Introduction
Mycoplasma are the smallest prokaryotic
microbes present in nature. These wall-less, malleable organisms
can pass through cell filters, and can grow and propagate under
cell-free conditions in vitro (1). Mycoplasma contain a 600–1,350
kbp genome and 23–35% GC. They reproduce predominantly via typical
binary fission and have a tendency to form ‘fried egg’ colonies in
solid culture media. At present, seven species of Mycoplasma
have been found to be pathogenic to humans, including M.
pneumoniae, M. urealytium, M. genitalium, M.
hominis, M. fermentation, M. penetrans and M.
pirum (1). M.
pneumoniae, which was initially separated, cultivated and named
by Chanock and Hayflick in 1962, has been examined the most
(2). In addition to primary
atypical pneumonia and community-acquired pneumonia, which induce
predominantly respiratory symptoms, M. pneumoniae can also
induce autoimmune hemolytic anemia and other diseases in the blood,
cardiovascular system, gastrointestinal tract, and skin, and can
induce pericarditis, myocarditis, nephritis and meningitis
(3–5).
M. pneumoniae infections are distributed
globally with local prevalence. As reported, its infection rate is
increasing annually, however, the specific pathogenic mechanism
remains to be fully elucidated (2). The pathogenesis of M.
pneumoniae infection is complex as it involves several
mechanisms, including adhesion damage, membrane fusion damage,
nutrition depletion, invasive damage, toxic damage, immune damage
and inflammatory damage (Fig. 1).
However, the specific mechanism underlying its effects remains to
be elucidated.
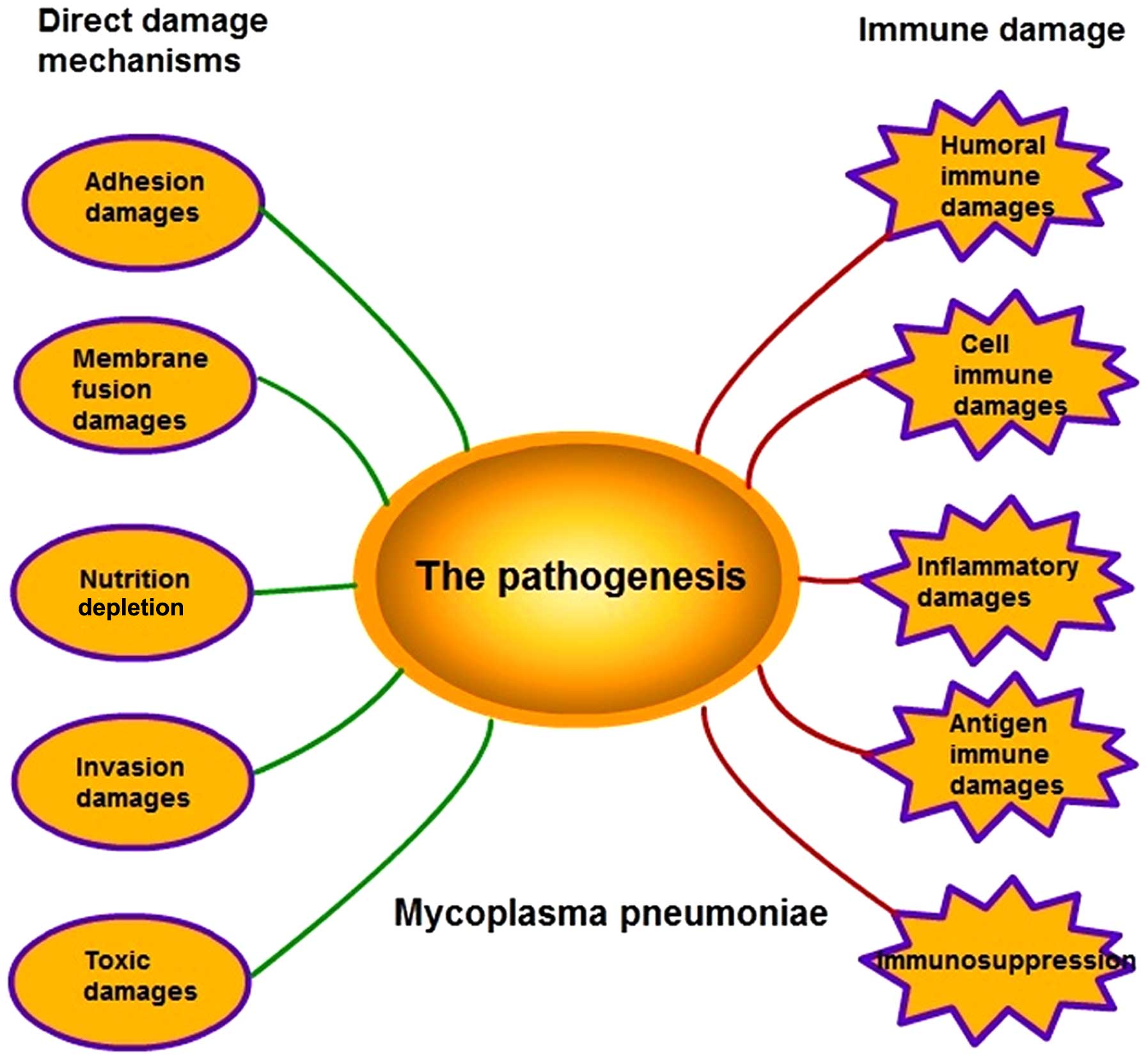 | Figure 1.Pathogenesis of M. pneumoniae.
The pathogenesis of M. pneumoniae comprises five direct
damage mechanisms, including adhesion damage, membrane fusion
damage, nutrition depletion, invasive damage, toxic damage, and
five types of immune damage, including humoral immune damage, cell
immune damage, inflammatory damage, antigen immune damage and
immunosuppression. |
Direct damage mechanisms
Adhesion damage
The adhesion of M. pneumoniae onto the
respiratory epithelia is a precondition dictating the propagation
and pathogenesis of M. pneumoniae (6). In addition to pseudo-stratified
columnar ciliated epithelia, M. pneumoniae can also adhere
to red blood cells, HeLa cells, fibroblasts, macrophages and
tracheal organ cultures in vitro, and can adhere to the
surfaces of glass or plastics (7).
M. pneumoniae is asymmetric under electron microscopy
(8). The cell membranes at one end
can extend outside to form a proline-rich top structure, also
termed the apical organ, and specifically adhere onto the
neuraminic acid receptors on the membranes of target cells.
Adhesion is an intricate process, as the adhesion
structure consists of an interactive adhesion network-like system
and adhesion auxiliary proteins. Specifically, the 170 kDa P1
protein functions as a key ligand during adhesion (9). Pulse-tracking tag experiments have
shown that 1 h following contact of M. pneumoniae with the
target cells, the P1 precursor proteins, which are scattered in the
cell membranes, rapidly shift to the apical organs, and the leading
peptide on their amino terminal is hydrolyzed to mature P1 proteins
(10). Due to its sole dependence
on the key P1 protein, M. pneumoniae is unable to adhere to
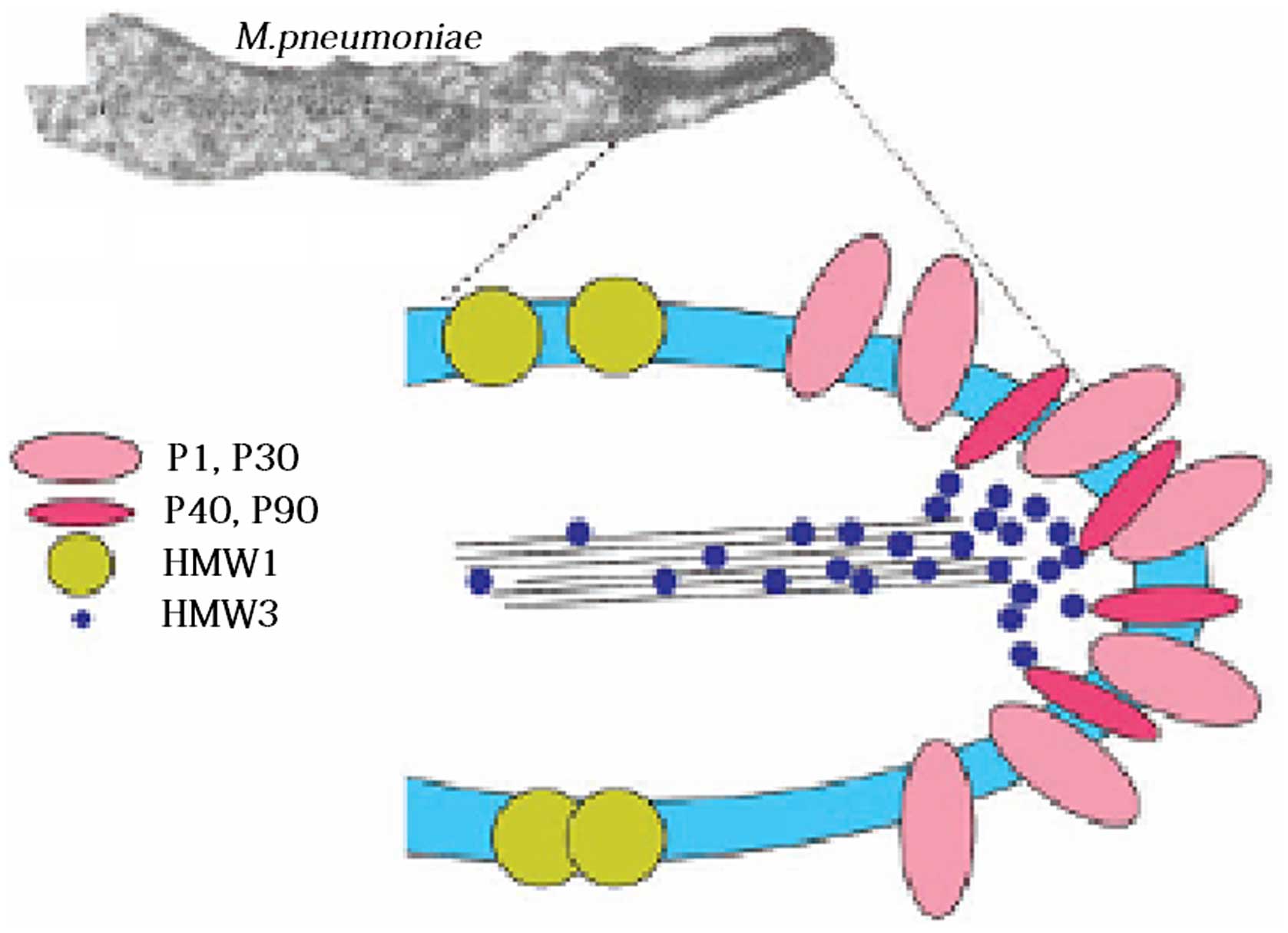
host cells, however, it can adhere with the assistance of several
collaborative auxiliary proteins, including P30 adhesion
factor-related protein A (72 KDa), B (85 KDa) and C (37 KDa), HMW
1–5 polypeptides, P40, P90 and P65; these components jointly
constitute a characteristic high-electron-density ‘adhesion protein
complex’ (Fig. 2) (11). This complex stabilizes the
integrity of the M. pneumoniae apical structure by forming a
cytoskeleton, anchoring the P1 protein into the cytoskeleton of the
adhesive organs, and allowing the P1 proteins on the adhesion cell
organs to adhere.
Marking experiments have shown that, in mutant
strains with loss of adhesion auxiliary proteins, the P1 protein is
chronically dispersed as a precursor in the cell membranes,
however, it cannot aggregate to the apical organs or convert into
mature P1 protein (9). Electron
microscopy has demonstrated that the adhesion of a M.
pneumoniae variant is concentrated in the adherend in the
following order: HMW1, HMW3, Pl, P30, P90, P40 and P65, which
indicates that these proteins have formed an interrelated adhesion
network (12). Specifically, HMW1,
HMW2 and HMW3 function as stable adherends and allow other
adhesions to locate onto the adherend, and, they are involved in
the adhesion onto the respiratory tract epithelia (13). As reported, the M.
pneumoniae mutant strains, HMW1 and HMW2, can prevent the P1
protein from correctly locating onto the apical structure, which
leads to irregular cell morphology, loss of toxicity and sliding
ability, and loss of adhering function (14). P30 does not directly affect the
positioning of the P1 protein onto the apical structure, however,
it interferes with the binding between P1 and its receptor
(15). The loss of P30 or
enzymatic cleavage of the carboxyl terminal leads to the complete
loss of adhesion function in M. pneumoniae, reduced sliding
ability, and marked changes in morphology and structure (16). For example, a bifurcate structure
appears in the apical tip, and numerous nucleoid-like substances
appear in the cytoplasm. When transposon Tn4001 from the genes of
an adhesion auxiliary protein C-mutant was used to transform M.
pneumoniae, the mutant strain showed reduced cell adhesion
ability. Following the loss of the P41 protein, the adherend in the
sliding process of M. pneumoniae was separated from the cell
(17). These adhesion auxiliary
proteins and adhesion molecules jointly form adhesion protein
complexes. The adsorption ability of host cells is decided by the
positioning of adhesion proteins and the interaction between the
components of the protein complexes.
M. pneumoniae can also utilize the MPN372
protein to combine with lung surfactant protein A (SP-A), pass
through the host barrier and permanently adhere to target cells
containing the SP-A receptor, including alveolar macrophages,
alveolar epithelial type II cells, and other histiocytes inside and
outside the lung (18). The
pretreatment of M. pneumoniae with low-dose proenzyme
reduces the binding between M. pneumoniae and SP-A by
80–90%, however, pretreatment with mannose does not inhibit the
binding between M. pneumoniae and SP-A, indicating that
M. pneumoniae protein components are involved in this
process (19).
Membrane fusion damage
The cell membranes of the Mycoplasma genus
are more durable, compared with those of other prokaryotes, and the
cytoskeletal protein network-like structure functions as a cell
walls in terms of maintaining cell integrity. Following M.
pneumoniae infection, the lipid bilayer of cell membranes is
susceptible to biomembrane fusion, and its structure involves the
transcription of specific genes, cytoskeletal changes and changes
in the nucleolus (20). Membrane
fusion can also cause changes in receptor-identifying sites in the
cell membranes, affecting the signal delivery between cells and the
production of cellular factors (21).
Nutrition depletion
The small-genome M. pneumoniae does not
possess the ability to self-synthesize amino acids, fatty acids,
cofactors or vitamins. Instead, following permanent adherence via
the adherend to the respiratory tract epithelia, M.
pneumoniae spreads microtubules and inserts them into host
cells, enabling oxygen consumption, use of glucose, absorption of
cholesterol, ingestion of amino acids and consumption of nutrients
in host cells, causing injury to the host cells (22,23).
Invasion damage
M. pneumoniae is usually regarded as an
extracellular parasite, however, certain studies have shown it can
also invade and damage cells. Studies have shown that M.
pneumoniae can invade A549 lung cancer cells, evidenced by its
detection in the cytoplasm and nucleus, and the invasive ability
depends on the duration and temperature of infection (24). In cell culture in vitro,
M. pneumoniae has been shown to invade non-phagocytes,
survive for >6 months and synthesize DNA inside cells (7). When the clinically isolated RYC15989
strain was utilized to infect human Hep-G2 cells and rat N2A cells,
intracellular Mycoplasma were observed under laser confocal
microscopy, and the intracellular invasion damaging ability of
M. pneumoniae was also confirmed (25). In addition, during invasion,
certain enzymes inside M. pneumoniae, including hydrolase,
nuclease and phosphoprotein phosphatase shift to the host cells.
Nuclease degrades DNA in host cells, whereas phosphoprotein
phosphatase interferes with the activity of serine/threonine and
tyrosine protein kinase (26,27).
Toxic damage
Adhesion provides conditions for M.
pneumoniae to induce regional cytotoxic effects, and M.
pneumoniae can directly induce damage via adhesion, auxiliary
proteins, capsular and invasive enzymes. M. pneumoniae also
exerts its toxin-like effects through its metabolites, exotoxin and
exotoxin-like toxic substances, lipids, lipopolysaccharides and
membrane lipoprotein (28).
Following the adherence of M. pneumoniae onto the surface of
bronchial cells, with the cytoskeleton rearrangement, M.
pneumoniae penetrates through the bronchial mucous membranes
and releases nuclease and H2O2, which result in swelling, necrosis
and a binding of bronchial epithelial cells, slower microvilli
movement, structural deformation, and the termination of swinging,
thereby inducing the infiltration of lymphocytes, plasma cells and
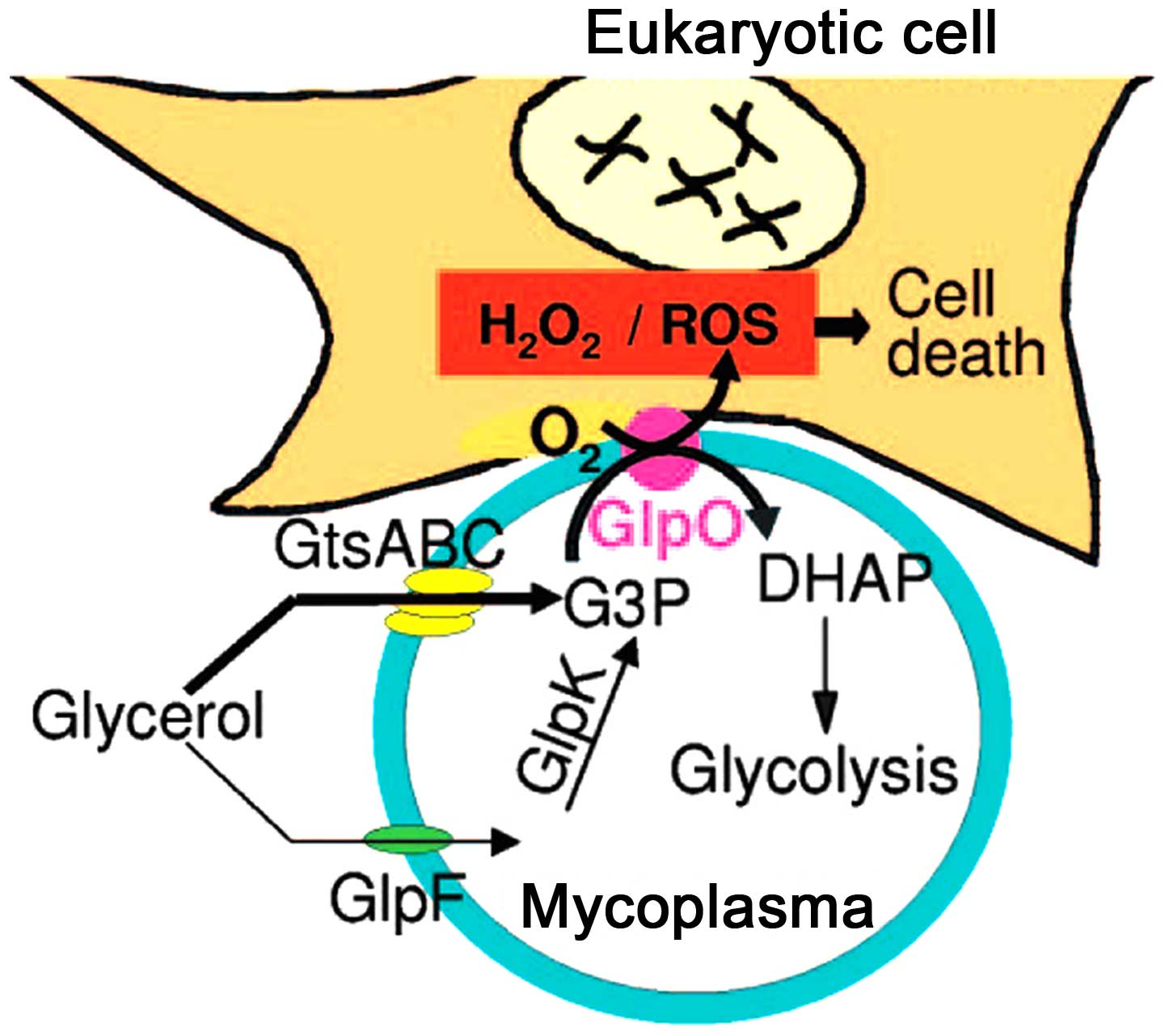
monocytes (22,29). With the lack of superoxide
dismutase and catalase in M. pneumoniae, the H2O2 and
superoxide groups synthesized by M. pneumoniae, and the
endogenous toxic oxygen molecules produced by the host cells,
increase the intracellular oxygen pressure in the epithelium, which
leads to oxidative stress and subsequent cell death (Fig. 3) (30). The major virulence factors
affecting the pathogenesis of M. pneumoniae include the
accumulation of H2O2 inside host cells and the effects of
superoxides on the ultrastructure of host cells (31). The ions of M.
pneumoniae-produced superoxides inhibit the activity and
degradation of catalases in the host cells, so that the host cells
become more sensitive to the toxic oxygen, resulting in
mitochondrial swelling, vascular degeneration, cilia destruction
and weakened cilia movement in the epithelium (32,33).
M. pneumoniae infection leads to the denaturation of red
blood cell hemoglobin, loss of reduced glutathione and cytolysis
(34,35).
M. pneumoniae is considered to be incapable
of secreting cytotoxin. The N-terminal of the M.
pneumoniae-associated pathogenic factor, MPN372, contains
ADP-ribose transferase activity and its structure is similar to the
S1 subunit of pertussis toxin, which induces extensive vascular
degeneration and can cause the death of mammalian cells, thereby
inducing chincough-like clinical symptoms; these are termed
community acquired respiratory distress syndrome (CARDS) toxin
(36). M. pneumoniae CARDS
toxin is internalized via clathrin-mediated endocytosis (37), and the CARDS toxin induces
pulmonary eosinophilic and lymphocytic inflammation (38,39).
Cellular vacuoles induced by M. pneumoniae CARDS toxin
originate from Rab9-associated compartments (40).
Immune damage
Clinical epidemiological findings show that the
symptoms of M. pneumoniae infection are not observed at
infancy, and that the pathogenic peak occurs in children >10
years old (41). In patients with
reduced immune function, M. pneumoniae infection does not
induce notable pathological changes in the lung. Experiments in
thymus-excised animals have shown that M. pneumoniae
infection does not readily induce pneumonia (42). Animal experiments have shown that
the histopathologic response occurs 10–14 days following primary
M. pneumoniae infection, but within 3 days following
secondary infection, indicating that the body responds via immune
cell accumulation following M. pneumoniae infection, but
produces a more marked immune response to a second infection
(43). These findings indicate
that the host immune response is important during the onset of
M. pneumoniae-induced pneumonia.
Humoral immune damage
The glycolipid antigen on the cell membranes of
M. pneumoniae induces humoral immunity, and the antibody
response is fundamental during the response against M.
pneumoniae infection. At an early stage of M. pneumoniae
infection, the body resists Mycoplasma settlement
predominantly via a non-specific defense mechanism by secreting
inhibitors, alexin and phagocytes (6). Animal experiments have shown that,
following infection of the body with M. pneumoniae, the
levels of complement components C1, C2, C3 and C4 in the bronchial
secretions are significantly improved (44). After 2 weeks, the level of alexin
begins to decline, whereas the antibody level increases, which
indicates the non-specific protective effect of alexin at an early
stage of M. pneumoniae infection (45). In children infected with M.
pneumoniae, the contents of C1q, C3, C4 and B in the serum
increase to varying degrees in the acute phase and recovery phase,
indicating that the alexin classical and bypass activation pathways
are involved during M. pneumoniae infection (46). With the lack of alexin, the surface
of neutrophils have been shown to adhere with and engulf M.
pneumoniae under electron microscopy, and the M.
pneumoniae in their phagocytosis vesicles remains active
(47). The specific sIgA produced
during M. pneumoniae infection can protect against infection
of respiratory mucous membranes, and its action is key in
indigenous resistance (48). It
was previously reported, that, 28 days following M.
pneumoniae infection in pigs, the numbers of B cells in the
alveolar lavage fluid and lung parenchyma increased 25-fold
(49). In addition, in the acute
phase and recovery phase of mycoplasma pneumonia, the contents of
IgG, IgM, IgA and immune complex in the serum increase
significantly, particularly in severely affected patients (50). Following M. pneumoniae
infection, the IgM level has been shown to markedly increase in
normal children, which usually occurred 7–14 days following
infection, peaked in weeks 3–4 and persisted for months (51). M. pneumoniae infection can
cause an increase in the level of total IgE in the serum, whereas
delayed-type and anaphylactic-type allergic reactions induce asthma
as an immediate reaction and delayed-phase reaction or a dual-phase
reaction, which induce the IgE-mediated airway inflammation and
airway hyper-reactivity (52).
However, there is no direct evidence that M. pneumoniae is
the direct cause of asthma. These previous studies indicate that
various specific and nonspecific immunoglobulins and complement
components are involved during M. pneumoniae infection,
which assist with the recovery and immunity.
Cell immune damage
Cellular immunity is required by the protein
antigens on the cell membranes of M. pneumoniae. Following
inoculation with M. pneumoniae antibody in patients infected
with M. pneumoniae, a tuberculin-like, delayed-type allergic
reaction occurs to differing degrees; the reaction is more severe
in severely-affected patients, however, this reaction can be
inhibited by anti-thymocyte serum (53). Tuberculin tests in patients
infected with M. pneumoniae show that the reaction intensity
directly affects the degree of lung damage, indicating that
cellular immunity is vital during the pathogenesis of M.
pneumoniae (32). In patients
with M. pneumoniae, the CD4+ T cell count is decreased, the
CD8+ T cell count is markedly increased and the ratio of CD4+T/CD8+
is reduced, and these changes are more marked in severely affected
patients (53). In adults with
M. pneumoniae infection, the peripheral blood CD4+ T count
is decreased, however, the ratio of T-lymphocytes to CD4+/CD8+
cells in the bronchoalveolar lavage fluid increases, possibly due
to abundant CD4+ T cells being involved in the inflammatory
reaction (54).
During M. pneumoniae infection, the Th1/Th2
ratio is unbalanced, although which type of cell is dominant
remains controversial. It was previously reported that, following
M. pneumoniae infection, Th1-dominated rats exhibit
aggregation of peribronchial lymphocytes, whereas Th2-dominated
rats exhibit hyperplasia of alveolus mesenchymal cells, which
indicate that the imbalance in auxiliary T lymphocyte subgroups is
associated with the type of lung damage (53). The mechanism underlying M.
pneumoniae-induced asthma may be correlated with the enhanced
secretion of Th2 cell factors (55).
Inflammatory damage
Inflammatory factors are important during the M.
pneumoniae-induced inflammatory reaction. Polymerase chain
reaction analysis has shown that, following primary M.
pneumoniae infection in BALB/C rats, the mRNA expression levels
of tumor necrosis factor-α (TNF-α), interleukin (IL)-1β and IL-6 in
the lungs were markedly increased, whereas the mRNA expression
levels of IL-2 and its receptor were not increased (56). Following the second infection, the
mRNA expression levels of TNF-α and IL-6 increased 10-fold, whereas
the mRNA expression of IL-2 decreased rapidly within 24 h, and that
of IL-10 increased markedly (56).
It has been reported that M. pneumoniae can induce the
production of IL-1β, which is extensively involved in several types
of damage, including tissue destruction and edema formation
(57). Following M.
pneumoniae infection, the serum level of IL-8 increases
markedly, whereas white blood cells locate to the site of
inflammation and infiltrate, accumulate and release active
substances in the affected tissues, causing damage (58). The serum level of TNF-α following
M. pneumoniae infection in the respiratory tract is
significantly increased, and is positively correlated with the
severity of illness (59). Serum
levels of soluble IL-2 receptor (sIL-2R) can be an important
indicator. In children with M. pneumoniae infection, the
increase in the level of sIL-2R can reactivate the mononuclear
cells in the circulation, and is involved in T lymphocyte
dysfunction (60). In children
with M. pneumoniae infection, the level of soluble
intercellular adhesion molecule-1 is also markedly increased, which
induces the increased bronchial reaction (61). M. pneumoniae antigens induce
a potent immune reaction and enhance the Th17 cell response in
vivo and in-vitro, with Treg and IL-10 being associated
with the suppression of the production of IL-17A (62). The cytadherence of M.
pneumoniae induces inflammatory responses through TLR4 and
autophagy (6). M.
pneumoniae infection has been shown to increase inflammatory
factors in a rat model of atherosclerosis and aggravate the state
of atherosclerosis (4).
Antigen immune damage
Antigenic variation
The M. pneumoniae membrane protein is
associated with invasiveness, and its variation directly affects
the toxicity of M. pneumoniae. The molecular weight of the
M. pneumoniae membrane V-1 antigen can change and is
associated with virulence. No toxicity or pathogenesis occurs when
its molecular weight is 100–200 kDa, however, toxicity and
pathogenesis are observed when its molecular weight is 30 kDa. The
gene mutation in V-1 antigen occurs at a 17-amino-acid repetitive
sequence at the C terminal of the 94.2 kDa antigen, whereas the
27.4 kDa antigen contains two adjacent, but discontinuous,
nine-amino-acid repetitive sequences, and variation is induced by
site-specific DNA inversion (63).
In addition, the adsorption of RBCs by M. pneumoniae can
alter the antigenicity of RBC membranes and induce autoantibody
against RBC membrane I antigen, namely the cold agglutinin of RBCs,
which induces autoimmune hemolytic anemia (64).
Immune evasion
The Mycoplasma-induced viscous polysaccharide
capsule, as with other bacteria, is readily formed inside the host,
however, it disappears rapidly in vitro, indicating
phagocytosis by the host cells. M. pneumoniae readily
induces variation in surface membrane antigens, in order to evade
attacks from the host immune system. M. pneumoniae may
tightly adsorb onto the surface of the host cells, depending on the
specific adhesion structure, to avoid phagocytosis prior to exact
antibody adjustment (65). The
polymorphism of M. pneumoniae adhesion antigens also weakens
the effects of specific antibodies (66). The glycerophosphatide on M.
pneumoniae cell membranes shares certain antigenic components
with the host cells, and thus can also evade the host's immune
surveillance. The invasion of intracellular parasitism assists in
enabling M. pneumoniae to evade the host's immune clearance
and drug effect. Therefore subjecting the patients to chronically
infected persons or asymptomatic carriers. Thus, the various immune
evasion mechanisms of M. pneumoniae constitute the
predominant factor explaining why M. pneumoniae can survive
chronically inside the host.
Cross-reacting antigen
The M. pneumoniae membrane antigen is in
antigen mimic of the RBC-membrane I antigen, and shares certain
antigenic components with Streptococcus pneumoniae 23 or 32
and M. genitalium (67). As
with several plants and bacteria, the membrane glycolipids of M.
pneumoniae share a common antigen in the brain and lung
tissues, which induce cross reaction. The carboxyl end of the P1
and P30 proteins in the adhesive organs of M. pneumoniae
show high levels of homology to the cytoskeletal proteins,
fibrinogen, keratin and troponin in eukaryotes (68). Thus, during infection,
autoantibodies in the brain, lung, RBC-membrane, lymphocytes and
myocardial cells commonly occur, which form immune complexes and
magnify the autoimmune response, leading to multisystem immune
damage.
Superantigen
M. pneumoniae membranes are full of
Mycoplasma lipid-associated membrane proteins. At least
three types of functional protein have been identified, including
M. pneumoniae N602 (b subfamily of F0F1-ATPase), M.
pneumoniae N162 and M. pneumoniae N611 (69). Specifically, the inflammatory
capacity of M. pneumoniae N602 is higher (~100-fold),
compared with that of M. pneumoniae N161 and M.
pneumoniae N162, indicating that M. pneumoniae N602 is a
potential superantigen component (70,71).
Immunosuppression
M. pneumoniae infection can induce
immunosuppression in the body and cause maladjustment of T cell
subgroups. Experiments have revealed that M. pneumoniae
infection causes severe destruction of B cells and T cells
(72). At 13–18 weeks in patients
infected with M. pneumoniae, the serum level of IgG declines
(73). Certain children infected
with M. pneumoniae suffer from hypoglobulinemia, decreased
chemoattraction in neutrophils, lower reactivity to
phytohemagglutin phytolectin and reduced resistance against
combined infections with other pathogens, including S.
pneumoniae (72). These
changes indicate that M. pneumoniae infection may induce
immunosuppression.
Conclusion and perspective
As summarized in the present review, it has been
demonstrated over several years that the pathogenesis of M.
pneumoniae infection is complex; the natural synergy between
the various factors involved is summarized in Fig. 1. There is no one factor alone,
which is involved. As increased efforts have focussed on
investigating M. pneumoniae gene structures and functions,
and in sequencing, the various pathogenic factors of M.
pneumoniae membrane proteins, invasive proteins and adhesive
proteins can be investigated at the molecular level. This
development not only assists with the treatment and prevention of
M. pneumoniae infection, but is also meaningful for the
development of Mycoplasma vaccines.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 31100137), the Natural
Science Foundation of Hunan Proince (grant no. 14JJ7044), the
Project Foundation of Health Department of Hunan Province (grant
no. B2011-058) and the 12th Five-Year Technology Innovation Team at
the University of South China.
References
|
1
|
Roca B: Mycoplasma infections. Rev Clin
Esp. 206:239–242. 2006.(In Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ubukata K: Mycoplasma pneumoniae. Nihon
Yakurigaku Zasshi. 141:287–289. 2013.(In Japanese). View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Özel C, Dafotakis M, Nikoubashman O,
Litmathe J, Matz O and Schöne U: Mycoplasma pneumoniae-induced
meningoencephalitis. Fortschr Neurol Psychiatr. 83:392–396.
2015.(In German). View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Atkinson TP and Waites KB: Mycoplasma
pneumoniae infections in childhood. Pediatr Infect Dis J. 33:92–94.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fan Q, Meng J, Li P, Liu Z, Sun Y and Yan
P: Pathogenesis and association of Mycoplasma pneumoniae infection
with cardiac and hepatic damage. Microbiol Immunol. 59:375–380.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shimizu T, Kimura Y, Kida Y, Kuwano K,
Tachibana M, Hashino M and Watarai M: Cytadherence of Mycoplasma
pneumoniae induces inflammatory responses through autophagy and
toll-like receptor 4. Infect Immun. 82:3076–3086. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Prince OA, Krunkosky TM and Krause DC: In
vitro spatial and temporal analysis of Mycoplasma pneumoniae
colonization of human airway epithelium. Infect Immun. 82:579–586.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Balish MF: Mycoplasma pneumoniae, an
underutilized model for bacterial cell biology. J Bacteriol.
196:3675–3682. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chourasia BK, Chaudhry R and Malhotra P:
Delineation of immunodominant and cytadherence segment(s) of
Mycoplasma pneumoniae P1 gene. BMC Microbiol. 14:1082014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Waldo RH III and Krause DC: Synthesis,
stability, and function of cytadhesin P1 and accessory protein B/C
complex of Mycoplasma pneumoniae. J Bacteriol. 188:569–575. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Seto S, Kenri T, Tomiyama T and Miyata M:
Involvement of P1 adhesin in gliding motility of Mycoplasma
pneumoniae as revealed by the inhibitory effects of antibody under
optimized gliding conditions. J Bacteriol. 187:1875–1877. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Willby MJ, Balish MF, Ross SM, Lee KK,
Jordan JL and Krause DC: HMW1 is required for stability and
localization of HMW2 to the attachment organelle of Mycoplasma
pneumoniae. J Bacteriol. 186:8221–8228. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Page CA and Krause DC: Protein
kinase/phosphatase function correlates with gliding motility in
Mycoplasma pneumoniae. J Bacteriol. 195:1750–1757. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chaudhry R, Varshney AK and Malhotra P:
Adhesion proteins of Mycoplasma pneumoniae. Front Biosci.
12:690–699. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chang HY, Prince OA, Sheppard ES and
Krause DC: Processing is required for a fully functional protein
P30 in Mycoplasma pneumoniae gliding and cytadherence. J Bacteriol.
193:5841–5846. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chang HY, Jordan JL and Krause DC: Domain
analysis of protein P30 in Mycoplasma pneumoniae cytadherence and
gliding motility. J Bacteriol. 193:1726–1733. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cloward JM and Krause DC: Loss of
co-chaperone TopJ impacts adhesin P1 presentation and terminal
organelle maturation in Mycoplasma pneumoniae. Mol Microbiol.
81:528–539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kannan TR, Musatovova O, Balasubramanian
S, Cagle M, Jordan JL, Krunkosky TM, Davis A, Hardy RD and Baseman
JB: Mycoplasma pneumoniae community acquired respiratory distress
syndrome toxin expression reveals growth phase and
infection-dependent regulation. Mol Microbiol. 76:1127–1141. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ledford JG, Goto H, Potts EN, Degan S, Chu
HW, Voelker DR, Sunday ME, Cianciolo GJ, Foster WM, Kraft M and
Wright JR: SP-A preserves airway homeostasis during Mycoplasma
pneumoniae infection in mice. J Immunol. 182:7818–7827. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Balish MF, Santurri RT, Ricci AM, Lee KK
and Krause DC: Localization of Mycoplasma pneumoniae
cytadherence-associated protein HMW2 by fusion with green
fluorescent protein: Implications for attachment organelle
structure. Mol Microbiol. 47:49–60. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bao S, Yu S, Guo X, Zhang F, Sun Y, Tan L,
Duan Y, Lu F, Qiu X and Ding C: Construction of a cell-surface
display system based on the N-terminal domain of ice nucleation
protein and its application in identification of Mycoplasma
adhesion proteins. J Appl Microbiol. 119:236–244. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Großhennig S, Schmidl SR, Schmeisky G,
Busse J and Stülke J: Implication of glycerol and phospholipid
transporters in Mycoplasma pneumoniae growth and virulence. Infect
Immun. 81:896–904. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schomburg J and Vogel M: A 12-year-old boy
with severe mucositis: Extrapulmonary manifestation of Mycoplasma
pneumoniae infection. Klin Padiatr. 224:94–95. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li S, Li X, Wang Y, Yang J, Chen Z and
Shan S: Global secretome characterization of A549 human alveolar
epithelial carcinoma cells during Mycoplasma pneumoniae infection.
BMC Microbiol. 14:272014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Meseguer MA, Alvarez A, Rejas MT, Sánchez
C, Pérez-Diaz JC and Baquero F: Mycoplasma pneumoniae: A
reduced-genome intracellular bacterial pathogen. Infect Genet Evol.
3:47–55. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sauteur PM Meyer, Huber BM and Goetschel
P: Neuroinvasive Mycoplasma pneumoniae infection without
intrathecal antibody response. Pediatr Infect Dis J. 31:1199–1200.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rhodes RH, Monastersky BT, Tyagi R and
Coyne T: Mycoplasmal cerebral vasculopathy in a lymphoma patient:
Presumptive evidence of Mycoplasma pneumoniae microvascular
endothelial cell invasion in a brain biopsy. J Neurol Sci.
309:18–25. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
McDermott AJ, Taylor BM and Bernstein KM:
Toxic epidermal necrolysis from suspected Mycoplasma pneumoniae
infection. Mil Med. 178:e1048–e1050. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Calvano RA, Scacchi MF, Sojo MM, Diaz SM,
Volonteri VI and Giachetti AC: Toxic epidermal necrolysis
associated with acute infection by Mycoplasma pneumoniae. Arch
Argent Pediatr. 111:e24–e27. 2013.(In Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Elkhal CK, Kean KM, Parsonage D, Maenpuen
S, Chaiyen P, Claiborne A and Karplus PA: Structure and proposed
mechanism of L-α-glycerophosphate oxidase from Mycoplasma
pneumoniae. FEBS J. 282:3030–3042. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Maenpuen S, Watthaisong P, Supon P,
Sucharitakul J, Parsonage D, Karplus PA, Claiborne A and Chaiyen P:
Kinetic mechanism of L-α-glycerophosphate oxidase from Mycoplasma
pneumoniae. FEBS J. 282:3043–3059. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ledford JG, Mukherjee S, Kislan MM, Nugent
JL, Hollingsworth JW and Wright JR: Surfactant protein-A suppresses
eosinophil-mediated killing of Mycoplasma pneumoniae in allergic
lungs. PLoS One. 7:e324362012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sun G, Xu X, Wang Y, Shen X, Chen Z and
Yang J: Mycoplasma pneumoniae infection induces reactive oxygen
species and DNA damage in A549 human lung carcinoma cells. Infect
Immun. 76:4405–4413. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kariya C, Chu HW, Huang J, Leitner H,
Martin RJ and Day BJ: Mycoplasma pneumoniae infection and
environmental tobacco smoke inhibit lung glutathione adaptive
responses and increase oxidative stress. Infect Immun.
76:4455–4462. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Inaba H, Geiger TL, Lasater OE and Wang
WC: A case of hemoglobin SC disease with cold agglutinin-induced
hemolysis. Am J Hematol. 78:37–40. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hardy RD, Coalson JJ, Peters J, Chaparro
A, Techasaensiri C, Cantwell AM, Kannan TR, Baseman JB and Dube PH:
Analysis of pulmonary inflammation and function in the mouse and
baboon after exposure to Mycoplasma pneumoniae CARDS toxin. PLoS
One. 4:e75622009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Techasaensiri C, Tagliabue C, Cagle M,
Iranpour P, Katz K, Kannan TR, Coalson JJ, Baseman JB and Hardy RD:
Variation in colonization, ADP-ribosylating and vacuolating
cytotoxin, and pulmonary disease severity among Mycoplasma
pneumoniaestrains. Am J Respir Crit Care Med. 182:797–804. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Somarajan SR, Al-Asadi F, Ramasamy K,
Pandranki L, Baseman JB and Kannan TR: Annexin A2 mediates
Mycoplasma pneumoniae community-acquired respiratory distress
syndrome toxin binding to eukaryotic cells. MBio. 5(pii):
e01497-142014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Medina JL, Coalson JJ, Brooks EG, Le Saux
CJ, Winter VT, Chaparro A, Principe MF, Solis L, Kannan TR, Baseman
JB and Dube PH: Mycoplasma pneumoniae CARDS toxin exacerbates
ovalbumin-induced asthma-like inflammation in BALB/c mice. PLoS
One. 9:e1026132014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Johnson C, Kannan TR and Baseman JB:
Cellular vacuoles induced by Mycoplasma pneumoniae CARDS toxin
originate from Rab9-associated compartments. PLoS One.
6:e228772011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim EK, Youn YS, Rhim JW, Shin MS, Kang JH
and Lee KY: Epidemiological comparison of three Mycoplasma
pneumoniaepneumonia epidemics in a single hospital over 10 years.
Korean J Pediatr. 58:172–177. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lynch M, Taylor TK, Duignan PJ, Swingler
J, Marenda M, Arnould JP and Kirkwood R: Mycoplasmas in Australian
fur seals (Arctocephalus pusillus doriferus): Identification and
association with abortion. J Vet Diagn Invest. 23:1123–1130. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lai JF, Zindl CL, Duffy LB, Atkinson TP,
Jung YW, van Rooijen N, Waites KB, Krause DC and Chaplin DD:
Critical role of macrophages and their activation via MyD88-NFκB
signaling in lung innate immunity to Mycoplasma pneumoniae. PLoS
One. 5:e144172010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Halbedel S, Hames C and Stülke J: In vivo
activity of enzymatic and regulatory components of the
phosphoenolpyruvate: Sugar phosphotransferase system in Mycoplasma
pneumoniae. J Bacteriol. 186:7936–7943. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Beersma MF, Dirven K, van Dam AP,
Templeton KE, Claas EC and Goossens H: Evaluation of 12 commercial
tests and the complement fixation test for Mycoplasma
pneumoniae-specific immunoglobulin G (IgG) and IgM antibodies, with
PCR used as the ‘gold standard’. J Clin Microbiol. 43:2277–2285.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Loos M and Brunner H: Complement
components (C1, C2, C3, C4) in bronchial secretions after
intranasal infection of guinea pigs with Mycoplasma pneumoniae:
Dissociation of unspecific and specific defense mechanisms. Infect
Immun. 25:583–585. 1979.PubMed/NCBI
|
|
47
|
Thacker WL and Talkington DF: Analysis of
complement fixation and commercial enzyme immunoassays for
detection of antibodies to Mycoplasma pneumoniae in human serum.
Clin Diagn Lab Immunol. 7:778–780. 2000.PubMed/NCBI
|
|
48
|
Tuuminen T and Vainionpää R: Development
of enzyme immunoassays to detect salivary sIgA to Chlamydia
pneumoniae and Mycoplasma pneumoniae. Scand J Clin Lab Invest.
61:357–362. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Daxböck F, Brunner G, Popper H, Krause R,
Schmid K, Krejs GJ and Wenisch C: A case of lung transplantation
following Mycoplasma pneumoniae infection. Eur J Clin Microbiol
Infect Dis. 21:318–322. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Csángó PA, Pedersen JE and Hess RD:
Comparison of four Mycoplasma pneumoniae IgM-, IgG- and
IgA-specific enzyme immunoassays in blood donors and patients. Clin
Microbiol Infect. 10:1094–1098. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Venancio P, Brito MJ, Pereira G and Vieira
JP: Anti-N-methyl-D-aspartate receptor encephalitis with positive
serum antithyroid antibodies, IgM antibodies against Mycoplasma
pneumoniae and human herpesvirus 7 PCR in the CSF. Pediatr Infect
Dis J. 33:882–883. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Smith-Norowitz TA, Silverberg JI,
Kusonruksa M, Weaver D, Ginsburg D, Norowitz KB, Durkin HG,
Hammerschlag MR, Bluth MH and Kohlhoff SA: Asthmatic children have
increased specific anti-Mycoplasma pneumoniae IgM but not IgG or
IgE-values independent of history of respiratory tract infection.
Pediatr Infect Dis J. 32:599–603. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ye Q, Xu XJ, Shao WX, Pan YX and Chen XJ:
Mycoplasma pneumoniae infection in children is a risk factor for
developing allergic diseases. ScientificWorldJournal.
2014:9865272014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Xin LH, Wang J, Wang Z, Cheng W and Zhang
W: Effect of Mycoplasma pneumoniae infection on function of T
lymphocytes in bronchoalveolar lavage fluid of asthmatic children.
Zhongguo Dang Dai Er Ke Za Zhi. 16:277–280. 2014.(In Chinese).
PubMed/NCBI
|
|
55
|
Kang YM, Ding MJ, Han YL, Wang SF, Ma X
and Li H: Th1/Th2 immune response in bronchoalveolar lavage fluid
in children with severe Mycoplasma pneumoniae pneumonia. Zhongguo
Dang Dai Er Ke Za Zhi. 13:188–190. 2011.(In Chinese). PubMed/NCBI
|
|
56
|
Pang HX, Qiao HM, Cheng HJ, Zhang YF, Liu
XJ and Li JZ: Levels of TNF-alpha, IL-6 and IL-10 in
bronchoalveolar lavage fluid in children with Mycoplasma pneumoniae
pneumonia. Zhongguo Dang Dai Er Ke Za Zhi. 13:808–810. 2011.(In
Chinese). PubMed/NCBI
|
|
57
|
Yang J, Hooper WC, Phillips DJ and
Talkington DF: Regulation of proinflammatory cytokines in human
lung epithelial cells infected with Mycoplasma pneumoniae. Infect
Immun. 70:3649–3655. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Lee KE, Kim KW, Hong JY, Kim KE and Sohn
MH: Modulation of IL-8 boosted by Mycoplasma pneumoniae lysate in
human airway epithelial cells. J Clin Immunol. 33:1117–1125. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
He JE, Gao CY and Li HR: Effect of
low-dose methylprednisolone on serum TNF-α level in children with
Mycoplasma pneumoniae pneumonia. Zhongguo Dang Dai Er Ke Za Zhi.
15:850–853. 2013.(In Chinese). PubMed/NCBI
|
|
60
|
Tanaka H, Narita M, Teramoto S, Saikai T,
Oashi K, Igarashi T and Abe S: Role of interleukin-18 and T-helper
type 1 cytokines in the development of Mycoplasma pneumoniae
pneumonia in adults. Chest. 121:1493–1497. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Krunkosky TM, Jordan JL, Chambers E and
Krause DC: Mycoplasma pneumoniae host-pathogen studies in an
air-liquid culture of differentiated human airway epithelial cells.
Microb Pathog. 42:98–103. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Kurata S, Osaki T, Yonezawa H, Arae K,
Taguchi H and Kamiya S: Role of IL-17A and IL-10 in the antigen
induced inflammation model by Mycoplasma pneumoniae. BMC Microbiol.
14:1562014. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Teig N, Anders A, Schmidt C, Rieger C and
Gatermann S: Chlamydophila pneumoniae and Mycoplasma pneumoniae in
respiratory specimens of children with chronic lung diseases.
Thorax. 60:962–966. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Fink FM, Dengg K, Kilga-Nogler S,
Schönitzer D and Berger H: Cold haemagglutinin disease complicating
Mycoplasma pneumoniae infection in a child under cytotoxic cancer
treatment. Eur J Pediatr. 151:435–437. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Busolo F, Tonellato L, Scremin L, Tonin E,
Bertoloni G and Franceschi C: Phagocytosis of Mycoplasma pneumoniae
and Acholeplasma laidlawii measured as inhibition of [3H] uridine
uptake by macrophages. J Immunol Methods. 90:235–240. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kornspan JD, Tarshis M and Rottem S:
Adhesion and biofilm formation of Mycoplasma pneumoniae on an
abiotic surface. Arch Microbiol. 193:833–836. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Hon KL, Ip M, Chu WC and Wong W:
Megapneumonia coinfection: Pneumococcus, Mycoplasma pneumoniae, and
Metapneumovirus. Case Rep Med. 2012:3101042012.PubMed/NCBI
|
|
68
|
Hausner M, Schamberger A, Naumann W,
Jacobs E and Dumke R: Development of protective anti-Mycoplasma
pneumoniae antibodies after immunization of guinea pigs with the
combination of a P1-P30 chimeric recombinant protein and chitosan.
Microb Pathog. 64:23–32. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Shimizu T, Kida Y and Kuwano K: A
dipalmitoylated lipoprotein from Mycoplasma pneumoniae activates
NF-kappa B through TLR1, TLR2, and TLR6. J Immunol. 175:4641–4646.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Into T, Dohkan J, Inomata M, Nakashima M,
Shibata K and Matsushita K: Synthesis and characterization of a
dipalmitoylated lipopeptide derived from paralogous lipoproteins of
Mycoplasma pneumoniae. Infect Immun. 75:2253–2259. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Shimizu T, Kida Y and Kuwano K:
Triacylated lipoproteins derived from Mycoplasma pneumoniae
activate nuclear factor-kappaB through toll-like receptors 1 and 2.
Immunology. 121:473–483. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Okoli K, Gupta A, Irani F and Kasmani R:
Immune thrombocytopenia associated with Mycoplasma pneumoniae
infection: A case report and review of literature. Blood Coagul
Fibrinolysis. 20:595–598. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Rastawicki W, Rokosz N and Jagielski M:
Subclass distribution of human IgG antibodies to Mycoplasma
pneumoniae in the course of mycoplasmosis. Med Dosw Mikrobiol.
61:375–379. 2009.(In Polish). PubMed/NCBI
|