Introduction
Ischemic stroke is an acute vascular incident that
occurs when blood supply to the brain is obstructed, resulting in
irreversible brain damage. It has previously been reported that
chronic microglial activation may lead to inflammation-mediated
development of ischemic stroke, via the release of neurotoxic and
inflammatory molecules (1).
Overproduction of inflammatory mediators, such as nitric oxide (NO)
and prostaglandin E2 (PGE2), from activated
microglia may contribute to uncontrolled inflammation. Therefore,
agents that inhibit activated microglia are important potential
candidate drugs that may delay the progression of neurodegeneration
in disorders such as stroke. Our previous study indicated that Gua
Lou Gui Zhi decoction (GLGZD) inhibits the production of
proinflammatory mediators in vitro and vivo via
related signaling pathways (2,3).
However, the mechanism via which GLGZD inhibits neuroinflammation
and exerts its neuroprotective effects remain to be completely
elucidated.
The present study measured NO production in a rat
middle cerebral artery occlusion (MCAO) model of
experimentally-induced ischemic brain damage. Behavioral defects
and infarct volume were detected to confirm the generation of a
successful model. Subsequently, the expression levels of inducible
nitric oxide synthase (iNOS) and cylooxygenase-2 (COX-2) were
detected by reverse transcription-quantitative polymerase chain
reaction (RT-qPCR) and immunohistochemistry (IHC). In addition, the
present study aimed to investigate the mechanisms by which GLGZD
inhibits the production of proinflammatory mediators. The present
study demonstrated that GLGZD significantly inhibited the excessive
release of NO and PGE2, and simultaneously attenuated
the mRNA and protein expression levels of iNOS and COX-2. The
underlying mechanisms were shown to be associated with inhibition
of the phosphorylation of three members of the mitogen-activated
protein kinases (MAPK) family: Extracellular signal-regulated
kinase 1/2 (ERK1/2), p38 MAPK and c-Jun N-terminal kinase (JNK),
and of nuclear factor-κB (NF-κB) activation. These results
indicated that GLGZD exerts anti-inflammatory effects and
suppresses the expression of inflammatory mediators via the MAPK
and NF-κB signaling pathways. Therefore, these molecular mechanisms
may be exploited for the clinical treatment of ischemic stroke.
Materials and methods
Reagents and animals
2,3,5-Triphenyltetrazolium chloride (TTC) was
obtained from Sigma-Aldrich (Merck Millipore, Darmstadt, Germany).
RT-qPCR reagents were purchased from Takara Bio, Inc. (Otsu,
Japan). The PGE2 ELISA kit (cat. no. PKGE004B) was
purchased from R&D Systems, Inc. (Minneapolis, MN, USA). The
primary polyclonal antibodies targeting iNOS (cat. no. sc-49055)
and COX-2 (cat. no. sc-23983), and the secondary horseradish
peroxidase (HRP)-conjugated immunoglobulin G antibody were obtained
from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Specific
antibodies against ERK1/2 (cat. no. 9102), phosphorylated
(p)-ERK1/2 (cat. no. 9101), p38 (cat. no. 9212), p-p38 (cat. no.
9211), JNK (cat. no. 9252), p-JNK (cat. no. 9251) and β-actin (cat.
no. 4967) were purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA).
Male Sprague-Dawley rats (weight, 200–250 g; age, 6
weeks) were purchased from Shanghai SLAC Laboratory Animal Co.,
Ltd. (Shanghai, China). The rats were maintained under
specific-pathogen-free conditions, and were housed in a
temperature- (21±1°C) and humidity-controlled environment with a 12
h/12 h light/dark cycle and fed a standard rodent diet. The present
study was conducted in accordance with the Animal Facility
Guidelines of the University (Fujian University of Traditional
Chinese Medicine, Fuzhou, China). All experiments were performed in
a randomized manner. Rats were randomly distributed into three
groups (n=15/group), as follows: Sham-operated group, MCAO model
group and MCAO model + GLGZD treatment group (0.15 mg/kg, orally),
in which rats were orally administered GLGZD daily for 7 days. The
study was approved by the Institutional Animal Care and Use
Committee of Fujian University of Traditional Chinese Medicine
(Fuzhou, China).
Preparation of GLGZD extracts
Medicinal materials (Trichosanthis radix,
Ramulus cinnamomi, Paeonia lactiflora, Glycyrrhiza
radix, Zingiber officinale Roscoe and Fructus
jujubae) were purchased from Guo Yi Tang Chinese Herbal
Medicine Store (Fujian, China). GLGZD extract was prepared for
treatment, according to a previously described method (4). Briefly, fresh GLGZD was washed three
times with tap water to remove salt. The crude plant mixture was
soaked in double-distilled water for 30 min, and was extracted
twice for 2 h. The obtained solution was then filtered and
concentrated in a rotary evaporator to a final concentration of
1.16 g/ml for further use (5).
MCAO model development
Rats were subjected to MCAO, according to a
previously described method (5).
Briefly, after anesthetizing with 10% chloral hydrate, the left
common carotid artery and external carotid artery were exposed. A
poly-L-lysine-coated monofilament nylon suture was inserted into
the internal carotid artery for occlusion; the suture was
maintained intraluminally for 2 h, after which it was withdrawn to
restore cerebral blood flow. Finally, the chest cavity was closed
and sutured. In the sham group, the MCO of rats was isolated
without ligation and occlusion. Body temperature was monitored and
maintained at 37°C during the whole surgical procedure.
Behavioral testing
Neurological behavior in the groups was measured and
scored according to a previously described method (6). Briefly, neurological deficits were
assessed blindly at 2 h and 7 days post-reperfusion. The
neurological function score ranged between 0 and 4, as follows: 0,
Exhibition of normal, spontaneous movements; 1, unable to
completely extend the right forepaw; 2, repetitive circling to the
right; 3, unable to move to the right; 4, incapable of walking
unimpeded.
TTC staining
Six rats from each of the three groups were
decapitated following anesthesis with 10% chloral hydrate (0.3
ml/100 g) 7 days after MCAO, and coronal brain tissues were placed
on ice and maintained at −80°C. Subsequently, the tissues were
sliced into 2-mm sections and were immediately stained with 1% TTC
(20 g/l) at 37°C for 30 min. Images of the staining were captured
using a digital camera (Canon Oxus 950IS; Canon, Inc., Tokyo,
Japan) and the infarcted areas of each section were measured using
image analysis software (ImageJ 1.37; National Institutes of
Health, Bethesda, MD, USA). Infarct volume was presented as the
percentage of total brain volume that was damaged.
NO assay
Blood was collected from the abdominal aorta and was
centrifuged at 1,625 × g for 20 min at room temperature, in
order to obtain plasma for subsequent measurements. The
concentration of NO was assessed by measuring the amount of
accumulated nitrite, which is an indicator of NO production, using
a colorimetric assay with Griess reaction, as previously described
(7). Plasma (100 µl) from three
rats from each group was mixed with the same volume of Griess
reagent [0.1% N-(1-naphthyl)-ethylenediamine, 1% sulfanilamide in
5% phosphoric acid] in a 96-well microtiter plate. Absorbance
values were determined at 540 nm using a microplate absorbance
reader (BioTek Germany, Bad Friedrichshall, Germany). NO
concentration was determined following generation of a sodium
nitrite standard curve.
Determination of PGE2 by
ELISA
PGE2 levels were measured in harvested plasma from
each group (n=3) using an ELISA kit, according to the
manufacturer's protocol. The concentration of PGE2 was measured at
an absorbance of 450 nm using a microplate reader.
Western blot analysis
The cortex was dissected and immersed in lysis
buffer containing protease inhibitor (Roche Diagnostics GmbH,
Mannheim, Germany) for 30 min on ice. Lysates were then centrifuged
at 12,000 × g for 10 min at 4°C and the supernatants were
collected for analysis. Total protein concentrations were
determined using the bicinchoninic acid (BCA) method. Equal amounts
of protein (50 µg) were separated by 10% SDS-PAGE and were
transferred to polyvinylidene fluoride membranes (EMD Millipore,
Billerica, MA, USA). Membranes were then incubated with blocking
solution (5% non-fat milk) for 1 h at room temperature to block
non-specific binding, and were probed with primary antibodies
overnight at 4°C, including the phosphorylated and total forms of
ERK1/2, p38 MAPK and JNK, and β-actin (1:1,000). Subsequently, the
membranes were incubated with HRP-conjugated secondary antibody for
1 h at room temperature. The protein immunoreactive bands were
detected using an enhanced chemiluminescence reagent (RPN2132; GE
Healthcare Bio-Sciences, Pittsburgh, PA, USA) and ChemiDoc
XRS+ System imaging system (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The band intensity was normalized to the
β-actin band and quantitative analysis was performed using ImageJ
software.
RT-qPCR
Rats were anesthetized with 10% chloral hydrate (0.3
ml/100 g) and the brain cortex was removed for RNA isolation.
Briefly, total RNA was extracted using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Total RNA (2 µg) was reverse transcribed to produce cDNA using the
First Strand cDNA Synthesis kit (Takara Bio, Inc.) according to the
manufacturer's instructions. qPCR was performed using a SYBR Green
I qPCR kit (Takara Bio, Inc.) according to the manufacturer's
protocol. The following primers were used for qPCR: iNOS, forward
5′-CCTCGTTCAGCTCACCTTCG-3′, reverse 5′-GCCGCTCTCATCCAGAACCT-3′;
COX-2, forward 5′-ACTGACTATGAAGACCTATG-3′, reverse
5′-TTAATATACGGATTGGAAGT-3′; and GAP DH, forward
5′-TGGAGTCTACTGGCGTCTT-3′ and reverse 5′-TGTCATATTTCTCGTGGTTCA-3′.
Amplification was performed using Applied Biosystems Prism 7500
(7500 software v2.0.5; Applied Biosystems; Thermo Fisher
Scientific, Inc.) with the following cycling conditions:
Pre-denaturation at 95°C for 30 sec, followed by 40 cycles at 95°C
for 5 sec and 60°C for 30 sec, and final melt curve at 95°C for 15
sec, 60°C for 1 min, 95°C for 15 sec. The results were analyzed
using the 2−ΔΔCq method (8). Quantitative gene expression levels
were assessed relative to reference gene levels (GAPDH).
IHC
Rats were anesthetized with 10% chloral hydrate (0.3
ml/100 g) and perfused transcardially with saline and 4%
paraformaldehyde. The brains were then dissected and fixed in 4%
paraformaldehyde for 30 min. All tissues sections were transferred
to graded ethanol (70, 80, 90, 95, 95 and 100% for 30 min each),
then dehydrated tissues were cleared in xylene (30 min) and
embedded in paraffin. Subsequently, tissues were cut into 5-µm
coronal slices, and dried for 1 h at 60°C, deparaffinized in xylene
and rehydrated with an ethanol gradient (100, 95, 90, 85 and 75%),
and washed twice in PBS. The sections were removed and incubated in
0.3% H2O2 for 10 min and washed in distilled
water. Nonspecific binding of paraffin-embedded sections was
blocked with goat serum, and the sections were then incubated with
rat anti-iNOS and anti-COX-2 primary antibodies (1:500) overnight
at 4°C. After washing three times with PBS, the sections were
exposed to secondary antibodies (1:200) for 30 min at room
temperature and were visualized with diaminobenzidine. Images of
each cerebral cortex section were acquired using a light microscope
(Leica DMI4000B, Leica Microsystems GmbH, Wetzlar, Germany) at ×200
magnification. Semi-quantitative analysis was conducted by
determining the percentage of positively stained cells using ImageJ
software.
Electrophoretic mobility shift assay
(EMSA)
Nuclear protein was extracted from the cerebral
cortex for EMSA using a nuclear extraction kit (78833; Thermo
Fisher Scientific, Inc.). Nuclear protein concentrations were
determined using the BCA method (PICPI23223; Thermo Fisher
Scientific, Inc.). EMSAs were performed using the EMSA/Gel-Shift
kit (20148; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. Briefly, a double-stranded biotin-labeled
DNA oligonucleotide corresponding to the NF-κB p65 binding sequence
(Cell Signaling Technology, Inc.; forward
5′-AGTTGAGGGGACTTTCCCAGGC-3′ and reverse
3′-TCAACTCCCCTGAAAGGGTCCG-5′) was used for gel shift assays.
Nuclear protein (4.5 µg) from each sample was incubated with a
biotin-labeled NF-κB probe for 30 min at room temperature, in a
final volume of 20 µl. Subsequently, samples were subjected to
nondenaturing gel electrophoresis (5% acrylamide, 0.5 X TBE) and
were transferred to a nylon membrane (EMD Millipore) followed by
crosslinked for 2 min. Finally, the membrane was visualized by
chemiluminescence. Densitometry of the gel bands was analyzed using
ImageJ software.
Statistical analysis
Data are presented as the mean ± standard error of
the mean of three independent experiments. Statistical analysis was
performed on SPPS 15.0 (SPSS, Inc., Chicago, IL, USA) using one-way
analysis of variance and Dunnett's post-test. P<0.05 was
considered to indicate a statistically significant difference.
Results
GLGZD reduces infarct volume in
ischemic brain tissues
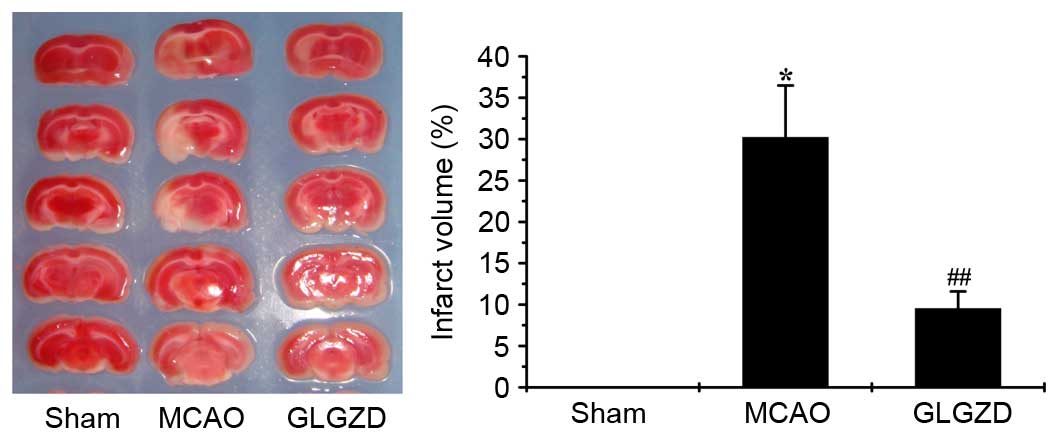
As shown in Fig. 1,
the infarct volume was increased in the MCAO group compared with in
the sham group. However, following GLGZD treatment for 7 days,
ischemic infarction was significantly inhibited compared with in
the MCAO group (P<0.01). This result suggests that GLGZD exerts
a therapeutic effect on ischemic brain tissue by decreasing
cerebral infarction.
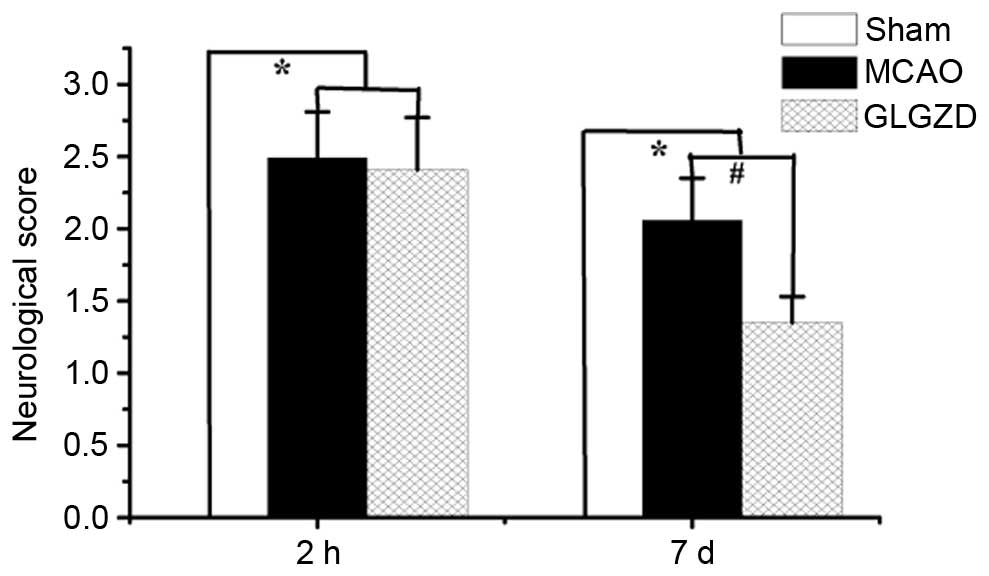
GLGZD improves the neurological
deficit in an MCAO rat model
To determine the ameliorative effects of GLGZD on
motor function, such as spasticity, in the postischemic brain, the
present study evaluated neurological deficits 2 h and 7 days
post-MCAO. Treatment with GLGZD markedly improved behavioral
deficits caused by MCAO, and these deficits were not observed in
the rats of the sham group (Fig.
2, P<0.05).
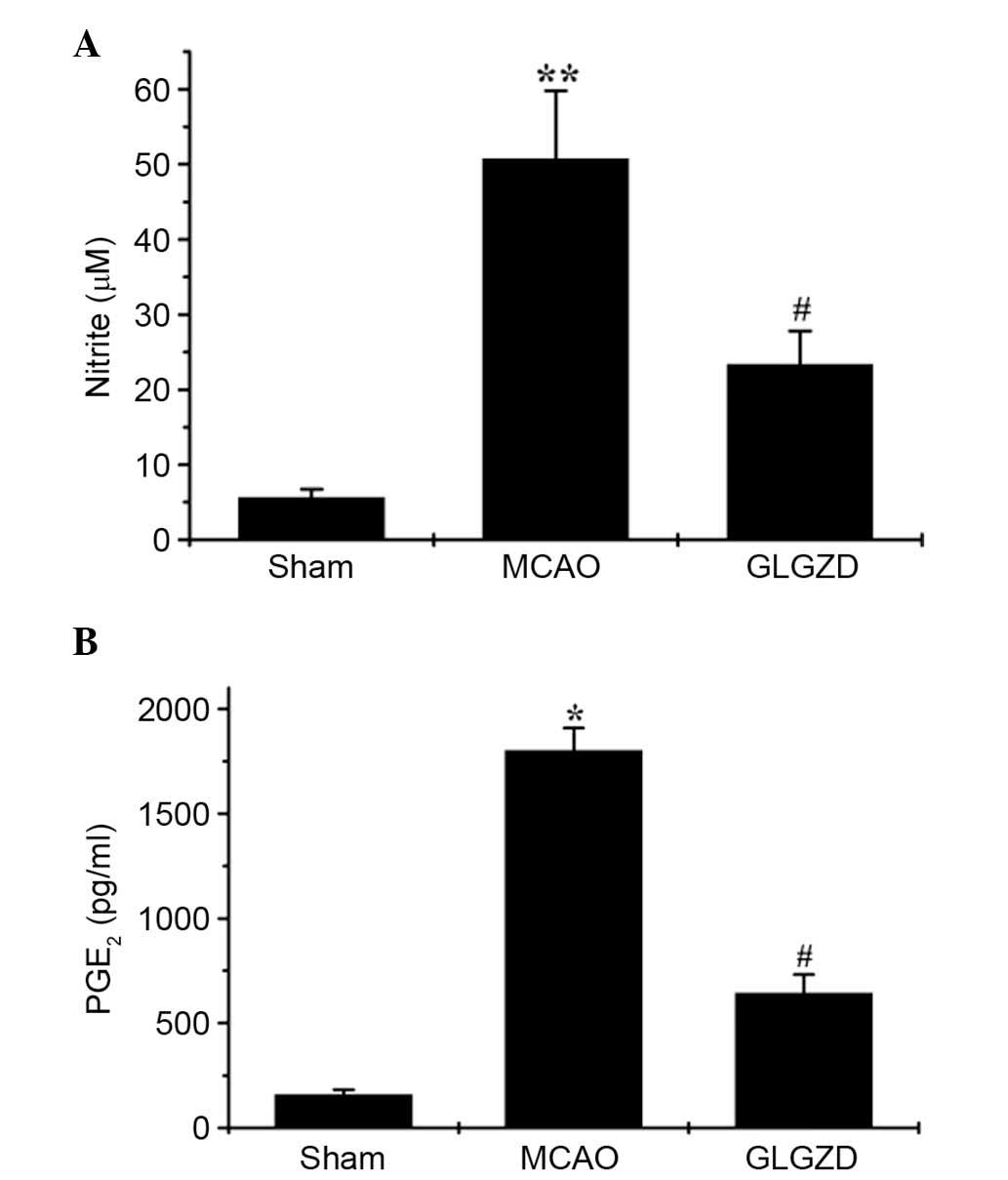
GLGZD suppresses NO and PGE2 release
in plasma samples from the MCAO rat model
The effects of GLGZD on NO and PGE2
production were investigated in all three groups using the Griess
reagent assay and an ELISA, respectively. MCAO-induced elevation of
NO and PGE2 levels in the rats was significantly
decreased following GLGZD administration (Fig. 3A and B, P<0.05). NO and
PGE2 release remained at basal levels in the sham group.
These results indicate that GLGZD exerts marked inhibitory effects
on the production of proinflammatory mediators (NO and
PGE2).
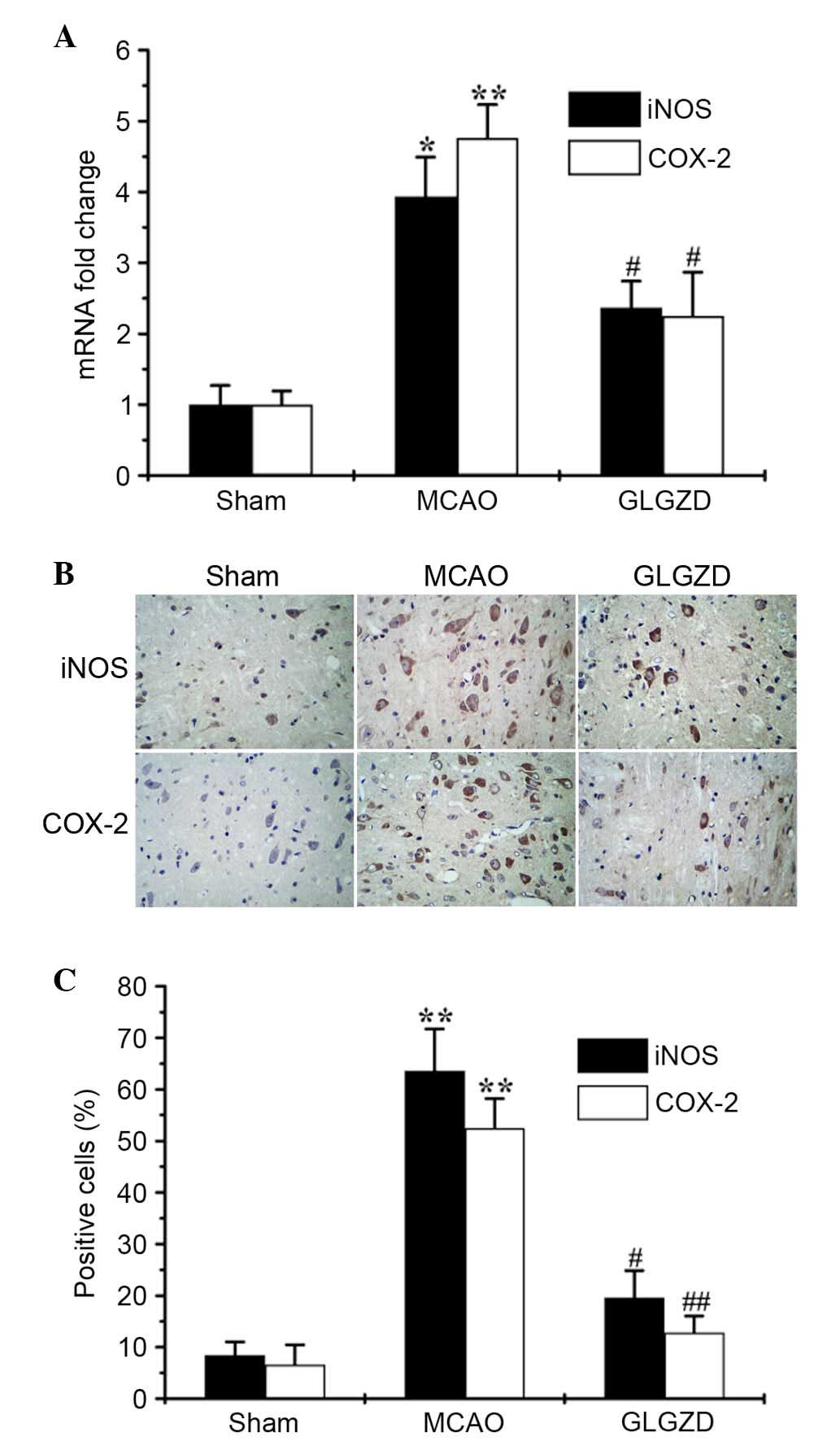
GLGZD inhibits the mRNA and protein
expression levels of iNOS and COX-2 in an MCAO model
It is well-known that NO is produced by iNOS, and
PGE2 is produced through induction of the
proinflammatory enzyme COX-2 (9).
The present study analyzed the expression levels of iNOS and COX-2
using qPCR and IHC. The results revealed that GLGZD significantly
reduced iNOS and COX-2 transcriptional (Fig. 4A) and translational levels
(Fig. 4B and C), which were
increased in MCAO rats. The results of the IHC analysis of iNOS and
COX-2 expression were consistent with those from the qPCR. These
results indicate that treatment with GLGZD suppresses NO and
PGE2 production by inhibiting iNOS and COX-2
expression.
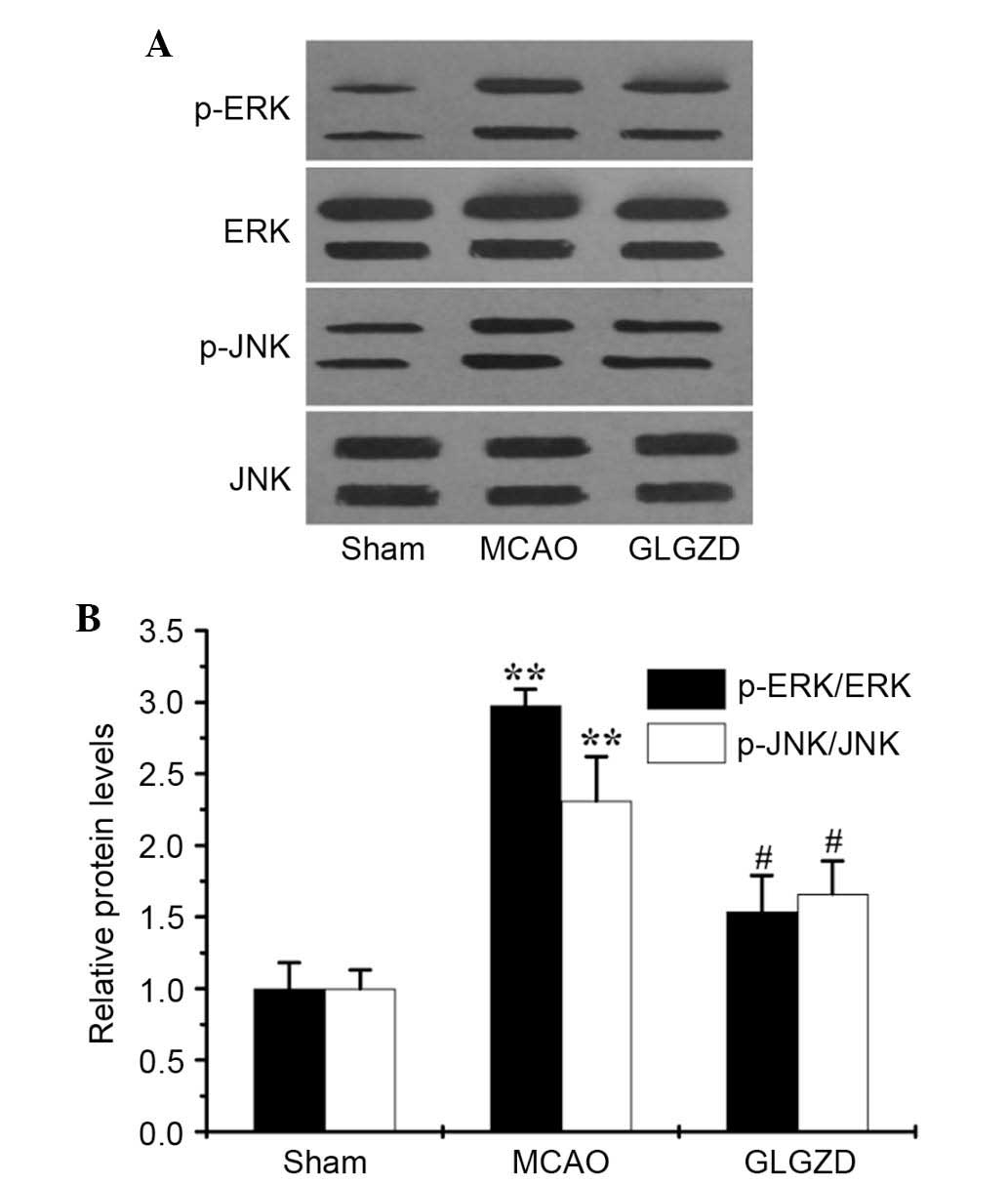
GLGZD downregulates phosphorylation of MAPKs and
activation of NF-κB in an MCAO rat model. To examine
whether GLGZD exerted inhibitory effects on proinflammatory
mediators by regulating activation of the MAPK and NF-κB pathways,
the phosphorylation of MAPKs (ERK1/2, JNK and p38 MAPK) were
determined by western blot analysis, and NF-κB activity was
detected by EMSA. Western blot analysis revealed that the levels of
p-ERK1/2 and p-JNK, but not p-p38 MAPK, were enhanced in the MCAO
group compared with the sham group (data not shown). However,
treatment with GLGZD markedly reduced p-ERK1/2 and p-JNK levels in
the ischemic brain (Fig. 5A and
B), whereas total protein levels remained unchanged.
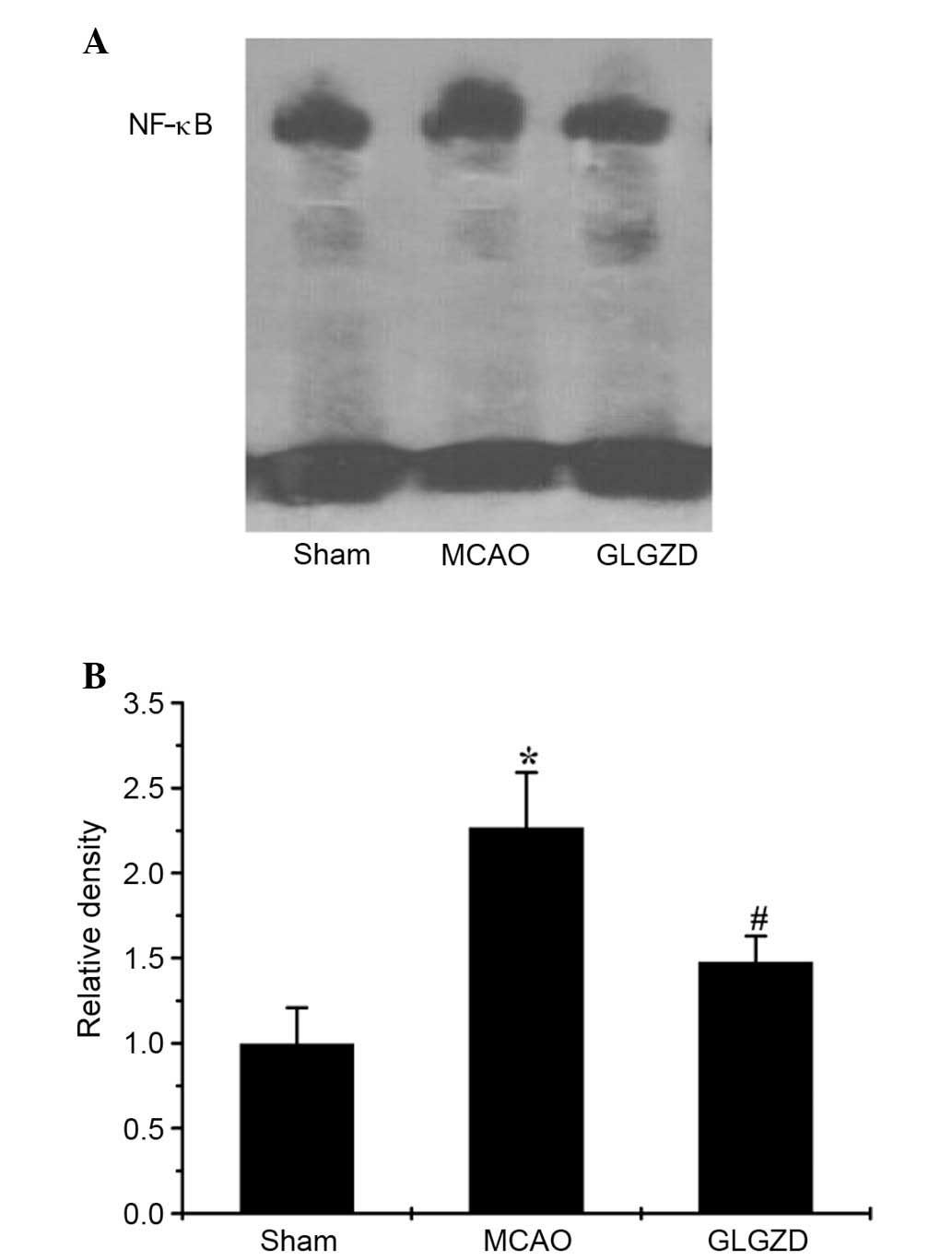
Following administration of GLGZD, NF-κB activation
was also measured to determine the effects of GLGZD on the NF-κB
pathway. As shown in Fig. 6A and
B, a significant increase in the DNA-binding activity of NF-κB
was observed in the MCAO group, as determined using an EMSA.
Conversely, GLGZD markedly attenuated the DNA binding activity of
NF-κB (P<0.05). These results suggest that signal transduction
via MAPK and NF-κB is associated with neuroinflammation, and may be
reduced by GLGZD in a rat model of cerebral ischemia.
Discussion
Neuroinflammation within the brain following stroke
has been implicated in the pathogenesis of cerebral ischemia, and
exacerbates secondary brain injury (10–12).
Stroke induces an increase in the release of inflammatory mediators
into the circulation. Microglia are innate immune cells in the
central nervous system, which serve a crucial role in
neuroinflammation. Hyperactivated microglia produce large amounts
of inflammatory molecules and promote neuronal death (13–15).
Furthermore, microglial activation is correlated with sequential
signaling pathways, including NF-κB and MAPK cascades, thus leading
to the expression and production of proinflammatory mediators
(16).
Among the inflammatory mediators released after
ischemic stroke, released NO and PGE2 from activated
microglia serve an important role in the pathogenesis of
neuroinflammation (17,18). NO is generated by iNOS (19) in microglial cells, and excessive
production of NO produced from L-arginine by iNOS is detrimental
(20). COX-2 is the key enzyme in
the formation of PGE2, which is produced by activated
microglial cells and mediates neuropathological processes during
neuroinflammation (21).
Therefore, inhibition of inflammatory mediators that are
responsible for the symptoms of neurodegenerative diseases is a
potential therapeutic strategy for the treatment of
neuroinflammation-associated ischemic stroke.
Various intracellular signaling pathways are
involved in the modulation of neuroinflammatory mediators. The
transcription factor NF-κB has a critical role in the inflammatory
response to stroke (22). It has
previously been reported that NF-κB activation can lead to the
marked upregulation of iNOS and COX-2 (23–25).
Furthermore, it has been reported that other signal transduction
molecules, such as MAPK, are important upstream modulators for the
production of inflammatory mediators (26). Therefore, the present study
investigated the NF-κB and MAPK signaling pathways in order to
clarify the underlying mechanism of GLGZD in neuroinflammation.
Our previous studies have revealed the
neuroprotective effects of the traditional Chinese medicine GLGZD,
and its effects have been suggested to have therapeutic potential
for the treatment of spasticity following ischemic stroke (27,28).
GLGZD is widely used in the treatment of spasticity after ischemic
stroke in clinical practice. The present study examined the
inhibitory effects of GLGZD on MCAO-induced production of
proinflammatory mediators, including iNOS and COX-2, and the
underlying mechanisms, including associated genes and signaling
pathways.
As shown as the present study, infarct volume was
markedly increased in the MCAO model group compared with in the
sham group; however, it was decreased following treatment with
GLGZD (Fig. 1, P<0.01).
Similarly, GLGZD significantly alleviated the neurological deficit
caused by MCAO (Fig. 2,
P<0.05). In addition, GLGZD markedly inhibited MCAO-induced NO
and PGE2 production, as measured by Griess reagent assay
and ELISA (Fig. 3A and B).
Simultaneously, the relevant expression levels of iNOS and COX-2
were markedly increased in the MCAO model; however, treatment with
GLGZD significantly reduced gene expression, as determined by
RT-qPCR, and protein expression, as detected by IHC (P<0.05,
Fig. 4A-C). To investigate the
possible signal transduction mechanism associated with the
inhibitory effects of GLGZD, the total and phosphorylated levels of
the MAPK pathway proteins (ERK1/2, JNK and p38) were determined by
western blotting. As presented in Fig.
5A and B, treatment with GLGZD attenuated the phosphorylation
of ERK-1/2 and JNK, which was enhanced in MCAO rats (P<0.05);
however, no influence was detected on p38 activation. The present
study further explored the activation of NF-κB using EMSA; the
results indicated that the NF-κB DNA-binding activity was
significantly suppressed by GLGZD as compared with in the MCAO
group (Fig. 6A and B, P<0.01).
Taken together, these findings indicated that GLGZD may act as a
potent inhibitor of MCAO-induced MAPK and NF-κB signaling.
In conclusion, the present study demonstrated that
GLGZD exerted novel anti-inflammatory mechanisms in MCAO-mediated
neuroinflammation. These results provide evidence of the
ameliorative effects of GLGZD on NO and PGE2 release and
the suppression of related gene expression, including iNOS and
COX-2, via the concomitant downregulation of MAPK and NF-κB
signaling. Further studies are required to investigate the
mechanism associated with the neuroprotective clinical effects of
GLGZD.
Acknowledgements
The present study was supported by a grant from the
National Natural Science Foundation of China (grant no.
81403265).
Glossary
Abbreviations
Abbreviations:
|
GLGZD
|
Gua Lou Gui Zhi decoction
|
|
MCAO
|
middle cerebral artery occlusion
|
|
NF-κB
|
nuclear factor-κB
|
|
MAPK
|
mitogen-activated protein kinases
|
|
NO
|
nitric oxide
|
|
iNOS
|
inducible nitric oxide synthase
|
|
PGE2
|
prostaglandin E2
|
|
ELISA
|
enzyme-linked immunosorbent assay
|
|
EMSA
|
electrophoretic mobility shift
assay
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
GAPDH
|
glyceraldehyde 3-phosphate
dehydrogenase
|
|
JNK
|
c-Jun N-terminal kinase
|
|
RT-qPCR
|
reverse transcription-quantitative
polymerase chain reaction
|
References
|
1
|
Brown GC and Neher JJ: Inflammatory
neurodegeneration and mechanisms of microglial killing of neurons.
Mol Neurobiol. 41:242–247. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hu H, Li Z, Zhu X, Lin R, Lin J, Peng J,
Tao J and Chen L: Gua Lou Gui Zhi decoction suppresses LPS-induced
activation of the TLR4/NF-κB pathway in BV-2 murine microglial
cells. Int J Mol Med. 31:1327–1332. 2013.PubMed/NCBI
|
|
3
|
Hu H, Li Z, Zhu X, Lin R, Peng J, Tao J
and Chen L: GuaLou GuiZhi decoction inhibits LPS-induced microglial
cell motility through the MAPK signaling pathway. Int J Mol Med.
32:1281–1286. 2013.PubMed/NCBI
|
|
4
|
Hu H, Lin R, Zhu X, Li Z and Chen L:
Anti-inflammatory effects of Gualou Guizhi decoction in transient
focal cerebral ischemic brains. Mol Med Rep. 12:1321–1327.
2015.PubMed/NCBI
|
|
5
|
Huang J, Tao J, Xue X, Yang S, Han P, Lin
Z, Xu W, Lin J, Peng J and Chen L: Gua Lou Gui Zhi decoction exerts
neuroprotective effects on post-stroke spasticity via the
modulation of glutamate levels and AMPA receptor expression. Int J
Mol Med. 31:841–848. 2013.PubMed/NCBI
|
|
6
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniectomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kovac A, Erickson MA and Banks WA: Brain
microvascular pericytes are immunoactive in culture: Cytokine,
chemokine, nitric oxide and LRP-1 expression in response to
lipopolysaccharide. J Neuroinflammation. 8:1392011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Egger T, Schuligoi R, Wintersperger A,
Amann R, Malle E and Sattler W: Vitamin E (alpha-tocopherol)
attenuates cyclo-oxygenase 2 transcription and synthesis in
immortalized murine BV-2 microglia. Biochem J. 370:459–467. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Glezer I, Simard AR and Rivest S:
Neuroprotective role of the innate immune system by microglia.
Neuroscience. 29:867–883. 2007. View Article : Google Scholar
|
|
11
|
Perry VH, Nicoll JA and Holmes C:
Microglia in neurodegenerative disease. Nat Rev Neurol. 6:193–201.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rock RB and Peterson PK: Microglia as a
pharmacological target in infectious and inflammatory diseases of
the brain. J Neuroimmune Pharmacol. 1:117–126. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Minghetti L: Cyclooxygenase-2 (COX-2) in
inflammatory and degenerative brain diseases. J Neuropathol Exp
Neurol. 63:901–910. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Griffiths MR, Gasque P and Neal JW: The
multiple roles of the innate immune system in the regulation of
apoptosis and inflammation in the brain. J Neuropathol Exp Neurol.
68:217–226. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Amor S, Puentes F, Baker D and van der
Valk P: Inflammation in neurodegenerative diseases. Immunology.
129:154–169. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lo JY, Kamarudin MN, Hamdi OA, Awang K and
Kadir HA: Curcumenol isolated from Curcuma zedoaria suppresses
Akt-mediated NF-κB activation and p38 MAPK signaling pathway in
LPS-stimulated BV-2 microglial cells. Food Funct. 6:3550–3559.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Banati RB, Gehrmann J, Schubert P and
Kreutzberg GW: Cytotoxicity of microglia. Glia. 7:111–118. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rock RB and Peterson PK: Microglia as a
pharmacological target in infectious and inflammatory diseases of
the brain. J Neuroimmune Pharmacol. 1:117–126. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Abramson SB, Amin AR, Clancy RM and Attur
M: The role of nitric oxide in tissue destruction. Best Pract Res
Clin Rheumatol. 15:831–845. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Boje KM: Nitric oxide neurotoxicity in
neurodegenerative diseases. Front Biosci. 9:763–776. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Minghetti L: Cyclooxygenase-2 (COX-2) in
inflammatory and degenerative brain diseases. J Neuropathol Exp
Neurol. 63:901–910. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Harari OA and Liao JK: NF-kB and innate
immunity in ischemic stroke. Ann N Y Acad Sci. 1207:32–40. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Baldwin AS Jr: The NF-kappa B and I kappa
B proteins: New discoveries and insights. Annu Rev Immunol.
14:649–683. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee AK, Sung SH, Kim YC and Kim SG:
Inhibition of lipopolysaccharide-inducible nitric oxide synthase,
TNF-alpha and COX-2 expression by sauchinone effects on
I-kappaBalpha phosphorylation, C/EBP and AP-1 activation. Br J
Pharmacol. 139:11–20. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Baeuerle PA and Henkel T: Function and
activation of NF-kappa B in the immune system. Annu Rev Immunol.
12:141–179. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim YJ, Hwang SY, Oh ES, Oh S and Han IO:
IL-1beta, an immediate early protein secreted by activated
microglia, induces iNOS/NO in C6 astrocytoma cells through p38 MAPK
and NF-kappaB pathways. J Neurosci Res. 84:1037–1046. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang L and Ai H: Effects of Gua Lou Gui
Zhi decoction on c-fos and c-jun on epileptic Rats. Sichuan Journal
of Traditional Chinese Medicine. 23:21–22. 2005.
|
|
28
|
Yang C, Chen L and Tao J: New usage of a
classical formula-Gua Lou Gui Zhi Decoction. Liaoning Journal of
Traditional Chinese Medicine. 8:166–167. 2012.
|