Introduction
β-N-methylamino-L-alanine (BMAA) is a non-protein
amino acid neurotoxin that attracts much attention since it
associates with the incidence of neurodegenerative diseases of
amyotrophic lateral sclerosis (ALS), Alzheimer's disease (AD) and
Parkinson's disease (PD), or combination of them (1,2).
BMAA is known to be produced by cyanobacteria (3). By means of biomagnification, BMAA has
been identified in aquatic and terrestrial environments (4). A previous study demonstrated that
BMAA can be taken up into the brain across the blood-brain barrier
via large neutral amino acid carriers, and this intake may be
augmented in response to factors, including diet, metabolism,
disease and age (5).
The neurotoxicity of BMAA was first concerned with
the high incidence of amyotrophic lateral
sclerosis/parkinsonism-dementia complex (ALS/PDC), a combination of
ALS, PD and dementia (6).
Significant levels of BMAA were detected in the brain and spinal
cord tissues of patients succumbing to ALS/PDC, whereas no or few
quantities of BMAA were detected in those controls (7). A large quantity of in vivo
studies have demonstrated memory and behavior disabilities
following exposure to BMAA, together with the degeneration and loss
of neurons in the central nervous system, and these changes appear
proportional to the dose of BMAA administered or the concentration
of BMAA present in the nervous tissue (8,9).
Endoplasmic reticulum (ER) stress has been suggested
to be involved in human neurodegenerative diseases, including ALS,
AD and PD, as well as other disorders (10). Under the condition of stress
circumstances, the normal function of the ER is disturbed to induce
protein misfolding and aggregation in the lumen of the ER. The ER
responds to the accumulation of misfolded protein with the unfolded
protein response (UPR) (11).
Binding immunoglobulin protein (Bip/Grp78) senses and transfers to
the misfolded protein, which makes it dissociate from the
ER-transmembrane receptor protein kinase RNA-like endoplasmic
reticulum kinase (PERK), inositol-requiring kinase 1 (IRE1) and
transcription factor 6 (ATF6), leading to their activation. PERK
phosphorylates the alpha subunit of eukaryotic initiation factor 2
(eIF2), which attenuates protein translation. IRE1α initiates the
splicing of transcription factor X-box binding protein (XBP1) mRNA,
which upregulates the ER chaperon genes, as well as the genes
involved in protein degradation. ATF6 (90 kDa) translocates to the
Golgi apparatus and is cleaved there to form a 50 kDa transcription
factor that translocates to the nucleus, where it activates the
transcription of XBP1 and ER chaperon genes. The aim of these
responses is to mitigate protein load and restore ER function.
If the disruption to ER is prolonged or aggravated,
the UPR develops towards apoptosis. The activation of IRE1 recruits
tumor-necrosis factor receptor associated factor 2 (TRAF2) and
apoptosis signal-regulating kinase 1 (ASK1), and triggers the c-JUN
N-terminal kinase (JNK) signaling pathway. JNK triggers human
procaspase-4 (or murine procaspase-12) and downstream caspases
(12). The phosphorylation of PERK
enhances the translation of activating transcription factor 4
(ATF4) via eIF2α phosphorylation, which induces the expression of
ER stress-specific CCAAT/-enhancer-binding protein homologous
protein (CHOP) (13). CHOP
downregulates the antiapoptotic mitochondrial B-cell lymphoma
(Bcl)-2 family, favoring caspase activation.
Understanding the intracellular pathway of BMAA
toxicity is important as it not only renders the target for
treating BMAA-induced neurotoxicity, but also facilitates the
exploration of the pathogenesis of neurodegenerative disorders.
Previous studies have indicated that ER stress is involved in the
pathogenesis of ALS, PD and AD, and serves a pivotal role in the
processing of neuronal degeneration and death (10). In the present study, the
intracellular molecular mechanism of BMAA-induced ER stress and
apoptosis was assessed in the cultured HT22 hippocampal cells,
Neuro-2a and SH-SY5Y neuroblastoma cells.
Materials and methods
Materials
BMAA, 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl
tetrazolium bromide (MTT), propidium iodide (PI) and salubrinal
(Sal) were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Thapsigargin (Tg) and tunicamycin (Tm) was obtained from Biomol
Research Labs (Plymouth Meeting, PA, USA). Antibodies specific to
Bcl-2 (cat. no. sc7382), Bcl-xl (cat. no. sc7195), Bcl-2-associated
X protein (Bax; cat. no. sc493), CHOP (cat. no. sc7351), GRP78 cat.
no. sc1050), GRP94 (cat. no. sc120647), ATF6α (p90; cat. no.
sc22799), eIF2α (cat. no. sc133132) and sXBP-1 (cat. no. sc8015)
were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA). Antibodies specific to cleaved JNK (cat. no. 9252), p38 (cat.
no. 9212), ERK (cat. no. 9102), caspase-9 (cat. no. 9508), cleaved
caspase-3 (cat. no. 9664), poly-ADP ribose polymerase (PARP; cat.
no. 9532), phosphorylated (P-)ASK1 (cat. no. 3761), P-eIF2α (cat.
no. 3597), P-JNK (cat. no. 9255), P-p38 (cat. no. 9211) and P-ERK
(cat. no. 9101) were purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA). Antibodies against cyclophilin B (CypB; cat.
no. ab16045) and protein disulfide isomerase (PDI; cat. no. ab2792)
were purchased from Abcam (Cambridge, UK). Antibodies against
β-tubulin (cat. no. sc9104), α-actinin (cat. no. sc15336) and actin
(cat. no. sc1616) were purchased from Santa Cruz Biotechnology,
Inc. GAPDH antibody (cat. no. CSA-335) was purchased from Enzo Life
Sciences, Inc. (Farmingdale, NY, USA). Lipofectamine 2000 was
purchased from Gibco (Thermo Fisher Scientific, Inc., Waltham, MA,
USA). Small interfering (si)RNAs against control scrambled and CHOP
were obtained from Santa Cruz Biotechnology, Inc. Gene Silencer
siRNA transfection reagent was purchased from Gene Therapy System
(San Diego, CA, USA). DAPI nucleic acid stain was purchased from
Molecular Probes (Eugene, OR, USA). Phosphate-buffered saline
(PBS), 4% paraformaldehyde, ethanol, methanol, sodium dodecyl
sulfate (SDS), Triton-X-100 and NaHCO3 were purchased from
Sigma-Aldrich.
Cell culture
The human neuroblastoma cell line, SH-SY5Y, mouse
hippocampal cell line, HT22 and mouse neuroblastoma cell line,
Neuro-2a, were purchased from the American Type Culture Collection
(Manassas, VA, USA). Cells were cultured in Dulbecco's modified
Eagle's medium (DMEM), supplemented with 10% (v/v) fetal bovine
serum (Hyclone; GE Healthcare Life Sciences, Logan, UT, USA) 100
U/ml penicillin and 100 µg/ml streptomycin (Gibco; Thermo Fisher
Scientific, Inc.), in a humidified 5% (v/v) CO2
incubator at 37°C. For the treatment, BMAA was dissolved in a
physiological concentration (10 mM) of NaHCO3, as it has been
reported that HCO3 is required for mediating BMAA toxicity
(14,15). The cells were serum-starved for 1 h
and incubated with BMAA, as previously described (14,15).
On completion of incubation, the cells were washed thoroughly with
PBS and subjected to various analyses.
MTT assay
Cell viability was evaluated using the conversion of
MTT to MTT-formazan by mitochondrial enzymes. Neuronal cells were
seeded into 12-well plates at a density of 4×105 cells/well in
growth medium and cultured to ~70% confluence. Following serum
starvation for 3 h, the cells were treated with various
concentrations of BMAA. After 24 or 48 h incubation at 37°C, the
cells were washed three times with PBS, and 30 µl MTT solution (5
mg/ml) was added to the cells and subsequently incubated for 1 h at
37°C. The medium was removed carefully and 300 µl dimethyl
sulfoxide was added to dissolve the blue formazan in living cells.
Finally, the absorbance at 540 nm was measured using an ELISA
reader (Multiskan EX; Thermo Lab systems, Beverly, MA, USA).
Western blot analysis
For western blot analysis, neuronal cells (3×105
cells/well) were seeded for 24 h into a 6-cm culture dish. The
cells were washed twice with ice-cold PBS and total cell lysates
were prepared in a lysis buffer containing 50 mM Tris-HCl (pH 7.4),
150 mM NaCl, 1% Triton X-100, 50 mM NaF, 5 mM sodium pyrophosphate,
1 mM EDTA, 1 mM EGTA, 1 mM DTT, 0.1 mM PMSF and protease inhibitor
cocktail. The whole cell lysates were centrifuged (12,000 × g for
10 min at 4°C) for removal of cellular debris. The protein
concentration was determined by the Lowry method using a Bio-Rad DC
protein assay kit (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Cell lysates containing equal quantities of protein (50 µg) were
resolved by 8–15% SDS-polyacrylamide gel electrophoresis and
transferred onto nitrocellulose membranes. Each membrane was
blocked with a solution containing 5% non-fat milk in Tris-buffered
saline with 0.05% Tween-20 (TBST) for 1 h at room temperature.
Following blocking, the membranes were incubated with the primary
antibodies (dilution, 1:2,000) in TBST overnight at 4°C. Following
incubation, the membranes were washed for 1 h with TBST and were
further probed with secondary HRP-conjugated anti-rabbit,
anti-mouse or anti-goat immunoglobulin G (dilution, 1:2,000;
AbFrontier, Seoul, South Korea) in TBST for 1 h at room
temperature. The immune complexes were visualized using an enhanced
chemiluminescence detection system (Pierce; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol.
siRNA
siRNA transfections were performed using
Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific,
Inc.). CHOP siRNA target sequences, purchased from Dharmacon (GE
Healthcare Life Sciences, Chalfont, UK) were as follows: Sense,
5′-CUGGGAAACAGCGCAUGAA-3′ and antisense, 5′-UUCAUGCGCUGUUUCCCAG-3′.
The universal negative control (Dharmacon; GE Healthcare Life
Sciences) was used as the scrambled siRNA. HT22 cells were seeded
into 6-well plates overnight and transferred into 1 ml serum-free
DMEM prior to transfection. Scrambled control and CHOP siRNA were
incubated with transfection reagent for 5 min at room temperature,
and the mixtures were subsequently added to the cells. Following 12
h incubation, 1 ml DMEM containing 20% calf serum was added to each
well. For experiments, the cells were transfected with siRNA for 24
h and were subsequently exposed to BMAA for 24 h. The efficiency of
siRNA-based interference of CHOP was monitored by western blot
analysis.
Flow cytometry for DNA content
The cells were seeded at 2×105 cells/ml density in
100-mm dishes. Following incubation for 24 h, the cells were
harvested, washed twice with PBS, and fixed with ice-cold 75%
ethanol at 4°C for 24 h. The cells were subsequently pelleted by
centrifugation at 1,000 × g for 5 min at 4°C and the ethanol layer
was discarded. Following washing with PBS, the fixed cells were
treated with 0.5 µg/ml RNase A in PI buffer for 30 min at 37°C. At
the end of the treatment, PI (20 µg/ml) was added and the cells
were stained for 30 min in the dark. The cell cycle was then
analyzed for DNA content using Kaluza flow cytometry software
(Beckman Coulter, Orange County, CA, USA).
Plasmid construction and
transfection
Full-length cDNA of Hsp70 was obtained by reverse
transcription-polymerase chain reaction (PCR). The primers,
synthesized by Macrogen, Inc. (Seoul, South Korea), used for PCR
amplification were 5′-CACCACCTACTCCGACAACCA-3′ and
5′-GCCCCTAATCTACCTCCTCAATG-3′. The PCR reaction mixture (40 µl)
contained 2 mM MgCl2, 0.2 mM dNTP, 1 µM primers and 1 unit Taq DNA
polymerase. The samples were amplified using 30 cycles of 60 sec
denaturation at 94°C and 30 sec annealing at 62°C. The product was
subsequently cloned into pcDNA3.1-HA plasmid (Invitrogen; Thermo
Fisher Scientific, Inc.) using BamHI and KpnI enzymes. The plasmids
were transfected into cells with Lipofectamine 2000 reagent,
according to the manufacturer's protocol. The cells were treated
with BMAA 24 h after transfection. The efficiency of transfection
was monitored by western blot analysis with anti-HA antibody (cat.
no. sc138; Santa Cruz Biotechnology, Inc.).
DAPI staining and confocal
microscopy
Equal numbers of neuronal cells were seeded onto
glass coverslips in a 12-well microplate. Following harvesting, the
cells were washed in PBS and fixed in 4% paraformaldehyde
containing 0.25% Triton X-100 for 10 min at 22°C. Cell nuclei were
stained with 6.5 µg/ml DAPI, 16% polyvinyl alcohol and 40% glycerol
C for 30 min at 37°C. The cells on the glass coverslips were
covered with histological mounting medium (National Diagnostics,
Atlanta, GA, USA). Fluorescence was evaluated in three independent
high-power fields (magnification, ×400) using an LSM510 confocal
laser microscope (Carl Zeiss, Oberkochen, Germany). The
excitation/emission wavelengths for DAPI was 358/461 nm. Apoptotic
cells were identified with nuclear morphology of apoptosis,
involving chromatin condensation and DNA fragmentation.
Statistical analyses
The results were expressed as the mean ± standard
deviation from at least three independent experiments. Statistical
analyses were performed using Student's t-test. Unless indicated
otherwise, P<0.05 was considered to indicate a statistically
significant difference.
Results
BMAA induces apoptotic neuronal death
in a dose- and time-dependent manner
Previous studies have demonstrated that the
induction of neuronal degeneration and death, rather than of
excitotoxicity or oxidative stress, requires high concentrations of
BMAA (≥1.0–3.0 mM) (14–16). The present study first estimated
the neurotoxic effect of BMAA on the viability of neuronal cells
using an MTT reduction assay. HT22, Neuro-2a and SH-SY5Y cells were
treated with increasing doses of BMAA (1.0, 2.0 and 3.0 mM). At 24
h after the beginning of exposure to BMAA, no significant injury
occurred with BMAA concentrations <1.0 mΜ, whereas a mild cell
death occurred at the 1.0 mΜ level, a moderate cell death occurred
at 2.0 mM and a marked cell death occurred at 3.0 mΜ (Fig. 1A). Next, to detect cell injury in
the different stages, cell viability was measured at 12, 24 and 48
h, and the results revealed a time-dependent increase in cell death
(Fig. 1B).
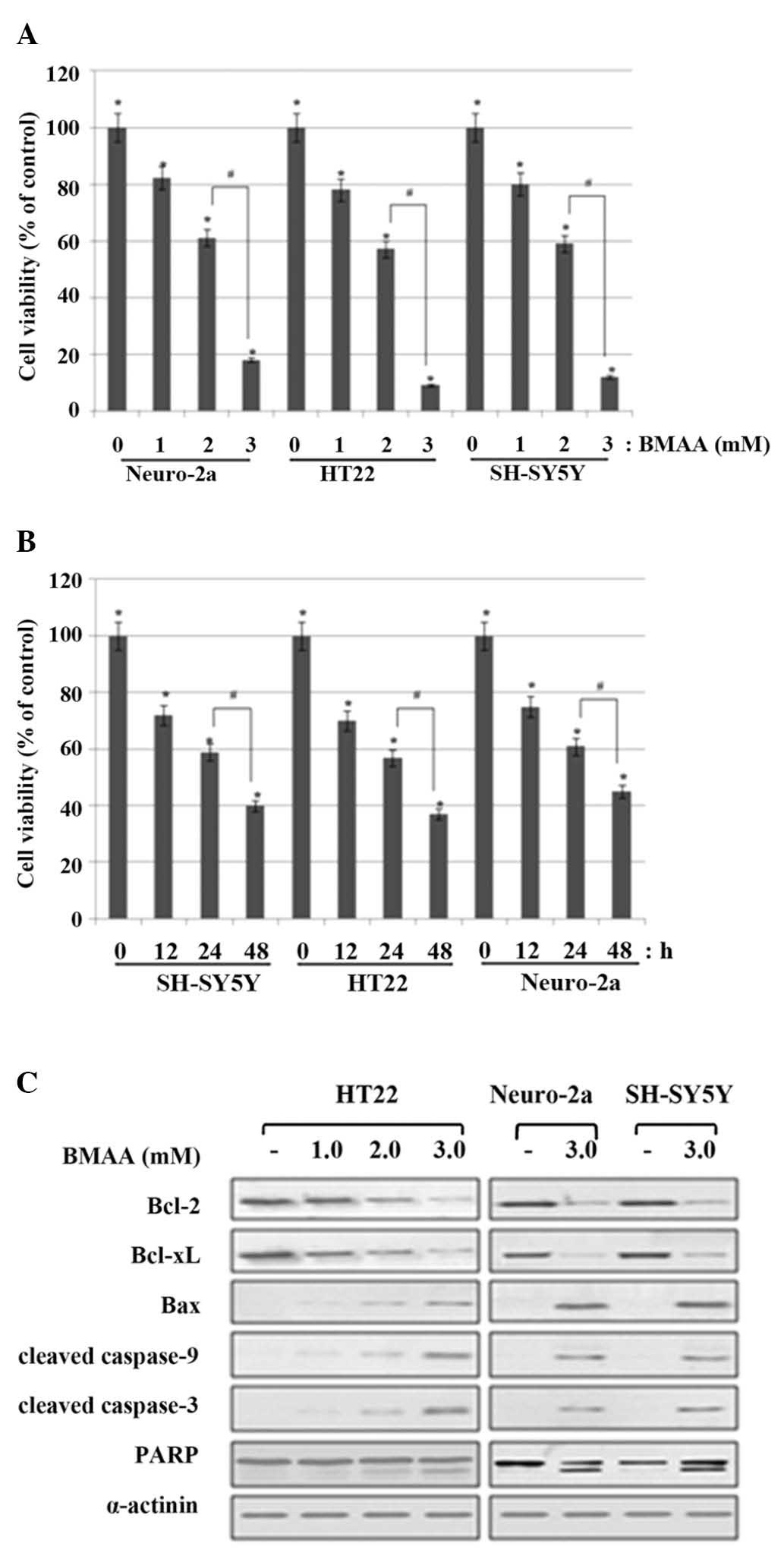 | Figure 1.Effects of BMAA on cell viability and
apoptosis of neuronal cells. (A) Neuronal cells were treated with
1.0, 2.0, or 3.0 mM BMAA and were analyzed at 24 h after the
beginning of exposure to BMAA. The percentage cell viabilities are
representative of at least three different experiments and are
expressed as the mean ± standard deviation (*P<0.05 vs.
untreated cells; #P<0.05 vs. 2.0 mM-treated cells). (B) The cell
viabilities were measured at 12, 24 and 48 h after treatment with
2.0 mΜ BMAA. The data are expressed as the mean ± standard
deviation obtained from at least three independent experiments
(*P<0.05 vs. untreated cells; #P<0.05 vs. 24 h-treated
cells). (C) The cells were treated with increasing doses of BMAA
(1.0, 2.0 and 3.0 mM) for 24 h. The protein expression levels of
Bcl-2 family members, and the cleavage of caspase and PARP, were
assessed by western blot analysis. α-actinin was used as a loading
control. The data are representative of at least three different
experiments. BMAA, β-N-methylamino-L-alanine; Bcl, B-cell lymphoma;
Bax, Bcl-2-associated X protein; PARP, poly-ADP ribose
polymerase. |
In response to BMAA exposure, the expression of
anti-apoptotic Bcl-2 and Bcl-xL revealed much lessened levels
compared with the control; however, the level of pro-apoptotic Bax
was notably increased, which shifted the Bax/Bcl-2 ratio to
withdraw the survival signal and favor apoptosis in a
dose-dependent manner. Cytochrome c is released by the regulation
of the Bcl-2 family proteins and binds to pro-caspase-9, which
cleaves initiator caspase-9 and in turn cleaves the effector
caspase-3. The cleavage of poly (ADP-ribose) polymerase (PARP) is
also involved in DNA repair and programmed cell death. The cleavage
of the caspases and PARP was detected in BMAA-treated cells,
however, not in control cells (Fig.
1C). Collectively, these data indicated that BMAA treatment
induces apoptotic neuronal injury and death in a dose- and
time-dependant manner.
Activation of UPR signaling and UPR
signaling-evoked apoptotic pathway
In response to BMAA exposure, the phosphorylation of
PERK and eIF2α, and the splicing of XBP1 were increased in a BMAA
dose-dependent manner. By contrast to the increased level of the
above proteins, the expression of full length ATF6 (ATF6 p90) was
detected with decreasing levels in the cytosol, suggesting the
cleavage and translocation of ATF6 into the nucleus (Fig. 2A). ER chaperons have been
demonstrated as monitors of ER stress. The predominant ER chaperon
proteins involve Bip/Grp78, protein disulfide isomerase, Grp94 and
Cyp B (17–19). The much increased levels of the ER
chaperons Bip/Grp78, Grp94, PDI, and CypB were represented in
BMAA-treated cells compared with control (Fig. 2B), suggesting the result in
response to the accumulation of misfolding proteins in the ER. The
persistent ER stress initiates UPR signaling-mediated pro-apoptotic
signaling. The phosphorylated ASK1 and CHOP were detected in
BMAA-treated cells in a dose-dependent manner, however, not in the
control cells, suggesting the activation of ER stress-mediated
apoptotic pathway in response to BMAA (Fig. 2C).
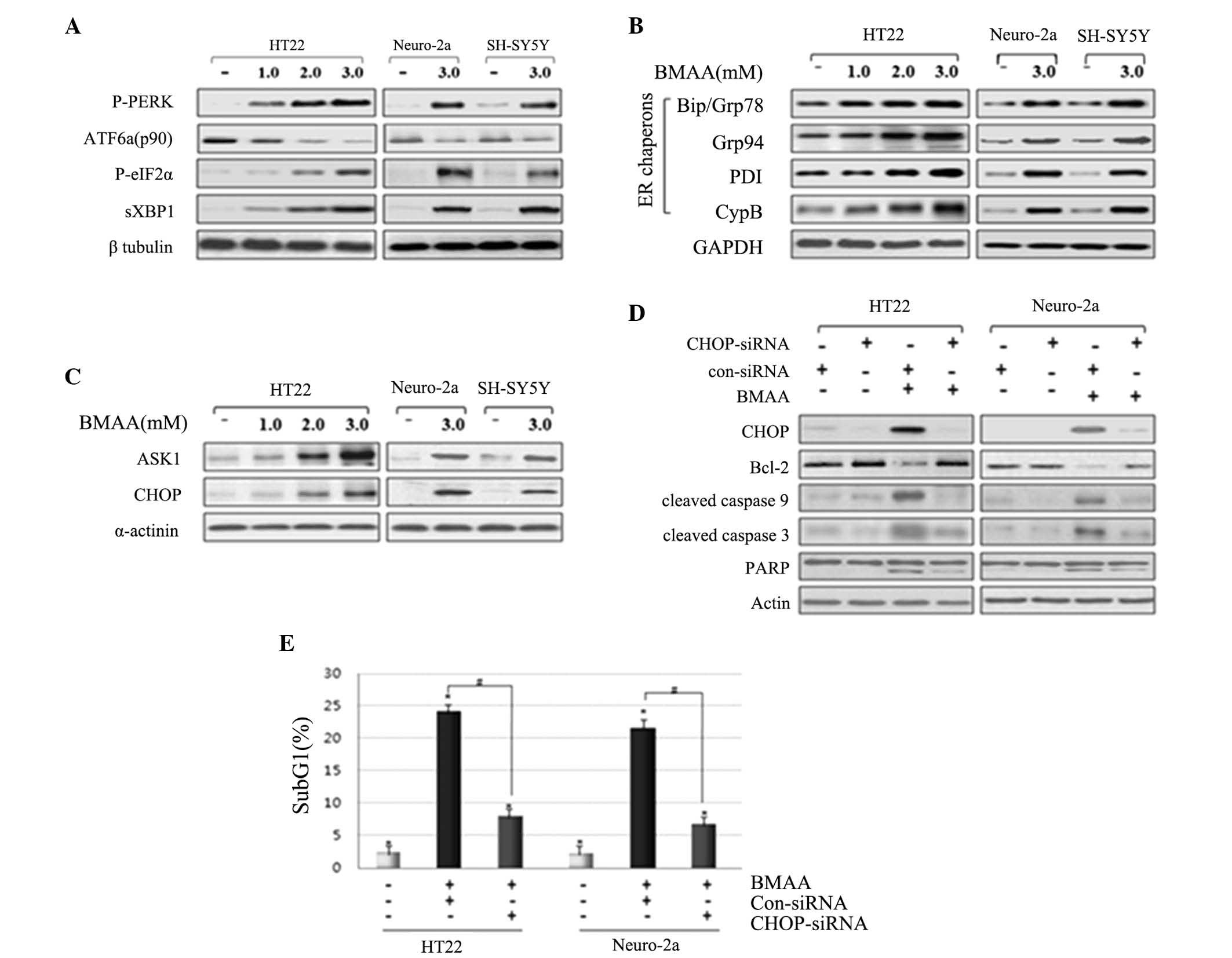 | Figure 2.Treating BMAA induces the activation
of UPR signaling and the UPR signaling-evoked apoptotic pathway.
Neuronal cells were exposed to increasing concentrations of BMAA
and detected at 24 h. Western blot analysis was performed with
antibodies specific to (A) P-PERK and eIF2α, spliced XBP1, IRE1α
and full length ATF6α, (B) ER chaperon, and (C) ASK1 and CHOP. (D)
Neuronal cells were transfected with con-siRNA and CHOP-siRNA for
24 h and were subsequently treated with 3.0 mM BMAA for 24 h.
Knockdown efficiency of CHOP-siRNA, the protein expression levels
of the Bcl-2 family, the cleaved caspases and PARP were assessed by
western blot analysis. β-tubulin, GAPDH α-actinin or actin were
used as a loading control. The results are representative of at
least three independent experiments. (E) The cells were transfected
with con-siRNA and CHOP-siRNA, and the percentage of subG1 cells
was determined by flow cytometry. The data are expressed as the
mean ± standard deviation from at least three independent
experiments (*P<0.05 vs. untreated cells; #P<0.05 vs.
BMAA-treated cells transfected with con-siRNA). BMAA,
β-N-methylamino-L-alanine; Bcl, B-cell lymphoma; Bax,
Bcl-2-associated X protein; PARP, poly-ADP ribose polymerase; P-,
phosphorylated; PERK, protein kinase RNA-like endoplasmic reticulum
kinase; ATF, transcription factor 6; XBP, X-box binding protein;
ER, endoplasmic reticulum; PDI, protein disulfide isomerase; CypB,
cyclophilin B; ASK, apoptosis signal-regulating kinase; CHOP,
CCAAT/-enhancer-binding protein homologous protein; con, control;
si, small interfering; eIF2, eukaryotic initiation factor 2. |
In order to confirm the pro-apoptotic action of
CHOP, the present study performed RNA interference experiments
using siRNA to reduce the levels of CHOP. HT22 and Neuro-2a cells
were transfected with CHOP siRNA, followed by BMAA treatment.
Western blot analysis revealed that the cells transfected with CHOP
siRNA exhibit enhanced expression of Bcl-2, whereas this reduces
the expression of cleaved-PARP compared with those untreated,
suggesting that the inhibition of ER-specific CHOP pathway inhibits
pro-apoptotic signaling (Fig. 2D).
To further evaluate DNA fragmentation in each neuronal cell, the
percentage of subG1 cells was analyzed via flow cytometry as a
measure of apoptotic index. Cells transfected with CHOP siRNA
exhibited a reduced population with cell cycle arrest in the sub-G1
phase compared with those without transfection or transfected with
control scrambled siRNA (Fig. 2E).
Taken together, these findings notably indicate that the UPR
signaling proteins and UPR-mediated apoptotic pathway are activated
as a result of ER stress in BMAA-treated neuronal cells.
Activation of the ERK, JNK and p38
MAPK pathway
Extracellular signal-regulated kinase (ERK), JNK and
p38 mitogen-activated protein kinase (MAPK) are the three major
members of the MAPK family. These MAPK members have been suggested
to be involved in the signaling pathway of UPR-mediated apoptosis
(20). Activated IRE1 recruits
TRAF2 to trigger the JNK-induced apoptotic signaling pathway
(12). IRE1 also leads to
apoptosis by stimulating the phosphorylation of ERK and p38 MAPK
during ER stress (21). CHOP is
also post-translationally regulated upon phosphorylation on serine
residues 78 and 81 by p38 MAPK, which increases its activity and
confers p38 as a substrate of ASK1 along the IRE1-ASK1-p38 pathway
(22). Western blot analysis
revealed that the phosphorylation of ERK, JNK and p38 MAPK were
markedly activated in BMAA-treated neurons (Fig. 3A). The findings revealed that ERK,
JNK and p38 MAPK are involved in the UPR-evoked apoptotic pathway
to contribute to the induction of neuronal death.
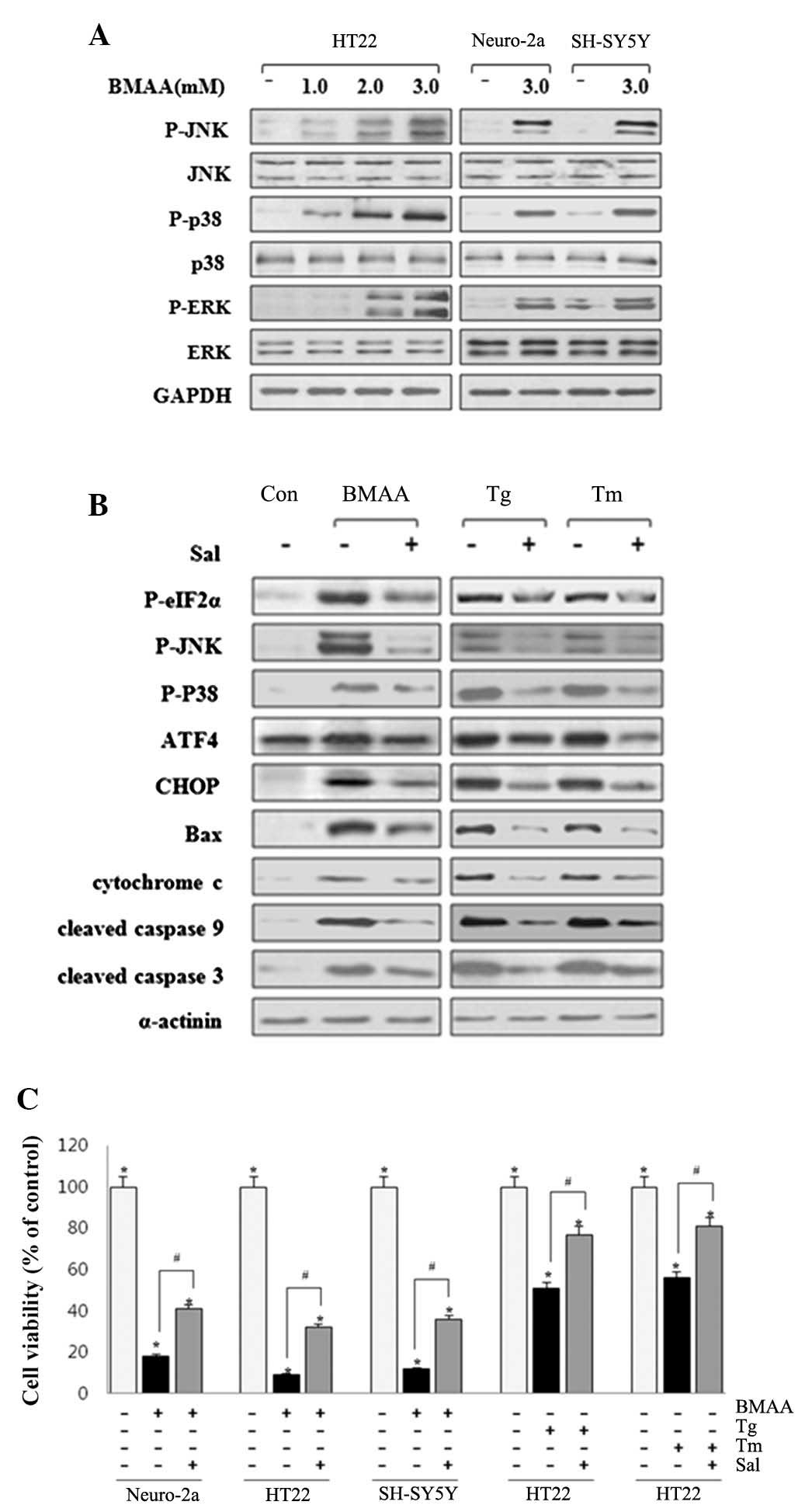 | Figure 3.BMAA initiates ER stress-induced
phosphorylation of p38, JNK, and ERK. (A) HT22, Neuro-2a and
SH-SY5Y cells were treated with BMAA at 1.0, 2.0 or 3.0 mM for 24
h. The protein expression levels of P- and total p38, JNK and ERK
were measured by western blotting. (B) The cells were pretreated
with 20 µM Sal for 30 min, followed by 3.0 mM BMAA, 1 µM Tg or 10
µg/ml Tm for 24 h, respectively. Sal inhibits ER stress-induced
phosphorylation of eIF2α, p38, and JNK, and the expression levels
of CHOP, Bax and cleaved caspases. The results shown are
representative of at least three independent experiments. (C) Cell
viability was measured using an MTT assay under the above
conditions. The data are expressed as the mean ± standard deviation
obtained from at least five independent experiments (*P<0.05 vs.
untreated cells. #P<0.05 vs. Sal-treated cells). BMAA,
β-N-methylamino-L-alanine; Bax, B-cell lymphoma-2-associated X
protein; P-, phosphorylated; ATF, transcription factor 6; ER,
endoplasmic reticulum; CHOP, CCAAT/-enhancer-binding protein
homologous protein; JNK, c-Jun N-terminal kinases; ERK,
extracellular signal-regulated kinase; Sal, salubrinal; Tg,
thapsigargin; Tm, tunicamycin. |
Sal is a selective inhibitor of phosphatase
complexes that dephosphorylate eIF-2α and has been primarily used
to investigate ER stress-induced apoptosis (23). Neuronal cells were pretreated with
20 µΜ Sal for 30 min. As a positive control, cells were exposed to
UPR inducers, Tg (an inhibitor of Sarco-ER Ca2+ ATPase pump) and Tm
(an inhibitor of N-linked glycosylation). BMAA-induced ER stress
was significantly inhibited by Sal treatment, as assessed by the
phosphorylation of eIF2α, JNK and p38 MAPK, and protein expression
levels of Bip/Grp78, GRP94 and CHOP. Similar suppression effect of
Sal also occurred in ER stress induced by Tg and Tm (Fig. 3B). Pretreatment of Sal notably
increases neuronal survival, as monitored by MTT assay, while the
cell death rate appears to remain higher compared with the control
(Fig. 3C). These findings
indicated that suppressing ER stress can effectively restrain
apoptotic neuronal death induced by BMAA treatment and facilitate
cell survival with protective effect.
Overexpression of Hsp70 suppresses
UPR-evoked apoptotic signaling
Molecular chaperons have been demonstrated to serve
the role of correcting misfolded protein and preventing them from
aggregation (24). A previous
study suggested that Hsp70 takes multiple protective effects on ER
stress and stress-induced apoptosis (25). Neuronal cells were treated with
BMAA following transfection with Hsp70. We first confirmed the
overexpression of Hsp70 by western blot assay. Under the condition
of Hsp70 overexpression, the phosphorylation of IRE1 and PERK, and
the cleavage of ATF6 were significantly attenuated compared with
the cells without transfection. The expression of ER chaperon GRP94
and PDI were lowered in Hsp70 transfected cells compared with
non-transfected cells. Bcl-2 and Bcl-xL exhibited an increased
level, while CHOP and Bim/Bax, and cleavage of caspases and PARP
were at lower levels in Hsp70-transfected cells (Fig. 4A). Hsp70 transfection markedly
increases neuronal survival, as measured using an MTT reduction
assay, and the defensive role of HSP70 is not limited to cell type
(Fig. 4B). The present study
subsequently examined the protective effect using confocal
microscopy. As visualized by DAPI nuclear staining, Hsp70 protected
neuronal cells against nuclear condensation and fragmentation
induced by BMAA, when compared with those without transfection
(Fig. 4C). To further evaluate DNA
fragmentation in each neuronal cell, flow cytometry was performed
as an apoptotic index. Cells transfected with HSP70 represented a
lower population of cell cycle arrest in the sub-G1 phase,
indicating that HSP70 effectively inhibits BMAA-induced apoptosis
(Fig. 4D). Taken together, these
data clearly demonstrated that HSP70 overexpression suppresses the
activation of UPR sensors and UPR-evoked apoptotic signaling as a
protective effect.
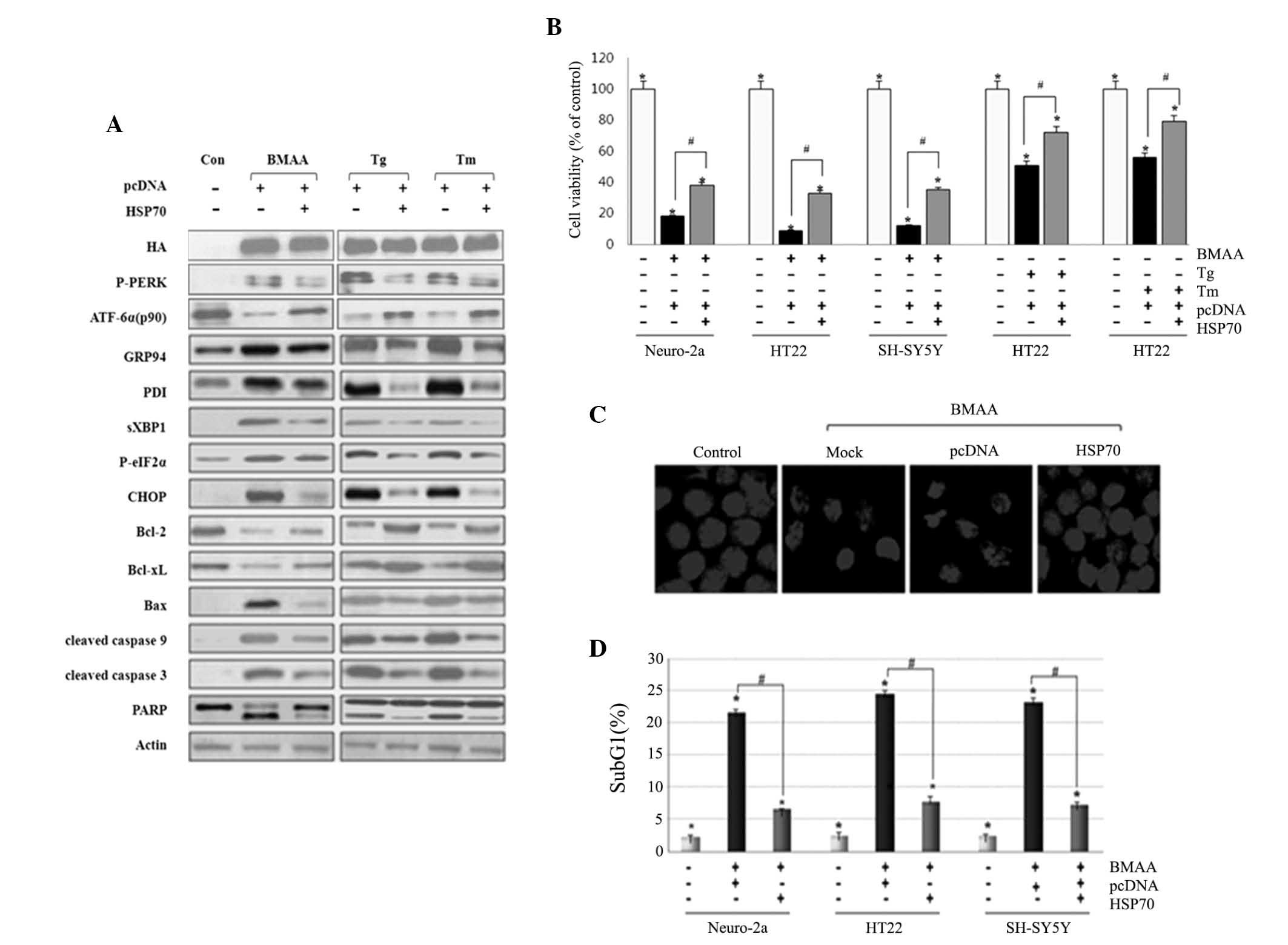 | Figure 4.Overexpression of HSP70 suppresses ER
stress-mediated neuronal death induced by BMAA, Tg and Tm.
Following transfection with DNA constructs of pcDNA and HSP70 for
24 h, the cells were treated with 3.0 mM BMAA, 1 µM Tg, or 10 µg/ml
Tm for 24 h. Con cells are untreated cells. (A) Western blotting
for ER stress and apoptosis markers was performed. HSP70 was tagged
with HA and the expression of HSP70 in the indicated transfectants
was analyzed by western blotting. Consistent results are
representative of at least three different experiments. (B) Cell
viability was measured using an MTT assay. The percentage viability
was plotted as the mean ± standard deviation of at least five
experiments (*P<0.05 vs. untreated cells; #P<0.05 vs.
BMAA-treated cells transfected with pcDNA only). (C) Nuclear
morphology was visualized by DAPI nuclear staining. Mock are
untransfected cells and con are untreated cells. The experiments
were performed in triplicate and visualized under a confocal
microscope. (D) The percentage of subG1 cells was analyzed via flow
cytometry. The data are expressed as the mean ± standard deviation
obtained from at least three independent experiments (*P<0.05
vs. untreated cells; #P<0.05 vs. BMAA-treated cells transfected
with pcDNA only). BMAA, β-N-methylamino-L-alanine; Bcl, B-cell
lymphoma; Bax, Bcl-2-associated X protein; P-, phosphorylated; ATF,
transcription factor 6; ER, endoplasmic reticulum; CHOP,
CCAAT/-enhancer-binding protein homologous protein; Tg,
thapsigargin; Tm, tunicamycin; HSP, heat shock protein; HA,
hemagglutinin; XBP, X-box binding protein; PARP, poly-ADP ribose
polymerase; PDI, protein disulfide isomerase; PERK, protein kinase
RNA-like endoplasmic reticulum kinase; con, control; eIF2,
eukaryotic initiation factor 2. |
Discussion
An important sign of neurodegenerative diseases is
the accumulation and aggregation of misfolded proteins (26). Recent studies suggested that BMAA
is incorporated into proteins by mischarged transfer RNA synthetase
(tRNAs), and this misincorporation causes protein misfolding and
aggregation in neuronal cells (27). A previous study also showed that
BMAA can be assembled into protein chains via binding to serine
transfer RNA, resulting in improper folding of the protein
(28). These findings were
consistent with the fact that a large fraction of BMAA was detected
from protein precipitates in ALS, PD and AD samples.
Previous studies also implicated that the
excitotoxicity of BMAA is mediated by the activation of
α-amino-3-hydroxy-5-isoxazolepropionic acid (AMPA) receptors
(29), N-methyl-D-aspartate (NMDA)
receptors and metabotropic glutamate receptors (mGluR) (30). This activation causes the increase
of intracellular calcium levels (31), and results in reactive oxygen
species (ROS) production and oxidative stress state (32). Misfolded protein accumulation,
glutamate-induced excitotoxicity, calcium dyshomeostasis and
oxidative stress are involved in the factors that induce ER stress
(33).
ER is the important organelle that facilitates
protein folding and an accumulation of misfolded proteins in the
lumen of the ER is activated in response to ER stress (34). The hallmark of this response is the
transcriptional upregulation of ER chaperone proteins. The present
study detected increased expression of the ER chaperon protein
Bip/Grp78, Grp94, PDI and CypB in response to BMAA-exposure. These
ER-resident chaperons and folding enzymes serve multiple roles in
promoting protein folding and endoplasmic reticulum-associated
protein degradation (ERAD). Their upregulation indicates the
disturbance of protein folding and the accumulation of misfolded
proteins in the ER. The present results revealed the
phosphorylation of PERK and eIF2α, the activation of IRE1, and the
splicing of XBP1, as well as the reduction of ATF6 p90. The
response of the three UPR pathways aims to mitigate the overload of
misfolded protein in the ER. It indicates that BMAA treatment
causes the dysfunction of ER and the aggregation of misfolded
proteins in the ER.
CHOP is activated to initiate the ER stress-induced
apoptotic response of cells, which amplifies the pro-apoptotic
signal by altering the balance between Bcl-2 and Bax (35). The expression of CHOP was markedly
induced in response to BMAA treatment. The role of CHOP was
confirmed by siRNA-mediated knockdown of the protein, which
significantly diminished apoptosis. This suggested that CHOP serves
a critical role in BMAA-induced cell death. Previous studies
demonstrated that the MAPKs are activated by ER stress inducers and
are involved in ER stress-induced apoptosis (20). Activated JNK and p38, which mainly
contribute to stress-induced apoptosis, are suggested serve a role
of increasing the expression and transcriptional activity of CHOP
(35). The phosphorylation of ERK,
JNK and p38 MAPK was observed, indicating the intense cellular
stress caused by BMAA-exposure.
Sal has been reported to protect against neuronal
injury in the rat brain through inhibiting ER stress, and Sal is
capable of penetrating into brain tissue in vivo (36). Sal, which inhibits eIF2a
dephosphorylation, alleviates the phosphorylation of JNK and p38
MAPK, and the translation of ATF4, thus suppressing the expression
of CHOP. Pretreatment with Sal alleviates BMAA-induced ER stress
and expression of CHOP, and enhances neuronal survival. Hsp70
prevents the misfolded proteins from aggregation, and allows them
to refold (37). Hsp70 also
affects the apoptotic pathway at the levels of cytochrome c release
and caspase activation, and protects cells against stress-induced
apoptosis (25). Overexpression of
Hsp70 reduces the expression of ER chaperons and UPR receptors,
suggesting that the initial UPR signaling is suppressed. The
pro-apoptotic signaling proteins, CHOP and Bax, are downregulated,
and the cleaved caspase-9 and −3 are attenuated, indicating that ER
stress-mediated apoptotic cell death is inhibited. These findings
indicated that ER stress is a highly promising therapeutic target
for BMAA-induced neuronal injury and death, and implicated ER
stress inhibitors and molecular chaperons as potential
pharmacological agents.
The present findings demonstrated that BMAA induces
ER stress-mediated apoptosis in the cultured neuronal cells. BMAA
induces the upregulation of ER chaperons and the activation of UPR
receptors of PERK, IRE1 and ATF6, as well as the phosphorylation of
MAPK member JNK, p38 and ERK. The ER stress-specific protein CHOP
is activated and drives pro-apoptotic responses that causes
mitochondrial damage and caspase activation. Furthermore, the
present study demonstrated that inhibition of ER stress using ER
stress antiagonist Sal and Hsp70 protein mitigates neuronal damage
and apoptosis as a protective effect.
Acknowledgements
The present study was supported by a grant from the
Kyung Hee University in 2012 (no. KHU-20121733) and a grant from
the Basic Science Research Program through the National Research
Foundation of Korea, which is funded by the Ministry of Education
(no. NRF-2013R1A1A2060694).
References
|
1
|
Murch SJ, Cox PA, Banack SA, Steele JC and
Sacks OW: Occurrence of beta-methylamino-l-alanine (BMAA) in
ALS/PDC patients from Guam. Acta Neurol Scand. 110:267–269. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pablo J, Banack SA, Cox PA, Johnson TE,
Papapetropoulos S, Bradley WG, Buck A and Mash DC: Cyanobacterial
neurotoxin BMAA in ALS and Alzheimer's disease. Acta Neurol Scand.
120:216–225. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Banack SA, Johnson HE, Cheng R and Cox PA:
Production of the neurotoxin BMAA by a marine cyanobacterium. Mar
Drugs. 5:180–196. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Banack SA and Cox PA: Biomagnification of
cycad neurotoxins in flying foxes: Implications for ALS-PDC in
Guam. Neurology. 61:387–389. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Smith QR, Nagura H, Takada Y and Duncan
MW: Facilitated transport of the neurotoxin,
beta-N-methylamino-L-alanine, across the blood-brain barrier. J
Neurochem. 58:1330–1337. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Plato CC, Cruz MT and Kurland LT:
Amyotrophic lateral sclerosis-Parkinsonism dementia complex of
Guam: Further genetic investigations. Am J Hum Genet. 21:133–141.
1969.PubMed/NCBI
|
|
7
|
Spencer PS, Palmer VS, Herman A and Asmedi
A: Cycad use and motor neurone disease in Irian Jaya. Lancet.
2:1273–1274. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Karamyan VT and Speth RC: Animal models of
BMAA neurotoxicity: A critical review. Life Sci. 82:233–246. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Holtcamp W: The emerging science of BMAA:
Do cyanobacteria contribute to neurodegenerative disease? Environ
Health Perspect. 120:A110–A116. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lindholm D, Wootz H and Korhonen L: ER
stress and neurodegenerative diseases. Cell Death Differ.
13:385–392. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rasheva VI and Domingos PM: Cellular
responses to endoplasmic reticulum stress and apoptosis. Apoptosis.
14:996–1007. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Urano F, Wang X, Bertolotti A, Zhang Y,
Chung P, Harding HP and Ron D: Coupling of stress in the ER to
activation of JNK protein kinases by transmembrane protein kinase
IRE1. Science. 287:664–666. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Boyce M and Yuan J: Cellular response to
endoplasmic reticulum stress: A matter of life or death. Cell Death
Differ. 13:363–373. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Weiss JH and Choi DW:
Beta-N-methylamino-L-alanine neurotoxicity: Requirement for
bicarbonate as a cofactor. Science. 241:973–975. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lobner D, Piana PM, Salous AK and Peoples
RW: Beta-N-methylamino-L-alanine enhances neurotoxicity through
multiple mechanisms. Neurobiol Dis. 25:360–366. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Okle O, Stemmer K, Deschl U and Dietrich
DR: L-BMAA induced ER stress and enhanced caspase 12 cleavage in
human neuroblastoma SH-SY5Y cells at low nonexcitotoxic
concentrations. Toxicol Sci. 131:217–224. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee AS: The ER chaperone and signaling
regulator GRP78/BiP as a monitor of endoplasmic reticulum stress.
Methods. 35:373–381. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim I, Xu W and Reed JC: Cell death and
endoplasmic reticulum stress: Disease relevance and therapeutic
opportunities. Nat Rev Drug Discov. 7:1013–1030. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Walker AK, Farg MA, Bye CR, McLean CA,
Horne MK and Atkin JD: Protein disulphide isomerase protects
against protein aggregation and is S-nitrosylated in amyotrophic
lateral sclerosis. Brain. 133:105–116. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nagai H, Noguchi T, Takeda K and Ichijo H:
Pathophysiological roles of ASK1-MAP kinase signaling pathways. J
Biochem Mol Biol. 40:1–6. 2007.PubMed/NCBI
|
|
21
|
Hung JH, Su IJ, Lei HY, Wang HC, Lin WC,
Chang WT, Huang W, Chang WC, Chang YS, Chen CC and Lai MD:
Endoplasmic reticulum stress stimulates the expression of
cyclooxygenase-2 through activation of NF-kappaB and pp38
mitogen-activated protein kinase. J Biol Chem. 279:46384–46392.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang XZ and Ron D: Stress-induced
phosphorylation and activation of the transcription factor CHOP
(GADD153) by p38 MAP Kinase. Science. 272:1347–1349. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Boyce M, Bryant KF, Jousse C, Long K,
Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D and Yuan
J: A selective inhibitor of eIF2alpha dephosphorylation protects
cells from ER stress. Science. 307:935–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim YE, Hipp MS, Bracher A, Hayer-Hartl M
and Hartl FU: Molecular chaperone functions in protein folding and
proteostasis. Annu Rev Biochem. 82:323–355. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mosser DD, Caron AW, Bourget L, Meriin AB,
Sherman MY, Morimoto RI and Massie B: The chaperone function of
hsp70 is required for protection against stress-induced apoptosis.
Mol Cell Biol. 20:7146–7159. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ross CA and Poirier MA: Protein
aggregation and neurodegenerative disease. Nat Med. 10:(Suppl).
S10–S17. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee JW, Beebe K, Nangle LA, Jang J,
Longo-Guess CM, Cook SA, Davisson MT, Sundberg JP, Schimmel P and
Ackerman SL: Editing-defective tRNA synthetase causes protein
misfolding and neurodegeneration. Nature. 443:50–55. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dunlop RA, Cox PA, Banack SA and Rodgers
KJ: The non-protein amino acid BMAA is misincorporated into human
proteins in place of L-serine causing protein misfolding and
aggregation. PLoS One. 8:e753762013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rao SD, Banack SA, Cox PA and Weiss JH:
BMAA selectively injures motor neurons via AMPA/kainate receptor
activation. Exp Neurol. 201:244–252. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cucchiaroni ML, Viscomi MT, Bernardi G,
Molinari M, Guatteo E and Mercuri NB: Metabotropic glutamate
receptor 1 mediates the electrophysiological and toxic actions of
the cycad derivative beta-N-Methylamino-L-alanine on substantia
nigra pars compacta DAergic neurons. J Neurosci. 30:5176–5188.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Brownson DM, Mabry TJ and Leslie SW: The
cycad neurotoxic amino acid, beta-N-methylamino-L-alanine (BMAA),
elevates intracellular calcium levels in dissociated rat brain
cells. J Ethnopharmacol. 82:159–167. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu X, Rush T, Zapata J and Lobner D:
beta-N-methylamino-l- alanine induces oxidative stress and
glutamate release through action on system Xc(−). Exp Neurol.
217:429–433. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Görlach A, Klappa P and Kietzmann T: The
endoplasmic reticulum: Folding, calcium homeostasis, signaling and
redox control. Antioxid Redox Signal. 8:1391–1418. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Doyle KM, Kennedy D, Gorman AM, Gupta S,
Healy SJ and Samali A: Unfolded proteins and endoplasmic reticulum
stress in neurodegenerative disorders. J Cell Mol Med.
15:2025–2039. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sokka AL, Putkonen N, Mudo G, Pryazhnikov
E, Reijonen S, Khiroug L, Belluardo N, Lindholm D and Korhonen L:
Endoplasmic reticulum stress inhibition protects against
excitotoxic neuronal injury in the rat brain. J Neurosci.
27:901–908. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chaudhuri TK and Paul S:
Protein-misfolding diseases and chaperone-based therapeutic
approaches. FEBS J. 273:1331–1349. 2006. View Article : Google Scholar : PubMed/NCBI
|














