold following treatment with LPS for 24 h, which
was inhibited by NAC, a ROS inhibitor (Fig. 1B). NAC also inhibited the
LPS-induced IL-1β and caspase-1 cleavage (Fig. 1A), indicating that the LPS-induced
inflammasome activation was ROS-dependent.
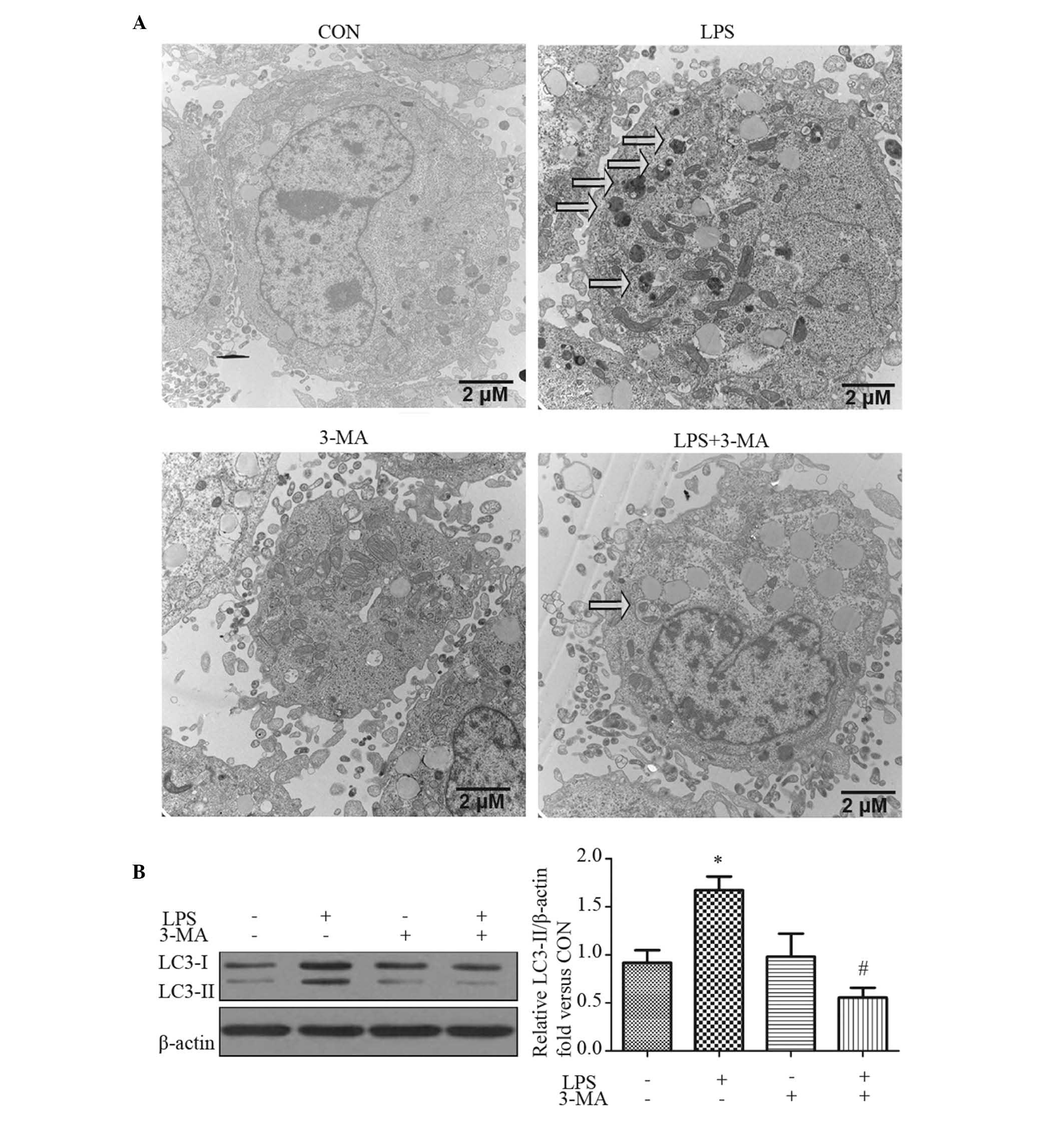
LPS induces autophagy in HepG2 cells
Autophagosomes are recognized at the ultrastructural
level as double-membrane vacuolar structures containing visible
cytoplasmic contents, including glycogen, mitochondria and
endoplasmic reticulum (28). TEM
of the HepG2 cells treated with LPS showed several autophagsomes
and autolysosomes (Fig. 2A). The
cells were also pre-treated with the autophagy inhibitor, 3-MA, for
1 h prior to LPS treatment for 24 h. 3-MA effectively inhibited the
formation of autophagosomes by LPS (Fig. 2A).
A reliable marker of autophagy is the conversion of
the ATG protein, LC3, from a soluble form (LC3-I) to a lipidized
form (LC3-II), which stably associates with the membranes of
autophagosomes (29). This
conversion can be detected by either observing the formation of
punctuate structures or by measuring the accumulation of LC3-II. In
the present study, the expression of LC3-II increased in the
LPS-stimulated hepatocytes with 24 h treatment (Fig. 2B). 3-MA significantly inhibited the
conversion of LC3-I to LC3-II, which was consistent with the
results of the TEM (Fig. 2B).
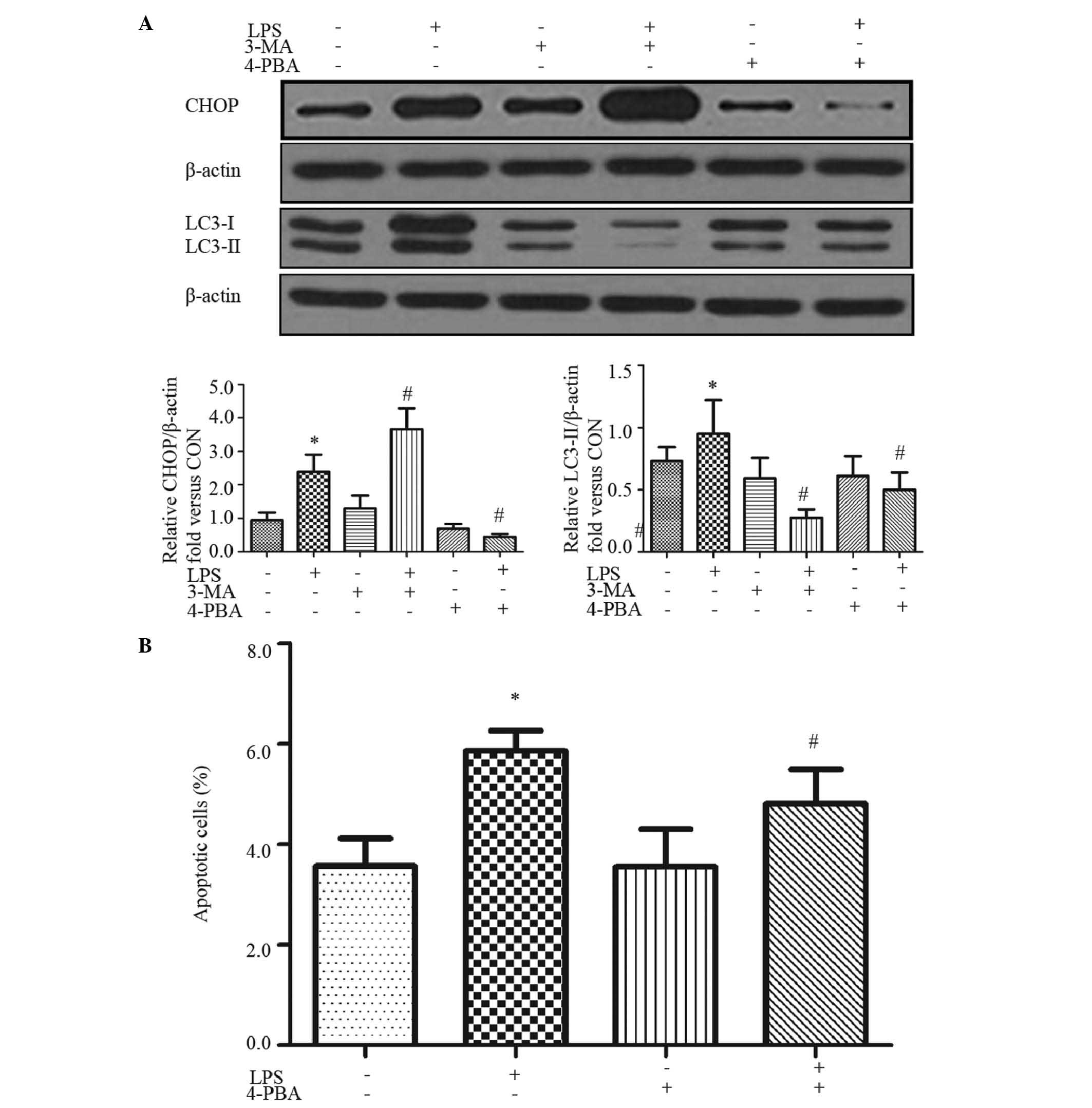
ER stress mediates LPS-induced apoptosis
and autophagy in HepG2 cells
Accumulating evidence suggests that ROS directly or
indirectly affects ER homeostasis and protein folding (30). Therefore, the present study further
analyzed CHOP, which is an ER stress marker and contributes to ER
stress-induced apoptosis (31). An
increase in the expression of CHOP was found following LPS
treatment for 24 h (Fig. 3A),
which was significantly inhibited by the ER stress inhibitor, 4-PBA
(Fig. 3A). Furthermore,
LPS-induced apoptosis was reversed partially by 4-PBA (Fig. 3B), suggesting that LPS-induced ER
stress contributed to HepG2 cell apoptosis.
In mammalian cells, ER stress has been shown to
facilitate the formation of autophagosomes, and the induction of
autophagy enables the removal of toxic misfolded proteins (32). Therefore, the present study aimed
to investigate whether ER stress is involved in LPS-induced
autophagy. The levels of microtubule-associated protein LC3 were
analyzed. As shown in Fig. 3A, LPS
significantly induced the levels of LC3B-II, and this effect was
abrogated by pretreatment of the cells with 4-PBA (Fig. 3A).
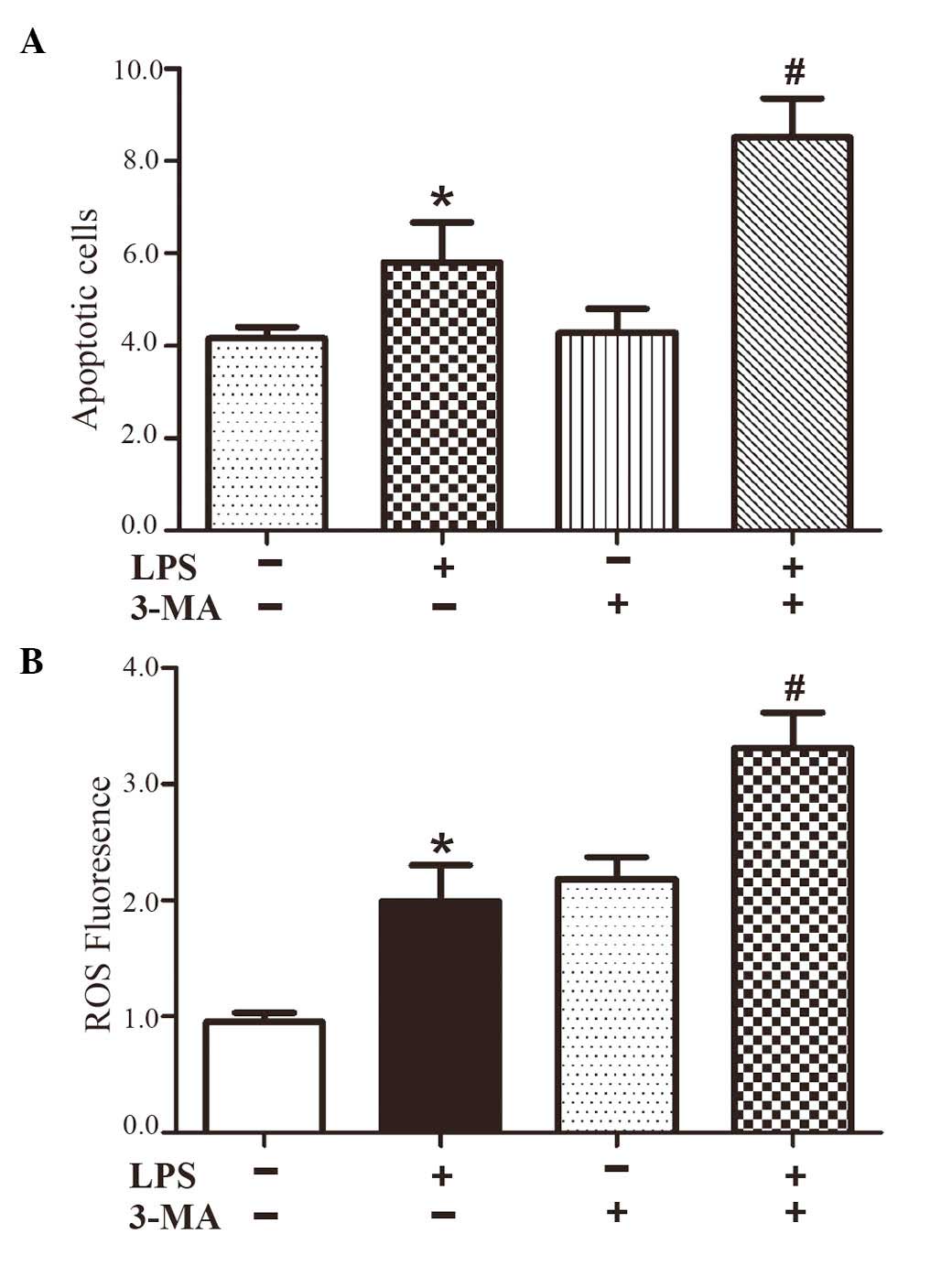
Inhibiting autophagy increases ER stress
and inflammatory cytokines
Autophagy is a physiological process for the
clearance of undesired injurious material, which protects the
cells. However, persistent and excessive autophagy leads to cell
death. To confirm the contribution of LPS in the induction of
autophagy, the HepG2 cells were pretreated with 3-MA for 1 h,
followed by treatment with LPS for 24 h. The results showed that
3-MA caused a marked increase in apoptosis of the LPS-treated HepG2
cells (Fig. 4A), which indicated
that the induction of autophagy by LPS was primarily protective.
Furthermore, 3-MA increased LPS-induced ROS generation and the
ROS-dependent expression of IL-1β (Fig. 1A), and increased the expression of
the ER stress marker, CHOP (Fig.
3A). These results suggested that the protective role of
autophagy in LPS-induced HepG2 cells may have contributed to
ameliorating ER stress and the ROS-dependent expression of
inflammatory cytokines.
Discussion
In the model of hepatic insulin resistance, its
primary pathological consequence, involving the substantial loss of
hepatocytes, originates from a complex crosstalk between ER stress,
oxidative stress and inflammation during ischemia (11). Prominent signals leading to the
substantial decline of hepatocytes, including tumor necrosis
factor-α (TNF-α), the overgeneration of ROS and the accumulation of
misfolded proteins in the ER. These stimuli also mediate potent
pro-inflammatory effects by promoting the activation of NF-κB or
the NLRP3 inflammasome (11).
Although several inflammatory cytokines have been indicated as
pro-diabetic mediators, anti-inflammation-based therepeutic
strategies targeting these cytokines in insulin resistance and T2D
have been suboptimal (33,34). The findings of the present study
provided evidence for the activation of NLRP3-dependent
inflammatory cytokines via ROS, induced by LPS in HepG2 cells, and
that autophagy inhibition was responsible for the elevated levels
of inflammation and ER stress induced by LPS, which contributed to
HepG2 cell death.
A wide variety of signals activate the NLRP3
inflammsome, and these include pathogen-associated molecular
patterns (35) and host-derived
molecules (9). The mechanisms by
which these structurally distinct molecules trigger NLRP3
inflammasome activation remain to be fully elucidated. One of the
suggested models states that NLRP3 is activated by a common pathway
of ROS (36). The source of ROS
remains to be fully elucidated, however, a previous study suggested
the involvement of one or several of the seven known NADPH oxidases
(36). In the present study, it
was demonstrated that ROS was required for inflammasome activation
in the HepG2 cells. During the progress of vascular inflammation,
oxidized-LDL induces the production of ROS (37) and causes lysosomal damage (38), which are implicated in the
mechanisms of NLRP3 inflammasome activation.
Products of the inflammasome are essential in the
impairment of insulin signaling. Hepatocytes treated with IL-1β or
TNF-α show impaired Akt activation in response to insulin (26), consistent with clinical findings
indicating that the IL-1β antagonist is promising for insulin
resistance and T2D (39). In the
present study, IL-1β was significantly increased following LPS
treatment. The liver has a marked capacity to degrade LPS to which
it is exposed almost continuously. The levels of LPS increase
during hepatic injury and inflammation. Gandhi et al
(40) demonstrated that LPS
administration to rats caused mild liver injury and weight loss,
however, all animals survived the endotoxin challenge. These
observations suggested that the cell death-inducing and survival
signals are stimulated by LPS in hepatocytes, with a predominance
of the latter (28). The present
study examined two responses, autophagy and ER stress, to LPS
stimulation in HepG2 cells.
Persistent ER stress results in cell death and
contributes to insulin resistance. The ER stress inhibitor, TUDCA,
inhibits apoptosis by ameliorating ER stress through the modulation
of intracellular calcium and thus attenuating liver cell death
(41). By contrast, a previous
study suggested that ER stress may be a source of the membranes
during the formation of autophagic vesicles. In the present study,
4-PBA significant inhibited LPS-induced LC3-II conversion,
suggesting that the induced autophagy is, at least partly,
dependent on ER stress. ER stress mediates polyglumaine-induced LC3
conversion, as essential step in formation of autophagy (42). ER stress negatively regulates the
AKT/tuberous sclerosis complex/mammalian target of rapamycin
pathway to enhance autophagy (43). In addition, ER stress leads to the
release of calcium and subsequent activation of AMPK, which
inhibits mROS, thereby promoting autophagy (44). In addition, ER stress-induced
autophagy may have evolved as a mechanism used by cells to dispose
of misfolded proteins, which cannot be degraded by ER-associated
degradation, consequently assisting ER homeostasis (15). In the present study, the inhibition
of autophagy by 3-MA significantly increased the expression of
CHOP. Autophagy, as a cell survival mechanism, allows cells to
remove damaged cytoplasmic proteins and organelles through
lysosomal degradation and thus improving survival under metabolic
stress (45). In the present
study, the role of autophagy as a protective mechanism of cell
survival was further confirmed by the autophagy inhibitor, 3-MA.
The results demonstrated that 3-MA contributed to LPS-induced cell
death, suggesting that autophagy had a protective role in this
system
Substantial evidence indicates that autophagy is a
potent suppressor of inflammation. Previously, Atg16L1 was
demonstrated to control the production of inflammatory cytokines,
including, TNF-α, IL-6 and IL-1β, in response to LPS in macrophages
(23). In the present study,
inhbiting autophagy in HepG2 cells caused aberrant LPS-induced
production of IL-1β and ROS. ROS may be accumulated in
autophagy-deficient cells undergoing apoptosis and trigger the
activation of caspase-1 following LPS stimulation. In myeloid cells
and fatty acid metabolism in non-immune cells, inhibition of
autophagy by pharmacological inhibitors or ATG deletion results in
mitochondrial ROS generation, which activates NLRP3 (46,47).
Inhibiting autophagy with 3-MA in THP1 macrophages results in the
accumulation of damaged mitochondria and increased concentrations
of mitochondrial ROS (46). By
preserving the functional pool of mitochondria, autophagy minimizes
ROS generation, which inhibits the activation of intracellular
pro-inflammatory factors, including the NFRP3 inflammasome and
NF-κB (46). Autophagy acts as a
tumor suppressor, likely by selectively removing damaged proteins
and organelles, particularly damaged and senescent mitochondria,
which are major cellular sources of ROS (48). However, reports have suggested that
autophagy may promote the expression of pro-inflammatory cytokines
and inflammation in certain biological contexts. For example,
Hepatitis B virus-induced activation of NF-κB and release of
inflammatory cytokines in hepatocytes is autophagy-mediated
(49,50). Further investigations on the
mechanisms of autophagy during inflammation are required.
In conclusion, the data obtained in the present
study showed that LPS induced the NLRP3-dependent proinflammatory
response via ROS accumulation, as well as the activation of ER
stress. Inhibiting autophagy increases ER stress and inflammatory
cytokines. These findings provide evidence for the association of
inflammation and ER stress with autophagy, and the protective role
of autophagy in LPS-induced cell death and ER stress in HepG2
cells. However, it is possible that HepG2 cells vulnerable to
death-inducing stimuli may be promoted by autophagy. Autophagy was
shown to have a protective effect by ameliorating ER stress and
NLRP3-dependent secretion of inflammatory cytokines, and further
decreased cell death.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (grant nos. 81370929 and 81400823), the
Novo Nordisk China Diabetes Young Scientific Talent Research
Funding (2013), the Research Fund for the Clinical Medicine of
Chinese Medical Association (grant no. 13040670452) and the Science
Foundation of the Education Department of Heilongjiang Province
(grant no. 12531316).
Abbreviations:
|
ATG
|
autophagy-related gene
|
|
CHOP
|
C/EPB homologous protein
|
|
DCFH-DA
|
2′,7′-dichlorofluorescin diacetate
|
|
ER
|
endoplasmic reticulum
|
|
FBS
|
fetal bovine serum
|
|
IL-1β
|
interleukin-1β
|
|
LC3
|
light chain 3
|
|
LDL
|
low density lipoprotein
|
|
LPS
|
lipopolysaccharide
|
|
NAC
|
N-acetyl-L-cysteine
|
|
NLRP3
|
nod-like receptor pyrin
domain-containing protein 3
|
|
ROS
|
reactive oxygen species
|
|
T2D
|
type 2 diabetes
|
|
TLR-4
|
toll-like receptor-4
|
|
TNF-α
|
tumor necrosis factor-α
|
|
UPR
|
unfolded response
|
|
4-PBA
|
4-phenyl butyrate
|
|
3-MA
|
3-methyladenine
|
References
|
1
|
Hotamisligil GS: Inflammation and
metabolic disorders. Nature. 444:860–867. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jager J, Grémeaux T, Cormont M, Le
Marchand-Brustel Y and Tanti JF: Interleukin-1beta-induced insulin
resistance in adipocytes through down-regulation of insulin
receptor substrate-1 expression. Endocrinology. 148:241–251. 2007.
View Article : Google Scholar :
|
|
3
|
Lagathu C, Yvan-Charvet L, Bastard JP,
Maachi M, Quignard-Boulangé A, Capeau J and Caron M: Long-term
treatment with interleukin-1beta induces insulin resistance in
murine and human adipocytes. Diabetologia. 49:2162–2173. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kubes P and Mehal WZ: Sterile inflammation
in the liver. Gastroenterology. 143:1158–1172. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Navab M, Gharavi N and Watson AD:
Inflammation and metabolic disorders. Curr Opin Clin Nutr Metab
Care. 11:459–464. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Amyot J, Semache M, Ferdaoussi M, Fontés G
and Poitout V: Lipopolysaccharides impair insulin gene expression
in isolated islets of Langerhans via toll-like receptor-4 and NF-κB
signalling. PloS one. 7:e362002012. View Article : Google Scholar
|
|
7
|
De Nardo D and Latz E: NLRP3 inflammasomes
link inflammation and metabolic disease. Trends Immunol.
32:373–379. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ting JP, Willingham SB and Bergstralh DT:
NLRs at the intersection of cell death and immunity. Nat Rev
Immunol. 8:372–379. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schroder K and Tschopp J: The
inflammasomes. Cell. 140:821–832. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vandanmagsar B, Youm YH, Ravussin A,
Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM and Dixit
VD: The NLRP3 inflammasome instigates obesity-induced inflammation
and insulin resistance. Nat Med. 17:179–188. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brenner C, Galluzzi L, Kepp O and Kroemer
G: Decoding cell death signals in liver inflammation. J Hepatol.
59:583–594. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ogata M, Hino S, Saito A, Morikawa K,
Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K,
et al: Autophagy is activated for cell survival after endoplasmic
reticulum stress. Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nishitoh H: CHOP is a multifunctional
transcription factor in the ER stress response. J Biochem.
151:217–219. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang L, Ren F, Zhang X, Wang X, Shi H,
Zhou L, Zheng S, Chen Y, Chen D, Li L, Zhao C and Duan Z:
Peroxisome proliferator-activated receptor alpha acts as a mediator
of endoplasmic reticulum stress-induced hepatocyte apoptosis in
acute liver failure. Dis Model Mech. 9:799–809. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
González-Rodríguez A, Mayoral R, Agra N,
Valdecantos MP, Pardo V, Miquilena-Colina ME, Vargas-Castrillón J,
Lo Iacono O, Corazzari M, Fimia GM, et al: Impaired autophagic flux
is associated with increased endoplasmic reticulum stress during
the development of NAFLD. Cell Death Dis. 5:e11792014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Green DR, Galluzzi L and Kroemer G:
Mitochondria and the autophagy-inflammation-cell death axis in
organismal aging. Science. 333:1109–1112. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Singh R, Kaushik S, Wang Y, Xiang Y, Novak
I, Komatsu M, Tanaka K, Cuervo AM and Czaja MJ: Autophagy regulates
lipid metabolism. Nature. 458:1131–1135. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Goldman SJ, Taylor R, Zhang Y and Jin S:
Autophagy and the degradation of mitochondria. Mitochondrion.
10:309–315. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang Y, Li YB, Yin JJ, Wang Y, Zhu LB, Xie
GY and Pan SH: Autophagy regulates inflammation following oxidative
injury in diabetes. Autophagy. 9:272–277. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kuballa P, Nolte WM, Castoreno AB and
Xavier RJ: Autophagy and the immune system. Annu Rev Immunol.
30:611–646. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang L, Li P, Fu S, Calay ES and
Hotamisligil GS: Defective hepatic autophagy in obesity promotes ER
stress and causes insulin resistance. Cell Metab. 11:467–478. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Saitoh T, Fujita N, Jang MH, Uematsu S,
Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, et al:
Loss of the autophagy protein Atg16L1 enhances endotoxin-induced
IL-1beta production. Nature. 456:264–268. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hotamisligil GS: Endoplasmic reticulum
stress and the inflammatory basis of metabolic disease. Cell.
140:900–917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wree A, Eguchi A, McGeough MD, Pena CA,
Johnson CD, Canbay A, Hoffman HM and Feldstein AE: NLRP3
inflammasome activation results in hepatocyte pyroptosis, liver
inflammation and fibrosis in mice. Hepatology. 59:898–910. 2014.
View Article : Google Scholar :
|
|
26
|
Wen H, Gris D, Lei Y, Jha S, Zhang L,
Huang MT, Brickey WJ and Ting JP: Fatty acid-induced NLRP3-ASC
inflammasome activation interferes with insulin signaling. Nat
Immunol. 12:408–415. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
LeBel CP, Ischiropoulos H and Bondy SC:
Evaluation of the probe 2′,7′-dichlorofluorescin as an indicator of
reactive oxygen species formation and oxidative stress. Chem Res
Toxicol. 5:227–231. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dangi A, Huang C, Tandon A, Stolz D, Wu T
and Gandhi CR: Endotoxin-stimulated rat hepatic stellate cells
induce autophagy in hepatocytes as a survival mechanism. J Cell
Physiol. 231:94–105. 2016. View Article : Google Scholar
|
|
29
|
Czaja MJ, Ding WX, Donohue TM Jr, Friedman
SL, Kim JS, Komatsu M, Lemasters JJ, Lemoine A, Lin JD, Ou JH, et
al: Functions of autophagy in normal and diseased liver. Autophagy.
9:1131–1158. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Malhotra JD and Kaufman RJ: Endoplasmic
reticulum stress and oxidative stress: A vicious cycle or a
double-edged sword? Antioxid Redox Signal. 9:2277–2293. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Scull CM and Tabas I: Mechanisms of ER
stress-induced apoptosis in atherosclerosis. Arterioscler Thromb
Vasc Biol. 31:2792–2797. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ding WX, Ni HM, Gao W, Yoshimori T, Stolz
DB, Ron D and Yin XM: Linking of autophagy to ubiquitin-proteasome
system is important for the regulation of endoplasmic reticulum
stress and cell viability. Am J Pathol. 171:513–524. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Uysal KT, Wiesbrock SM, Marino MW and
Hotamisligil GS: Protection from obesity-induced insulin resistance
in mice lacking TNF-alpha function. Nature. 389:610–614. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sabio G, Das M, Mora A, Zhang Z, Jun JY,
Ko HJ, Barrett T, Kim JK and Davis RJ: A stress signaling pathway
in adipose tissue regulates hepatic insulin resistance. Science.
322:1539–1543. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Franchi L, Muñoz-Planillo R, Reimer T,
Eigenbrod T and Núñez G: Inflammasomes as microbial sensors. Eur J
Immunol. 40:611–615. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dostert C, Pétrilli V, Van Bruggen R,
Steele C, Mossman BT and Tschopp J: Innate immune activation
through Nalp3 inflammasome sensing of asbestos and silica. Science.
320:674–677. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Napoli C, de Nigris F and Palinski W:
Multiple role of reactive oxygen species in the arterial wall. J
Cell Biochem. 82:674–682. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yuan XM, Li W, Olsson AG and Brunk UT: The
toxicity to macrophages of oxidized low-density lipoprotein is
mediated through lysosomal damage. Atherosclerosis. 133:153–161.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Larsen CM, Faulenbach M, Vaag A, Vølund A,
Ehses JA, Seifer t B, Mandr up-Poulsen T and Donath MY:
Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N
Engl J Med. 356:1517–1526. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gandhi CR, Kuddus RH, Nemoto EM and Murase
N: Endotoxin treatment causes an upregulation of the endothelin
system in the liver: Amelioration of increased portal resistance by
endothelin receptor antagonism. J Gastroenterol Hepatol. 16:61–69.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xie Q, Khaoustov VI, Chung CC, Sohn J,
Krishnan B, Lewis DE and Yoffe B: Effect of tauroursodeoxycholic
acid on endoplasmic reticulum stress-induced caspase-12 activation.
Hepatology. 36:592–601. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kouroku Y, Fujita E, Tanida I, Ueno T,
Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E and Momoi T: ER
stress (PERK/eIF2alpha phosphorylation) mediates the
polyglutamine-induced LC3 conversion, an essential step for
autophagy formation. Cell Death Differ. 14:230–239. 2007.
View Article : Google Scholar
|
|
43
|
Qin L, Wang Z, Tao L and Wang Y: ER stress
negatively regulates AKT/TSC/mTOR pathway to enhance autophagy.
Autophagy. 6:239–247. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Høyer-Hansen M and Jäättelä M: Connecting
endoplasmic reticulum stress to autophagy by unfolded protein
response and calcium. Cell Death Differ. 14:1576–1582. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Corcelle EA, Puustinen P and Jäättelä M:
Apoptosis and autophagy: Targeting autophagy signalling in cancer
cells-'trick or treats'? FEBS J. 276:6084–6096. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhou R, Yazdi AS, Menu P and Tschopp J: A
role for mitochondria in NLRP3 inflammasome activation. Nature.
469:221–225. 2011. View Article : Google Scholar
|
|
47
|
Nakahira K, Haspel JA, Rathinam VA, Lee
SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim
HP, et al: Autophagy proteins regulate innate immune responses by
inhibiting the release of mitochondrial DNA mediated by the NALP3
inflammasome. Nat Immunol. 12:222–230. 2011. View Article : Google Scholar
|
|
48
|
Degenhardt K, Mathew R, Beaudoin B, Bray
K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al:
Autophagy promotes tumor cell survival and restricts necrosis,
inflammation, and tumorigenesis. Cancer Cell. 10:51–64. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Luo MX, Wong SH, Chan MT, Yu L, Yu SS, Wu
F, Xiao Z, Wang X, Zhang L, Cheng AS, et al: Autophagy mediates
HBx-induced nuclear factor-κB activation and release of IL-6, IL-8
and CXCL2 in hepatocytes. J Cell Physiol. 230:2382–2389. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Talero E, Alcaide A, Ávila-Román J,
García-Mauriño S, Vendramini-Costa D and Motilva V: Expression
patterns of sirtuin 1-AMPK-autophagy pathway in chronic colitis and
inflammation-associated colon neoplasia in IL-10-deficient mice.
Int Immunopharmacol. 35:248–256. 2016. View Article : Google Scholar : PubMed/NCBI
|