Introduction
Atherosclerosis (AS), which is a common disorder
with increasing prevalence worldwide, results in the progressive
decline in the function of multiple organs, with the most serious
effect being an increased risk of cardiovascular events and
mortality (1). However, the
mechanisms underlying development of AS remain unclear. Endothelial
dysfunction (ED) resulting from inflammation is crucial throughout
the development of AS (2,3). Previous studies using experimental
models of AS and diabetes revealed that the accumulation of
cholesterol-rich lipoproteins in the artery wall results in ED
(4–7). ED is predominantly mediated by
endothelin-1 (ET-1) (4–7), which is a potent endothelial injury
factor secreted by human umbilical vein endothelial cells
(HUVECs).
The renin-angiotensin system (RAS) is important for
vascular homeostasis. Activation of the RAS product, angiotensin II
(AngII) is associated with arterial wall remodeling (8), and AngII is one of the central
vasoactive signaling molecules in ED progression (9). AngII is involved in various
pathological conditions, including hypertension, diabetes and AS
(10–12). In addition, high concentrations of
AngII result in necrosis and apoptosis of HUVECs (13).
Protein translation, folding and trafficking occur
in the endoplasmic reticulum (ER), and this organelle responds
early to cellular stress (14). ER
stress is caused by numerous factors, including oxidative stress,
hypoxia and exposure to chemicals, including AngII (15). Numerous studies have revealed that
AS, diabetes mellitus, inflammatory conditions and cardiovascular
disorders are associated with ER stress induction (16,17).
The signal transduction pathway associated with
AngII-induced inflammation in endothelial cells has been
investigated. Guo et al (18) demonstrated that the nuclear factor
κB (NF-κB) signaling pathway is involved in AngII-induced HUVEC
dysfunction. Therefore, molecules that inhibit activation of the
NF-κB signaling pathway may be beneficial for the treatment of
AngII-induced ED.
Hydrogen sulfide (H2S) was originally
identified as a gasotransmitter in the cardiovascular system
(19); however, several biological
functions of H2S have now been demonstrated, including a
potential role in endothelial cell protection (19). H2S is primarily produced
by three constitutively expressed enzymes: Cystathionine-γ-lyase
(CSE), cystathionine β-synthase and 3-mercaptopyruvate
sulphurtransferase (20).
Endogenous H2S has important regulatory roles in
cardiovascular function. CSE-knockout mice have markedly reduced
H2S production in the aorta, heart and serum, and
develop pronounced hypertension (21). Evidence suggests that
H2S is an important regulator of inflammation, and may
inhibit H2O2-induced production of inflammatory mediators by
endothelial cells (22), including
interleukin-6 and tumor necrosis factor-α (23,24).
Certain studies have demonstrated that H2S exerts
antiatherogenic (25) and
antioxidant (26) effects.
However, the mechanisms underlying H2S regulation of
inflammatory molecule production remain unclear.
In the present study, HUVECs were treated with AngII
to establish an in vitro cytotoxicity model. The aim of the
present study was to investigate the following: i) Whether ER
stress, NF-κB and ET-1 were involved in AngII-induced cytotoxicity;
ii) whether inhibition of the CSE/H2S pathway was
associated with AngII-induced cytotoxicity; and, if so, iii)
whether supplementation of exogenous H2S suppressed
AngII-induced cytotoxicity via inhibition of ER stress, inducible
nitric oxide synthase (iNOS)/nitric oxide (NO) and ET-1; and iv)
whether supplementation of exogenous H2S inhibited
AngII-induced ER stress via suppression of the ET-1/NF-κB signaling
pathway.
Materials and methods
Materials
Dulbecco's modified Eagle's medium (DMEM) and fetal
bovine serum (FBS) were obtained from Gibco; Thermo Fisher
Scientific, Inc. (Waltham, MA, USA). Sodium hydrosulfide (NaHS),
AngII, pyrrolidinedithiocarbamic acid (PDTC), BQ788, Nitrite
Detection kit, Hoechst 33258 and terminal deoxynucleotidyl
transferase mediated dUTP nick end labeling (TUNEL) In Situ
Cell Death Detection kit were purchased from Sigma-Aldrich (St.
Louis, MO, USA). Antibodies against ET-1 (cat. no. ab2786), iNOS
(cat. no. ab3523), total p65 (cat. no. ab16502), phosphorylated
(p)-p65 (cat. no. ab86299), caspase-12 (cat. no. ab62484),
glucose-regulated protein 78 (GRP78; cat. no. ab21685) and
CCAAT-enhancer-binding protein homologous protein (CHOP; cat. no.
ab11419) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; cat.
no. ab8245) were provided by Abcam (Cambridge, UK). The anti-CSE
antibody was provided by ProteinTech Group, Inc. (Chicago, IL, USA;
cat. no. 12,032-1-AP). Horseradish peroxidase-conjugated IgG (H+L)
secondary antibodies were purchased from the Beyotime Institute of
Biotechnology (Shanghai, China; cat. nos. A0192 and A0208). Cell
Counting kit-8 (CCK-8) was purchased from Dojindo Molecular
Technologies, Inc. (Shanghai, China). All other reagents, unless
specified, were purchased from Beyotime Institute of
Biotechnology.
Cell culture and treatments
HUVECs were obtained from The Cell Bank of Type
Culture Collection of Chinese Academy of Sciences (Shanghai,
China), and cultured in DMEM supplemented with 10% FBS at 37°C in a
humidified atmosphere containing 5% CO2. Prior to every experiment,
the medium was replaced with fresh serum-containing medium, unless
indicated.
Cells were divided into the following treatment
groups: Untreated; AngII, consisting of 1×106 HUVEC cells treated
with 10–6 M AngII for 0–24 h; PDTC+AngII, consisting of 1×106 cells
exposed to 100 µM PDTC for 60 min prior to treatment with 10-6 M
AngII for 24 h; NAHS+AngII, consisting of 1×106 cells exposed to
200–400 µmol/l NAHS for 60 min prior to treatment with 10–6 M AngII
for 24 h; AngII+BQ788, consisting of 1×106 cells exposed to 1 nM or
1 µM BQ788 for 24 h and 60 min, respectively, prior to treatment
with 10-6 M AngII for 24 h; and AngII+ET-1, consisting of 1×106
cells exposed to 1 nM ET-1 for 0–24 h prior to treatment with 10-6
M AngII for 24 h.
Cell viability assay
HUVECs cells were seeded in 96-well plates at a
density of 1×106 cells/well prior to treatment with various agents.
Following this, a solution of 100 µl CCK-8 was added into each well
at a 1:10 dilution and the treated plates were incubated for a
further 2 h at 37°C. Absorbance was measured at 450 nm with a
microplate reader. The mean optical density (OD) of four wells for
each group was used to calculate the percentage cell viability
according to the following formula: Percentage cell viability = (OD
treatment group / OD control group) × 100.
NO determination in culture
supernatants
Nitrite, a marker for the production of NO, was
measured in culture supernatants using a Nitrite Detection kit.
Briefly, 50 µl aliquots of cell culture medium from each dish were
collected and mixed with 100 µl Griess reagent (50 µl 1%
sulfanilamide 50 µl 0.1% naphthylethylenediamine dihydrochloride in
2.5% H3PD4) in 96-well microtiter plates. The absorbance of NO2 was
measured at 520 nm with a microplate reader.
Measurement of CSE activity
Following treatment, the HUVECs were collected and
homogenized in 50 mM ice-cold potassium phosphate buffer (pH 6.8).
The reaction mixture [100 mmol/l potassium phosphate buffer (pH
7.4), 10 mmol/l L-cysteine, 2 mmol/l pyridoxal 5′-phosphate, and
10% (w/v) homogenate] was added to Erlenmeyer flasks, along with
the center wells (2 ml cryovial test tubes containing 0.5 ml 1%
zinc acetate as the trapping solution and a 2×2.5 cm filter paper
to increase the air/liquid contact surface). The flasks were then
flushed with N2 and sealed with a double layer of parafilm. The
reactions were initiated by transferring the flasks from ice to a
37°C shaking water bath. After 90 min, 0.5 ml 50% trichloroacetic
acid was added to terminate the reaction. The flasks were resealed
and incubated at 37°C for a further 60 min to ensure the complete
trapping of H2S released from the reaction mixture.
Following this, the contents of the center wells were transferred
to test tubes containing 3.5 ml water. Subsequently, 0.5 ml 20 mM
N, N-dimethyl-p-phenylenediamine sulfate in 7.2 M HCl
was added, immediately followed by 0.5 ml 30 mM FeCl3 in 1.2 M HCl.
The absorbance of the resulting solution was measured at 670 nm 20
min later using a spectrophotometer.
Western blot analysis
Following treatment, the cells were washed three
times with phosphate-buffered saline (PBS) and lysed in lysis
buffer consisting of 1 ml radioimmunopreciptation assay lysis
buffer, 25 µg/l phenylmethylsulfonyl fluoride and 110 mU PhosSTOP
phosphatase inhibitor (Sigma-Aldrich) on ice for 30 min. The
resulting cell lysates were centrifuged at 10,943 × g for 15
min at 4°C. Proteins were quantified using the bicinchoninic acid
method according to the manufacturer's protocol (Beyotime Institute
of Biotechnology).
Total proteins (300 ng) were separated by 10% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis and were
transferred to polyvinylidene difluoride membranes via
electroblotting. The membranes were blocked with 5% (w/v) non-fat
milk powder and 0.1% (v/v) Tween 20 for 1 h, and were then
incubated with ET-1 (1:200), CSE (1:1,000), p-p65 (1:2,000), iNOS
(1:2,000), CHOP (1:2,000), caspase-12 (1:2,000), GRP78 (1:800),
GAPDH (1:1,000) and total p65 (1:1,000) primary antibodies
overnight at 4°C. Following three washes in Tris-buffered saline
with 0.1% Tween 20, the membranes were incubated with the
corresponding secondary antibodies (dilution, 1:1,000) for 1 h at
room temperature. Membranes were visualized using an enhanced
chemiluminescence kit according to the manufacturer's instructions
(Beyotime Institute of Biotechnology). Relative protein expression
levels were semi-quantitatively analyzed by densitometry using
Quantity One software (version, 4.62; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
Hoechst 33258 and TUNEL staining to
assess HUVEC apoptosis
HUVEC apoptosis following treatment was detected by
TUNEL and Hoechst 33258 staining. TUNEL staining was performed
using an In Situ Cell Death Detection kit according to the
manufacturer's protocol. Cells (1×106) in culture plates were fixed
with 4% paraformaldehyde in PBS for 10 min. Following three washes
in PBS, cells were stained with 50 µl TUNEL dye for 1 h, rinsed
briefly with PBS and air-dried. Cells were visualized under a
florescence microscope. Apoptotic cells with condensed nuclei
fluoresced green fluorescence, whereas viable cells exhibited
normal nuclear size and were not florescent. Quantitative analysis
of the mean fluorescence intensity of each group was performed
using ImageJ software (version, 1.41o; National Institutes of
Health, Bethesda, MD, USA).
In addition, Hoechst 33258 staining was performed
using an In Situ Cell Death Detection kit according to the
manufacturer's protocol. Cells were fixed with 4% paraformaldehyde
in PBS for 10 min. Following three washes in PBS, cells were
stained with 5 mg/l Hoechst 33258 dye for 10 min, rinsed briefly
with PBS and air-dried. Cells were visualized under a florescence
microscope. Apoptotic cells with condensed nuclei fluoresced blue,
whereas viable cells exhibited normal nuclear size and weak
florescence. Quantitative analysis of the mean fluorescence
intensity of each group was performed as for TUNEL staining.
Statistical analysis
All data are presented as the mean ± standard error
of the mean. Differences between groups were analyzed using one-way
analysis of variance followed by the Bonferroni correction method,
which was performed using the SPSS software program (version, 15.0;
SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to indicate
a statistically significant difference.
Results
AngII induces cytotoxicity via
triggering ER stress and inducing the expression of NF-κB and ET-1
in HUVECs
Since iNOS, NO, ET-1 and CSE have been demonstrated
to be involved in AngII-induced cytotoxicity (27–29)
and increased levels of p-p65 (30), western blotting was performed to
analyze the expression levels of these proteins in AngII-treated
HUVECs. As presented in Fig. 1,
untreated HUVECs produced low levels of ET-1 (Fig. 1A), iNOS (Fig. 1B), p-p65 (Fig. 1C), CHOP (Fig. 1D) and GRP78 (Fig. 1E), which increased significantly
following AngII treatment in a time-dependent manner. The
expression levels (Fig. 1F) and
activity (Fig. 1G) of CSE
decreased significantly in a time-dependent manner following AngII
treatment, whereas nitrite production (Fig. 1H) was significantly increased.
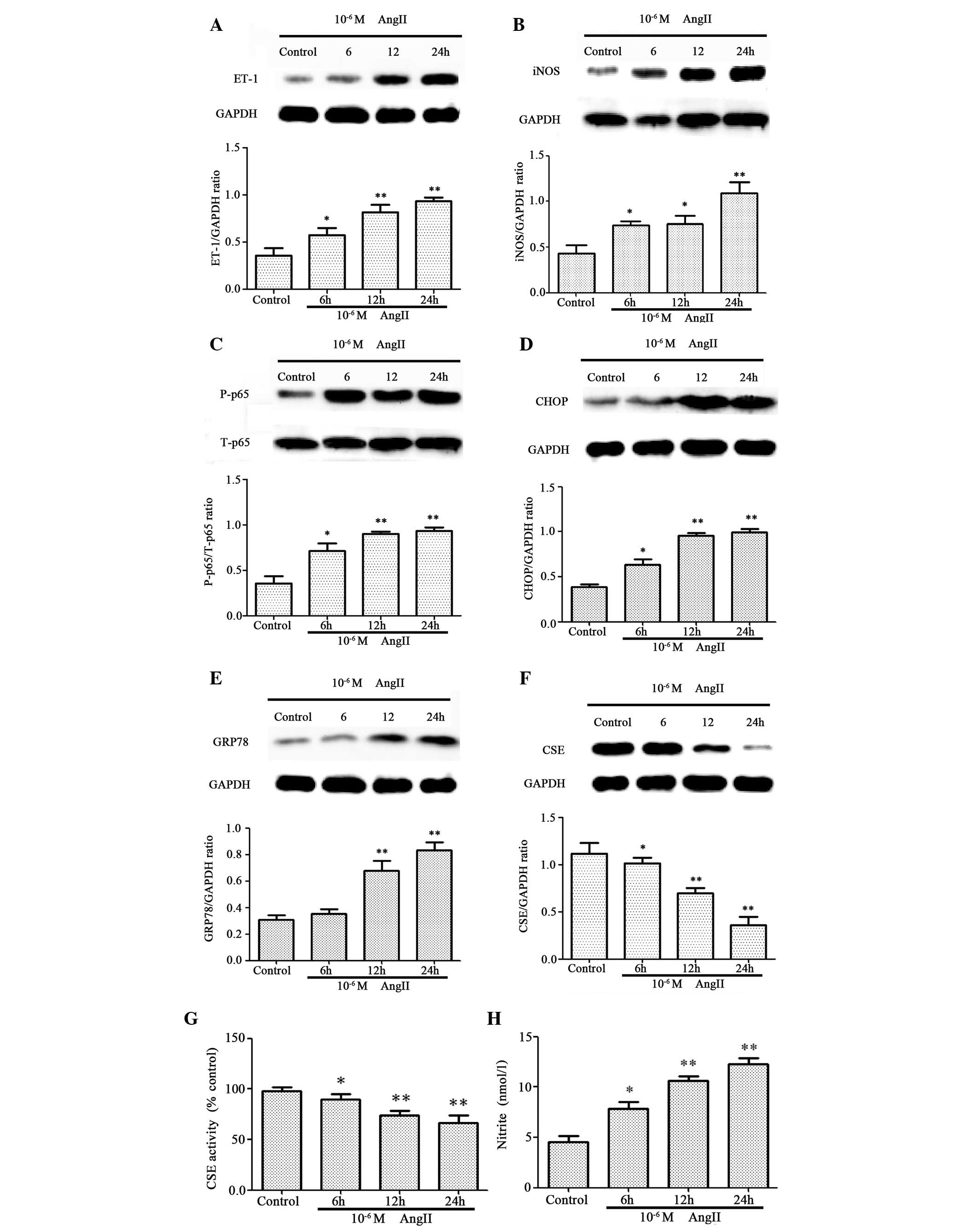 | Figure 1.AngII induces cytotoxicity by
triggering endoplasmic reticulum stress and inducing the expression
of nuclear factor-κB and ET-1 in HUVECs. HUVECs were exposed to
10-6 M AngII for 6, 12 or 24 h. Protein expression levels of (A)
ET-1, (B) iNOS, (C) p-p65, (D) CHOP, (E) GRP78 and (F) CSE were
assessed by western blot analysis. (G) CSE activity was assessed by
methylene blue spectrophotometry. (H) Nitrite, a marker of nitric
oxide production, was detected in culture supernatants using a
Nitrite Detection kit. Data are presented as the mean ± standard
error of the mean (n=3). *P<0.05 and **P<0.01 vs. control
group. AngII, angiotensin II; ET-1, endothelin-1; HUVECs, human
umbilical vein endothelial cells; iNOS, inducible nitric oxide
synthase; p-p65, phosphorylated p65; T-p65, total p65; CHOP,
CCAAT-enhancer-binding protein homologous protein; GRP78,
glucose-regulated protein 78; CSE, cystathionine-γ-lyase; GAPDH,
glyceraldehyde 3-phosphate dehydrogenase. |
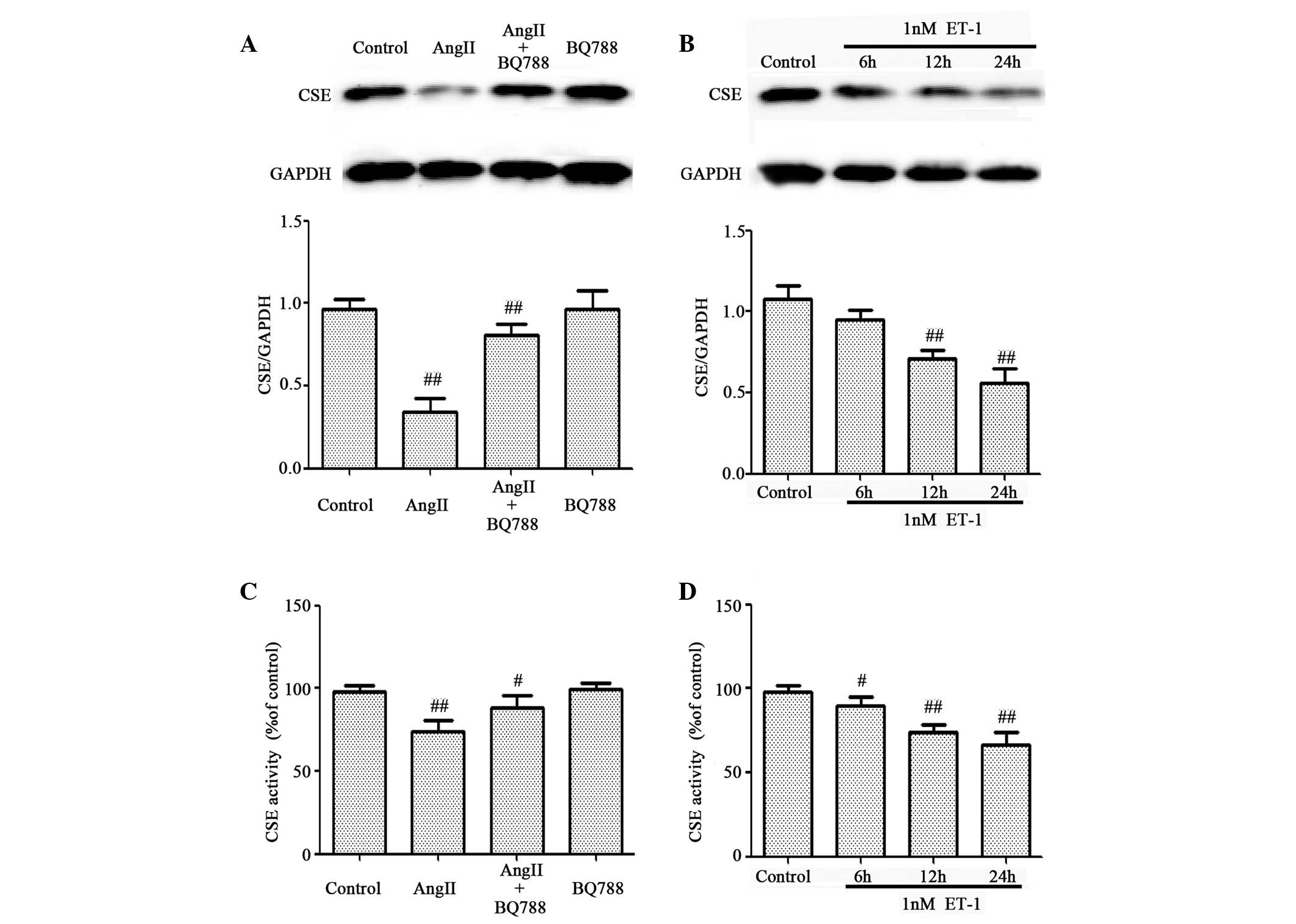
ET-1 mediates AngII-induced inhibitory
effects on the expression level and activity of CSE in HUVECs
AngII affects endothelial cells primarily via the
activation of genes associated with inflammation, such as ET-1
(31). The present study
investigated whether ET-1 activity contributed to the inhibition of
CSE expression and activity mediated by AngII (Fig. 2). HUVECs were preconditioned with a
well-known ET-1 receptor antagonist, BQ788, prior to AngII
treatment. Pretreatment of cells with 1 µM BQ788 significantly
attenuated AngII-induced downregulation of CSE expression (Fig. 2A) and activity (Fig. 2C). In addition, similar to the
AngII results, treatment of HUVECs with ET-1 significantly
suppressed CSE expression (Fig.
2B) and activity (Fig. 2D) in
a time-dependent manner. These data suggest that ET-1 contributes
to AngII-induced downregulation of the expression and activity of
CSE in HUVECs.
Exogenous treatment with
H2S and ET-1 inhibitor reduces cytotoxicity via
suppressing ER stress, iNOS/NO and ET-1 in AngII-treated
HUVECs
Since AngII-induced cytotoxicity results in a
decrease in CSE expression and activity, and therefore a decrease
in endogenous H2S production, the protective effects of
exogenous H2S on AngII-induced inflammatory responses
were investigated. As presented in Fig. 3, pretreatment of HUVECs with NaHS
for 1 h prior to AngII exposure significantly attenuated the
increased levels of ET-1 (Fig.
3A), iNOS (Fig. 3B), GRP78
(Fig. 3C), CHOP (Fig. 3D) and nitrite (Fig. 3E) induced by 10-6 M AngII treatment
for 24 h, in a dose-dependent manner. The decrease in cell
viability induced by 10-6 M AngII treatment for 24 h was
significantly abrogated, in a dose-dependent manner, by
pretreatment with NaHS for 1 h prior to AngII exposure (Fig. 3F).
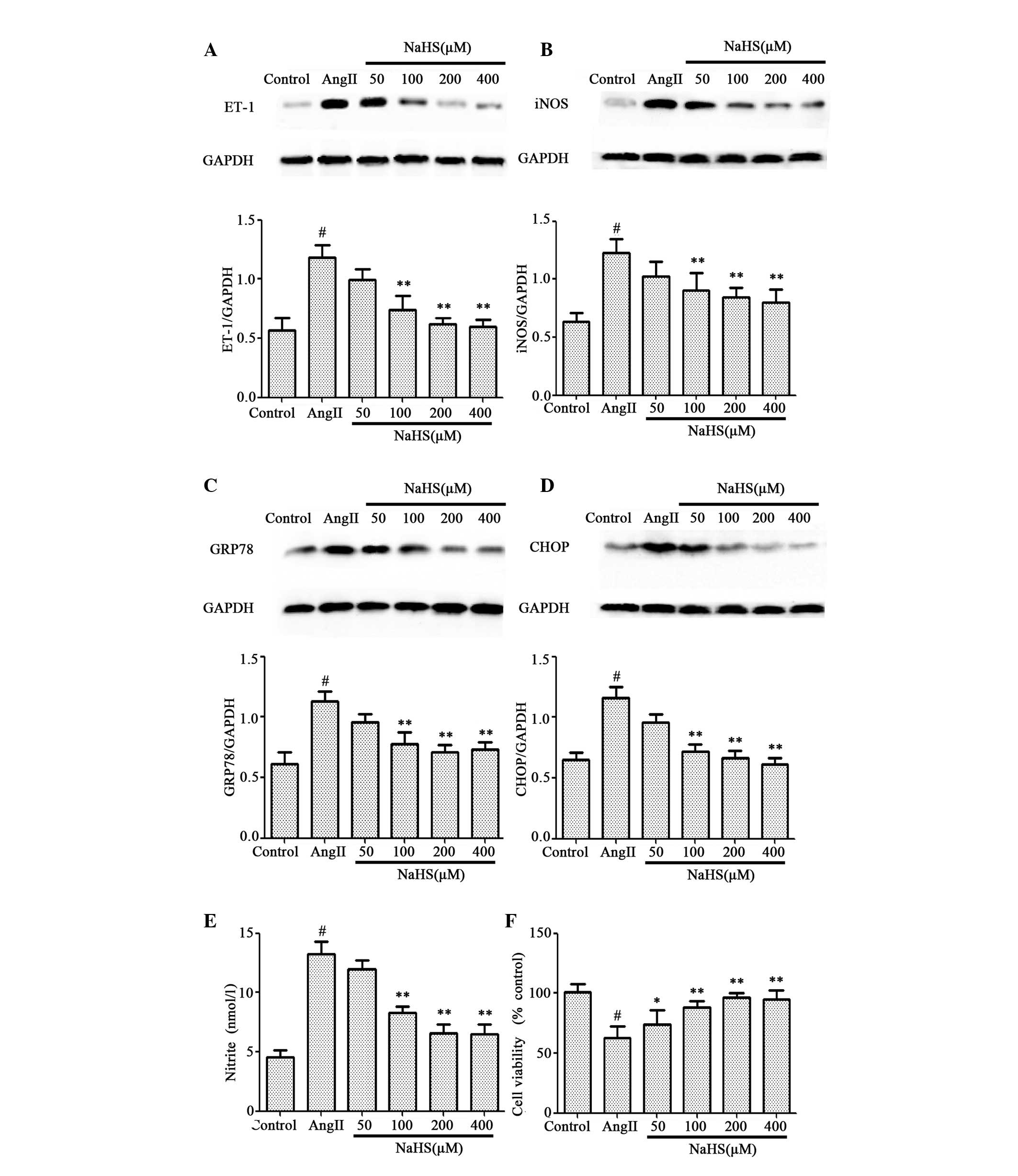 | Figure 3.Effects of NaHS pretreatment on
AngII-treated HUVECs. HUVECs were treated with 10-6 M AngII for 24
h following pretreatment with NaHS at concentrations from 50 to 400
µM for 1 h. Protein expression levels of (A) ET-1, (B) iNOS, (C)
GRP78 and (D) CHOP were assessed by western blot analysis. (E)
Nitrite was detected in the culture supernatant using a Nitrite
Detection kit. (F) Cell viability was measured using the Cell
Counting kit-8 assay. Data are presented as the mean ± standard
error of the mean (n=3). *P<0.05 and **P<0.01 vs. AngII
group; #P<0.01 vs. control group. NaHS, sodium hydrosulfide;
AngII, angiotensin II; HUVECs, human umbilical vein endothelial
cells; ET-1, endothelin-1; iNOS, inducible nitric oxide synthase;
GRP78, glucose-regulated protein 78; CHOP, CCAAT-enhancer-binding
protein homologous protein; GAPDH, glyceraldehyde 3-phosphate
dehydrogenase. |
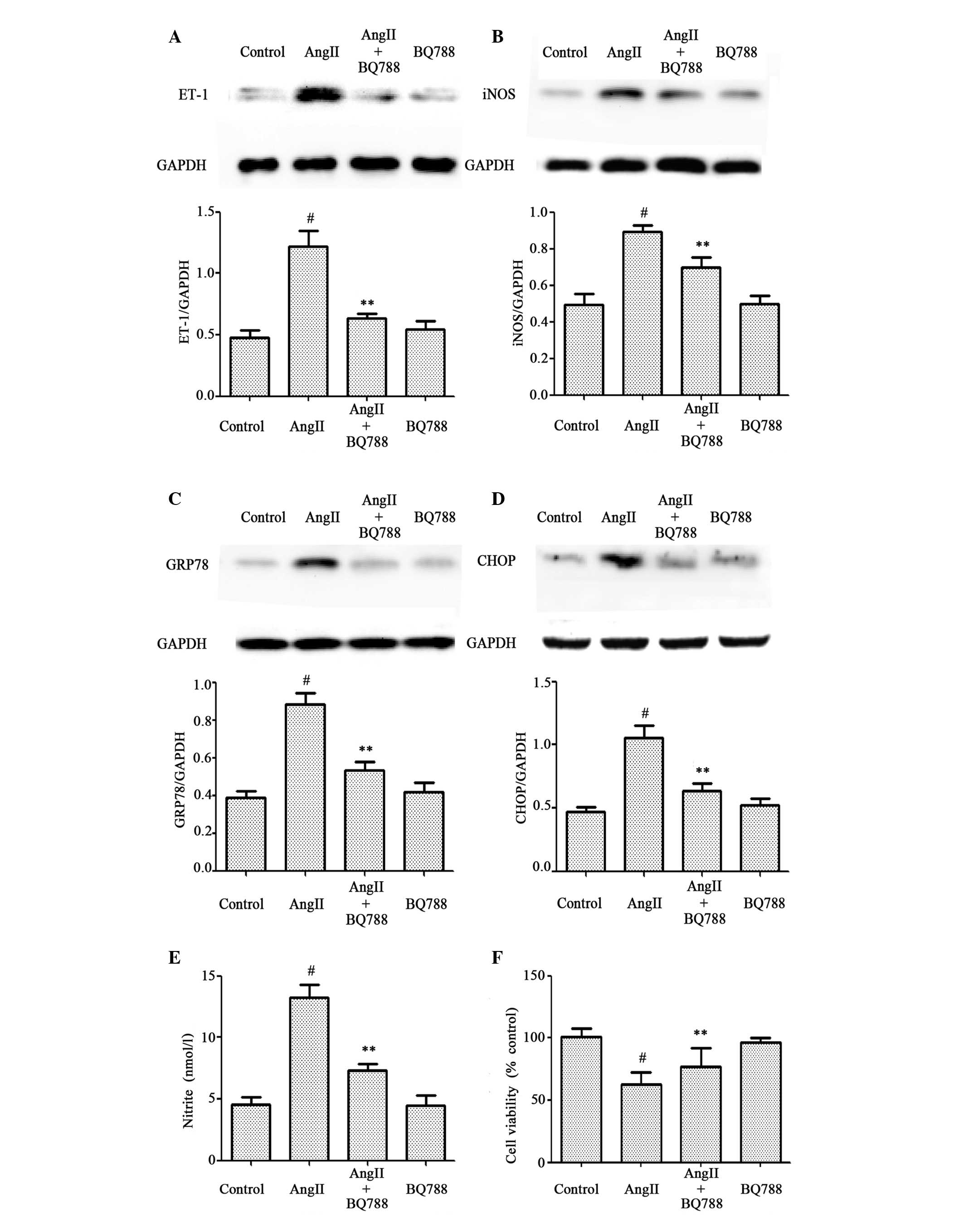
Similarly, as presented in Fig. 4, pretreatment of cells with 1 µM
BQ788 for 1 h prior to 10-6 M AngII exposure abrogated the
increased production of ET-1 (Fig.
4A), iNOS (Fig. 4B), GRP78
(Fig. 4C), CHOP (Fig. 4D) and nitrite (Fig. 4E), and significantly attenuated the
decreased cell viability induced by 10-6 M AngII treatment for 24 h
(Fig. 4F). BQ788 treatment alone
did not affect the basal levels of ET-1, iNOS, GRP78, CHOP or
nitrite in HUVECs. These findings suggest that pretreatment with an
ET-1 inhibitor had a similar cytoprotective effect to the
H2S donor NaHS on AngII-induced cytotoxicity. Therefore,
the activation of ET-1, GRP78, CHOP, iNOS, enhancement of nitrite
production and decrease in cell viability may be involved in
AngII-induced cytotoxicity in HUVECs.
Exogenous H2S and an NF-κB
inhibitor attenuate AngII-induced cytotoxicity in HUVECs
As presented in Fig.
1, exposure of cells to 10-6 M AngII for 24 h markedly enhanced
the expression levels of p-p65. As presented in Fig. 3, pretreatment of cells with NaHS
for 1 h prior to AngII exposure significantly abrogated
AngII-induced cytotoxicity. Therefore, the potential involvement of
the p-p65 signaling pathway in AngII-induced cytotoxicity was
examined. HUVECs were pretreated with 100 µM PDTC for 1 h prior to
10-6 M AngII exposure for 24 h. As presented in Fig. 5, pretreatment with PDTC had a
similar cytoprotective effect to NaHS on AngII-induced
overexpression of GRP78 (Fig. 5A),
CHOP (Fig. 5B), iNOS (Fig. 5C) and nitrite (Fig. 5D), and AngII-induced decrease in
cell viability (Fig. 5E),
suggesting involvement of p-p65 activation in AngII-induced
cytotoxicity in HUVECs. NaHS or PDTC treatment alone had no
effect.
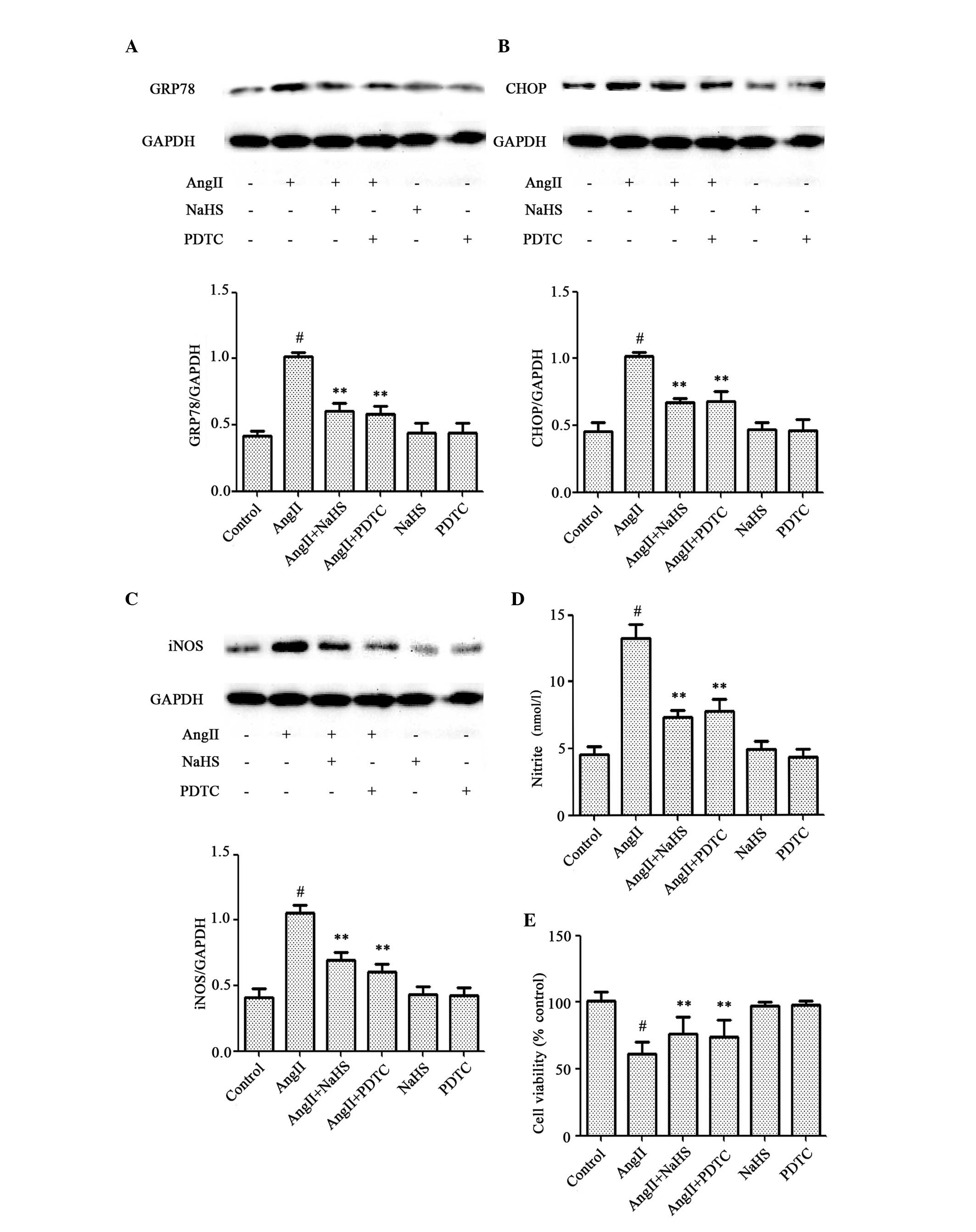 | Figure 5.Exogenous H2S and a
nuclear factor-κB inhibitor attenuate AngII-induced cytotoxicity in
HUVECs. HUVECs were treated with 10-6 M AngII for 24 h following
pretreatment with 200 µM NaHS or 100 µM PDTC for 1 h prior to AngII
exposure. Protein expression levels of (A) GRP78, (B) CHOP and (C)
iNOS were assessed by western blot analysis. (D) Nitrite was
detected in the culture supernatant using a Nitrite Detection kit.
(E) Cell viability was measured using the Cell Counting kit-8
assay. Data are presented as the mean ± standard error of the mean
(n=3). **P<0.01 vs. the AngII-treated group; #P<0.01 vs. the
control group. H2S, hydrogen sulfide; AngII, angiotensin
II; HUVECs, human umbilical vein endothelial cells; NaHS, sodium
hydrosulfide; PDTC, pyrrolidinedithiocarbamic acid; GRP78,
glucose-regulated protein 78; CHOP, CCAAT-enhancer-binding protein
homologous protein; iNOS, inducible nitric oxide synthase; GAPDH,
glyceraldehyde 3-phosphate dehydrogenase. |
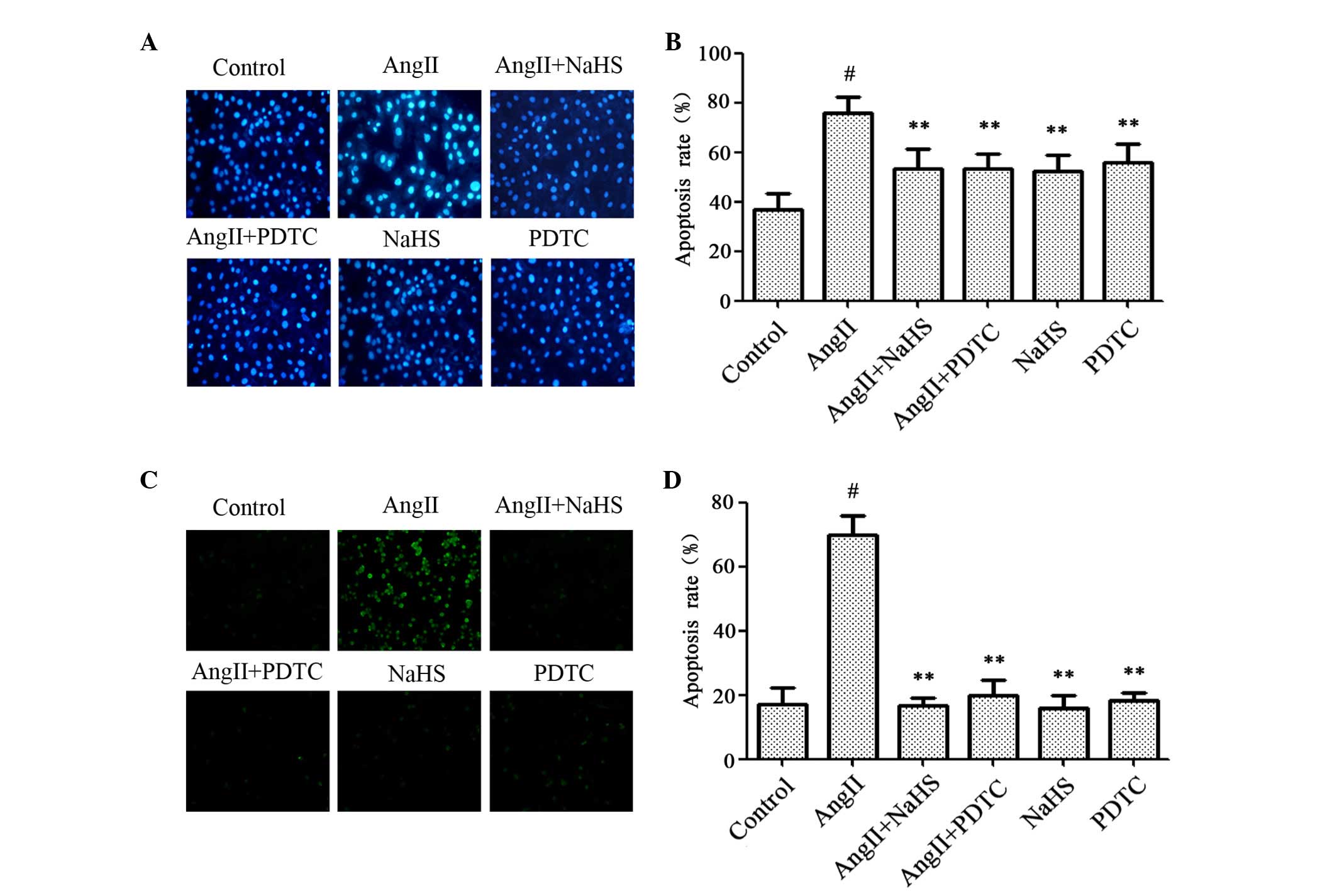
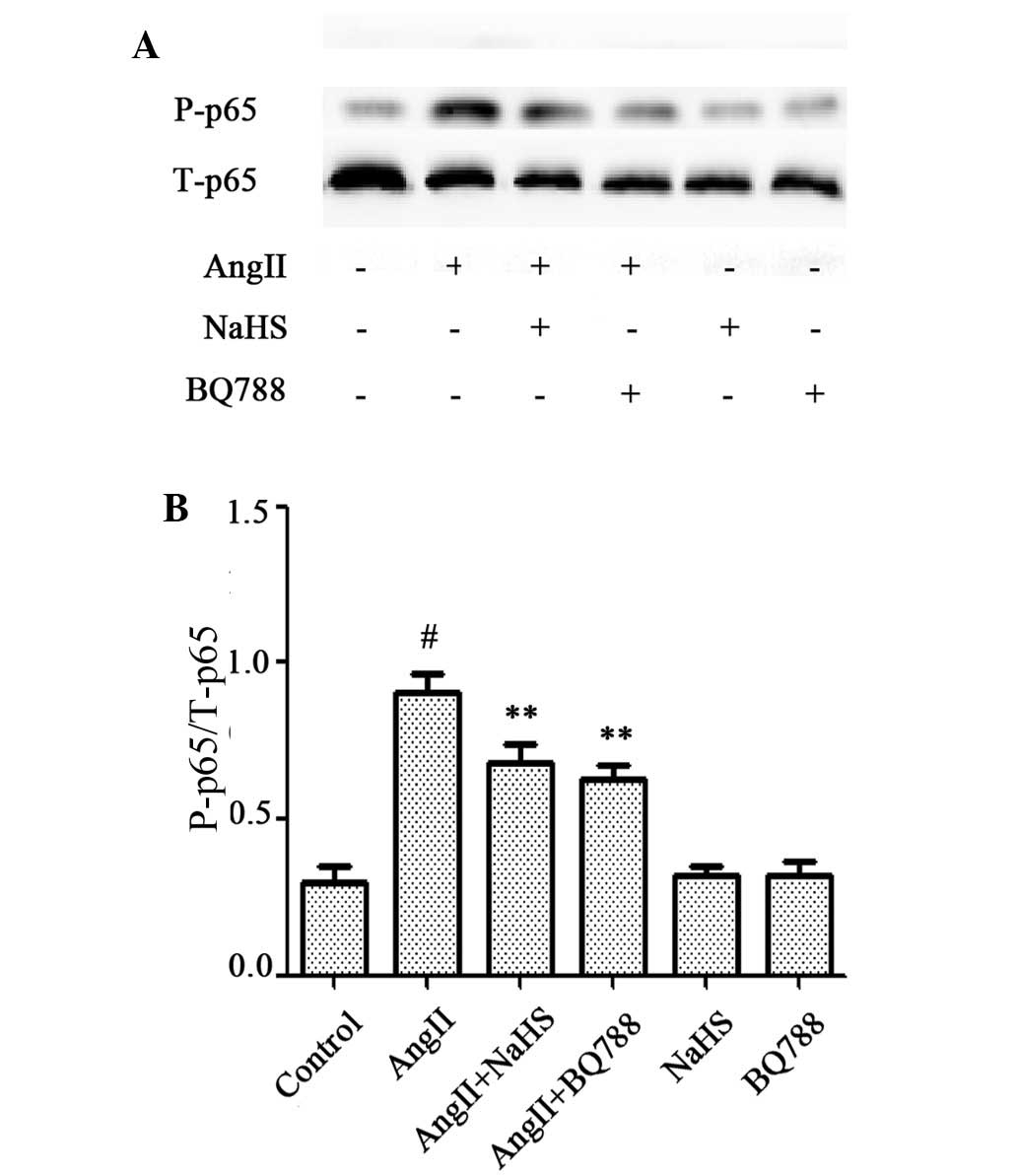
Exogenous H-2S and NF-κB inhibitor
reduce AngII-induced apoptosis in HUVECs
The effects of NaHS and PDTC on AngII-induced
apoptosis were investigated. As presented in Fig. 6, HUVECs treated with 10-6 M AngII
for 24 h exhibited typical apoptotic characteristics, including
chromatin condensation, shrinkage of nuclei, and the presence of
apoptotic bodies, and increased Hoechst (Fig. 6A and B) and TUNEL (Fig. 6C and D) staining. However,
pretreatment of cells with 200 µM NaHS or 100 µM PDTC for 1 h prior
to AngII exposure markedly decreased the number of cells exhibiting
nuclear condensation and fragmentation. NaHS or PDTC alone did not
markedly alter cell morphology or the percentage of apoptotic
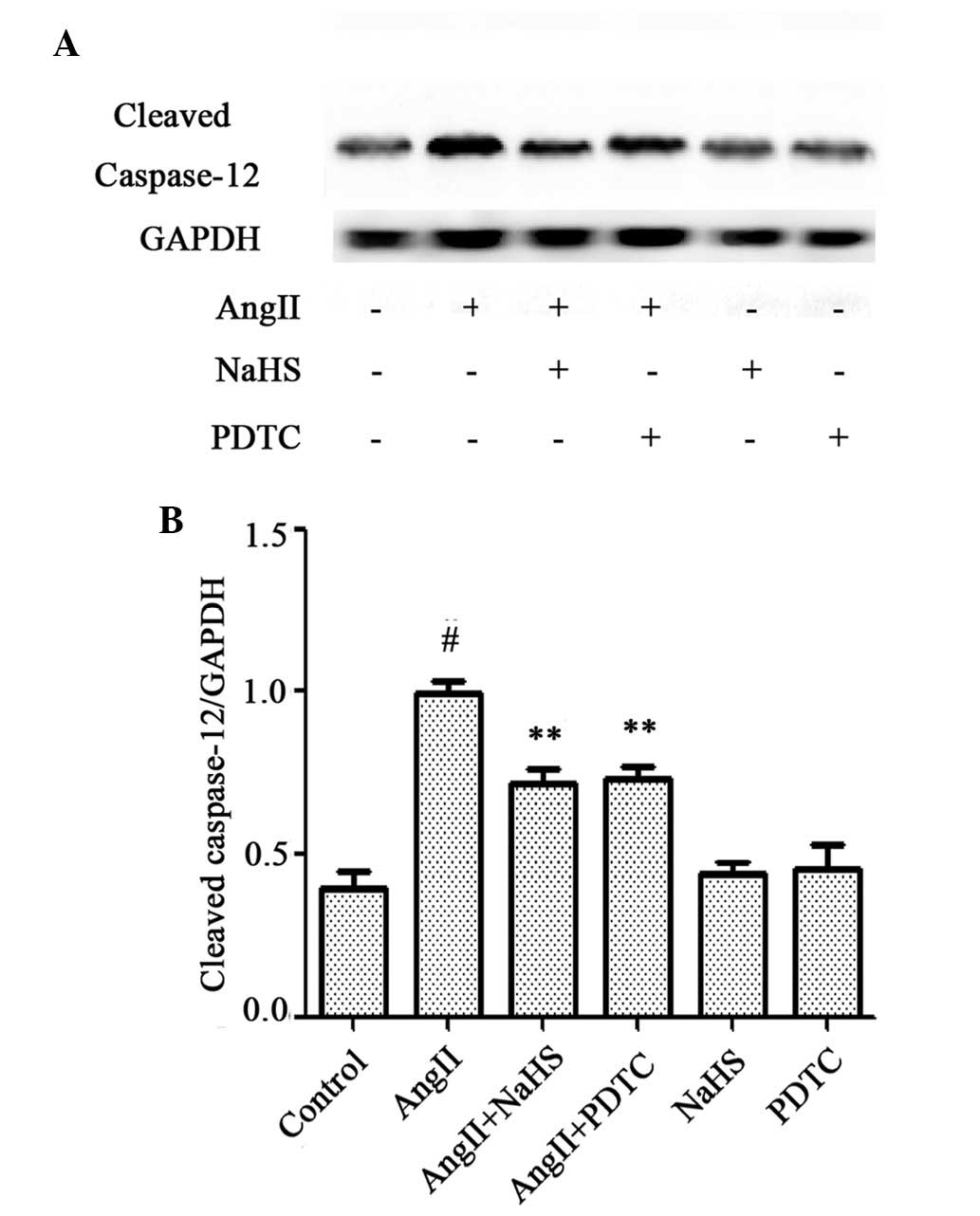
HUVECs. In addition, western blot analysis (Fig. 7A and B) revealed that exposure of
cells to 10-6 M AngII for 24 h significantly upregulated the
expression of cleaved caspase-12, which is considered to be one of
the primary effectors of apoptosis (21); this effect was significantly
abrogated by the pretreatment of cells with NaHS or PDTC for 1 h.
NaHS or PDTC alone did not affect the basal expression level of
cleaved caspase-12 in HUVECs. These findings suggest that exogenous
H2S protects HUVECs against AngII-induced apoptosis, and
that the NF-κB signaling pathway contributes to the AngII-induced
apoptosis of HUVECs.
Exogenous H2S and an ET-1
inhibitor attenuate the phosphorylation of p65 induced by AngII in
HUVECs
It has previously been demonstrated that exogenous
H2S suppresses p-p65 in HUVECs (24), whereas AngII and ET-1 induce
activation of p-p65 (18,32). Since it has been demonstrated that
p-p65-mediated endothelial cell insult contributes to ET-1-induced
cytotoxicity (20), it was
hypothesized that exogenous H2S may inhibit activation
of the ET-1/NF-κB signaling pathway during AngII-induced
cytotoxicity in HUVECs. The effects of H2S and an ET-1
inhibitor on the AngII-induced overexpression of p-p65 were
therefore investigated. As presented in Fig. 8, following treatment with 10-6 M
AngII for 24 h, HUVECs significantly overexpressed p-p65. Notably,
this effect was attenuated by pretreatment with 200 µM NaHS for 1 h
or 1 µM BQ788 for 1 h prior to AngII exposure. These data suggest
that exogenous H2S and ET-1 inhibition protect HUVECs
against AngII-induced cytotoxicity via the ET-1/NF-κB signaling
pathway.
Discussion
RAS, particularly AngII, has been demonstrated to be
crucial for the progression of AS (33); however, the underlying mechanisms
remain to be fully elucidated. Evidence suggests that ER stress and
ED are key contributors to AngII-induced cytotoxicity (15,34).
Consistent with previous studies (15,34,35),
the present study revealed that AngII markedly induced HUVEC
injury, including a decrease in cell viability, and increases in NO
production, protein expression levels of iNOS, ET-1, GRP78 and
CHOP, as well as phosphorylation of p65.
Previous studies have demonstrated that AngII
induces numerous inflammatory mediators, including ET-1 (36–38),
which is implicated in the progression of endothelial cell injury
(39–41). Therefore, the present study
confirmed that the activation of ET-1 is crucial for AngII-induced
cytotoxicity. The findings of the present study revealed that
pretreatment of HUVECs with BQ788, a specific inhibitor of ET-1,
significantly abrogated the inhibition of CSE expression and
activity levels induced by AngII. Furthermore, administration of
exogenous ET-1 imitated the inhibitory effect of AngII on the
expression and activity of CSE in HUVECs. Therefore, ET-1 may
affect the function of endothelial cells by inhibiting the
expression and activity of CSE.
Features of H2S, an active
gasotransmitter, include low molecular weight, continuous
production, quick diffusion and extensive biological effects
(42). It has previously been
demonstrated that H2S is cytoprotective, and its
generation in vessels was decreased upon RAS activation (26), which has been revealed to be an
important factor in the development of AS (43). The majority of studies using
H2S donors, including NaHS and GYY4137 have demonstrated
that exogenous H2S attenuates a multitude of
pathophysiological processes, such as vascular endothelium
dysfunction and smooth muscle cell proliferation (44–46).
The present study demonstrated that AngII-induced cytotoxicity
significantly suppressed the expression and activity of CSE in
HUVECs; therefore, it was hypothesized that supplementation of
H2S may protect HUVECs against AngII-induced ED. This
hypothesis was supported by the results of the present study. When
HUVECs were pretreated with NaHS for 1 h prior to AngII exposure
for 24 h, AngII-stimulated cytotoxicity was markedly abrogated,
evidenced by increased cell viability. These data are consistent
with a previous study, which demonstrated that NaHS markedly
improves ED induced by AngII (28). Since H2S is known to be
protective of endothelial cells, the present study pretreated
HUVECs with NaHS for 1 h prior to incubation with AngII for 24 h.
Pretreated cells had almost undetectable levels of CHOP, GRP78,
iNOS and nitrite, and significantly improved cell viability. These
results suggest that H-2S exerts protective effects against
AngII-induced cytotoxicity in HUVECs. In addition, HUVECs
pretreated with PDTC for 1 h prior to incubation with AngII for 24
h had almost undetectable levels of CHOP, GRP78, iNOS and nitrite,
and significantly improved cell viability; mimicking the effects of
NaHS.
A previous study revealed that the NF-κB signaling
pathway is involved in AngII-induced HUVEC dysfunction (18), and it is widely accepted that NF-κB
is required for full induction of ET-1 (47). NF-κB is expressed in the majority
of cell types and is crucial for transcription. The p65 protein is
the primary transcriptionally active component of NF-κB (48). It was hypothesized that NF-κB is
involved in the mechanism underlying H2S suppression of
AngII-stimulated cytotoxicity in HUVECs. Therefore, the present
study examined the effects of H2S on p-p65 protein
expression levels in HUVECs exposed to AngII. P-p65 was
significantly increased in HUVECs exposed to AngII. Conversely,
pretreatment with 200 µM NaHS or 1 µM BQ788 for 1 h significantly
decreased the p-p65 protein expression level in HUVECs exposed to
AngII.
To investigate whether the protective effect of
exogenous H2S on AngII-stimulated cytotoxicity via the
NF-κB signaling pathway in HUVECs is associated with its
endothelial cell protective function, the effects of NaHS and PDTC
on HUVEC apoptosis and caspase-12 cleavage induced by AngII were
examined. NaHS and PDTC attenuated apoptosis in HUVECs, which is a
key process in ED during the development of AS. Consistent with
this, Qabazard et al (49)
demonstrated that supplementation with GYY4137 markedly decreased
oxidative stress-induced overexpression of CHOP, GRP78 and
caspase-12 in endothelial cells. In addition, this study
demonstrated that p38 mitogen-activated protein kinases (MAPK)
demonstrated a key role in the regulation of AngII-induced ED
(49). Therefore, p38 MAPK may be
a good target for NaHS to intervene in AngII-induced ED.
Qabazard et al (49) investigated whether the
AngII-stimulated ET-1 generation is crucial for the regulation of
p38 MAPK expression. In addition, Touyz et al (50) revealed that pretreatment with BQ788
1 h prior to incubation with AngII for 24 h may inhibit expression
of p-p38 MAPK induced by AngII. Since NaHS attenuated AngII-induced
p-p38MAPK expression, this may provide, at least partially, the
mechanism underlying the increased expression of ET-1 and AngII
during the development of AS.
In conclusion, the present study, to the best of our
knowledge, is the first to demonstrate that ET-1-mediated
inhibition of CSE expression and activity is involved in
AngII-induced cytotoxicity. H2S supplementation may
protect against AngII-stimulated ET-1 generation and subsequent
cytotoxicity in HUVECs via the inhibition of p-NF-κB expression.
The findings of the present study suggested that exogenous
H2S may provide a potential novel therapeutic strategy
for the prevention and treatment of AS.
References
|
1
|
Feinstein M, Ning H, Kang J, Bertoni A,
Carnethon M and Lloyd-Jones DM: Racial differences in risks for
first cardiovascular events and noncardiovascular death: The
atherosclerosis risk in communities study, the cardiovascular
health study, and the multi-ethnic study of atherosclerosis.
Circulation. 126:50–59. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Davignon J and Ganz P: Role of endothelial
dysfunction in atherosclerosis. Circulation. 109(23): Suppl 1.
III27–III32. 2004.PubMed/NCBI
|
|
3
|
Loyer X, Potteaux S, Vion AC, Guérin CL,
Boulkroun S, Rautou PE, Ramkhelawon B, Esposito B, Dalloz M, Paul
JL, et al: Inhibition of microRNA-92a prevents endothelial
dysfunction and atherosclerosis in mice. Circ Res. 114:434–443.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Barton M, Haudenschild CC, d'Uscio LV,
Shaw S, Münter K and Luscher TF: Endothelin ETA receptor blockade
restores NO-mediated endothelial function and inhibits
atherosclerosis in apolipoprotein E-deficient mice. Proc Natl Acad
Sci USA. 95:14367–14372. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Brunner F, Brás-Silva C, Cerdeira AS and
Leite-Moreira AF: Cardiovascular endothelins: Essential regulators
of cardiovascular homeostasis. Pharmacol Ther. 111:508–531. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kamata K, Kanie N, Matsumoto T and
Kobayashi T: Endothelin-1-induced impairment of
endothelium-dependent relaxation in aortas isolated from controls
and diabetic rats. J Cardiovasc Pharmacol. 1(44): Suppl. S186–S190.
2004. View Article : Google Scholar
|
|
7
|
Barton M, Traupe T and Haudenschild CC:
Endothelin, hypercholesterolemia and atherosclerosis. Coron Artery
Dis. 14:477–490. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tummala PE, Chen XL, Sundell CL, Laursen
JB, Hammes CP, Alexander RW, Harrison DG and Medford RM:
Angiotensin II induces vascular cell adhesion molecule-1 expression
in rat vasculature: A potential link between the renin-angiotensin
system and atherosclerosis. Circulation. 100:1223–1229. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Marampon F, Gravina GL, Scarsella L,
Festuccia C, Lovat F, Ciccarelli C, Zani BM, Polidoro L, Grassi D,
Desideri G, et al: Angiotensin-converting-enzyme inhibition
counteracts angiotensin II-mediated endothelial cell dysfunction by
modulating the p38/SirT1 axis. J Hypertens. 31:1972–1983. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ayari H: Respective roles of cortisol,
aldosterone and angiotensin II during pathophysiology of
atherosclerosis. Ann Biol Clin (Paris). 71:381–388. 2013.(In
French). PubMed/NCBI
|
|
11
|
Nishida Y, Takahashi Y, Susa N, Kanou N,
Nakayama T and Asai S: Comparative effect of angiotensin II type I
receptor blockers on serum uric acid in hypertensive patients with
type 2 diabetes mellitus: A retrospective observational study.
Cardiovasc Diabetol. 12:1592013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ocaranza MP, Michea L, Chiong M, Lagos CF,
Lavandero S and Jalil JE: Recent insights and therapeutic
perspectives of angiotensin-(1–9) in the cardiovascular system.
Clin Sci (Lond). 127:549–557. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang HY, Bian YF, Zhang HP, Gao F, Xiao
CS, Liang B, Li J, Zhang NN and Yang ZM: Angiotensin-(1–7)
treatment ameliorates angiotensin II-induced apoptosis of human
umbilical vein endothelial cells. Clin Exp Pharmacol Physiol.
39:1004–1010. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Biol. 8:519–529. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Uhal BD, Nguyen H, Dang M, Gopallawa I,
Jiang J, Dang V, Ono S and Morimoto K: Abrogation of ER
stress-induced apoptosis of alveolar epithelial cells by
angiotensin 1–7. Am J Physiol Lung Cell Mol Physiol. 305:L33–L41.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chistiakov DA, Sobenin IA, Orekhov AN and
Bobryshev YV: Role of endoplasmic reticulum stress in
atherosclerosis and diabetic macrovascular complications. Biomed
Res Int. 2014:6101402014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chaudhari N, Talwar P, Parimisetty A,
Lefebvre d'Hellencourt C and Ravanan P: A molecular web:
Endoplasmic reticulum stress, inflammation, and oxidative stress.
Front Cell Neurosci. 8:2132014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guo RW, Yang LX, Li MQ, Liu B and Wang XM:
Angiotensin II induces NF-kappa B activation in HUVEC via the
p38MAPK pathway. Peptides. 27:3269–3275. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pushpakumar SB, Kundu S and Sen U:
Endothelial dysfunction: The link between homocysteine and hydrogen
sulfide. Curr Med Chem. 21:3662–3672. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Łowicka E and Beltowski J: Hydrogen
sulfide (H2S)-the third gas of interest for pharmacologists.
Pharmacol Rep. 59:4–24. 2007.PubMed/NCBI
|
|
21
|
Yang G, Wu L, Jiang B, Yang W, Qi J, Cao
K, Meng Q, Mustafa AK, Mu W, Zhang S, et al: H2S as a physiologic
vasorelaxant: Hypertension in mice with deletion of cystathionine
gamma-lyase. Science. 322:587–590. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Suo R, Zhao ZZ, Tang ZH, Ren Z, Liu X, Liu
LS, Wang Z, Tang CK, Wei DH and Jiang ZS: Hydrogen sulfide prevents
H2O2-induced senescence in human umbilical vein endothelial cells
through SIRT1 activation. Mol Med Rep. 7:1865–1870. 2013.PubMed/NCBI
|
|
23
|
Ng DS, Peh MT, Anwar AB, Atan MSB
Mohammed, Tsai C-Yi, Kumar SD and Moore PK: P27 Changes in the
hydrogen sulfide (H2S) system arising from administration of high
fat diet. Nitric Oxide. 31:(Suppl 2). S46–S47. 2013. View Article : Google Scholar
|
|
24
|
Pan LL, Liu XH, Gong QH, Wu D and Zhu YZ:
Hydrogen sulfide attenuated tumor necrosis factor-α-induced
inflammatory signaling and dysfunction in vascular endothelial
cells. PLoS One. 6:e197662011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mani S, Untereiner A, Wu L and Wang R:
Hydrogen sulfide and the pathogenesis of atherosclerosis. Antioxid
Redox Signal. 20:805–817. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xue H, Yuan P, Ni J, Li C, Shao D, Liu J,
Shen Y, Wang Z, Zhou L, Zhang W, et al: H(2)S inhibits
hyperglycemia-induced intrarenal renin-angiotensin system
activation via attenuation of reactive oxygen species generation.
PLoS One. 8:e743662013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lin YJ, Kwok CF, Juan CC, Hsu YP, Shih KC,
Chen CC and Ho LT: Angiotensin II enhances endothelin-1-induced
vasoconstriction through upregulating endothelin type A receptor.
Biochem Biophys Res Commun. 451:263–269. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Al-Magableh MR, Kemp-Harper BK and Hart
JL: Hydrogen sulfide treatment reduces blood pressure and oxidative
stress in angiotensin II-induced hypertensive mice. Hypertens Res.
38:13–20. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kossmann S, Hu H, Steven S, Schönfelder T,
Fraccarollo D, Mikhed Y, Brähler M, Knorr M, Brandt M, Karbach SH,
et al: Inflammatory monocytes determine endothelial nitric oxide
synthase uncoupling and nitro-oxidative stress induced by
angiotensin II. J Biol Chem. 289:27540–27550. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Miyata K, Satou R, Shao W, Prieto MC,
Urushihara M, Kobori H and Navar LG: ROCK/NF-κB axis-dependent
augmentation of angiotensinogen by angiotensin II in
primary-cultured preglomerular vascular smooth muscle cells. Am J
Physiol Renal Physiol. 306:F608–F618. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Adiarto S, Heiden S, Vignon-Zellweger N,
Nakayama K, Yagi K, Yanagisawa M and Emoto N: ET-1 from endothelial
cells is required for complete angiotensin II-induced cardiac
fibrosis and hypertrophy. Life Sci. 91:651–657. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Browatzki M, Schmidt J, Kübler W and
Kranzhöfer R: Endothelin-1 induces interleukin-6 release via
activation of the transcriptio n factor NF-kappaB in human vascular
smooth muscle cells. Basic Res Cardiol. 95:98–105. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen X, Rateri DL, Howatt DA, Balakrishnan
A, Moorleghen JJ, Morris AJ, Charnigo R, Cassis LA and Daugherty A:
Amlodipine reduces AngII-induced aortic aneurysms and
atherosclerosis in hypercholesterolemic mice. PLoS One.
8:e817432013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Murdoch CE, Chaubey S, Zeng L, Yu B,
Ivetic A, Walker SJ, Vanhoutte D, Heymans S, Grieve DJ, Cave AC, et
al: Endothelial NADPH oxidase-2 promotes interstitial cardiac
fibrosis and diastolic dysfunction through proinflammatory effects
and endothelial-mesenchymal transition. J Am Coll Cardiol.
63:2734–2741. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wei LH, Huang XR, Zhang Y, Li YQ, Chen HY,
Heuchel R, Yan BP, Yu CM and Lan HY: Deficiency of Smad7 enhances
cardiac remodeling induced by angiotensin II infusion in a mouse
model of hypertension. PLoS One. 8:e701952013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lin CC, Hsieh HL, Chi PL, Yang CC, Hsiao
LD and Yang CM: Upregulation of COX-2/PGE2 by ET-1 mediated through
Ca2+-dependent signals in mouse brain microvascular endothelial
cells. Mol Neurobiol. 49:1256–1269. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shiraki A, Oyama J, Komoda H, Asaka M,
Komatsu A, Sakuma M, Kodama K, Sakamoto Y, Kotooka N, Hirase T and
Node K: The glucagon-like peptide 1 analog liraglutide reduces
TNF-α-induced oxidative stress and inflammation in endothelial
cells. Atherosclerosis. 221:375–382. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee W, Ku SK and Bae JS: Factor Xa
inhibits HMGB1-induced septic responses in human umbilical vein
endothelial cells and in mice. Thromb Haemost. 112:757–769. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Schrader LI, Kinzenbaw DA, Johnson AW,
Faraci FM and Didion SP: IL-6 deficiency protects against
angiotensin II induced endothelial dysfunction and hypertrophy.
Arterioscler Thromb Vasc Biol. 27:2576–2581. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Rojas E, Rodríguez-Molina D, Bolli P,
Israili ZH, Faría J, Fidilio E, Bermúdez V and Velasco M: The role
of adiponectin in endothelial dysfunction and hypertension. Curr
Hypertens Rep. 16:4632014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lamarca B: Endothelial dysfunction. An
important mediator in the pathophysiology of hypertension during
pre-eclampsia. Minerva Ginecol. 64:309–320. 2012.PubMed/NCBI
|
|
42
|
Du J, Huang Y, Yan H, Zhang Q, Zhao M, Zhu
M, Liu J, Chen SX, Bu D, Tang C and Jin H: Hydrogen sulfide
suppresses oxidized low-density lipoprotein (ox-LDL)-stimulated
monocyte chemoattractant protein 1 generation from macrophages via
the nuclear factor kappaB (NF-κB) pathway. J Biol Chem.
289:9741–9753. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sata M and Fukuda D: Crucial role of
renin-angiotensin system in the pathogenesis of atherosclerosis. J
Med Invest. 57:12–25. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lynn EG and Austin RC: Hydrogen sulfide in
the pathogenesis of atherosclerosis and its therapeutic potential.
Expert Rev Clin Pharmacol. 4:97–108. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu Z, Han Y, Li L, Lu H, Meng G, Li X,
Shirhan M, Peh MT, Xie L, Zhou S, et al: The hydrogen sulfide
donor, GYY4137, exhibits anti-atherosclerotic activity in high fat
fed apolipoprotein E(−/−) mice. Br J Pharmacol. 169:1795–1809.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Meng G, Zhu J, Xiao Y, Huang Z, Zhang Y,
Tang X, Xie L, Chen Y, Shao Y, Ferro A, et al: Hydrogen sulfide
donor GYY4137 protects against myocardial fibrosis. Oxid Med Cell
Longev. 2015:6910702015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Piechota-Polanczyk A, Kleniewska P and
Goraca A: The influence of ETA and ETB receptor blockers on
LPS-induced oxidative stress and NF-κB signaling pathway in heart.
Gen Physiol Biophys. 31:271–278. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Guo R, Wu K, Chen J, Mo L, Hua X, Zheng D,
Chen P, Chen G, Xu W and Feng J: Exogenous hydrogen sulfide
protects against doxorubicin-induced inflammation and cytotoxicity
by inhibiting p38MAPK/NFκB pathway in H9c2 cardiac cells. Cell
Physiol Biochem. 32:1668–1680. 2013.PubMed/NCBI
|
|
49
|
Qabazard B, Ahmed S, Li L, Arlt VM, Moore
PK and Stürzenbaum SR: C. elegans aging is modulated by hydrogen
sulfide and the sulfhydrylase/cysteine synthase cysl-2. PLoS One.
8:e801352013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Touyz RM, Yao G, Viel E, Amiri F and
Schiffrin EL: Angiotensin II and endothelin-1 regulate MAP kinases
through different redox-dependent mechanisms in human vascular
smooth muscle cells. J Hypertens. 22:1141–1149. 2004. View Article : Google Scholar : PubMed/NCBI
|